

Abstract
Histone modifications are epigenetic marks that play fundamental roles in many biological processes including the control of chromatin-mediated regulation of gene expression. Little is known about interindividual variability of histone modification levels across the genome and to what extent they are influenced by genetic variation. We annotated the rat genome with histone modification maps, identified differences in histone trimethyl-lysine levels among strains, and described their underlying genetic basis at the genome-wide scale using ChIP-seq in heart and liver tissues in a panel of rat recombinant inbred and their progenitor strains. We identified extensive variation of histone methylation levels among individuals and mapped hundreds of underlying cis- and trans-acting loci throughout the genome that regulate histone methylation levels in an allele-specific manner. Interestingly, most histone methylation level variation was trans-linked and the most prominent QTL identified influenced H3K4me3 levels at 899 putative promoters throughout the genome in the heart. Cis- acting variation was enriched in binding sites of distinct transcription factors in heart and liver. The integrated analysis of DNA variation together with histone methylation and gene expression levels showed that histoneQTLs are an important predictor of gene expression and that a joint analysis significantly enhanced the prediction of gene expression traits (eQTLs). Our data suggest that genetic variation has a widespread impact on histone trimethylation marks that may help to uncover novel genotype–phenotype relationships.
Histones are the main component of chromatin and undergo several post-translational modifications (PTMs) (Kouzarides 2007). These PTMs play an important role in genome organization, stability, and the control of gene expression. In the past five years, extensive chromatin signatures have been characterized in various cell types (Barski et al. 2007; Mikkelsen et al. 2007; Ernst et al. 2011). Using recurrent combinations of histone marks, Ernst and colleagues could then define numerous chromatin states corresponding to repressed, poised, and active promoters, strong and weak enhancers, as well as transcribed and repressed regions. Several studies addressed the complex interaction between genetic variation and chromatin status and its heritability (Gaulton et al. 2010; Kasowski et al. 2010; McDaniell et al. 2010; Degner et al. 2012). These genome-wide studies in human lymphoblastoid cell lines (LCLs) reported pronounced allele-specific differences of open chromatin and transcription-factor (TF) binding. Kadota et al. (2007) were the first to show allele-specific histone marks in a limited set of LCLs and suggested a DNA sequence-dependent influence on post-translational histone marks. Recently, two groups identified genetic variants affecting histone modifications in human cells and incorporated TF binding, RNA polymerase II, and DNase I hypersensitvity data to show that in many cases the same variant affects multiple phenotypes (Kasowski et al. 2013; McVicker et al. 2013). However, many molecular aspects, such as the extent to which cis- and trans-acting factors affect the chromatin status and to what extent they are tissue specific, still remain elusive. To study the magnitude and impact of genetic variation on histone modification patterns, we annotated the rat genome with histone modification maps, identified quantitative differences in histone trimethyl-lysine levels among strains, and described their underlying genetic basis using cosegregation analysis in the BXH/HXB panel of rat recombinant inbred (RI) strains, derived from a cross of Brown Norway (BN) and spontaneously hypertensive rats (SHR). This panel has previously been used for genome-wide mapping of expression traits, which guided the identification of several genes implicated in common complex cardiovascular and metabolic diseases (Hubner et al. 2005; Petretto et al. 2008; Pravenec et al. 2008; Heinig et al. 2010). The genomes of both parental strains have been sequenced at more than 20× coverage (Atanur et al. 2010; Simonis et al. 2012), which revealed 3.6 million SNPs and thousands of short indels and larger deletions as well as hundreds of copy-number variations. Furthermore, ∼20,000 SNPs have been genotyped across the 30 RI strains (Saar et al. 2008), leading to the identification of 1384 distinct strain distribution patterns (SDPs).
Results
Strain specificity of histone modification marks
To obtain an overall picture of the differences of chromatin states between two distinct rat strains, BN and SHR, we generated chromatin immunoprecipitation (ChIP)-seq data of histone PTMs in the heart and liver tissue of three BN and three SHR male animals at 6 wk of age. Methylations are among the most stable PTMs of histones (Zee et al. 2010) and have been suggested to contribute to the inheritance of epigenetic traits (Greer and Shi 2012). We therefore studied four well-characterized histone methylation marks: H3K4me3, which is associated with active promoters; H3K4me1, preferentially associated with promoters and enhancers; H4K20me1, associated with transcribed regions; and H3K27me3, associated with Polycomb-repressed regions (Barski et al. 2007; Mikkelsen et al. 2007). We further collected data for mRNA expression levels in five biological replicates of these progenitor rat strains. Subsequently, we collected genome-wide H3K4me3, H3K27me3, and RNA-seq profiles across a panel of 30 BXH/HXB RI rats. We then combined these comprehensive chromatin and expression profiles and analyzed them computationally (see Supplemental Fig. 1). We defined regions of interest based on peak calling algorithms or known gene models and quantified histone modification levels as normalized ChIP-seq read counts. The high quality of all ChIP-seq data is in line with recently published standard measurements (Landt et al. 2012; Supplemental Table 1), and typical histone occupancy profiles (Barski et al. 2007) were observed for expressed and nonexpressed genes (Supplemental Fig. 2). The genome sequence of both parental strains allowed us to exclude any SNP-related effect on the alignment of the short ChIP-sequencing reads and on the quantification of modified regions (Supplemental Fig. 3). As expected, the histone marks showed little interindividual variation of histone methylation levels between biological replicates of the two strains (correlation coefficient R between 0.90 and 0.99), but we noted pronounced differences between the strains (Fig. 1). In heart tissue, 7% of identified peaks showed differential H3K4me3 methylation levels (FDR < 0.05) and 16% of H3K4me1 peaks were significantly different between the two strains (Table 1). The fraction of genes with differential methylation levels for the broader marks H4K20me1 and H3K27me3 were similar (15% and 14%). Nevertheless, we detected a large proportion of significant differences between BN and SHR individuals (15% of total H4K20me1 and 33% of total H3K27me3 peaks). Comparable numbers of strain-specific differences were found in liver tissue (Table 1). Overall, we observed differential histone marks that ranged from small effects to strong bimodal differences with some marks being completely absent in one strain while a high peak was detected in the other strain (Fig. 2; Supplemental Fig. 4). We validated the ChIP-seq results of three separate loci from each modification and tissue using quantitative PCR (Supplemental Fig. 5).
Figure 1.

Intra-strain and inter-strain correlations of histone marks. Pearson correlation coefficients of the normalized and log-transformed modification levels between three biological replicates of each parental strain shows globally high reproducibility within strains and higher levels of variation between strains.
Table 1.
Results of differential histone marks between progenitor strains in heart and liver tissue
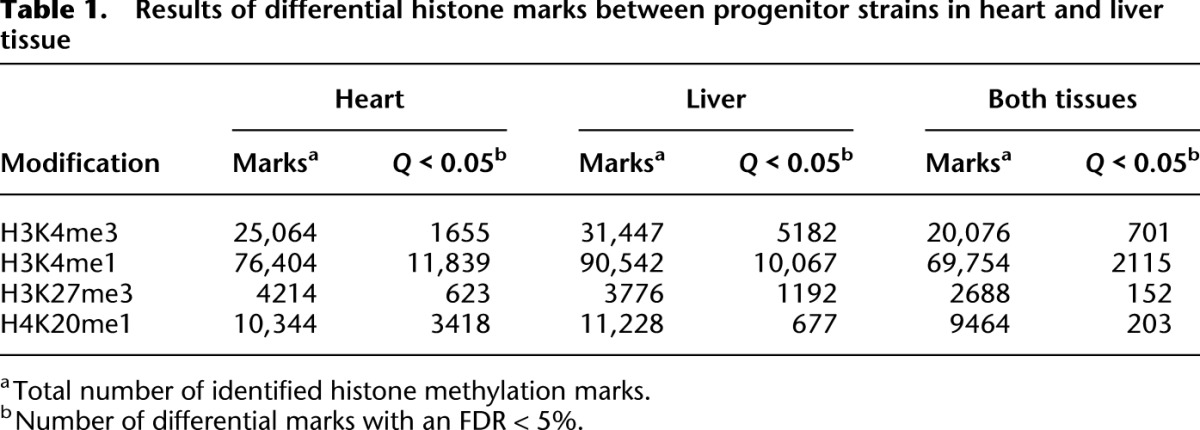
Figure 2.

Strain-specific and allele-dependent histone methylation marks in progenitor and RI rats. Example of strain-specific H3K4me1 peaks (A), H4K20me1 peaks (B), H3K4me3 peaks (C), and H3K27me3 peaks (D) in three BN and three SHR rats. In C and D, RI strains were split according to their genotype at the position of the histone mark and are depicted in blue (BN genotype) or orange (SHR genotype). Genomic positions, Ensembl genes, and their direction of transcription are indicated by arrows. In C, strain-specific H3K4me3 marks colocalized with an alternative TSS, which was detected using RNA-seq and is depicted in red.
Tissue specificity of histone modification marks
The epigenome is unique for each tissue and cell type in a multicellular organism. Thus, we explored to what extent histone marks are shared between two tissues. We compared all detected histone peaks in heart and liver tissue and found that 55% of H3K4me3 marks were shared between tissues. Similarly, 51% of H3K27me3, 72% of H3K4me1, and 78% of H4K20me1 marks were observed in both tissues (Table 1). As expected, genes occupied by heart-specific H3K4me3 marks were enriched for functional categories such as muscle system processes, heart contraction, and cardiovascular system development (Supplemental Table 2). Genes in the vicinity of liver-specific marks were enriched for functional terms of the immune system and several metabolic processes.
Genetic determinants of histone modifications
Having identified extensive strain-specific differences between chromatin states of all analyzed histone marks, we were interested in how many differential histone marks can be explained through genetic variability. We used a panel of 30 BXH/HXB RI strains and analyzed the cosegregation of histone modification levels with sequence variants using linkage analysis. We chose to study the histone marks H3K4me3 and H3K27me3, because we wanted to study both an active as well as a repressive mark. H3K4me3 is strongly associated with transcriptionally competent chromatin, whereas H3K27me3 is associated with gene silencing (Bernstein et al. 2005; Barski et al. 2007). In addition, changes of H3K4me3 levels can have stable long-lasting effects. Greer et al. (2011) have shown that perturbation of H3K4me3 levels in parental C. elegans can alter longevity in third-generation offspring even after the initial source of perturbation is no longer present. Furthermore, both histone methylation marks are involved in mediating mitotic inheritance of lineage-specific gene expression patterns (Ringrose and Paro 2004), rendering both modifications excellent candidates to study the impact of genetic variability on histone marks.
We performed a quantitative genetic analysis using histone modification levels of identified peaks as quantitative traits. For each histone methylation trait and each of the 1384 SDPs in the RI panel, we applied a negative binomial regression model to determine linkage. In heart we found 5142 significant locus-trait associations at an FDR of 5% (Table 2; Fig 3A; Supplemental Fig. 6). In line with our previous observation in parental rats, H3K27me3 peaks were much broader with lower sequencing coverage and thus, only 117 QTLs were observed for this modification. Furthermore, 252 H3K4me3 peaks showed linkage to two or more quantitative trait loci (QTLs), emphasizing the complex nature of genetic factors that impact histone modifications (Supplemental Table 3). Similar results were obtained for liver tissue; 11.9% of H3K4me3 and 11.4% of H3K27me histoneQTLs that were detected independently in heart and liver tissue were common to both tissues, indicating not only a high reproducibility of the detected QTLs but also a common regulation in both tissue types (Table 2). Of all histone H3K4me3 marks with parental differences, 68% (46%) showed linkage to at least one locus in heart (liver) tissue (Supplemental Fig. 7), suggesting a strong genetic regulation. For the remaining H3K4me3 marks with parental differences, we found no QTL indicating that these histone marks may be affected by multiple loci with small effect sizes. Furthermore, 73% of H3K4me3 histone marks with histoneQTL were not called different between the parents, but showed linkage in the RI panel. This was previously shown in segregating crosses of yeast (Brem et al. 2002) and may be explained as common transgressive effects (Tanksley 1993; Rieseberg et al. 1999) or by limited statistical power in the parental comparisons.
Table 2.
HistoneQTL mapping results in heart and liver tissue
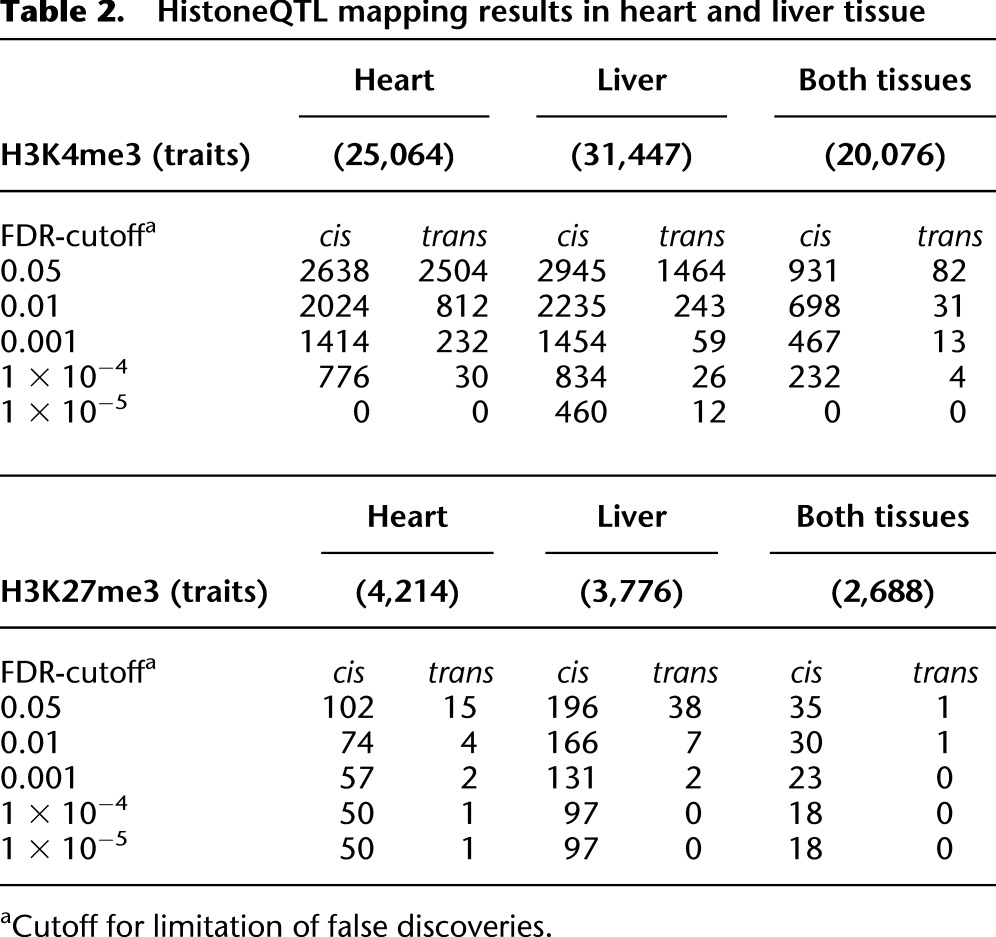
Figure 3.

QTL mapping of histone modifications. (A) Quantile-quantile plots for the QTL analyses of histone modification traits. For each trait and each tissue, we show the observed quantiles of the association statistic plotted against the quantiles of the permutation-based null distribution. The traits are occupancy levels of H3K4me3 regions defined by the peak calling analysis (MACS), as well as H3K4me3 and H3K27me3 regions defined by annotations of known protein coding genes (Ensembl). (B) Boxplot of SNP frequency in H3K4me3 regions with cisQTLs compared with all regions. (C) Example of an altered TF-binding site between BN and SHR rats and its binding motif. (D) Boxplot of differential H3K4me3 modification at the same locus. (E) Genomic distribution of all identified QTLs is shown for H3K4me3 modification in heart tissue. One large QTL hotspot was identified at chromosome 3 regulating 899 histone marks. (F) Overlap of chr3-regulated H3K4me3 histoneQTLs and differentially regulated histone marks between SHR.BN-chr3 congenic and SHR control rats. The x-axis shows the logarithmic fold change of H3K4me3 marks with QTL (BN/SHR). The y-axis shows the logarithmic fold change of the same H3K4me3 marks in SHR.BN-chr3 rats compared with SHR controls. (Blue dots) Trans-regulated QTLs that have been validated using SHR and SHR.BN-chr3 congenic rats. (Blue triangles) Validated cis-regulated QTLs in the chr3 hotspot.
We next determined whether the loci found by linkage acted locally or distally (Rockman and Kruglyak 2006). In keeping with QTL studies of gene expression (Brem et al. 2002; Schadt et al. 2003; Morley et al. 2004; Hubner et al. 2005), we used the terminology cis-acting or cisQTL when the QTL was located within a distance of <10 Mb of the regulated trait, or transQTL when the regulated trait was found elsewhere in the genome. We found that differential H3K4me3 signatures were characterized by a high proportion of trans-acting loci (49% in heart, 33% in liver) (Table 2). In addition, trans-acting loci tended to show a moderate level of significance, whereas cis-acting loci were detected with higher significance. Similar observations have been made for many gene expression QTL studies. For H3K27me3 marks, we observed less QTLs in total and nearly all of them were cis-regulated. Since cis-acting QTLs point to regulatory sequence elements in close proximity to the regulated trait, we investigated whether cis-regulated histone regions contain a higher number of sequence polymorphisms than trans-regulated or nonregulated regions. We found that cis-regulated H3K4me3 regions contain ∼2.5-fold more SNPs than the average H3K4me3 region, which is evidence for functional changes of cis-regulatory elements in these regions (Fig. 3B). Transcription factors (TF) recruit the transcription machinery as well as chromatin-modifying enzymes to DNA sequences surrounding their binding motifs (Li et al. 2007; Fuda et al. 2009). Therefore, we computationally scored how strong sequence variants in cis-regulated H3K4me3 regions affect the affinity of a TF by altering the sequence of its binding site (Manke et al. 2010). For each motif of the TRANSFAC database (Wingender et al. 1996; http://www.gene-regulation.com), we tested whether high scores for differential TF binding occurred more often in cis-regulated regions compared with the remaining regions. In heart tissue we found 14 motifs of TF, whose binding affinity to cis-regulated regions was significantly altered (FDR < 0.1; Supplemental Table 4). Among these factors was T-box 5 (TBX5), which is involved in heart development (Fig. 3C,D). In liver there were eight motifs of differentially occupied TF. One of the TFs was hepatocyte nuclear factor 4, alpha (HNF4A), which is essential for liver development and maintenance of liver-specific gene expression. These results suggest a novel role of the identified transcription factors to directly or indirectly recruit H3K4 methyltransferase or demethylase complexes to the promoters of the differentially modified genes in heart and liver.
Trans-acting histoneQTLs represent loci that influence trimethylation of histones remote from the histone trait itself. We tested whether trans-acting loci with FDR < 0.01 share distinct gene functions using GO enrichment analysis of all expressed genes that are encoded in trans-acting loci and compared them with the remaining expressed genes that are not in trans-acting loci. In heart and liver we found a significant overrepresentation of the GO term “nucleosome assembly” (P = 8.5 × 10−7 heart and P = 3.4 × 10−7 liver). These results have to be interpreted with caution, since this GO term also comprises histone genes, which occur in clusters on the genome. In heart tissue, we observed 899 H3K4me3 traits with a colocalizing trans-acting histoneQTLs (QTL hotspot) at chromosome 3, suggestive of coregulation by a common gene in the region (Fig. 3E). To validate our findings in vivo, we performed ChIP-seq of SHR.BN-chr3 congenic rats, in which the region of the QTL hotspot at chromosome 3 was transferred from the BN donor strain into the genetic background of the SHR recipient strain. We then compared differential data from congenic rats and QTLs from BXH/HXB RI rats and detected a significant overlap of differentially regulated histone marks in the congenic rats with those found to be coregulated by the QTL hotspot at chromosome 3 (Fig. 3F). Using the congenic rats, we confirmed 768 (85%) of the trans-linked H3K4me3 marks, and interestingly, higher histone modification levels were always associated with the BN allele, indicating a common regulation of these marks. Genes corresponding to trans-linked promoter regions (<4 kb from TSS) were enriched for GO terms chromatin organization (P = 3.1 × 10−6) and histone modification (P = 3.0 × 10−5). Target genes showed no genotype-dependent gene expression; however, we observed an overrepresentation of target genes of known heart development TFs (Schlesinger et al. 2011) (P = 0.002), suggesting that the trans-QTL hotspot might be a chromatin footprint of expression differences earlier in development.
Several interesting candidate genes are located in the QTL region; for example, mediator complex subunit 22 (Med22), which is differentially expressed between BN and SHR rats. It is part of the head of the mediator complex that is essential for the formation of the preinitiation complex (Soutourina et al. 2011). Similarly intriguing, WD repeat domain 5 (Wdr5)—a protein-coding gene required for global and gene-specific K4 trimethylation (Wysocka et al. 2005)—resides within this QTL hotspot, but it contains neither a nonsynonymous sequence variation nor a cis-regulated gene expression QTL. However, as depletion of WDR5 can change the H3K4me3 levels over an extended period of time (Greer et al. 2011), altered gene expression at an early stage of development could have an outlasting effect on the adolescent heart. There are several SNPs and indels in the upstream and intronic region of Wdr5 with unknown consequences and it merits further investigation in genetic and functional assays. All candidate genes exhibiting either cis-regulated differential gene expression or with nonsynonymous SNPs and indels in the protein-coding region are listed in Supplemental Table 5.
Impact of epigenetic variation on gene expression
Histone modifications are strongly correlated with gene expression. As a matter of fact, there is not only a qualitative but also a quantitative relationship between expression and histone modifications (Karlić et al. 2010). Hence, we would expect quantitative trait loci that influence histone modifications to also affect gene expression levels. We generated deep RNA-seq data in the heart and liver tissue of sex and age matched animals of all 30 RI lines as well as their parental strains, resulting in >160 million reads per sample on average (Supplemental Table 1). First we performed an analysis similar to Degner et al. (2012) (see Methods). We used all significant gene-histoneQTL pairs and estimated the FDR for eQTL for this set. We found that 18.1% and 14.5% of all H3K4me3 and H3K27me3 QTL were also eQTL. Vice versa, 20% of all eQTL were also QTL for a histone mark. This is in line with findings from Degner et al. (2012), where 16% of DNase I sensitivity QTL (dsQTL) were also eQTL and 23% of eQTL were also dsQTL.
Then we performed an integrated analysis of all three quantitative traits that were profiled across the genome in the 30 RI strains (H3K4me3, H3K27me3, and RNA-seq data). We followed a two-step procedure. In the first step, we performed separate QTL analyses for each of the three traits representing a gene. When any of the three revealed a genome-wide significant association with a locus, we proceeded to a detailed analysis of all three traits and the genotype at the locus in step two. Here we applied a likelihood based model selection technique (Schadt et al. 2003) to identify the best model from a set of competing graphical models (Fig. 4A–D; Supplemental Fig. 8). For each gene-locus pair, we selected the model that best explains the observed data using Akaike’s information criterion and assessed the robustness of our findings by bootstrapping (Supplemental Fig. 9). This approach allowed us to significantly increase the number of gene expression traits that can be attributed to genetic variation (eQTLs) by 37% and 5% in liver and heart tissue, respectively (Fig. 4E,F; Supplemental Table 6). Overall, we analyzed 2194 genes in both tissues, of which 15% had a genome-wide significant eQTL. In contrast, using a model selection approach that included histone trimethylation levels, we were able to link the expression of 27% of the analyzed genes to a genetic marker. This means that 12% of eQTL (265) could not have been detected without using the information from the histone modifications, demonstrating how the integrated analysis helps to identify novel genotype–phenotype relations. For the vast majority (84%) of genes, expression levels were positively correlated with H3K4me3 levels and negatively correlated with H3K27me3 levels, as previously described (Karlić et al. 2010). Loci affecting both histone marks of the same gene were predominantly (76%) acting synergistically, with opposing effects on the two antagonistic marks, whereas only 24% were acting in a buffering way.
Figure 4.

Integrated analysis of histone modifications and gene expression. We determined the most likely model of how genetic variants influence histone modification levels and gene expression using a likelihood-based model selection procedure. The competing models are shown in the bottom right panel of each subfigure. (A full list can be found in Supplemental Fig. 8.) The boxplots in the top row show that levels of both histone marks of Cbln1 (A) are genotype dependent in opposite directions and predict gene expression levels with high correlation (bottom left). Histone modification levels of only one lysine residue, either H3K4me3 or H3K27me3, are genotype dependent for Pparg (B) and Nov (C). We also observe instances such as Dpysl5 (D) where modification levels of both histone marks are genotype dependent but in the same direction, which leads to a buffering and no effect of the genotype on gene expression. A summary of the integrated analysis for heart (E) and liver (F) shows how many genes were analyzed and how often we were able to link gene expression and genetic variation either directly or indirectly via an intermediate histone mark. In some cases we are not able to distinguish direct and indirect models, but nevertheless the set of models that contained a path from the genetic variant to gene expression was selected against all other competing models (bootstrap P > 0.95). The total number of genes linked by the integrated analysis is substantially larger than the number of genome-wide significant eQTLs.
Finally, we were interested in whether the effect of genetic variation on histone modification levels is mediated by transcription or also occurs independently. First we focused on the intergenic H3K4me3 peaks (Supplemental Fig. 10) that might tag regulatory elements of unknown function, of which 3431 (15%) were associated with genetic variations. Since no transcription is occurring in intergenic regions and 75% of genetic effects on H3K4me3 marks are intergenic, we concluded that they occur independently of active transcription. Then we turned to genes with histoneQTL and estimated the number of genes for which RNA expression and histone modification levels are not correlated. We computed correlation coefficients between expression and histone modification levels and applied the Q-value method (Storey 2003) on the resulting P-values to estimate the proportion of true null hypotheses (Supplemental Fig. 11), which was 36% (90% CI: 26%–48%) in liver and 75% (90% CI: 67%–83%) in heart. Assuming that differences in RNA-seq expression levels reflect differences of active transcription, we thus estimate that 36% of genes show a genotype-dependent effect on histone modifications that cannot be attributed to transcription.
Discussion
We identified extensive variation of histone methylation levels among the parental and recombinant inbred strains. Many of the detected differences showed a strong genetic regulation. Overall, we found more than 4000 significant locus-trait associations both in heart and in liver tissue for the H3K4me3 modification. This is ∼10 times more than what a recent study in a small set of human lymphoblastoid cells has found (McVicker et al. 2013). For H3K27me3 peaks, only as few as 100 QTLs were observed for this modification. Similar observations were made by McVicker and colleagues, who identified 469 QTLs for H3K4me3 and only two QTLs for H3K27me3. This is most likely due to the difference in genomic distribution of both histone modifications. While the H3K4me3 modification is located in small genomic regions such as promoters and forms narrow and sharp sequencing peaks, the identified peaks of H3K27me3 are much broader, resulting in lower sequencing coverage. Another indicator of the correlation between sequencing depths and the number of detected histoneQTLs arises from the fact that we identified twice as many H3K27me3 QTLs in liver tissue compared with heart tissue, in which we generated and analyzed only half as many nonredundant sequencing reads. The genomic distribution and the resulting differences in read coverage of the studied histone marks may also partially account for the observed differences in correlation coefficients between biological replicates and the two different strains when comparing different histone marks. For the narrow peak marks H3K4me3 and H3K4me1 we noticed very high correlation factors between biological replicates within the same strain and much lower correlation factors between individuals of different strains. For the broad histone marks H3K27me3 and H4K20me1 we observed overall less strain differences. We cannot attribute this to reagents or technical factors except that the experiments were done subsequently at different time points, which may or may not have had an impact on the results. However, we believe that the biggest difference is attributed to the architecture of the peaks (narrow or broad).
We also determined whether the loci found by linkage acted locally (≤10 Mb) in cis or distally (>10 Mb) in trans. We found that differential H3K4me3 signatures were characterized by a high proportion of trans-acting loci. In addition, trans-acting loci tend to show a moderate level of significance, whereas cis-acting loci were detected with higher significance. Similar observations have been made for many expression QTLs in previous studies (Brem et al. 2002; Schadt et al. 2003; Morley et al. 2004; Hubner et al. 2005). For H3K27me3 marks, we observed less QTLs in total and nearly all of them were cis-regulated. This is again probably caused by the nature of the modification and the higher detection threshold in general.
At rat chromosome 3, we observed a large QTL hotspot that regulates nearly 900 different H3K4me3 traits. Notably, this hotspot was only observed in heart but not in liver tissue, suggesting a tissue-specific regulation of the underlying genetic factor. Several interesting candidate genes are located in the QTL region, for example, mediator complex subunit 22 (Med22) that is differentially expressed between BN and SHR rats. Although Med22 is expressed in both liver and heart tissue, differential expression is only observed in heart tissue, which is concordant with the tissue specificity of the QTL hotspot. MED22 is part of the head of the mediator complex that is essential for the formation of the preinitiation complex (Soutourina et al. 2011). However, its role in the recruitment of histone H3K4 methylase complexes is still unknown. Similarly intriguing, WD repeat domain 5 (Wdr5)—a protein-coding gene required for global and gene-specific K4 trimethylation (Wysocka et al. 2005)—resides within this QTL hotspot, but it contains neither a nonsynonymous sequence variation nor a cis-regulated gene expression QTL. However, as depletion of WDR5 can change the H3K4me3 levels over an extended period of time (Greer et al. 2011), altered gene expression at an early stage of development could have an outlasting effect on the adolescent heart. There are several SNPs and indels in the upstream and intronic region of Wdr5 with unknown consequences and it merits further investigation in genetic and functional assays.
We also analyzed the enrichment of TF-binding motifs in differential histone marks. In heart tissue we found 14 TF motifs whose binding affinity to cis-regulated regions was significantly altered. Although there were many general TFs, such as MYC, MAX, and E2F1, we also identified some interesting tissue-specific TFs. Among these factors was TBX5, which is involved in heart development. In liver there were eight motifs of differentially occupied TFs. One of the TFs was HNF4A that is essential for liver development and maintenance of liver-specific gene expression. These results suggest a novel role of the identified transcription factors to directly or indirectly recruit H3K4 methyltransferase or demethylase complexes to the promoters of the differentially modified genes in heart and liver. However, further experiments are required to establish the possible interaction between these TFs and the histone methyltransferase or demethylase complexes.
The integrated analysis of histone modification and gene expression data led to an increased discovery of gene expression traits whose variation was attributed to genetic factors. It is important to note that the analysis of graphical models was used to infer correlation between genotype, histone, and expression traits and not to determine their causal relations. Although it is clear that the genotypic variation cannot be caused by any of the other traits, we are not attempting to distinguish whether changes of histone modification levels cause changes of gene expression or vice versa.
In conclusion, our study demonstrates that genetic variation has widespread effects on post-translational histone modifications with functional consequences on gene expression levels. We identified many trans-regulated histoneQTLs and showed how genetic variation shapes the landscape of transcriptionally competent chromatin. It is tempting to speculate that this may alter the cellular response to internal or external stressors. Our large-scale publicly available data sets are available for the identification of the underlying genes in novel loci involved in chromatin control and may help to pinpoint functional regulatory polymorphisms influencing susceptibility to disease in this model of human cardiovascular and metabolic disorders. We demonstrated new avenues to find novel genotype—phenotype relationships by integrating histone modification data with genotype and gene expression data.
Methods
Rat strains and tissues
The BXH/HXB recombinant inbred (RI) strain panel has been described in detail elsewhere (Hubner et al. 2005). The full set comprises 30 RI strains and was used >F60. A selective breeding regime was utilized to produce SHR.BN-chr3 congenic rats, harboring a BN-derived fragment (0–60 Mb) at chromosome 3 (McDermott-Roe et al. 2011). Rats were kept in the animal facility of the Institute of Physiology, Czech Academy of Science (Prague, Czech Republic) in a climate-controlled environment with 12-h light/dark cycles. Rats were housed in polystyrene cages containing wood shavings and fed standard rodent chow and water ad libitum. Six-week-old male rats were killed by cervical dislocation, and tissue (left ventricle of the heart and liver) was collected between 8 and 10 am, snap frozen in liquid nitrogen, and stored at −80°C. All animal procedures were in accordance with the Animal Protection Law of the Czech Republic.
Chromatin immunoprecipitation and sequencing
We performed 168 ChIP-seq experiments to determine histone methylation levels in 30 RI strains and three biological replicates of their progenitor strains. ChIP samples were prepared as previously described with the following changes (Barski et al. 2007): Frozen left ventricular heart and liver tissue was crunched using pestle and mortar; 125 mg tissue was homogenized in a Potter-Elvehjem tissue grinder, nuclei were isolated by centrifugation through a dense sucrose cushion and digested with micrococcal nuclease (Sigma-Aldrich) to generate native chromatin templates consisting mainly of mononucleosomes. Chromatin was precipitated with anti-H3K4me3 (NEB, 9751 S), anti-H3K27me3 (Millipore, 07-449), anti-H3K4me1 (Abcam, ab8895), or anti-H4K20me1 (Abcam, ab9051) and antibody-bound DNA fragments were purified by MinElute PCR Purification Kit (Qiagen). Specificity of immunoprecipitation was confirmed with known genes by qPCR. A total of 50 ng of DNA was used for construction of ChIP-seq libraries according to the manufacturer’s protocol (Illumina). After cluster generation, sequencing was performed using the Illumina Genome Analyzer IIx (GAIIx) or the HiSeq 2000 platform. The total number of sequencing tags obtained for each sample is listed in Supplemental Table 1. In addition, ChIP-seq was performed for three biological replicates of SHR.BN-chr3 congenic rats and three SHR control rats for heart tissue with anti-H3K4me3 (NEB, 9751 S).
mRNA preparation and RNA-seq data generation
We performed 80 RNA-seq experiments to determine gene expression and alternative promoter usage of 30 RI strains and five biological replicates of their progenitor strains. Total RNA was extracted from heart and liver tissue and RNA quality was assessed (2100 Bioanalyzer, Agilent) to assure a RIN score >9.5 for all samples. After heat fragmentation, rRNA-depleted poly(A)+ mRNA was used to generate random-primed double-stranded cDNA. Adapters were ligated using the SPRI bead system and the final library was amplified with 15 PCR cycles. We then sequenced one sample per lane on a HiSeq 2000 instrument from Illumina with TruSeq 2× 100 bp PE chemistry. Reads were mapped to the BN reference genome RGSC 3.4 using TopHat v1.2.0 (Trapnell et al. 2009). This approach allows alignment of reads across known and predicted splice junctions. We supplied known splice junctions annotated in the Ensembl reference database (Flicek et al. 2012) to the pipeline and also enabled de novo splice junction detection.
Gene expression levels were estimated using read counts within gene bodies. We counted all alignments that were overlapping at least one exon of the gene and normalized the counts to the length of the union of all exonic regions of the gene. Gene expression values were normalized across samples using a quantile-based scaling method (Schulte et al. 2010). Differential analysis and QTL mapping of gene expression levels were performed using the methods described in the sections “Differential testing of histone marks in progenitor strains” and “Differential testing of histone marks in RI strains and genetic mapping.”
Data analysis of ChIP-seq data
A schematic overview of the various steps of the data analysis process is provided in Supplemental Figure 1.
Alignment of ChIP-seq data
Short reads were mapped to the BN reference genome (RGSC-3.4) using ELANDv2 algorithm (Illumina CASAVA 1.7) that used multiseed and gapped alignment, with a maximum of two mismatches in the default seed length of 32 bases. In cases where we sequenced two technical replicates of the same sample to increase the number of sequenced reads, we pooled all mapped reads after mapping to the BN reference genome. For each sample we removed duplicated reads that are likely PCR amplification artifacts using SAMtools (Li et al. 2009). In order to make sure that sequence variants in the SHR genome do not influence the results of the short read alignment and the quantification, we additionally aligned all reads against a modified reference sequence where we substituted all reference SNP alleles by SHR alleles.
Peak finding
Based on the genomic distribution of each histone modification, we used two different peak calling strategies to identify ChIP-enriched regions. The profiles of H3K4me3 and H3K4me1 show sharp peaks, therefore we used MACS (Zhang et al. 2008) to call peaks with FDR < 0.05 in each sample independently against input control using default parameters.
H3K27me3 or H4K20me1 peaks are very broad and span several kilobases. Therefore, we developed an approach based on a Hidden Markov Model (HMM) to identify peaks that carry either modification. Since the coverage for these modifications is generally low, we quantified the modification signal in 2000-bp bins and used this as the sequence of observations. The model consists of two hidden states—one for unmodified and one for modified regions. We fitted a two-component mixture of lognormal distributions
with
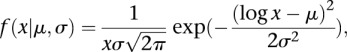 |
for the two states using an expectation maximization algorithm (Dempster et al. 1977). Initial probabilities and transition probabilities of the HMM were estimated using the Baum-Welch algorithm, while the parameters of the emission densities were fixed. The model was implemented as part of the R package (http://histonehmm.molgen.mpg.de). We called regions with a posterior probability of the modified state >0.5 modified. In order to reduce artifacts of the arbitrary binning, we averaged posterior probabilities of four different binnings that are each shifted by an offset of 500 bp. We evaluated and compared the performance of the HMM based peak caller with MACS2 (Zhang et al. 2008) and SICER (Zang et al. 2009) using a data set of qPCR validated H3K27me3 sites (Micsinai et al. 2012). The ROC curves in Supplemental Figure 12 show that our method performs best in terms of area under the curve. We then applied our model to ChIP-seq data from two progenitor strains and used the identified regions in order to select genes for a gene-based differential analysis.
The modified regions of each sample were then compared and a region was considered for subsequent differential analysis if it was detected independently in more than five distinct RI strains and in three parental rats of the same strain. Finally, we overlapped the regions found in both tissues in order to define the set of all possible histone modification sites for the tissue specificity analysis.
Quantification and data normalization
For each sample, we counted aligned reads that fall in a region with an identified peak. In order to be able to compare these values across samples, we have applied quantile normalization (Kasowski et al. 2010). We selected only reproducible regions that had a minimum average coverage of 1× in all three replicates of one of the parental strains. For H3K4me3 and H3K27me3 we also require regions to have more than 1× average coverage in at least 15% of the recombinant inbred strains.
Quality assessment of ChIP-seq data
We assessed the quality of our ChIP-seq data using metrics developed by the ENCODE Consortium (Landt et al. 2012), which is summarized in Supplemental Table 1. For H3K4me3 we compared the MACS peak regions to genome annotations from Ensembl release 56 (Supplemental Fig. 10). In addition we generated read coverage plots (Supplemental Fig. 2) similar to Barski et al. (2007). For H3K4me3 and H3K4me1 we plotted the average read coverage 2000 bp upstream of and downstream from the TSS for Ensembl-annotated expressed and nonexpressed genes. For H3K27me3 and H4K20me1 we plotted the average read coverage 2000 bp upstream of and downstream from the gene combined with the average coverage per base in 10 bins across the length of the gene body for expressed and nonexpressed genes. In order to assess the reproducibility of the quantitative measurements we compared biological replicates of the parental strains using pairwise correlations (Fig. 1).
Differential testing of histone marks in progenitor strains
We compared three biological replicates of each progenitor strain in order to determine differentially modified regions. The normalized read count data was analyzed using a negative binomial (NB) regression model. The dispersion parameter of the NB model was estimated by mean-dependent local regression using DESeq, which was shown to be more appropriate for small numbers of biological replicates than maximum likelihood estimation (Anders and Huber 2010). Subsequently, we adjusted P-values from the analysis for multiple testing using the Benjamini-Hochberg method (Benjamini and Hochberg 1995).
Differential testing of histone marks in RI strains and genetic mapping
QTLs were defined by trait-marker regression also using a NB regression model. The sample size of 30 RI strains is sufficiently large to estimate the dispersion by maximum likelihood as described (Venables and Ripley 2002). The genetic map contained a total of M = 1384 nonredundant SNP genotype profiles. For each pair of trait i and marker j we computed the likelihood ratio statistic (LRS) of the full model containing the genotype variable against a null model containing only an intercept term and stored the results in a matrix Z = (zij). In order to determine the significance of LRS scores while accounting for the presence of correlated genotype variables due to linkage disequilibrium we used a permutation strategy. Since we also aimed to assess the significance of trans-QTL hotspots, we applied the same B = 100 permutations of sample labels to the complete trait matrix, also preserving the correlation between traits (Breitling et al. 2008). Permutation results were stored in the array Z′ = (zijb). Depending on the number of traits N, we obtained between 692 × 106 and 1.6 × 109 statistics under the null hypothesis which we used to assign P-values to the original LRS scores: P(z) = Σijb I(z′ijb > z)/(NMB), where I is an indicator function. The significance of the size of trans-QTL hotspots s at LRS threshold z* was assessed by computing the maximal size of trans-hotspots in each permutation sb = maxj Σib I(z′ijb > z*). The P-value was obtained by P(s) = Σb I(sb > s)/B. Finally, we adjusted the QTL P-values for multiple testing using the Benjamini-Hochberg method (Benjamin and Hochberg 1995). QTL regions for each trait were defined by merging adjacent significant markers into larger regions. QTLs were classified as cis-acting if the QTL was located within a distance of <10 Mb of the trait or as trans-acting if it was further away or on a different chromosome.
Differential transcription factor binding
In order to determine whether the binding sites of particular transcription factors were preferentially altered by SNPs in cis-regulated H3K4me3 regions, we first obtained 50 bp of flanking sequences for all SNPs located in all H3K4me3 regions subject to QTL mapping in each respective tissue. For each PWM from the TRANSFAC database set (Wingender et al. 1996; http://www.gene-regulation.com) of nonredundant PWMs, we scored the difference of binding affinities of the reference and alternative alleles using the sTRAP method (Manke et al. 2010). We then classified the SNPs into two classes: SNPs in cis-regulated regions (FDR < 0.1) and SNPs in other regions. We tested for each TF whether the absolute value of the log ratio of binding P-values was larger in the cis-regulated group compared with the other group using a one-sided Wilcoxon-Mann-Whitney test. Finally, we adjusted these P-values for multiple testing using the Benjamini-Hochberg method.
Overlap of QTL studies
Similar to Degner et al. (2012), we used all significant gene-histoneQTL pairs and obtained nominal eQTL P-values for those trait marker combinations. Then we used the Benjamini-Hochberg method to adjust these P-values and called a locus a shared histone and expression QTL if the adjusted P-value was <0.05.
Integration with gene expression data
For integration of ChIP-seq and RNA-seq data sets, we used a gene-based analysis of Ensembl-annotated genes, as this is the more conservative approach, to minimize false-positive findings in our data. Since H3K4me3 is located in promoter regions, we quantified ChIP-seq read coverage in regions 2-kb upstream of and downstream from annotated TSS (Supplemental Fig. 2; Supplemental Table 7). For H3K27me3 we quantified reads in the entire gene body of Ensembl annotated genes. In order to assess the relation between genetic variants, histone modification levels, and gene expression, we followed a two-step procedure. Each gene is represented by three random variables, which correspond to each of the three traits. First we performed a QTL analysis for each of the three traits separately. If a significant QTL was detected for any of the three traits of a gene we performed a detailed analysis in step two. We used a method similar to Schadt et al. (2005) in order to determine the most likely graphical model. Competing models are shown in Supplemental Figure 8. Each model is specified by a tuple (G = [X, E], P), where G is a directed acyclic graph of dependency relations between the random variables X and P is a set of distributions corresponding to X. Random variables are only dependent on their parents in the graph. Random variables can be discrete or continuous valued and discrete variables can only have parents that are also discrete. Discrete variables are modeled using a multinomial distribution while continuous variables are distributed according to a normal distribution, where the mean is a linear function of the parent nodes: f[xi|pa(i)] = N(β0 + Σj in pa(i) βjxj, σ), where pa(i) denotes the set of parent nodes of node I, and N(μ, σ) denotes the density function of the normal distribution with mean μ and standard deviation σ. Genotypes are encoded as discrete variables with levels 0 and 1. The log-transformed histone and expression values are modeled as continuous variables. The likelihood function of the model can be factorized as L(G, P) = Πi f[xi|pa(i)]. All models were fitted using maximum likelihood estimators for the parameters. We have used the statistical software R in order to implement the graphical model analysis. The source code of the R package is available in the Supplemental Material and on our website http://www.molgen.mpg.de/∼heinig/histoneQTL/. In order to select the best model, we compute the Akaike information criterion [AIC = −2 log L(G,P) + 2 p, where p is the number of parameters of the distributions in P] and select the model with the smallest AIC. The robustness of the results is assessed by drawing 100 bootstrap samples and recording how many times a given model had the minimal AIC. The bootstrap sampling scheme maintains the ratio of the genotypes at the SNP included in the model. We accept a model as robust if the bootstrap probability P > 0.95. The analysis was carried out separately for genes where all three traits were available (i.e., the quantification level was above the threshold for analysis) as well as for genes where only two of the three traits were available. In these cases we used only the subset of models that did not contain the third variable.
Analysis of histoneQTL dependent and independent of transcription
We extended our analysis also to 22,216 intergenic H3K4me3 peaks that might tag regulatory elements of unknown function, of which 3431 (15%) were associated with genetic variations. For comparison, among the 6907 promoter regions there were 1085 (15%) that had a QTL. To rule out that any transcription is occurring in intergenic regions, we also checked for transcripts that were not part of the reference annotation by using cufflinks to create novel gene annotations from the RNA-seq data used in this study and an additional RNA-seq library, created from total RNA that was depleted of ribosomal RNA. In total, 75% of all genetic effects on histone marks are intergenic and therefore independent of transcription.
We estimated the number of genes for which RNA expression and histone modification levels are not correlated using the Q-value method of Storey (2003). For each gene that had a histoneQTL and gene expression levels above the detection limit (in total 2222, considering genes with two histoneQTL twice), we tested whether the Pearson correlation coefficients between the log transformed count data of expression and histone modification levels was equal to zero or not. We applied the Q-value method on the resulting P-values to estimate the proportion π0 of true null hypotheses. For comparison, we also showed the estimation of π0 using alternative methods (Efron 2004; Strimmer 2008) in Supplemental Figure 11. In order to obtain a confidence interval for this estimate, we drew 1000 bootstrap samples from the set of P-values, computed the estimate π0, and finally determined the 5% and 95% quantiles from the observed distribution. We estimated that π0 was 36% (90% CI: 26%–48%) in liver and 75% (90% CI: 68%–83%) in heart. Assuming that differences in RNA-seq expression levels reflect differences of active transcription, we thus estimate that 36% of genes show a genotype-dependent effect on histone modifications that cannot be attributed to transcription.
Quantitative real-time PCR
The findings of ChIP-seq data were confirmed by ChIP followed by regular qPCR on the ABI 7900HT detection system (Applied Biosystems). PCR primers were designed to amplify designated genomic regions using Primer Express software (Applied Biosystems). qPCR assays were carried out in 384-well plates with a final volume of 20 µL each for 40 cycles. We used Power SYBR Green PCR Master Mix (Applied Biosystems) with 1/10 diluted ChIPed DNA or unenriched input DNA as template. Enrichment ratios were calculated according to the 2−▵▵Ct method with endogenous controls (Gapdh, Myod1). Primer and probe sequences can be found in Supplemental Table 8.
Data access
Raw sequencing and processed data are available via EBI’s ArrayExpress (http://www.ebi.ac.uk/arrayexpress) (Parkinson et al. 2007) under accession number E-MTAB-1102 and via the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) (Leinonen et al. 2011) under accession number ERP001430. The processed data set is also available at http://www.molgen.mpg.de/∼heinig/histoneQTL/.
Acknowledgments
This work was supported by funding from the European Union EURATRANS award (HEALTH-F4-2010-241504 to N.H., M.V., E.C., S.C., R.C.J., M.W., M.P.), the Helmholtz Alliance ICEMED, and the Deutsche Forschungsgemeinschaft (Forschergruppe 1054, HU 1522/1-1) to N.H. C.R. was supported by an EMBO long-term fellowship (ALTF 1156-2009). M.P. was supported by grants P301/10/0290 and LL1204 from the Grant Agency of the Czech Republic. C.J., M.C.-T., and F.J. were supported by grants from the Netherlands Organization for Scientific Research (NWO).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.169029.113.
References
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanur SS, Birol I, Guryev V, Hirst M, Hummel O, Morrissey C, Behmoaras J, Fernandez-Suarez XM, Johnson MD, McLaren WM, et al. 2010. The genome sequence of the spontaneously hypertensive rat: analysis and functional significance. Genome Res 20: 791–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, Gingeras TR, et al. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181 [DOI] [PubMed] [Google Scholar]
- Breitling R, Li Y, Tesson BM, Fu J, Wu C, Wiltshire T, Gerrits A, Bystrykh LV, de Haan G, Su AI, et al. 2008. Genetical genomics: spotlight on QTL hotspots. PLoS Genet 4: e1000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755 [DOI] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. 2012. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482: 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster A, Laird N, Rubin D 1977. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B Methodol 39: 1–38 [Google Scholar]
- Efron B 2004. Large-scale simultaneous hypothesis testing: the choice of a null hypothesis. J Am Stat Assoc 99: 96–104 [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. 2012. Ensembl 2012. Nucleic Acids Res 40: D84–D90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda NJ, Ardehali MB, Lis JT 2009. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461: 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, et al. 2010. A map of open chromatin in human pancreatic islets. Nat Genet 42: 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Shi Y 2012. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13: 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, Brunet A 2011. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479: 365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A, et al. 2010. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 467: 460–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, Maciver F, Mueller M, Hummel O, Monti J, Zidek V, et al. 2005. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet 37: 243–253 [DOI] [PubMed] [Google Scholar]
- Kadota M, Yang HH, Hu N, Wang C, Hu Y, Taylor PR, Buetow KH, Lee MP 2007. Allele-specific chromatin immunoprecipitation studies show genetic influence on chromatin state in human genome. PLoS Genet 3: e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlić R, Chung H-R, Lasserre J, Vlahovicek K, Vingron M 2010. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci 107: 2926–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, et al. 2010. Variation in transcription factor binding among humans. Science 328: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, Boyle AP, Zhang QC, Zakharia F, Spacek DV, et al. 2013. Extensive variation in chromatin states across humans. Science 342: 750–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T 2007. Chromatin modifications and their function. Cell 128: 693–705 [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22: 1813–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M 2011. The sequence read archive. Nucleic Acids Res 39: D19–D21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL 2007. The role of chromatin during transcription. Cell 128: 707–719 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke T, Heinig M, Vingron M 2010. Quantifying the effect of sequence variation on regulatory interactions. Hum Mutat 31: 477–483 [DOI] [PubMed] [Google Scholar]
- McDaniell R, Lee B-K, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, et al. 2010. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328: 235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott-Roe C, Ye J, Ahmed R, Sun X-M, Serafín A, Ware J, Bottolo L, Muckett P, Cañas X, Zhang J, et al. 2011. Endonuclease G is a novel determinant of cardiac hypertrophy and mitochondrial function. Nature 478: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK 2013. Identification of genetic variants that affect histone modifications in human cells. Science 342: 747–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsinai M, Parisi F, Strino F, Asp P, Dynlacht BD, Kluger Y 2012. Picking ChIP-seq peak detectors for analyzing chromatin modification experiments. Nucleic Acids Res 40: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG 2004. Genetic analysis of genome-wide variation in human gene expression. Nature 430: 743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A, Holloway E, Kolesnykov N, Lilja P, Lukk M, et al. 2007. ArrayExpress–a public database of microarray experiments and gene expression profiles. Nucleic Acids Res 35: D747–D750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretto E, Sarwar R, Grieve I, Lu H, Kumaran MK, Muckett PJ, Mangion J, Schroen B, Benson M, Punjabi PP, et al. 2008. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nat Genet 40: 546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravenec M, Churchill PC, Churchill MC, Viklicky O, Kazdova L, Aitman TJ, Petretto E, Hubner N, Wallace CA, Zimdahl H, et al. 2008. Identification of renal Cd36 as a determinant of blood pressure and risk for hypertension. Nat Genet 40: 952–954 [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Archer MA, Wayne RK 1999. Transgressive segregation, adaptation and speciation. Heredity 83: 363–372 [DOI] [PubMed] [Google Scholar]
- Ringrose L, Paro R 2004. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38: 413–443 [DOI] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L 2006. Genetics of global gene expression. Nat Rev Genet 7: 862–872 [DOI] [PubMed] [Google Scholar]
- Saar K, Beck A, Bihoreau M-T, Birney E, Brocklebank D, Chen Y, Cuppen E, Demonchy S, Dopazo J, Flicek P, et al. 2008. SNP and haplotype mapping for genetic analysis in the rat. Nat Genet 40: 560–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, et al. 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302 [DOI] [PubMed] [Google Scholar]
- Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, Sieberts SK, Monks S, Reitman M, Zhang C, et al. 2005. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 37: 710–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger J, Schueler M, Grunert M, Fischer JJ, Zhang Q, Krueger T, Lange M, Tönjes M, Dunkel I, Sperling SR 2011. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet 7: e1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte JH, Marschall T, Martin M, Rosenstiel P, Mestdagh P, Schlierf S, Thor T, Vandesompele J, Eggert A, Schreiber S, et al. 2010. Deep sequencing reveals differential expression of microRNAs in favorable versus unfavorable neuroblastoma. Nucleic Acids Res 38: 5919–5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M, Atanur SS, Linsen S, Guryev V, Ruzius F-P, Game L, Lansu N, de Bruijn E, van Heesch S, Jones SJM, et al. 2012. Genetic basis of transcriptome differences between the founder strains of the rat HXB/BXH recombinant inbred panel. Genome Biol 13: r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutourina J, Wydau S, Ambroise Y, Boschiero C, Werner M 2011. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science 331: 1451–1454 [DOI] [PubMed] [Google Scholar]
- Storey JD 2003. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann Stat 31: 2013–2035 [Google Scholar]
- Strimmer K 2008. A unified approach to false discovery rate estimation. BMC Bioinformatics 9: 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanksley SD 1993. Mapping polygenes. Annu Rev Genet 27: 205–233 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD 2002. Modern applied statistics with S. Springer Science + Business Media, New York [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R 1996. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 24: 238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD 2005. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121: 859–872 [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W 2009. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25: 1952–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee BM, Levin RS, Xu B, LeRoy G, Wingreen NS, Garcia BA 2010. In vivo residue-specific histone methylation dynamics. J Biol Chem 285: 3341–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: r137. [DOI] [PMC free article] [PubMed] [Google Scholar]