

Abstract
What is the significance of the extensive variability observed in individual members of a single-cell phenotype? This question is particularly relevant to the highly differentiated organization of the brain. In this study, for the first time, we analyze the in vivo variability within a neuronal phenotype in terms of input type. We developed a large-scale gene-expression data set from several hundred single brainstem neurons selected on the basis of their specific synaptic input types. The results show a surprising organizational structure in which neuronal variability aligned with input type along a continuum of sub-phenotypes and corresponding gene regulatory modules. Correlations between these regulatory modules and specific cellular states were stratified by synaptic input type. Moreover, we found that the phenotype gradient and correlated regulatory modules were maintained across subjects. As these specific cellular states are a function of the inputs received, the stability of these states represents “attractor”-like states along a dynamic landscape that is influenced and shaped by inputs, enabling distinct state-dependent functional responses. We interpret the phenotype gradient as arising from analog tuning of underlying regulatory networks driven by distinct inputs to individual cells. Our results change the way we understand how a phenotypic population supports robust biological function by integrating the environmental experience of individual cells. Our results provide an explanation of the functional significance of the pervasive variability observed within a cell type and are broadly applicable to understanding the relationship between cellular input history and cell phenotype within all tissues.
What is a cell type? This question has been a central project of biology and molecular biology. Typically, we deconstruct a tissue or organ into its constituent cell types based on anatomical, physiological or biochemical features, and examine each distinct cell phenotype to understand its larger function. In this context, it has been a major biological aspiration to connect cell phenotype to the genome via gene expression. But elucidating the organization of cell types by linking cell phenotype analysis to transcriptional state has been largely elusive. This elusiveness is due to the variability seen in transcriptional data sets produced from what are expected to be homogeneous cell populations. As high-throughput data acquisition methods have now become highly precise, it has become obvious that the variability observed in the results is not a mere distribution around a mean, but reflects true heterogeneity, the activity of cells in a range of distinct states. Even when we take single-cell genomic measures, this variability within cell types persists (Guo et al. 2010; Eberwine and Bartfai 2011). This variability is not only present at the individual cell level, but extends to the levels of electrical and neural network function (Eberwine and Bartfai 2011; Marder 2011; Marder and Taylor 2011). Reconciling cell type in the face of such heterogeneity in the adult mammalian brain and accurately defining post-development diversity continue to be difficult challenges, as expressed by Birren and Marder (2013) and Wichterle et al. (2013). The significance of understanding and defining brain cell type is highlighted in the recently announced BRAIN Initiative, which seeks to understand how “cellular phenotypes based on transcriptional profiles may change as a function of developmental stage, age, cell state (e.g., cell cycle for mitotic cells), activity levels, and experience among other things” (NIH RFA-MH-14-215 2013). We believe our results are part of a solution to this problem.
Defining cell type within the highly differentiated and networked mammalian brain relies on location, connectivity, morphology, histochemistry, neurotransmitter type, and most recently on transcriptomic profiles. Significant efforts have detailed how coordinated transcriptional mechanisms lead to neuronal diversification and connectivity in the context of developmental dynamics (Chen et al. 2006b; Kramer et al. 2006; Luo et al. 2008; Friese et al. 2009). However, an increasing amount of evidence demonstrates significant heterogeneity and plasticity caused by further post-developmental, adaptive changes within developed lineages. Cells remain plastic and are able to change adaptively in response to inputs; rather than reaching a final stable state or cell fate they continue to acquire new response capabilities in the mature organism. Thus, the current state of a cell is a product of the cumulative influences or inputs received throughout its history. Recent results support the idea that this cumulative record is represented by the transcriptome, representing an essential “snapshot state memory” of the phenotype (e.g., Kim and Eberwine 2010). The cell’s transcriptome adapts to inputs to change the cell, in effect becoming a repository of the cell’s input history.
In the context of mature neurons, recent experiments demonstrate how cellular experience influences heterogeneity through “neurotransmitter respecification” in adult rat brains, which was accomplished by modifying the amount of light/dark stimulus received by these adult rats (Dulcis et al. 2013). Another example shows in vivo reprogramming of circuit connectivity in mature neocortical neurons in mice (De la Rossa et al. 2013). As cutting edge discovery of plasticity and diversity within and across neuron types continues, the causes of these phenomena remain unclear (De la Rossa et al. 2013; Dulcis et al. 2013). We suspect that adaptive responses to inputs of this kind may cause the variability that is observed in high-throughput studies of phenotypically similar cells (Eberwine and Bartfai 2011; Kim et al. 2011). In other words, a cell type that might have been expected to be homogenous, sharing a common end fate, might rather be heterogeneous due to each cell within the cell type adapting to a distinct input history. Therefore, we hypothesize that neuronal transcriptomic variability reflects synaptic input variability to the phenotype’s individual cell members. We test this hypothesis by examining gene expression differences within individual mature neurons of the same neuroanatomical phenotype. We analyze the potential organization of these differences in terms of neuronal input types. If such an organization were supported by the data, expression variability would be functionally meaningful by facilitating alternative responses within the phenotype.
We now have been able to investigate this, for the first time, by taking in vivo measures of each cell’s high-throughput transcriptional state in several hundred neurons taken from a single nucleus. Specifically, we investigated this hypothesis by studying the nucleus tractus solitarius (NTS), a brainstem nucleus consisting of the anatomical phenotype of medium sized, fusiform neurons forming a column in the dorsal medulla as the sensory nucleus for the viscera. The neurons surround and are innervated by the tractus solitarius (ts) that conveys afferent inputs of the glossopharyngeal and vagus cranial nerves from visceral organs, including blood pressure sensory baroreceptor afferents. The NTS plays an integrative role in autonomic homeostasis, receiving inputs that place demands on homeostasis such as inputs conveying visceral states, pain, posture, exercise, temperature, circadian time, and mood. As individual NTS neurons must integrate distinct combinatorial input sets, their variability may reflect their inputs. We examined the NTS phenotype in vivo in its native tissue context using microfluidic qPCR across several hundred NTS neurons (Supplemental Material and Methods). In these single neurons we assayed the expression of 96 key genes derived from a previous microarray study of the nucleus (Khan et al. 2008).
NTS neurons were identified by two cell markers signifying distinct inputs, FOS and tyrosine hydroxylase (TH) (Fig. 1). A very extensive amount of literature demonstrates the use of immediate early gene Fos as an indicator for the subset of NTS neurons responsive to acute hypertensive disturbances. FOS is an established indicator of cellular activation—in the present case identifying cells directly influenced by blood pressure baroreceptor afferent inputs (Li and Dampney 1992, 1994; Rogers et al. 1993; Chan and Sawchenko 1994, 1995, 1998; Glass et al. 2007). Carefully conducted control studies have shown that high Fos levels depend on neurons being directly influenced by the increased activity of the blood pressure baroreceptor afferent inputs (Potts et al. 1997; Chan et al. 2000). Simultaneously, the NTS population of norepinephrine cells, indicated by the expression of the catecholamine synthesis enzyme TH, receives “higher order” influences through one or more additional interneurons and integrative inputs rather than direct blood pressure inputs from baroreceptors afferents (Chan and Sawchenko 1994, 1995, 1998; Rinaman 2010, 2011). Thus, the markers for FOS and TH identify discrete NTS neuronal populations in term of their expected inputs. Using Fos and Th as markers for distinguishing the expected input types, we investigated the differences in transcriptional states of individual NTS neurons with respect to these inputs.
Figure 1.

Single neuron gene expression analysis. (A) Workflow summary of our experimental approach to obtain single cell samples and measure gene expression. (B) PCA of the gene expression data of single cells from hypertensive rats was performed to analyze variation within gene expression data. Projection of single cells (scores) along the first two principal components (PCs) is shown. Additional components were explored (Supplemental Information). (C) Loading values of the genes along the first two PCs. Genes with the five highest and lowest loading values along PC 1 and genes with the five highest loading values along PC 2 are labeled. The highest and lowest genes along the multiple PCs explored provided the basis for the selection of a subset of 48 genes with significant contributions to variability observed in the data. (D) Pairwise comparison of single cells based on Spearman rank correlation coefficients. Single cells are compared based on their respective 48-gene rank order. Red indicates a high correlation between cells; black represents no correlation between a pair of single cells. (E) The Spearman rank correlation coefficients are used to determine the similarity distance between each cell. The high-dimensional set of similarity values between all possible single cell pairs are then projected into three dimensions using MDS. Thus each sphere in this three-dimensional space represents a single neuron. The relative distance between two spheres in this 3D space corresponds to the relative similarity (or dissimilarity) between two cells.
Results
We obtained a high-dimensional single neuron gene expression data set comprised of 28,880 data points representing expression of 96 genes each in 300 single neurons lifted from the NTS of six rats (Supplemental Material and Methods). Gene expression levels were measured using a high-throughput qPCR platform (BioMark, Fluidigm), which has demonstrated the ability to reproducibly measure gene expression over five orders of magnitude with minimal technical variability (Supplemental Fig. S1). Our quality control workflow followed established procedures for minimizing nonspecific contamination of samples (Espina et al. 2006). In addition to visual inspection of tissue and captured cell body, we measured neuron, astrocyte, microglial, and endothelial specific gene expression in a separate set of single neurons and astrocytes collected from the NTS via laser capture microdissection (LCM) to test for potential cross-contamination from nontargeted cell types that may affect our single cell samples (Supplemental Material and Methods). Our results showed minimal to nonexistent crossover contamination (Fig. 2; Supplemental Figs. S2, S3) consistent with the repeated performance of LCM approaches used by a number of other groups (e.g., Wang et al. 2002; Ye et al. 2003; Zhang et al. 2003; Espina et al. 2006; Macdonald et al. 2008). We normalized the high-dimensional single neuron gene expression data using established approaches that evaluated multiple reference genes to select those suitable for data normalization and comparison across single cells (Supplemental Fig. S4; Vandesompele et al. 2002; Andersen et al. 2004).
Figure 2.

Single neuron and astrocyte LCM. (A) Tyrosine hydroxylase (TH) immunohistochemical staining and collection of TH+ single cells from a coronal section of a normotensive rat brainstem. Colored outline images represent magnified tissue sections from which TH+ single cells were captured. (B) Glial fibrillary acidic protein (GFAP) immunohistochemical staining and collection of GFAP+ single cells from an adjacent coronal section of a normotensive rat brainstem. Colored outline images represent magnified tissue sections from which GFAP+ single cells were captured. (C) Gel electrophoresis image of reverse-transcribed cDNA from whole-brain tissue (positive control) (lanes 1–3), a representative single neuron sample (lanes 5–7), a representative astrocyte sample (lanes 8–10), and a no-template control (NTC) (negative control) (lanes 11,12). All samples underwent 22 pre-amplification cycles prior to undergoing a 40-cycle PCR. Products from the 40-cycle PCR were placed on an E-Gel EX Agarose Gel 4% (Invitrogen). The rat whole brain positive control shows product bands for Gapdh (148bp), Th (68bp), and Gfap (93bp). Both single neuron and astrocyte samples show formation of Gapdh. However, the neuron sample does not show any Gfap product at the expected 93-bp size. A light band in lane 7 at <50 bp suggests a nonspecific product. Similar behavior is observed in lane 9, where the astrocyte sample shows no Th product at the expected 68-bp size. Only a light product band at <50 bp is present, suggesting a nonspecific product. The results indicate minimal to no crossover contamination occurring between astrocytes and neurons.
Our results revealed significant variability in normalized gene expression across all single cells (Fig. 3). Approximately two-thirds of the genes showed expression values spanning three orders of magnitude as measured over multiple high-throughput qPCR runs, multiple rats, and in hypertensive and baseline states. Initially we analyzed the variability in single cell gene expression using Principal Component Analysis (PCA). Our results revealed an unstructured scatter of cellular states, as shown by the PCA scores along the first five principal components that accounted for 48.94% of the variability in the data (Fig. 1B,C). We derived a subset of 48 genes that significantly contributed to the observed variability, using the five highest and lowest corresponding loading values along the first five principal components as a basis for gene selection (Fig. 1B; Supplemental Fig. S5; Supplemental Table S4; Supplemental Material and Methods). Next, we analyzed the single cell states, characterized by this 48-dimensional gene expression (i.e., transcriptional) profile per cell, to determine the presence of any structure or organization subtending these seemingly disparate cell states. We performed all possible pairwise comparisons of the single cells using a Spearman rank correlation. The high-dimensional data set of correlation coefficients (Fig. 1D) were then converted to corresponding similarity values, which were projected into three dimensions using multidimensional scaling (MDS) (Fig. 1E). MDS is a technique useful to visualize similarities and dissimilarities of high-dimensional data in a lower-dimensional space (Fuller et al. 2002; Ross et al. 2003; Taguchi and Oono 2005). In this context, the proximity between any two cells in the MDS space corresponds to how similar or dissimilar the rank correlation of gene expression is between that particular cell pairing. The single cells were distributed in a cloud in this MDS mapping (Fig. 1E), revealing no initially obvious structures or organization to cell states.
Figure 3.

Gene expression and variance distributions. Boxplots overlaid with in-line scatter plots showing the spread of expression data for all genes (−ΔCt). Each gray dot corresponds to a particular gene expression level in a particular single cell sample. (A) Baseline-normotensive cells. (B) Hypertensive cells.
We subsequently analyzed the single cell variability with respect to gene expression of the two input-type markers Fos (transcript of Fos) and Th. Both Fos and Th expression levels are surrogates for different neuronal response capacity to particular inputs; Fos expression denotes neurons directly receiving baroreceptor afferent inputs, and Th expression denotes catecholaminergic neurons receiving “higher order” integrative inputs. We first considered the extremes of the single cell multiplex gene expression distribution (Fig. 1E) with respect to the two input-type markers to identify and annotate two input-based subtypes (Fig. 4A): cells with Fos expression and minimal to no Th expression (Th−/Fos+) and cells with Th expression and minimal to no Fos expression (Th+/Fos−). Categorization of the same single cells based on mRNA expression or immunoreactivity to the respective markers was nearly identical. Only six single cells categorized as “Fos−” showed FOS immunoreactivity. Although a single cell may be labeled as “Fos−”, this annotation is simply an indicator of low Fos mRNA levels present in that particular cell. Given the dynamic and transient nature of Fos regulation, this slight discrepancy is unsurprising.
Figure 4.

Input-driven extreme phenotypes. (A) Input-based cell type identification. A bivariate plot of single cells obtained from hypertensive rats based on their normalized expression of Th and Fos. The extremes of the distributions for each gene were initially explored, resulting in two extreme classes of single cells: (1) cells with no Th expression and (2) cells with no Fos expression. The gray lines indicate the threshold criteria used to define the extreme subtypes Th−/Fos+ (blue filled circles) and Th+/Fos− (orange filled circles). The effect of various thresholds used for classifying extreme classes is shown in Supplemental Figures S8–S11. (B) Clustering of input-based cell types. Three-dimensional MDS projection of single cells with the extreme phenotype classifications applied. These projects are based on the similarity of single cells with respect to their ranked expression order of the 48 gene subset. Alternative viewing perspectives of the 3D MDS projections are included in Supplemental Figure S6A,C,E. (C) Gene expression correlation modules. Gene-to-gene pairwise Spearman rank correlation coefficients across the extreme subtype cells were calculated and are represented in the heat map. The highly variable genes show that the underlying gene expression in these extreme subtypes can be organized into two correlative groups, or transcription modules. These transcription modules group genes that show higher correlations (upper left quadrant and lower right quadrant of the heat map) with each other across single cells of the extreme subtypes. Columns and rows with the same index representing a particular gene follow the row annotation in panel D. (D) Gene expression gradients in input-based cell types. Heat map of normalized gene expression data. An overall gene expression gradient pattern can be observed in the gene expression profile of the 48 highly variable genes across the extreme subtypes. Focusing on the extreme regions to the left and right of the whited-out region on the heat map, opposite expression behaviors can be observed in the two transcription modules between the two extreme input-based subtypes. The upper set of genes in the heat map shows an overall decrease in gene expression in Th+/Fos− cells and an increase in Th−/Fos+ cells. The opposite behavior is observed in the lower set of genes. The expression patterns of these extreme subtypes occupy opposite ends of the gene expression gradient observed.
Applying the mRNA-based annotation of these single cell subtypes to the MDS visualization of cell states revealed a surprisingly structured organization. The two subtypes were distinctly clustered at the opposing extremes of the overall distribution of cells (Fig. 4B; Supplemental Fig. S6A,C,E). The separation of the two extreme subtypes was statistically significant as no such clustering was observed in randomized permutations of the data (Supplemental Fig. S7). Cells categorized by their input types (i.e., Fos or Th expression level) maintained close proximity to each other in the transcriptional space, indicating that individual cells receiving a particular input type share similar transcriptional profiles, an indicator of cell response.
This structured organization supports a novel perspective that differential inputs to individual cells may drive variation in the transcriptional profiles of NTS neurons. It is interesting to note that the highly variable genes identified using PCA were rank-correlated across many single cells categorized by the two input-type markers (Fig. 4C). The underlying gene expression was organized into two correlative modules with the Fos and Th expression profiles serving as exemplars for each group to distinctly separate the two populations of NTS cells (Fig. 4C,D). Note that the expression of other key genes relevant to catecholaminergic function (e.g., Dbh and Slc6a2) was most highly correlated with Th gene expression, consistent with a well-regarded expectation of co-regulation of these genes (Qadri et al. 1991; Stadler et al. 1992; Lu et al. 1996; Blume et al. 1999; Richards et al. 1999; Gallinat 2001; Dogan et al. 2004). This result serves as an internal validation of our analysis. The alignment of the two input-type markers with the variation seen in the measured transcriptional profiles of NTS cells implies a causal relationship where inputs to individual cells play a major role in shaping the transcriptional profiles. This relationship argues that inputs influence neuronal transcriptional states and is further substantiated by the quantitative nature of this relationship. Regardless of the defined threshold for Fos expression or Th expression, cells with the highest Fos or highest Th expression tended to be the same cells having extreme expression of the 48 variable genes highlighted by PCA (Supplemental Figs. S8–S11).
While the majority of the cells expressed either Fos or Th, there were smaller populations with lower expression of one or both input-type markers. We interpret these various expression levels as indications of different populations with respect to the two input types. For example, some subset of Th-expressing cells may respond weakly to baroreceptor inputs through interactions with interneurons, yielding variable Fos expression in those cells. If so, the NTS cell types may form a continuous distribution with respect to strength of input from different sources, and by implication a continuous distribution of expression patterns may result. With this expectation, we categorized cells with lower expression of Th and Fos based on median expression for each input-type marker to yield four “intermediate” subtypes (Fig. 5A; Supplemental Material and Methods, section “Single Cell Subtypes”). Mapping these annotations onto the MDS visualization of cell states revealed that the subtypes showing lower Th or Fos levels were located in between the two extreme cell types (Fig. 5B). Similarly, subtypes showing higher levels of Th or Fos aligned closer to the corresponding extreme input-based subtype. Additionally, these results indicate that the 48 highly variable genes show correlated expression within these intermediate subtypes (Fig. 5C). The gene expression in the intermediate subtypes was correlated based on the same modules observed in the cases of the extreme Th+/Fos− and Th−/Fos+ subtypes (Supplemental Fig. S12).
Figure 5.

Input-driven intermediate phenotypes. (A) Intermediate input-based cell types. Bivariate plot of single cells obtained from hypertensive rats (Fig. 2A) based on their expression of Th and Fos. Finer classifications of single cells that express both input-type markers are included. The gray line segments in the upper right quadrant of the plot represent the threshold limits used to define four intermediary cell subtypes: (1) Thhigh/Foslow (red circles); (2) Thhigh/Foshigh (yellow circles); (3) Thlow/Foslow (cyan circles); (4) Thlow/Foshigh (gray circles) cells. A 30% quantile limit of Th expression of the single cells from hypertensive rats was used to define which cells would be classified as Thlow and Thhigh. The median Fos expression value of the intermediary cells was used to define Foslow and Foshigh cells. (B) Locating intermediate cell types in multidimensional gene expression space. Three-dimensional MDS projection of single cells with the four intermediate subtype classifications applied. These intermediate subtypes lie in between the extreme groups (smaller more transparent spheres). Cells with higher Th expression are positioned closer to the extreme Th+/Fos− subtypes while cells with more dominant Fos expression are positioned closer to the extreme Th−/Fos+ subtypes group. Alternative viewing perspectives of the 3D MDS projections are included in Supplemental Figure S6B,D,F. (C) Gene expression gradients in intermediate cell types. Gene expression gradient pattern observed in heat map across intermediate cell groups. Focusing on the middle region in between the whited-out sections of the heat map, gene expression gradient patterns occur across the “intermediate” input-based cell groups. Moving from left to right, the overall expression patterns of genes in transcription module 1 and transcription module 2 transition from one extreme subtype to the other. Single cells that have more dominant Th expression have gene expression profiles more similar to cells within the Th+/Fos− subtypes, whereas cells with more dominant Fos expression have expression patterns similar to the Th−/Fos+ subtypes.
The coordinated gene expression patterns, or transcription modules (Fig. 4C), further differentiate the expression states of cells. The various active states that lie along the gradient structure are governed by underlying gene regulatory networks, which can be used to further distinguish these states. A comparison of rank correlative gene networks, a surrogate for the regulatory interactions occurring in single cells within the extreme states of Figure 4, shows distinct structures of correlative gene expression behavior (Fig. 6). In the baseline Th+/Fos− network, transcription factors (TFs) showed a high degree of connectivity, i.e., correlative relationships, with genes across both transcription modules. However, under the hypertensive challenge, the relationships between TFs and genes within the transcription modules were reduced and shift mainly to genes in transcription module 2 in the Th+/Fos− (higher-order input cell) and Th−/Fos+ (second-order input cell) networks (Fig. 6).
Figure 6.

Gene correlation networks. The correlative network structures represent correlative relationships shared between TFs and target genes of each module across the three cell types: baseline Th+, hypertension Th+, hypertension Fos+. Cytoscape software was used to visualize the correlative network relationships. Edge opacity represents the strength of the correlation shared between genes across the respective sample subset (e.g., Th+/Fos− single cells): the darker the edge, the higher the correlation coefficient values. These network structures illustrate the pairwise Spearman rank correlative relationships among the subset of 48 genes. TFs are separated from the subset while the remaining genes are organized into their respective transcription modules 1 and 2 (Fig. 4C). The correlation network is based on pairwise gene correlations across various subsets of single cells. Only pairwise Spearman correlation coefficients ≥0.4 were included. Node colors represent scaled −ΔCt values of a representative single cell sample from the respective neuronal subtype. (A) Pairwise gene correlation network across normotensive single cells. Note the high number of correlative relationships shared between TFs and genes from both modules 1 and 2. (B) Correlation network based on hypertensive Th+/Fos− single cells shows a significant change in the number of correlative relationships between TFs and downstream target genes, and the majority of these relationships exist between TFs and genes within module 2. Similarly, this same shift in pairwise relationships occurs in Th−/Fos+ single cells, shown in C. This shift in relationships suggests that a physiological perturbation, in this case acute hypertension, causes a shift in the correlative relationships between TFs and downstream genes.
These input-driven shifts in expression correlation and potential gene regulation effects were also reflected in the constrained space occupied by hypertensive Th+/Fos− cells relative to the Th+ cells from control animals at baseline blood pressure levels (Supplemental Fig. S13). Although there was some individual rat-to-rat variability within these transcriptional modules, the same pattern of structured variation across input classes was present in each animal (Supplemental Fig. S14).
Discussion
Having analyzed NTS neurons of the same neuroanatomical phenotype in vivo in the context of their specific input connectivity, we found that post-developmental neuronal cell type is strongly associated with the specificity of connections. Studying gene expression profiles of NTS neurons at the single cell level provided us with the appropriate resolution to distinguish cell types with respect to the inputs they received. Our results support the importance of connectivity in defining a cell type, through the transcriptional regulation of neurons by their inputs. Viewing the distribution of neuronal cell types as a function of specific inputs allowed us to interpret cell-to-cell variability as structured heterogeneity rather than noise around a mean.
This single cell variability likely reflects cellular functional heterogeneity (Enver et al. 2009), influencing a cell’s position along the gradient of the observed multiplex gene expression (Fig. 4D). This structure is evident in the MDS visualization where single cells fall into input-defined clusters of cells that are positioned along an expression pattern gradient (Figs. 4D, 5C). Since input history of an individual cell influences the cell’s transcriptomic state, we postulate that the cumulative input history of a cell provides a driving force for adjustment or analog tuning of the transcription modules, placing cells within interchangeable, stable states along the gradient of catecholaminergic (Th+/Fos−) and non-catecholaminergic (Th−/Fos+) hypertension responsive cell states.
Visualization, using MDS, of gene expression gradients, dynamic landscapes, and analog tuning of expression defining cell development and function is a recent application used most notably in hematopoietic and embryonic stem cells and cell signaling systems, such as NFKB signaling (Hough et al. 2009; Tay et al. 2010; Bendall et al. 2011). Our application of such techniques and concepts to ostensibly terminally differentiated single cells is novel as far as we are aware. The input-based ordered structure within the heterogeneous gene expression of single neurons in the MDS space now allows us to contextualize single cells along transcriptional module gradients, suggesting a plastic rather than a discrete cell phenotype. Finding correlated gene expression modules delineated by inputs is consistent with transcriptional phenotypes that result from combinatorial inputs. Subsequent variability within a given phenotype results from differences in input type and strength to each cell. In this context, any additional variability within a sub-phenotype, reflected in the spread of single cells of that particular group, may reflect variability of other inputs to the cell population. Additional input-driven analysis would be expected to further fractionate the phenotype.
Our results, which suggest an input-based organization of the NTS neuronal phenotype within a cloud of cellular states, raise intriguing possibilities as to the mechanisms through which such a gene expression gradient could be tuned in individual neurons. It is likely that combinatorial actions of transcriptional and post-transcriptional regulatory processes are involved in transducing cellular inputs into the downstream regulation of transcriptional states. Such regulatory network coordination to generate complex patterns of gene expression has been well described with respect to developmental dynamics, and typically involves a unique combination of regulatory factors for each cell type (Chen et al. 2006a,b; Kramer et al. 2006; Luo et al. 2008; Friese et al. 2009). It is possible that such formalism extends into post-developmental gene expression variability between neuronal phenotypes. We should also consider alternative regulatory schemes where graded gene expression spanning the spectrum of cellular states may be driven by a set of regulators in common with the NTS neuronal phenotypes, with inputs tuning cell-to-cell differences in regulatory activity and combinatorial action. Given the 1-h duration of the hypertension perturbation in our study, it is unlikely for the transcriptional regulatory network to influence neuronal network connectivity in such a short period. Hence, such feedback cannot serve as an alternative explanation of association between cellular inputs and correlated gene regulatory states.
These interchangeable cell states can be schematically represented as a dynamic gene expression landscape populated by individual cells based on their transcriptional response (Fig. 7A,B). The landscape figures and two-dimensional contour plots are used to help illustrate the distinct cell states and the influence of inputs. Such a conceptualization is an evolution of the Waddington “canalization” to describe developmental phenotypes (Waddington 1942) and was used to organize the interrelationships between various cell types that emerge through dynamic expression changes during development (Enver et al. 2009). The contour plots are a projection of the single cells in the 3D MDS space onto a 2D plane (Fig. 7A,B; Supplemental Material and Methods, section “Contour Plots and Dynamic Landscape”). The “depth” of a well along the landscape (Supplemental Material and Methods, section “Contour Plots and Dynamic Landscape”) at any given location was derived from the local density of cells so that a cluster of many cells is deeper and indicates a potential local “attractor” reflecting constrained gene expression in those particular cells. In this representation, these valleys and wells, or “attractor”-like states, correspond to dominant expression states of relatively stable expression modules (e.g., those corresponding to Th+/Fos− and Th−/Fos+ extreme subtypes). The remaining topography corresponds to potential intermediary states that may be transient in response to input histories of individual cells and physiological perturbations (Fig. 7A,B, color-coded groupings). The path that these cells take along the gene expression landscape is a function of the input(s) received and is likely to be as varied as the input(s) (Fig. 7C). The exposure to a hypertensive challenge changes the constraints (Supplemental Fig. S13) and distribution of cells within the gene expression landscape (Fig. 7A,B), consistent with phenotypes that are determined by distinct state-dependent responses. Ultimately, the type of inputs received alters the regulatory network, resulting in constrained cell states, akin to a phenotype being an adaptive product of cellular input.
Figure 7.

Reversible cell states within the phenotype landscape shaping the variability and organization of single cell states. Contour plots are used to illustrate the concepts of distinct cell states and the influence that inputs have in determining these states. The contour plots are a projection of the single cells in the 3D MDS space onto a 2D plane. The landscape topography is based on an inversion of the probability densities of single cells. Cell states are represented by wells in the landscape and ellipsoid regions in the 2D contour plot. The colored ellipsoids capture these states and symbolically represent potential “attractor”-like states within this landscape. These contour and landscape topographies were created for single cells collected from rats undergoing an acute hypertension challenge (A) and from baseline normotensive rats (B). Comparing these two landscapes (A and B) shows that the well in which catecholaminergic cells (orange spheres in A and B) lie is much more constrained and local under the hypertensive challenge than in the baseline state. The changing landscape between the two physiological states suggests that physiological perturbation (e.g., hypertension) influences not only the state of the single cells, but the very nature of the landscape in which they exist. Thus inputs have a significant impact on transcriptional behavior and ultimately the phenotypic state of a cell. (C) Input-based gene expression phenotypes in NTS. Schematic of the influence of various inputs into the NTS shaping the cellular state and organization within a “homogeneous” single cell phenotype. Integrative inputs place demands on homeostasis such as those conveying visceral states, pain, posture, exercise, temperature, circadian time, mood etc. NTS neurons must integrate distinct combinatorial input sets. Our data revealed that gene expression variability across single neurons reflects their combinatorial inputs.
Plausibly, NTS Fos+ cells receive particular combinatorial inputs beyond blood pressure and integrate variable sets of cardiovascular homeostasis modulators such as pain, temperature, exercise, or mood, all of which affect cellular state and input processing. The influence of various inputs on NTS cell states is symbolically represented in Figure 7C (Paton 1998; Dampney and Horiuchi 2003; Michelini 2007; Rinaman 2011; Grill and Hayes 2012). Such input-based influences imply that NTS neurons are individually gated in dynamic responses to combinatorial inputs, rather than behaving as a homogeneous population and integrating all inputs into a population rate code. NTS neurons dynamically responding to inputs shape a mechanism of blood pressure homeostasis based on the selection or gating of particular NTS neurons activated by combinatorial demands on blood pressure.
Similarly demonstrating a functional meaning to variability, Marder and Taylor (2011) have shown that variability extends to the levels of electrical and neural network function. In this mechanism, the “neural code” by which blood pressure regulation is performed would be based on molecular states of individual neurons. This novel explanation of blood pressure homeostasis in terms of parallel distinct functional response pathways is something not found when assuming a rate code control by a homogeneous neuronal population (Fig. 7C). A mechanism of this kind is consistent with the presence of variable activity and absence of a blood pressure rate code observed in NTS baroreceptor neurons (Rogers et al. 1993, 1996; Paton et al. 2001).
These principles of input-structured phenotype may extend to other central neuronal phenotypes. Large populations of neurons with multiple sources of inputs, adaptive response to inputs, and variable activity of single neurons are common in the brain. Measures of adaptive variability within a neuronal phenotype may enable development of a molecular physiology interacting with higher-level functions. This expectation of the influence of input history on neuronal cell type and function across the brain is supported by the emerging perspective reflected in the recently announced BRAIN Initiative. With the convergence of sophisticated experimental techniques and accurate and precise high-throughput technologies we have a unique opportunity to develop “…an integrated view of molecular identity (DNA sequence, single-cell transcriptomes, epigenomic information, and protein expression). This picture, in combination with information on anatomical connectivity and functional measures (e.g. physiology) will afford an unprecedented view of the vertebrate brain.” (NIH RFA-MH-14-215 2013)
This perspective, supported by our results, expands the definition of a neuronal cell type to include post-developmental plasticity and highlights the role of transcriptional regulation in shaping these phenotypes. Furthermore, an alternative hypothesis can now be proposed on how a cell population supports robust biological function: Functional robustness is achieved through the development of a graded set of cellular responses, rather than a uniform population response. Analyses of this type could also be extended outside the brain to other environments where cells clearly vary and receive different inputs. Finally, the identification of cell type specific gene network topologies may be facilitated by the organization of variability in the transcriptional identity and response of individual cells.
Methods
We collected 300 single neurons lifted from the NTS of six rats (hypertensive rats n = 4, normotensive rats n = 2), 220 of which were collected from hypertensive rats and the remaining 80 individual neurons collected from normotensive rats. Due to the absence of a perturbation in arterial blood pressure in normotensive rats, Fos+ cells responding to an acute hypertensive challenge were only collected in hypertensive rats. The anatomical distribution of collected Fos+ neurons within the NTS was consistent with the extensive literature characterizing Fos expression in the NTS (Li and Dampney 1992, 1994; Chan and Sawchenko 1994, 1995, 1998; Miura et al. 1994; Graham et al. 1995; Shih et al. 1996; Chan et al. 1998, 2000). Acute hypertension challenge, immunohistochemistry staining, and LCM are described in greater detail in Supplemental Material and Methods.
High-throughput qPCR
Gene expression levels were measured across four high-throughput qPCR assay chips on the BioMark (Fluidigm), a highly reproducible qPCR platform, which has demonstrated minimal technical variability over five orders of gene expression (Supplemental Fig. S1). Additional details regarding the high-throughput qPCR (Spurgeon et al. 2008) are described in Supplemental Material and Methods.
Data normalization
A total of 300 single cells and 96 gene assays were collected, which were reduced by rigorous quality control (QC) to 192 single cell samples (41 normotensive samples and 151 hypertensive samples) and 81 different gene assays that were included in the present analysis.
Raw Ct values for individual samples were normalized against an average expression level between Actb and Rpl19 to obtain a –ΔCt. A –ΔCt value (Spurgeon et al. 2008) was used in order to relate this value to actual gene expression (e.g., a −ΔCt value of 10 in one cell has higher gene expression than a cell with a −ΔCt value of 5 or −2 for a particular gene). The following equation was used to calculate −ΔCt:
Actb and Rpl19, included as part of a set of potential housekeeping genes, were selected based on previously developed methods (Vandesompele et al. 2002; Andersen et al. 2004). The –ΔCt values were used as a measure for relative gene expression and used as the basis for the analytical methods utilized in this report.
Principal Component Analysis (PCA)
The pcaMethods package (Stacklies et al. 2007) and associated functions in the R statistical software (R Development Core Team 2013) were used to perform PCA. A subset of 48 genes was derived as significantly contributing to the observed variability, using the five highest and lowest corresponding loading values along the first five principal components as a basis for gene identification. For further details please refer to Supplemental Material and Methods.
Gene correlation networks
The statistical software R was used to determine rank correlation coefficients between the subset of 48 genes for the six different single cell “sub-phenotypes” initially identified. A Spearman rank correlation coefficient cutoff of 0.4 was used to define whether or not two genes had a correlative relationship. Cytoscape (www.cytoscape.org) was used to visualize the correlative relationships.
Distance and multidimensional scaling (MDS)
Relative distances between single cells were determined using the Spearman rank correlations obtained for the set of hypertensive samples and subset of 48 genes identified from PCA. The following equations were used to determine distance:
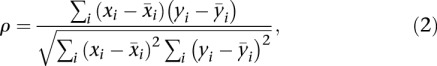 |
where 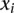 and
and 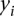 correspond to gene expression rank between two single cell samples;
correspond to gene expression rank between two single cell samples;
where 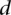 corresponds to the Spearman rank distance between two cells.
corresponds to the Spearman rank distance between two cells.
The pairwise relative distances between single cell samples was performed using the stats package provided through the R statistical software (R Development Core Team 2013).
Nonmetric MDS was performed on single cells in conjunction with PCA in order to analyze single cells that lie in an n-dimensional space (due to the nature of the multiplex gene expression data). MDS was performed on the single cells obtained from hypertensive rats since genes were rank-ordered for each sample (based on −ΔCt). The MDS would then map the relative distances between the samples onto a lower-dimensional plane, while minimizing the error observed between the actual distances in the n-dimensional space and the lower-dimensional space (i.e., minimizing the stress) (Van Deun and Delbeke 2000). The isoMDS function provided in the MASS package (Venables and Ripley 2002) for R platform was used to perform the MDS. Following MDS, single cell samples were plotted in the lower-dimensional MDS space with the input-type marker categorization (combinatorial expression levels of Th and Fos) overlaid on the samples. The first MDS axis discriminates samples based on the rank-ordering of expression levels of genes from transcription module 2. MDS axis 2 accounts for biological variability in both hypertensive and baseline samples (Supplemental Fig. S13), and MDS axis 3 discriminates cells based on rank expression levels of genes from transcription module 1. Both two-dimensional and three-dimensional plots were created via plotrix and rgl packages (Lemon 2006; Adler and Murdoch 2013) provided by the R statistical software (R Development Core Team 2013).
Data access
Both raw Ct and –ΔCt values of samples passing quality control are included as Supplemental Material (Supplemental Tables S1, S2).
Acknowledgments
The work presented here is funded through NIH NIGMS R01 GM083108 and NIH NHLBI R01HL111621. J.P. acknowledges Mrs. Sirisha Achanta for guidance and assistance with performing the LCM quality control experiments.
Author contributions: J.P. performed analysis and contributed to writing and figure design; A.B. performed analysis, writing, and figure design; K.K. was involved in design, initial analysis, and writing/editing; A.S. was involved in sample acquisition, experimental execution, and analysis of blood pressure data; S.G. contributed to design, single cell LCM, and staining methods; B.O. was involved in initial analysis and editing; J.S. designed the study and was involved in analysis, writing, and figure design; R.V. designed the study and was involved in analysis, writing, and figure design. J.P. and A.B. contributed equally to the study. All authors discussed the results and commented on the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.161802.113.
References
- Adler D, Murdoch D 2013. rgl: 3D visualization device system (OpenGL). http://cran.r-project.org/package=rgl
- Andersen CL, Jensen JL, Ørntoft TF 2004. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64: 5245–5250 [DOI] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. 2011. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332: 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birren SJ, Marder E 2013. Neuroscience. Plasticity in the neurotransmitter repertoire. Science 340: 436–437 [DOI] [PubMed] [Google Scholar]
- Blume A, Herdegen T, Unger T 1999. Angiotensin peptides and inducible transcription factors. J Mol Med 77: 339–357 [DOI] [PubMed] [Google Scholar]
- Chan RK, Sawchenko PE 1994. Spatially and temporally differentiated patterns of c-fos expression in brainstem catecholaminergic cell groups induced by cardiovascular challenges in the rat. J Comp Neurol 348: 433–460 [DOI] [PubMed] [Google Scholar]
- Chan RKW, Sawchenko PE 1995. Hemodynamic regulation of tyrosine hydroxylase messenger RNA in medullary catecholamine neurons: a c-fos-guided hybridization histochemical study. Neuroscience 66: 377–390 [DOI] [PubMed] [Google Scholar]
- Chan RK, Sawchenko PE 1998. Organization and transmitter specificity of medullary neurons activated by sustained hypertension: implications for understanding baroreceptor reflex circuitry. J Neurosci 18: 371–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JYH, Chen W-C, Lee H-Y, Chan SHH 1998. Elevated Fos expression in the nucleus tractus solitarii is associated with reduced baroreflex response in spontaneously hypertensive rats. Hypertension 32: 939–944 [DOI] [PubMed] [Google Scholar]
- Chan RKW, Jarvina EV, Sawchenko PE 2000. Effects of selective sinoaortic denervations on phenylephrine-induced activational responses in the nucleus of the solitary tract. Neuroscience 101: 165–178 [DOI] [PubMed] [Google Scholar]
- Chen AI, de Nooij JC, Jessell TM 2006a. Graded activity of transcription factor Runx3 specifies the laminar termination pattern of sensory axons in the developing spinal cord. Neuron 49: 395–408 [DOI] [PubMed] [Google Scholar]
- Chen C-L, Broom DC, Liu Y, de Nooij JC, Li Z, Cen C, Samad OA, Jessell TM, Woolf CJ, Ma Q 2006b. Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron 49: 365–377 [DOI] [PubMed] [Google Scholar]
- Dampney RA, Horiuchi J 2003. Functional organisation of central cardiovascular pathways: studies using c-fos gene expression. Prog Neurobiol 71: 359–384 [DOI] [PubMed] [Google Scholar]
- De la Rossa A, Bellone C, Golding B, Vitali I, Moss J, Toni N, Lüscher C, Jabaudon D 2013. In vivo reprogramming of circuit connectivity in postmitotic neocortical neurons. Nat Neurosci 16: 193–200 [DOI] [PubMed] [Google Scholar]
- Dogan MD, Sumners C, Broxson CS, Clark N, Tümer N 2004. Central angiotensin II increases biosynthesis of tyrosine hydroxylase in the rat adrenal medulla. Biochem Biophys Res Commun 313: 623–626 [DOI] [PubMed] [Google Scholar]
- Dulcis D, Jamshidi P, Leutgeb S, Spitzer NC 2013. Neurotransmitter switching in the adult brain regulates behavior. Science 340: 449–453 [DOI] [PubMed] [Google Scholar]
- Eberwine J, Bartfai T 2011. Single cell transcriptomics of hypothalamic warm sensitive neurons that control core body temperature and fever response signaling asymmetry and an extension of chemical neuroanatomy. Pharmacol Ther 129: 241–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enver T, Pera M, Peterson C, Andrews PW 2009. Stem cell states, fates, and the rules of attraction. Cell Stem Cell 4: 387–397 [DOI] [PubMed] [Google Scholar]
- Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, Geho DH, Petricoin E, Liotta L 2006. Laser-capture microdissection. Nat Protoc 1: 586–603 [DOI] [PubMed] [Google Scholar]
- Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S 2009. γ and α motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci 106: 13588–13593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller GN, Hess KR, Rhee CH, Yung WK, Sawaya RA, Bruner JM, Zhang W 2002. Molecular classification of human diffuse gliomas by multidimensional scaling analysis of gene expression profiles parallels morphology-based classification, correlates with survival, and reveals clinically-relevant novel glioma subsets. Brain Pathol 12: 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallinat S 2001. Gene expression profiling of rat brain neurons reveals angiotensin II-induced regulation of calmodulin and synapsin I: possible role in neuromodulation. Endocrinology 142: 1009–1016 [DOI] [PubMed] [Google Scholar]
- Glass MJ, Chan J, Frys KA, Oselkin M, Tarsitano MJ, Iadecola C, Pickel VM 2007. Changes in the subcellular distribution of NADPH oxidase subunit p47phox in dendrites of rat dorsomedial nucleus tractus solitarius neurons in response to chronic administration of hypertensive agents. Exp Neurol 205: 383–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JC, Hoffman GE, Sved AF 1995. c-Fos expression in brain in response to hypotension and hypertension in conscious rats. J Auton Nerv Syst 55: 92–104 [DOI] [PubMed] [Google Scholar]
- Grill H, Hayes M 2012. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 16: 296–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P 2010. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 18: 675–685 [DOI] [PubMed] [Google Scholar]
- Hough SR, Laslett AL, Grimmond SB, Kolle G, Pera MF 2009. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS ONE 4: e7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RL, Vadigepalli R, McDonald MK, Rogers RF, Gao GR, Schwaber JS 2008. Dynamic transcriptomic response to acute hypertension in the nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 295: R15–R27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eberwine J 2010. RNA: state memory and mediator of cellular phenotype. Trends Cell Biol 20: 311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Sul J-Y, Peternko NB, Lee JH, Lee M, Patel VV, Kim J, Eberwine JH 2011. Transcriptome transfer provides a model for understanding the phenotype of cardiomyocytes. Proc Natl Acad Sci 108: 11918–11923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer I, Sigrist M, de Nooij JC, Taniuchi I, Jessell TM, Arber S 2006. A role for Runx transcription factor signaling in dorsal root ganglion sensory neuron diversification. Neuron 49: 379–393 [DOI] [PubMed] [Google Scholar]
- Lemon J 2006. Plotrix: a package in the red light district of R. R-News 6: 8–12 [Google Scholar]
- Li Y-W, Dampney RA 1992. Expression of c-fos protein in the medulla oblongata of conscious rabbits in response to baroreceptor activation. Neurosci Lett 144: 70–74 [DOI] [PubMed] [Google Scholar]
- Li Y-W, Dampney RAL 1994. Expression of fos-like protein in brain following sustained hypertension and hypotension in conscious rabbits. Neuroscience 61: 613–634 [DOI] [PubMed] [Google Scholar]
- Lu D, Yang H, Raizada MK 1996. Angiotensin II regulation of neuromodulation: downstream signaling mechanism from activation of mitogen-activated protein kinase. J Cell Biol 135: 1609–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Fishell G, di Sanguinetto SADT, Dasen JS, Arber S 2008. Transcriptional mechanisms controlling motor neuron diversity and connectivity. Curr Opin Neurobiol 18: 36–43 [DOI] [PubMed] [Google Scholar]
- Macdonald JA, Murugesan N, Pachter JS 2008. Validation of immuno-laser capture microdissection coupled with quantitative RT-PCR to probe blood–brain barrier gene expression in situ. J Neurosci Methods 174: 219–226 [DOI] [PubMed] [Google Scholar]
- Marder E 2011. Variability, compensation, and modulation in neurons and circuits. Proc Natl Acad Sci (Suppl) 108: 15542–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Taylor AL 2011. Multiple models to capture the variability in biological neurons and networks. Nat Neurosci 14: 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini LC 2007. The NTS and integration of cardiovascular control during exercise in normotensive and hypertensive individuals. Curr Hypertens Rep 9: 214–221 [DOI] [PubMed] [Google Scholar]
- Miura M, Takayama K, Okada J 1994. Neuronal expression of Fos protein in the rat brain after baroreceptor stimulation. J Auton Nerv Syst 50: 31–43 [DOI] [PubMed] [Google Scholar]
- NIH RFA-MH-14-215. 2013. BRAIN Initiative: transformative approaches for cell-type classification in the brain (U01). http://grants.nih.gov/grants/guide/rfa-files/RFA-MH-14-215.html
- Paton JF 1998. Convergence properties of solitary tract neurones driven synaptically by cardiac vagal afferents in the mouse. J Physiol 508: 237–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JF, Li YW, Schwaber JS 2001. Response properties of baroreceptive NTS neurons. Ann N Y Acad Sci 940: 157–168 [DOI] [PubMed] [Google Scholar]
- Potts P, Polson J, Hirooka Y, Dampney RA 1997. Effects of sinoaortic denervation on Fos expression in the brain evoked by hypertension and hypotension in conscious rabbits. Neuroscience 77: 503–520 [DOI] [PubMed] [Google Scholar]
- Qadri F, Badoer E, Stadler T, Unger T 1991. Angiotensin II-induced noradrenaline release from anterior hypothalamus in conscious rats: a brain microdialysis study. Brain Res 563: 137–141 [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/
- Richards EM, Raizada MK, Gelband CH, Sumners C 1999. Angiotensin II type 1 receptor-modulated signaling pathways in neurons. Mol Neurobiol 19: 25–41 [DOI] [PubMed] [Google Scholar]
- Rinaman L 2010. Ascending projections from the caudal visceral nucleus of the solitary tract to brain regions involved in food intake and energy expenditure. Brain Res 1350: 18–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaman L 2011. Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions. Am J Physiol 300: R222–R235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RF, Paton JF, Schwaber JS 1993. NTS neuronal responses to arterial pressure and pressure changes in the rat. Am J Physiol 265: R1355–R1368 [DOI] [PubMed] [Google Scholar]
- Rogers RF, Rose WC, Schwaber JS, Rogers RF, Rose C, Schwaber JS 1996. Simultaneous encoding of carotid sinus pressure and dP/dt by NTS target neurons of myelinated baroreceptors. J Neurophysiol 76: 2644–2660 [DOI] [PubMed] [Google Scholar]
- Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, Liu HC, Mahfouz R, Raimondi SC, Lenny N, et al. 2003. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 102: 2951–2959 [DOI] [PubMed] [Google Scholar]
- Shih C-D, Chan SHH, Chan JYH 1996. Participation of Fos protein at the nucleus tractus solitarius in inhibitory modulation of baroreceptor reflex response in the rat. Brain Res 738: 39–47 [DOI] [PubMed] [Google Scholar]
- Spurgeon SL, Jones RC, Ramakrishnan R 2008. High throughput gene expression measurement with real time PCR in a microfluidic dynamic array. PLoS One 3: e1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacklies W, Redestig H, Scholz M, Walther D, Selbig J 2007. pcaMethods—a Bioconductor package providing PCA methods for incomplete data. Bioinformatics 23: 1164–1167 [DOI] [PubMed] [Google Scholar]
- Stadler T, Veltmar A, Qadri F, Unger T 1992. Angiotensin II evokes noradrenaline release from the paraventricular nucleus in conscious rats. Brain Res 569: 117–122 [DOI] [PubMed] [Google Scholar]
- Taguchi Y-H, Oono Y 2005. Relational patterns of gene expression via non-metric multidimensional scaling analysis. Bioinformatics 21: 730–740 [DOI] [PubMed] [Google Scholar]
- Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW 2010. Single-cell NF-κB dynamics reveal digital activation and analogue information processing. Nature 466: 267–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deun K, Delbeke L 2000. Multidimensional scaling. http://www.mathpsyc.uni-bonn.de/doc/delbeke/delbeke.htm
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD 2002. Modern applied statistics with S, 4th ed. Springer, New York [Google Scholar]
- Waddington CH 1942. Canalization of development and the inheritance of acquired characters. Nat Publ Gr 150: 563–565 [Google Scholar]
- Wang J, Zheng H, Ou X, Fink LM, Hauer-Jensen M 2002. Deficiency of microvascular thrombomodulin and up-regulation of protease-activated receptor-1 in irradiated rat intestine. Am J Pathol 160: 2063–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Gifford D, Mazzoni E 2013. Neuroscience. Mapping neuronal diversity one cell at a time. Science 341: 726–727 [DOI] [PubMed] [Google Scholar]
- Ye P, Bagnell R, D’Ercole AJ 2003. Mouse NG2+ oligodendrocyte precursors express mRNA for proteolipid protein but not its DM-20 variant: a study of laser microdissection-captured NG2+ cells. J Neurosci 23: 4401–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Mojsilovic-Petrovic J, Andrade MF, Zhang H, Ball M, Stanimirovic DB 2003. The expression and functional characterization of ABCG2 in brain endothelial cells and vessels. FASEB J 17: 2085–2087 [DOI] [PubMed] [Google Scholar]