

Abstract
Gene expression is regulated in a context-dependent, cell-type-specific manner. Condition-specific transcription is dependent on the presence of transcription factors (TFs) that can activate or inhibit its target genes (global context). Additional factors, such as chromatin structure, histone, or DNA modifications, also influence the activity of individual target genes (individual context). The role of the global and individual context for post-transcriptional regulation has not systematically been investigated on a large scale and is poorly understood. Here we show that global and individual context dependency is a pervasive feature of microRNA-mediated regulation. Our comprehensive and highly consistent data set from several high-throughput technologies (PAR-CLIP, RIP-chip, 4sU-tagging, and SILAC) provides strong evidence that context-dependent microRNA target sites (CDTS) are as frequent and functionally relevant as constitutive target sites (CTS). Furthermore, we found the global context to be insufficient to explain the CDTS, and that flanking sequence motifs provide individual context that is an equally important factor. Our results demonstrate that, similar to TF-mediated regulation, global and individual context dependency are prevalent in microRNA-mediated gene regulation, implying a much more complex post-transcriptional regulatory network than is currently known. The necessary tools to unravel post-transcriptional regulations and mechanisms need to be much more involved, and much more data will be needed for particular cell types and cellular conditions in order to understand microRNA-mediated regulation and the context-dependent post-transcriptional regulatory network.
Regulation of gene expression is highly context-specific. The ENCODE Project (The ENCODE Project Consortium 2012) provided convincing evidence that whether or not a specific transcription factor (TF) binds to a specific binding site (TFBS) is not only dependent on the sequence of the binding site but also on its chromatin state (Wang et al. 2012b), on DNA methylation (Wang et al. 2012a), on other DNA binding factors (Yáñez-Cuna et al. 2012), and numerous additional factors, which are difficult to measure and predict. All these factors form the so-called “cellular context” that influences the expression level of genes.
Expression of genes is not only regulated at the level of transcription but also post-transcriptionally in various ways, of which regulation mediated by microRNAs is one of the most prevalent (He and Hannon 2004). MicroRNAs are 20- to 24-nt long noncoding RNAs that have been found in animals and plants. They play a pivotal role in development, tumorigenesis, the immune system, and during viral infections (for a review, see Bartel 2004). Within the RNA-induced silencing complex (RISC), microRNAs are responsible for target recognition by binding to target sites, often located in the 3′-UTR of mRNAs. This is predominantly mediated by the so-called seed region (nucleotides 2–8 of the microRNA). In general, RISC causes down-regulation of the target mRNA either by inhibiting translation or promoting degradation (Bartel 2009). Neither the exact mode of binding nor the mechanisms of down-regulation are completely understood (Eulalio et al. 2008; Kozak 2008; Guo et al. 2010; Djuranovic et al. 2011; Mishima et al. 2012; Meijer et al. 2013).
Computational prediction of microRNA targets is a difficult task (Sethupathy et al. 2006; Ritchie et al. 2009; Thomas et al. 2010). This is a consequence of the low specificity of seed matches alone: There are several lines of evidence suggesting that additional factors such as target site location (Grimson et al. 2007), additional base-pairing at the microRNA 3′ end (Brennecke et al. 2005), target site accessibility (Kertesz et al. 2007), other RNA binding proteins (Jacobsen et al. 2010), microRNA and mRNA copy numbers (Ben-Moshe et al. 2012; Carroll et al. 2013), differential isoform usage (Sandberg et al. 2008; Boutet et al. 2012), and additional unknown factors or interplay between any of these play important roles in distinguishing functional from nonfunctional target sites. Interestingly, several of these additional factors are not static but may change dynamically, providing context for target recognition: For instance, depending on which RNA binding proteins (RBPs) are expressed at what level in a given cell-type, RISC may or may not bind at a certain binding site. This is an example of individual context, since an RBP is able to affect individual target sites of a microRNA. In contrast, dynamic microRNA expression determines a global context, i.e., if a microRNA is differentially expressed, all of its targets are affected. This important distinction is often neglected; and often, only the global context is considered (Carroll et al. 2013).
Several examples of individual context-specific microRNA-mediated regulation can also be found in the literature (for a review, see Pasquinelli 2012). Bhattacharyya et al. (2006) identified the RNA binding protein, ELAVL1, as a derepressor for miR-122 regulation of the SLC7A1 mRNA. In normal hepatocarcinoma cells, SLC7A1 is repressed by a miR-122 target site in its 3′-UTR. Under different stress conditions, ELAVL1 is released from the nucleus into the cytoplasm, which abolishes SLC7A1 repression. The exact mechanism, however, remains unclear. Intriguingly, ELAVL1 has also been implicated in activating a target site of the microRNA let-7 in the 3′-UTR of MYC (Kim et al. 2009), which indicates that ELAVL1 can both induce and prevent microRNA-mediated regulation. In addition to ELAVL1, DND1 (Kedde et al. 2007) and PUM1 (Kedde et al. 2010) have also been identified to influence microRNA regulation. There may be other RNA binding proteins that interfere with or facilitate microRNA/target interactions.
These examples illustrate that the presence of a functional target site is not sufficient for regulation. It may be active under certain conditions but nonfunctional in a different context. Presently, our knowledge about context-dependent microRNA-mediated regulation is only based on a few examples, and the underlying molecular mechanisms are poorly understood.
By immunoprecipitation of microRNA/target complexes using monoclonal antibodies to RISC complex components followed by high-throughput sequencing of the protein-protected microRNA target sites, the complete targetome of cellular and viral microRNAs has become accessible. More than 10,000 putative microRNA binding sites, so-called clusters, are obtained in a single HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation) or PAR-CLIP (photoactivated ribonucleotide-enhanced crosslinking immunoprecipitation) experiment. Although the annotation of the responsible microRNA to an identified cluster still leaves room for improvement, >75% of microRNA target interactions can be correctly annotated, thereby allowing in-depth analyses of microRNA regulatory networks (Gottwein et al. 2011; Haecker et al. 2012; Riley et al. 2012; Skalsky et al. 2012; Erhard et al. 2013a).
To study context-specific microRNA-mediated regulation, we generated AGO2-PAR-CLIP data from two human B-cell lines. In addition, we reanalyzed two recently published sets of AGO2-PAR-CLIP data from two different human B-cell lines (Gottwein et al. 2011). These four cell lines represent different stages of B-cell development and are either infected by Kaposi’s sarcoma-associated herpes virus (KSHV), coinfected by both KSHV and Epstein-Barr-Virus (EBV), or not infected. Thus, each cell line provides a distinct context for microRNA-mediated regulation. All data sets were reanalyzed using a new algorithm called PARma (Erhard et al. 2013a). PARma considers the topology of the microRNA/target interaction and the position of UV light-induced cross-links in more detail than state-of-the-art methods, and it provides quality control scores for both—the identification of microRNA target site clusters and the annotation of the interacting microRNA to these sites. For two of these four cell lines, we generated three additional data sets, including RIP-chip, 4sU-tagging-derived RNA half-lives, and large-scale SILAC-based proteomics. This allowed us to comprehensively analyze the effect of context-dependent microRNA/target interactions on the recruitment of the target mRNAs to AGO2 complexes, on target RNA stability, and on target protein levels. By considering viral as well as host microRNAs, we investigated both microRNA/target interactions that coevolved within a species as well as interactions of an exogenous microRNA with endogenous target sites. The results provide compelling evidence that context dependency of microRNA-mediated regulation is not restricted to a few examples but is a widespread and general feature of post-transcriptional regulation mediated by both cellular and viral microRNAs.
Results
Differential analysis of PAR-CLIP data
To comprehensively study regulation of cellular gene expression by both cellular and Kaposi’s sarcoma-associated herpes virus (KSHV)-encoded microRNAs, we applied AGO2-PAR-CLIP to two human B-cell lines—the body cavity-based lymphoma cell line BCBL1, which is latently infected with KSHV, and the Burkitt lymphoma cell line DG75, which is KSHV negative. Applying PARma (Erhard et al. 2013a) with stringent criteria (see Supplemental Methods), we identified 15,577 clusters, 12,333 of which mapped to known transcripts (Ensembl v60).
In order to assess the quality of the PAR-CLIP data sets, we first computed the positional distribution of all target sites in mRNAs (Fig. 1A). Target sites of viral microRNAs shared the well-described features of cellular microRNA target sites: They preferentially bind to the 3′ untranslated region (3′-UTR) and rarely to the 5′-UTR of transcripts (Grimson et al. 2007; Hafner et al. 2010). Within the 3′-UTR, target sites tend to accumulate at the very beginning, i.e., immediately after the stop codon, and at the transcript end, i.e., immediately upstream of the poly-A tail (Grimson et al. 2007). We furthermore checked the accuracy of the microRNA assignment to target sites by confirming that virtually no reads mapped to KSHV microRNA target sites in the KSHV negative cell line DG75 (a feature that is not used by PARma to assign microRNAs) (see Fig. 1B). The few instances with random reads in DG75 may nevertheless be bona fide KSHV microRNA target sites: As we observed random reads spread across a multitude of transcripts at low frequency, these reads presumably result from infrequent unspecific immunoprecipitates or insufficient removal of background total RNA rather than microRNA-specific signatures. This is further supported by a significantly lower frequency of T to C conversions and lower consistency across replicates for these reads (Fig. 1B; Supplemental Fig. S1).
Figure 1.

Validation of PAR-CLIP experiments. (A) The distribution of relative positions of target sites on mRNAs is shown. The x-axis represents the average length of 5′ untranslated regions (5′UTR), the coding regions (CDS), and the 3′ untranslated regions of all transcripts with at least one PAR-CLIP cluster. Each transcript was divided into 60 bins, and the relative frequency of target sites falling into each bin is shown on the y-axis. The data clearly illustrate the preferences of target sites in the 3′UTR as compared to CDS and 5′UTR. Viral microRNAs have the same preferences as cellular microRNAs. (B) The normalized number of reads in each cluster (rows) for each of the independent PAR-CLIP experiments (columns) is shown for KSHV microRNA target sites in the four PAR-CLIP libraries. KSHV-negative cell lines (columns 1 and 2) almost exclusively have no reads, whereas for KSHV-positive cell lines, dozens to hundreds of reads are observed per target site. Replicates are highly correlated, indicating high reproducibility. The additional annotations on the left side indicate the part of the transcript where a cluster is located (orange, 5′-UTR; yellow, coding; green, 3′-UTR; gray, not located on known mRNA) and the expression of the transcript in all experiments (red, at least twofold lower expression than the mean expression value for this transcript across all experiments; light red, at least 1.4-fold lower expression than the mean; light blue, at least 1.4-fold higher expression; blue, at least twofold higher expression). We also visualized and inspected individual target sites (Supplemental Fig. S1). (C,D) The log2 RIP-chip enrichment distributions of mRNAs only containing target sites of cellular microRNAs, only containing KSHV microRNA target sites, and containing target sites from both cellular and KSHV microRNAs in the uninfected cell line DG75 and the KSHV positive cell line BCBL1, respectively. KSHV targets are enriched in BCBL1 but not in DG75. (E) The mRNA half-life ratios are shown for the same sets of genes as in C and D. The half-life of mRNAs with KSHV target sites is significantly reduced in BCBL1.
We further validated our PAR-CLIP data set using published data for the same cell lines: (1) PAR-CLIP targets are highly consistent with RIP-chip data (Fig. 1C,D; Dölken et al. 2010); (2) KSHV microRNA targets are selectively enriched in BCBL1 and not DG75 in the RIP-chip experiments (cf. Fig. 1C,D); and (3) PAR-CLIP target sites lead to a measurable reduction of target mRNA half-lives (Fig. 1E; Supplemental Table S2).
To be able to perform a more in-depth analysis on KSHV microRNA targets in human B-cells, we also included recently published PAR-CLIP data from two additional B-cell lines, namely BC1 and BC3 (Gottwein et al. 2011). We reanalyzed all data sets using PARma, which yielded 21,628 clusters, 16,425 of which mapped to known transcripts (see Supplemental Table S1; see Supplemental Table S6 for individual read mapping statistics). Intriguingly, the overlaps of target sites of both ubiquitously expressed cellular and KSHV microRNAs were surprisingly small (Fig. 2A,B). Such extreme differences of called target sites may be due to experimental bias or context dependency, i.e., a major fraction of microRNA target sites is only active in some of the cell lines considered.
Figure 2.

Comparison of PAR-CLIP data sets. (A) The number of target sites observed only in individual cell lines (outermost labeled circles), in two cell lines (circles on the edges between cell lines), and in all three cell lines (center circle), for KSHV microRNA target sites. Relatively few target sites appear to be active in multiple cell lines (see Supplemental Fig. S2A for overlaps of cellular microRNA targets). (B) Summary of all pairwise overlaps for clusters of cellular and viral microRNAs in all data sets. The Jaccard index (J) is the number of clusters in the intersection divided by the total number of clusters in any of the two experiments. Jaccard indices of ∼70% for all replicate measurements indicate high reproducibility, whereas comparisons across cell lines show relatively low overlap (J < 40%) (see also Supplemental Fig. S2). (C) The PAR-CLIP read heatmap for target sites of the KSHV microRNA miR-K12-4-3p (see Fig. 1B for more information about PAR-CLIP read heatmaps). Between KSHV-positive cell lines, there is no correlation, but there are distinct clusters of target sites. No obvious dependency between clusters and mRNA expression level is observable.
There are some traces of batch effects (Fig. 2B), but experimental bias does not explain all differences: Replicate experiments for all four cell lines yielded highly reproducible results (Fig. 2B,C; Supplemental Fig. S1), and differences were also observable for inner-laboratory comparisons, i.e., there are target sites of constitutively expressed microRNAs that are reproducibly missing in DG75 and reproducibly observable in BCBL1 and vice versa, and these cell lines have been measured simultaneously, using the exact same protocol in the same laboratory (the same is true for BC1 and BC3) (see Supplemental Discussion for a detailed analysis of technical bias and further discussion). To further analyze potential context dependency of microRNA/target site interactions, we used a per-microRNA differential analysis of all PAR-CLIP experiments (Supplemental Figs. S4, S8–S11).
Intriguingly, when we considered all target sites of a single microRNA, there was no clear correlation of target sites across cell lines (Fig. 2C; Supplemental Fig. S2). Instead, distinct clusters of target sites emerged, for instance, several kshv-miR-K12-4-3p target sites that appear to be active in BCBL1 only and not in BC1 or BC3. This suggests that context-dependent microRNA-mediated regulation may be substantially more important than generally expected. An obvious explanation for this would be that all these targets are not expressed in BC1 and BC3 or are expressed at very low levels such that PAR-CLIP is not able to identify them. This would be one kind of global context. Low levels or absence of mRNA is detectable by expression measurements that are indicated on the left side of the heatmaps in Figure 2C and is, in fact, not a general feature of these target sites, arguing in favor of an individual context that determines the activity of many of these sites. Additionally, there are mutually exclusive target sites (MES): Target sites of the same microRNA that are missing in BCBL1 and active in BC1 or BC3 and target sites missing in BC1 and BC3 but active in BCBL1, i.e., the target sites in one condition are not a subset of the target sites in the other condition. Thus, the presence of MES cannot be explained by a higher expression or activity of the respective microRNA or mRNA, i.e., by global context.
Context-dependent target sites of KSHV microRNAs
Taken together, our differential analysis of PAR-CLIP data suggests that microRNA-mediated regulation is substantially and generally dependent on the cellular context. To experimentally test this hypothesis, we used three sets of additional high-throughput methods to investigate the consequences of context-dependent microRNA-mediated regulation. First, using RIP-chip, we tested whether context-dependent microRNA/target interactions, as found in the PAR-CLIP data, had a measurable impact on the recruitment of the target mRNA to RISC in their specific context only. Second, using microarray-based transcriptomics, including metabolic labeling of RNA, and SILAC-based proteomics experiments, we tested whether such context-dependent microRNA/target interactions also have a measurable impact on mRNA half-lives and on mRNA as well as on protein levels of their targets in their specific context only. All these experiments were performed by comparing DG75 to BCBL1. We selected all KSHV microRNAs that showed a KSHV-specific activity pattern, i.e., the set of target sites was depleted of reads in DG75 and included reproducible target sites of all three KSHV positive cell lines (Supplemental Figs. S3, S8, S9). Furthermore, all selected KSHV microRNAs are expressed in all KSHV-positive cell lines according to the microRNA reads from the PAR-CLIP experiment (see Supplemental Table S4) and showed a clear pattern of MES as introduced above.
Context-dependent microRNA targets are associated with RISC in a context-dependent manner
First, we looked at the recruitment of the mRNA targets of these KSHV microRNAs to AGO2-complexes. We recently used RIP-chip to identify KSHV and EBV microRNA targets in human B-cells (Dölken et al. 2010). Since then, we performed two additional RIP-chip replicates of the KSHV-positive cell line, BCBL1, to perform a more solid statistical analysis (Erhard et al. 2013b).
Data were normalized using principal component analysis as described (Erhard et al. 2013b) and differential enrichment values were computed for BCBL1 and DG75 as the second principal component (PC2), indicating whether an mRNA is more strongly associated with RISC in BCBL1 in comparison to DG75. All PAR-CLIP target sites were mapped to genes; and genes with any KSHV target site in BCBL1, with a constitutive target site in all KSHV-positive cell lines, and with exclusive sites in BCBL1 or BC1/BC3 were compared to all other genes with any PAR-CLIP target site as background (Fig. 3).
Figure 3.

PAR-CLIP targets in RIP-chip experiments, mRNA half-life measurements, and expression measurements. (A) Differential RIP-chip enrichment scores (PC2 scores; positive values indicate higher enrichment in BCBL1 than in DG75). Generally, KSHV microRNA targets active in BCBL1 are significantly shifted toward higher values as compared to all other genes with any PAR-CLIP target site, in contrast to KSHV target sites exclusively active in BC1 or BC3 and not in BCBL1. B illustrates this further: The enrichment of genes with any KSHV site, with a constitutive or a BCBL1 exclusive site over genes with BC1/BC3 exclusive sites among all genes with PC2 score > 2 is about twofold in all cases. (C) Distributions of half-life differences between BCBL1 and DG75 for all genes with PAR-CLIP target sites. Thus, positive values indicate a longer mRNA half-life in BCBL1 than in DG75. Genes with KSHV microRNA targets active in BCBL1 tend to have shorter half-lives in BCBL1 than in DG75. This is highly significant for all BCBL1 target genes as well as the constitutive targets but not for BCBL1-specific targets, even if their half-life is on average ∼20 min shorter in BCBL1 than in DG75. However, KSHV microRNA targets that are inactive in BCBL1 do not show any shift in their half-lives. (D) The difference between targets active exclusively in BCBL1 is statistically significantly different from targets active exclusively in BC1 or BC3, when their ranks among all PAR-CLIP targets are considered. (E) Genes are scattered according to their mRNA log2 fold changes between BCBL1 and DG75 on the x-axis and to their protein log2 fold changes on the y-axis. In both dimensions, none of the KSHV target sets is significantly down-regulated on either the mRNA or protein level (Supplemental Fig. S5). However, target sites active in BCBL1 appear to be shifted toward the bottom right. These sites correspond to genes whose protein level fold change between BCBL1 and DG75 is lower than expected from the mRNA level. (F) The ranks of protein fold changes normalized to their mRNA levels for all gene sets considered. Normalized protein fold changes are significantly lower for genes with BCBL1-specific target sites than for genes with target sites inactive in BCBL1 (P < 0.01, Wilcoxon rank sum test) (see also Supplemental Fig. S3).
The differential RIP-chip enrichment was significantly shifted toward higher values for genes with BCBL1 exclusive sites in comparison to the background (P < 0.0007, Kolmogorov-Smirnov test), indicating that BCBL1-exclusive target sites indeed lead to a stronger association of the target mRNA in BCBL1 to RISC. This was also true for constitutive KSHV target sites (P < 0.009) as well as for all KSHV target sites active in BCBL1 (P < 3.10−8). However, BC1/BC3-specific target sites, which were not active in BCBL1, were indistinguishable from the background (Fig. 3A; Supplemental Table 1). In particular, genes with active KSHV microRNA target sites in BCBL1 showed a twofold enrichment of genes that are significantly (PC2 score > 2) more associated with RISC in BCBL1 than in DG75 over background genes. In contrast, genes with KSHV microRNA target sites that are exclusively active in BC1 or BC3 and not in BCBL1 are indistinguishable from background genes (Fig. 3B).
This provides strong evidence that a major fraction of the KSHV microRNA target sites identified by PAR-CLIP exclusively in BC1/BC3 and not in BCBL1 do not mediate a strong recruitment of their target mRNA to RISC in BCBL1, i.e., they are indeed context-dependent target sites. Context-dependent microRNA/target interactions, as defined by differential analysis of PAR-CLIP data, can thus be confirmed using an independent RIP-chip experiment.
Target mRNA stability is affected in a context-dependent manner
Next, we analyzed the context-dependent effects of the KSHV microRNAs on target RNA stability. Since microRNAs can induce destabilization of the mRNA transcripts (Bartel 2009), microRNA/target interactions that are active in BCBL1 should decrease the target mRNA half-life in BCBL1 as compared to DG75. Target sites inactive in BCBL1 (and only active in BC1/BC3) in contrast should not decrease mRNA half-life.
Previously, we applied metabolic labeling of newly transcribed RNA followed by microarray analysis to separate newly synthesized and preexisting RNA (Dölken et al. 2008). We computed RNA half-lives based on the ratios of newly synthesized to total RNA for both DG75 and BCBL1 (Dölken et al. 2010) and considered the differences in target mRNA half-lives in between BCBL1 and DG75.
Intriguingly, the mRNA half-life of KSHV microRNA targets in BCBL1 was decreased by ∼20 min (P < 3.10−5) on average, whereas for KSHV microRNA targets not active in BCBL1, no significant decrease was observed (Fig. 3C; Supplemental Table 1). Furthermore, the half-life difference values of BCBL1-exclusive target genes were significantly smaller than half-life difference values of BC1- or BC3-exclusive target genes (P < 0.008, Wilcoxon rank sum test) (Fig. 3D). Thus, context-dependent microRNA/target interactions impact on mRNA stability in a context-dependent manner.
Interestingly, constitutive KSHV microRNA target sites showed an even stronger decrease in the mRNA half-life than for context-dependent target sites (>35 min on average, P < 10−5). A possible explanation is that constitutive microRNA/target interactions are less susceptible to the cellular context, resulting in more substantial target suppression. Therefore, constitutive interactions likely represent the most important targets for the virus.
Protein levels are differentially regulated for context-dependent microRNA targets
We now asked whether context-dependent microRNA targets are also reflected in steady-state mRNA or protein levels in two different contexts. It is important to note that protein levels in a cell depend on multiple factors, including protein half-lives and microRNA-independent post-transcriptional regulation, most of which are well described to have a substantially greater impact on protein levels than generally exerted by microRNAs. Therefore, targets of the viral microRNAs may not necessarily show differential expression between DG75 and BCBL1 on protein or mRNA levels (Supplemental Fig. S3; Dölken et al. 2010). Especially viral microRNAs are likely to counteract the cellular response to infection (Cullen 2011; Kincaid and Sullivan 2012), which is reflected by the fact that KSHV microRNAs target multiple induced genes (Dölken et al. 2010).
Indeed, when mRNA or protein levels were considered individually, no significant shift in expression fold changes was observed for any set of microRNA targets (Fig. 3E; Supplemental Fig. S3). Thus, in spite of the fact that mRNA half-lives are significantly decreased by KSHV microRNAs, there is no observable effect on steady-state levels of either mRNAs or proteins. However, if protein fold changes are normalized to mRNA fold changes, a small but statistically significant difference can be observed between BCBL1-specific targets and BC1/BC3-specific targets (P < 0.01, Wilcoxon rank sum test) (Fig. 3F). Since this normalization effectively removes all effects of mRNA levels and half-lives, this indicates that KSHV microRNAs not only have an impact on mRNA half-life in a context-dependent manner but also on how many proteins are produced per mRNA molecule. Constitutive targets of KSHV microRNAs did not show this pattern, presumably because of their strong impact on mRNA half-lives (Fig. 3).
Taken together, RIP-chip data, RNA half-life data, as well as mRNA and protein expression data, provide good evidence that a substantial amount of KSHV microRNA target sites as found by differential analysis of PAR-CLIP data is indeed context-dependent, resulting in a differential association with RISC and a context-dependent impact on target gene expression.
Context-dependent target sites of cellular microRNAs
We next selected context-dependent microRNA/target interactions that are either active in BCBL1 or DG75 but not in both. Thus, we first selected all microRNAs that are not differentially expressed between BCBL1 and DG75 (less than twofold) and are reliably detected in the PAR-CLIP experiments (at least 100 reads in all four data sets). Furthermore, all microRNAs had to have at least 20 target sites as identified by a 7-mer seed by PARma. All identified microRNAs showed a clear pattern of context dependency in their target sites (Supplemental Figs. S10, S11). Using the same criteria as in the analysis of KSHV microRNAs, context-dependent target sites were defined (Supplemental Fig. S4; see Supplemental Table S5).
Again, context-dependent microRNA/target interactions as defined by the differential PAR-CLIP analysis resulted in highly significant differential association with RISC (Fig. 4A; Supplemental Table 1). Specifically, context-dependent targets are more than twofold enriched in significantly differentially RISC-associated mRNAs (PC2 score > 2) for both cellular contexts. Furthermore, target mRNA half-lives are again significantly lowered by the context-dependent activity of the microRNA/target interactions (P < 0.0002, Wilcoxon rank sum test) (Fig. 4B). Thus, as in the analysis of KSHV microRNAs, context-dependent target sites of cellular microRNAs also lead to differential RISC-association and have functional impact on target mRNA half-lives in a context-dependent manner.
Figure 4.

Analysis of context-dependent targets of cellular microRNAs. (A) Distributions of the differential RIP-chip scores as compared to all genes with any PAR-CLIP target sites (see also Fig. 3A). Both targets exclusively active in DG75 as well as in BCBL1 are significantly shifted toward stronger association with RISC in their respective context. The vertical lines indicate a threshold for strongly differentially RISC-associated genes. In both cases, the respective context-dependent targets are more than twofold enriched over the background genes (∼10% of background genes in comparison to >20% of the target genes in both cases). (B) The rank distribution of half-life differences for both sets of context-dependent targets is shown (see also Fig. 3D). BCBL1-specific targets are significantly shifted toward lower half-life difference ranks in comparison to DG75-specific targets indicative for effects of context-dependent microRNA/target interactions in the respective context only. (C,D) The distributions of mRNA and protein fold changes between BCBL1 and DG75 for context-dependent targets of cellular microRNAs, respectively, as compared to the background of all genes with any PAR-CLIP target site. Clearly, based on mRNA as well as on protein levels, context-dependent targets are more highly expressed in their target context. This indicates that the target mRNA expression directly contributes to the cellular context of microRNA-mediated regulation. (E) Depiction of how many genes with context-dependent target sites (n = 311) are constitutively (less than twofold) expressed in both cell lines on an mRNA level and have a protein detected in both cell lines (n = 97), how many are differentially expressed but with a detected protein (n = 54), how many do not have a protein and are differential (n = 63), and how many without a detected protein are constitutive (n = 97). (F) Scatterplot of the microarray intensity measurements for all genes with a PAR-CLIP target site. Interestingly, there seem to be subpopulations of target genes that have extremely low expression values in one of the two contexts and high intensities in the other, where respective target sites are exclusively active. These may indeed correspond to not expressed genes in their inactive target context (see also Supplemental Fig. S4).
The analysis of steady-state expression levels revealed a clear pattern of context-dependent targets: Both sets of context-dependent targets are clearly shifted in comparison to the background with respect to both mRNA and protein fold changes (Fig. 4C,D). Specifically, genes tend to have higher expression in the context where the microRNA/target interactions are active, indicating that a significant number of targets are context-dependent because they are not expressed or are expressed at low levels in one condition, i.e., affected by the global context.
Importantly, this is not solely due to a completely abrogated expression in the nonactive context since proteins are detected for almost half of all context-dependent targets in both cell lines; and in more than two-thirds of the cases, the fold change is smaller than twofold (Fig. 4E). Thus, it is not the absence or presence of target mRNAs that lead to context dependency of target sites. Rather, this indicates a complex dependency of the target site activity on the exact target mRNA expression levels. However, there may be a subpopulation within both sets of context-dependent targets, where a missing activity of a target site may be explained by the complete absence of the target mRNA (Fig. 4F).
mRNA levels and flanking sequence motifs explain context-dependent microRNA/target interactions
Thus, we analyzed to which extent mRNA expression levels contribute to the cellular context and whether there are other factors that are necessary to explain the widespread context dependency of target sites. First, we tested whether the target mRNA level is the only contributor that constitutes the cellular context for microRNA-mediated gene regulation.
Read counts are not only subject to biological variance but also to a substantial amount of sampling noise since many clusters only have a few dozen reads. To compare PAR-CLIP read count fold changes with mRNA fold changes in a more robust manner, it is therefore important to estimate the extent of this sampling noise. We used a population-based estimate of variance using a conditional gamma distribution (see Supplemental Methods). This approach is similar to recent methods to estimate the significance of differential expression in RNA-seq data (Anders and Huber 2010; Robinson et al. 2010). Importantly, our noise model nicely reflects variation observed in replicate experiments (Supplemental Fig. S6).
If this noise model is applied to the comparison of mRNA fold change-corrected PAR-CLIP target sites, >50% of all context-dependent target sites, i.e., at least 14% of all target sites of the selected set of cellular microRNAs, cannot be explained as judged by the P-value distribution (Fig. 5). This means that in these cases, the PAR-CLIP read count fold change is significantly higher than expected from the corresponding mRNA fold change, and this difference also cannot be explained by sampling noise inherent to low-count data such as PAR-CLIP. Thus, target site activities are not simply linearly dependent on mRNA levels.
Figure 5.

Comparison of mRNA fold changes to PAR-CLIP read count fold changes. (A) Scatterplot comparing mRNA fold changes to PAR-CLIP read count fold changes of all target sites of the cellular microRNAs analyzed. For the PAR-CLIP data, a pseudocount of 1 was used. Green dots represent target sites that can be explained by the mRNA fold change while respecting sampling noise of the read counts, whereas orange and red dots correspond to significant outliers (P < 0.05 and P < 0.01, respectively). The P-value distribution in C of all these target sites suggests that at least 14.9% (363 instances with P < 0.01 of overall 2436 target sites after subtraction of baseline indicated by the horizontal line) of all differential target site activities cannot be explained by the mRNA fold change and sampling noise. B and D illustrate this for the context-dependent microRNA/target interactions only. Here, >50% of all sites cannot be explained by mRNA levels (see also Supplemental Fig. S5).
Furthermore, as illustrated in Figure 5B, there are several instances in which the target gene is not differentially expressed (i.e., data points around zero on the mRNA log2 fold change axis) but where the target sites show a >16-fold elevated activity. In these cases, mRNA expression alone clearly cannot explain target site activity. Thus, other factors contribute to context-specific microRNA function.
RNA binding proteins (RBPs) likely constitute such additional contributors. Thus, we performed a motif search in regions flanking context-dependent target sites (seed site ±80 bp). For motif discovery, we used MERCI (Vens et al. 2011), which is based on efficiently enumerating all discriminative k-mers of two sets of sequences. Specifically, we searched for k-mers that do not occur in the negative set and occur at least n times in the positive set, and we only considered target sites from mRNAs that are not differentially expressed. n was chosen according to the total number of sequences in the positive set. MERCI identified 20–30 k-mers when we compared target sites of cellular microRNAs exclusively present in BCBL1 to those exclusively present in DG75 and target sites of viral microRNAs exclusively present in BCBL1 to those in BC1/BC3 or vice versa (Fig. 6A; Table 1; Supplemental Table S3). These discriminative k-mers occur in 75%–90% of all context-dependent target sites that cannot be explained by the mRNA level, and as few as five motifs already can explain 30%–40% of all sites. In contrast, discriminative k-mers found by chance in randomized sequences only occur in a considerably lower number of sequences (Supplemental Fig. S7). Thus, these motifs are likely candidates of binding sites for RBPs contributing to context-dependent recognition of target sites by microRNAs.
Figure 6.
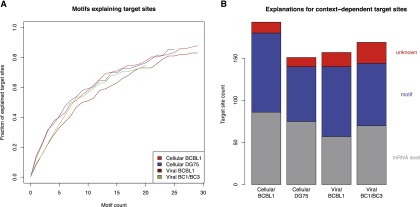
Role of sequence motifs for context-dependent target sites. (A) The fraction of context-dependent target sites that contain a certain number of discriminative k-mers. Only target sites that cannot be explained by mRNA levels were used. A k-mer is discriminative if it occurs n times in the positive set (e.g., cellular BCBL1-exclusive sites in red) and does not occur in the corresponding negative set (e.g., cellular DG75-exclusive sites) (see Table 1). We sorted discriminative k-mers according to their number of occurrences in decreasing order and chose a cutoff for n based on our randomization experiments (Supplemental Fig. S6; Supplemental Table S3). In all cases, between 75% and 90% of all context-dependent target sites can be explained by a discriminative k-mer. (B) Putative explanations for the full sets of context-dependent target sites are illustrated. On average, >90% can be explained by either differential mRNA levels or the presence of a discriminative k-mer.
Table 1.
Identified motifs by MERCI
In summary, from all context-dependent target sites identified by PAR-CLIP and validated by RIP-chip experiments, 4sU tagging-based mRNA half-lives, and mRNA and protein expression measurements, >90% can either be explained by differential mRNA levels or by the presence of a putative RBP binding motif (Fig. 6B).
Context-dependent target sites are less conserved than constitutive sites
Finally, we asked whether context-dependent target sites have distinct evolutionary conservation patterns as compared to constitutive target sites. Following the approach of Friedman et al. (2009), for each target site we computed the branch length along the phylogenetic tree of 46 vertebrates by summing all branches in which the seed of a cluster is fully conserved in the genome-wide multiple alignment of 46 vertebrate species. The branch length thus incorporates both the evolutionary age as well as the loss of a target site in specific lineages. Specifically, a target site that emerged in the last common ancestor of primates and rodents and has not been lost in any primate or rodent lineage, has a branch length of 2.342 (Fig. 7, shaded areas).
Figure 7.

Conservation of target sites. Distributions of branch lengths of target sites are illustrated (see main text for a definition of branch lengths). Shaded regions indicate the maximal branch lengths of target sites conserved in primates, in primates and rodents, in mammals, and in vertebrates. All cellular microRNAs considered here are conserved in vertebrates. Constitutive target sites of these microRNAs are significantly more conserved (P < 0.00304, two-sided Kolmogorov-Smirnov test) than context-dependent target sites. Moreover, neither context-dependent nor constitutive target sites of viral microRNAs show evidence for evolutionary conservation (see also Supplemental Fig. S7).
Intriguingly, constitutive target sites of conserved cellular microRNAs are significantly more strongly conserved than context-dependent sites (P < 0.003, two-sided Kolmogorov-Smirnov test). For instance, although >80% of constitutive sites are conserved beyond the last common ancestor of primates and rodents, only ∼65% of context-dependent sites are conserved beyond this clade; and 20% of context-dependent sites even show a signature of recent evolution within the primate lineage. Importantly, this does not reflect the overall conservation level of the respective 3′-UTRs, but is specific to the seed sites (Supplemental Fig. S5).
Target sites of viral microRNAs, independent of whether they are context-dependent or constitutive, show patterns of much weaker conservation. This can be expected because there are no conserved viral microRNAs, and pathogenicity of KSHV may rather induce positive selection of its microRNA target sites on host mRNAs.
Discussion
In this study, we analyzed PAR-CLIP data from four human B-cell lines, three of which are infected with Kaposi’s sarcoma-associated herpes virus (KSHV), using an improved computational approach to identify target sites of both cellular and viral microRNAs (PARma) (Erhard et al. 2013a). The overlap in target sites between the four cell lines was surprisingly low (∼40%), indicating a large set of context-dependent microRNA/target interactions. Three additional sets of high-throughput data (RIP-chip, 4sU-tagging-derived RNA half-lives, and SILAC proteomics data) supported this observation: Context-dependent microRNA targets are associated with RISC in a context-dependent manner and have a measurable functional impact on their targets in a context-dependent manner. This was observed for the targets of both cellular and viral microRNAs. The latter offered an important control as they were exclusively observed in the cells expressing the viral microRNAs. Thus, we propose a new layer of complexity in microRNA targeting: Depending on the cellular context, specific microRNA/target interactions may be active or not, even if both microRNA and target mRNA are expressed. Furthermore, we could show that the evolutionary conservation differs between context-dependent and constitutive target sites, indicating that selective pressure may be different for context-dependent and constitutive target sites or that they have different evolutionary ages.
Cellular context may be formed directly by the quantities of microRNAs and mRNAs: Dependent on the exact copy numbers of microRNAs and mRNAs in each cell, intricate regulatory mechanisms may emerge leading to highly complex patterns of regulation (Mukherji et al. 2011). Furthermore, due to the many-to-many relationship of regulators and targets, microRNAs and mRNAs are embedded in a highly complex regulatory network (Hobert 2008). Our analyses indicate that the quantities of microRNAs and target mRNAs are direct contributors to the cellular context. However, based on our results, >50% of all observed context-dependent microRNA/target interactions cannot be explained by microRNA or mRNA levels and, therefore, are likely dependent on indirect factors.
Differential isoform usage might constitute relevant contexts. It may lead to target sites that are active in one condition and inactive in another, even if the corresponding microRNA is constitutively expressed and the target appears constitutively expressed on the gene level. Especially differential usage of polyadenylation sites has been shown to be a major contributor to the cellular context during development and proliferation (Sandberg et al. 2008; Boutet et al. 2012). However, in our data sets, alternative polyadenylation sites cannot be distinguished, and more involved experiments would be necessary to identify context-dependent target sites due to possible alternative polyadenylation.
The presence of RNA binding proteins (RBPs) may prevent microRNA binding to nearby sites (Bhattacharyya et al. 2006) or also induce binding (Kim et al. 2009). In a recent study, the whole RNA binding proteome of a cell line was examined by PAR-CLIP coupled to high resolution mass spectrometry (Baltz et al. 2012). This study revealed two important aspects of RBPs: First, in a single cell type, about 800 different RBPs can be identified. This unexpectedly high number of RBPs allows for highly complex combinatorics of competitive or activating RBP–microRNA interactions. Second, crosslinking events were observed for almost 30% of all uridines in 3′-UTRs, suggesting that mRNAs are broadly covered by RBPs. Indeed, we could identify a handful of sequence motifs that are able to explain a large fraction of context-dependent target sites, indicating that RBPs may play important roles in shaping the cellular context for microRNA-mediated regulation.
Thus, there is an intriguing analogy of the transcriptional and post-transcriptional layer of regulation: DNA, which is the material for transcriptional regulation, is covered by histones, transcription factors, and other DNA binding proteins; and the composition and dynamics of these proteins contribute to the cellular context (The ENCODE Project Consortium 2012). This cellular context determines to which extent a certain transcription factor can bind to a specific target site and exerts its regulatory role. Context-dependent regulatory networks may differ dramatically across different cell types or conditions (Neph et al. 2012). Similarly, mRNAs, which are the units for post-transcriptional regulation, are covered by RBPs; and we argue that their composition and dynamics contribute to a cellular context for microRNA-mediated regulation. Additionally, factors other than these covering proteins may further shape the cellular context for both transcriptional and post-transcriptional regulation: For transcriptional regulation, distinct modifications of chromatin or the DNA may also determine context. Furthermore, chromosomal conformations may place distal binding sites of transcription factors to promotors of different genes in three-dimensional space and may therefore also be important.
mRNAs may even provide more opportunities for context-dependent regulation: Although DNA usually is restricted to a single cellular compartment, the nucleus, the life cycle of mRNAs may span multiple compartments and subcompartments. This cellular localization may itself be regulated, and depending on the localization, mRNAs may be translated or not. For instance, sequestering of mRNAs to P-bodies by microRNAs leads to a reduced translation and mRNA decay (Pasquinelli 2012). Furthermore, the single-stranded mRNA gives rise to complex secondary and tertiary structures, and it has been shown that the accessibility of target sites determines whether or not microRNAs can bind to the mRNA (Kertesz et al. 2007). Interestingly, the conformation of RNAs is highly flexible and may be reshaped in a context-dependent way: Kedde et al. (2010) have shown that the activation of the RNA binding protein, PUM1, induces a local change in a hairpin structure of the 3′-UTR of the CDKN1B mRNA. Upon PUM1 activation, an inaccessible binding site of miR-221/miR-222 is opened for binding, leading to an efficient repression of CDKN1B.
The differential analysis of a collection of high-quality large-scale experiments for microRNA target site discovery indicates that context-dependent microRNA targeting is not restricted to a few examples, but is a widespread phenomenon and a general feature of microRNA-mediated regulation. Under natural conditions, several factors may contribute to the cellular context that may determine which target sites of a microRNA are active and which are not. Thus, true target sites discovered in an experiment performed in a particular cell line may not be active in a different context. In addition, experiments that impose non-natural conditions on cells may identify target sites that are active only in such a non-natural context and at the same time may miss naturally occurring target sites.
Thus, additional experiments to unravel context-dependent microRNA targets are of great importance for both the identification of microRNA targets in particular contexts as well as the investigation of key contributors that determine cellular context.
Methods
Cell lines
DG75-eGFP (named DG75 throughout this paper) and BCBL1 were cultured in RPMI medium supplemented with 10% fetal calf serum and pen/strep.
PAR-CLIP and sequencing
PAR-CLIP on DG75 and BCBL1 was performed by the Zavolan laboratory as described (Kishore et al. 2011; Jaskiewicz et al. 2012), and the data are available at GEO (accession number: GSE43909; see Supplemental Table S6 for sequencing statistics). The PAR-CLIP sequencing data for BC1 and BC3 from Gottwein et al. (2011) have been downloaded from GEO (accession number: GSE32113). We applied PARma to the whole collection of all PAR-CLIP data sets as described (Erhard et al. 2013a).
SILAC-based proteomics
SILAC and LC-MS/MS were performed as described in the Mann laboratory at MPI for Biochemistry in Munich. The raw files from the mass spectrometer have been analyzed using MaxQuant (version 1.2.2.5) (Cox and Mann 2008), using standard parameters against all human proteins from Ensembl (v60).
RIP-chip analysis
For the RISC-IPs, 5 × 108 cells were taken for each replicate and processed as previously described (Dölken et al. 2010) using 6 μg purified monoclonal hAGO2 antibody (α-hAGO2; 11A9) or monoclonal BrdU-antibody (Abcam; used as control).
RNA half-life measurements by 4sU-tagging
The RNA half-life data for DG75 and BCBL1 have been published previously (Dölken et al. 2010). In brief, newly transcribed RNA was labeled for 1 h by adding 100 μM 4sU to the cell culture medium. Total RNA was prepared using TRIzol, and newly transcribed RNA was purified as described (Dölken et al. 2008). Three replicates of newly transcribed, total, and preexisting RNA were measured.
Acknowledgments
This work was supported by grants from BMBF (NGFN-Plus #01GS0801 to R.Z., L.D., and J.H.), MRC (G1002523 to L.D.), NHSBT (WP11-05 to L.D.), and Swiss Cancer League (#KFS 02477-08-2009 to M.Z.).
Author contributions: F.E. carried out the analyses, interpreted data, and wrote the manuscript. D.L. and G.M. carried out experiments. J.H. supervised the experiments. L.D. designed the experiments, contributed ideas, and helped to draft the manuscript. L.J. and M.Z. carried out the PAR-CLIP experiments. R.Z. supervised the project and helped to draft the manuscript. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.166702.113.
References
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690 [DOI] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Moshe NB, Avraham R, Kedmi M, Zeisel A, Yitzhaky A, Yarden Y, Domany E 2012. Context-specific microRNA analysis: identification of functional microRNAs and their mRNA targets. Nucleic Acids Res 40: 10614–10627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124 [DOI] [PubMed] [Google Scholar]
- Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, Iori K, Rando TA 2012. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 10: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM 2005. Principles of microRNA-target recognition. PLoS Biol 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll AP, Tooney PA, Cairns MJ 2013. Context-specific microRNA function in developmental complexity. J Mol Cell Biol 5: 73–84 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cullen BR 2011. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev 25: 1881–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R 2011. A parsimonious model for gene regulation by miRNAs. Science 331: 550–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölken L, Malterer G, Erhard F, Kothe S, Friedel CC, Suffert G, Marcinowski L, Motsch N, Barth S, Beitzinger M, et al. 2010. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human γ-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 7: 324–334 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhard F, Dölken L, Jaskiewicz L, Zimmer R 2013a. PARma: identification of microRNA target sites in AGO-PAR-CLIP data. Genome Biol 14: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhard F, Dölken L, Zimmer R 2013b. RIP-chip enrichment analysis. Bioinformatics 29: 77–83 [DOI] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Izaurralde E 2008. Getting to the root of miRNA-mediated gene silencing. Cell 132: 9–14 [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, et al. 2011. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10: 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R 2012. Ago HITS-CLIP expands understanding of Kaposi’s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog 8: e1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Hannon GJ 2004. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5: 522–531 [DOI] [PubMed] [Google Scholar]
- Hobert O 2008. Gene regulation by transcription factors and microRNAs. Science 319: 1785–1786 [DOI] [PubMed] [Google Scholar]
- Jacobsen A, Wen J, Marks DS, Krogh A 2010. Signatures of RNA binding proteins globally coupled to effective microRNA target sites. Genome Res 20: 1010–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskiewicz L, Bilen B, Hausser J, Zavolan M 2012. Argonaute CLIP—a method to identify in vivo targets of miRNAs. Methods 58: 106–112 [DOI] [PubMed] [Google Scholar]
- Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JA, Slanchev K, le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Ørom UA, et al. 2007. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131: 1273–1286 [DOI] [PubMed] [Google Scholar]
- Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, Agami R 2010. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol 12: 1014–1020 [DOI] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E 2007. The role of site accessibility in microRNA target recognition. Nat Genet 39: 1278–1284 [DOI] [PubMed] [Google Scholar]
- Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23: 1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid RP, Sullivan CS 2012. Virus-encoded microRNAs: an overview and a look to the future. PLoS Pathog 8: e1003018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M 2011. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods 8: 559–564 [DOI] [PubMed] [Google Scholar]
- Kozak M 2008. Faulty old ideas about translational regulation paved the way for current confusion about how microRNAs function. Gene 423: 108–115 [DOI] [PubMed] [Google Scholar]
- Meijer HA, Kong YW, Lu WT, Wilczynska A, Spriggs RV, Robinson SW, Godfrey JD, Willis AE, Bushell M 2013. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 340: 82–85 [DOI] [PubMed] [Google Scholar]
- Mishima Y, Fukao A, Kishimoto T, Sakamoto H, Fujiwara T, Inoue K 2012. Translational inhibition by deadenylation-independent mechanisms is central to microRNA-mediated silencing in zebrafish. Proc Natl Acad Sci 109: 1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A 2011. MicroRNAs can generate thresholds in target gene expression. Nat Genet 43: 854–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA 2012. Circuitry and dynamics of human transcription factor regulatory networks. Cell 150: 1274–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE 2012. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13: 271–282 [DOI] [PubMed] [Google Scholar]
- Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA 2012. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J 31: 2207–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie W, Flamant S, Rasko JE 2009. Predicting microRNA targets and functions: traps for the unwary. Nat Methods 6: 397–398 [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB 2008. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethupathy P, Megraw M, Hatzigeorgiou AG 2006. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods 3: 881–886 [DOI] [PubMed] [Google Scholar]
- Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, Nusbaum JD, Feederle R, Delecluse HJ, Luftig MA, et al. 2012. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog 8: e1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Lieberman J, Lal A 2010. Desperately seeking microRNA targets. Nat Struct Mol Biol 17: 1169–1174 [DOI] [PubMed] [Google Scholar]
- Vens C, Rosso MN, Danchin EG 2011. Identifying discriminative classification-based motifs in biological sequences. Bioinformatics 27: 1231–1238 [DOI] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. 2012a. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22: 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. 2012b. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22: 1798–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Cuna JO, Dinh HQ, Kvon EZ, Shlyueva D, Stark A 2012. Uncovering cis-regulatory sequence requirements for context specific transcription factor binding. Genome Res 22: 2018–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]