

Abstract
DNA lesions that block replication fork progression are drivers of cancer-associated genome alterations, but the error-prone DNA repair mechanisms acting on collapsed replication are incompletely understood, and their contribution to genome evolution largely unexplored. Here, through whole-genome sequencing of animal populations that were clonally propagated for more than 50 generations, we identify a distinct class of deletions that spontaneously accumulate in C. elegans strains lacking translesion synthesis (TLS) polymerases. Emerging DNA double-strand breaks are repaired via an error-prone mechanism in which the outermost nucleotide of one end serves to prime DNA synthesis on the other end. This pathway critically depends on the A-family polymerase theta, which protects the genome against gross chromosomal rearrangements. By comparing the genomes of isolates of C. elegans from different geographical regions, we found that in fact most spontaneously evolving structural variations match the signature of polymerase theta-mediated end joining (TMEJ), illustrating that this pathway is an important source of genetic diversification.
Identifying the mechanisms that fuel genome change is crucial for understanding evolution and carcinogenesis. Spontaneous mutagenesis is caused predominantly by misinsertions or slippage events of replicative polymerases that are missed by their proofreading domains and not corrected by mismatch repair (Lynch 2008). Less frequently, but with a potentially much more detrimental effect, mutations can arise when DNA damage obstructs progression of DNA replication; and stalled replication forks eventually collapse, resulting in highly mutagenic double-stranded breaks (DSBs). Although error-free homologous repair, in which the sister chromatid is used as a template, restores the original sequence, infrequent but highly mutagenic error-prone end joining processes can give rise to spontaneous deletions and tumor-promoting translocations (Mitelman et al. 2007).
To circumvent fork collapse at DNA damage, cells use various alternative polymerases that are capable of incorporating nucleotides across DNA lesions and are hence called translesion synthesis (TLS) polymerases. TLS acts on a wide variety of DNA lesions that can result from endogenous as well as exogenous genotoxic sources: DNA lesions that result from UV-light exposure, for instance, are efficiently bypassed by the well-conserved TLS polymerase eta (pol eta), inactivation of which in humans leads to the variant form of the skin cancer predisposition syndrome, Xeroderma Pigmentosum (Masutani et al. 1999b; Johnson et al. 2007). Abundant in vitro studies demonstrate the involvement of TLS polymerases pol eta and pol kappa in the bypass of lesions that are produced by endogenous reactive compounds, showing that these polymerases are also essential for protection of the genome under unchallenged conditions (Haracska et al. 2000; Fischhaber et al. 2002; Kusumoto et al. 2002).
Although error-prone while replicating, and thus potentially causing misinsertions, TLS polymerases are thought to protect cells against the more mutagenic effects of replication fork collapse (Knobel and Marti 2011). Here, we investigate the contribution of TLS polymerases on the maintenance of genome stability and the mechanisms acting on stalled DNA replication by characterizing C. elegans strains that are defective for the Y-family polymerases pol eta and pol kappa. Unexpectedly, we found that DSBs resulting from replication-blocking endogenous lesions are not repaired via canonical DSB repair pathways, but through an error-prone repair mechanism that critically depends on the A-family DNA polymerase theta (pol theta).
Results
TLS polymerases protect genomes against spontaneous deletions
In previous work, we established the role of the C. elegans homologs of TLS polymerases pol eta (POLH-1) and pol kappa (POLK-1) in the protection against a wide range of exogenous DNA damaging agents (Roerink et al. 2012). In these studies, we also sporadically observed readily recognizable mutant phenotypes during normal culturing of polh-1polk-1 double mutant animals, which prompted us to suspect a prominent role for these Y-family TLS polymerases in the prevention of spontaneous mutations (Supplemental Fig. S1). To address the nature of this increased mutagenesis in an unbiased way, we cultured populations of animals with specific defects in TLS for 60 generations, thus allowing spontaneous mutations to accumulate, and then sequenced their genomes (Fig. 1A; Supplemental Table S1; Supplemental Data File). Mutation accumulation (MA) lines of a wild-type strain (Bristol N2) and of the mismatch repair deficient strain, msh-6—for which an ∼100-fold higher mutation frequency has been reported (Tijsterman et al. 2002)—were sequenced as references. All genomes have been sequenced with a minimal 12-fold base coverage (Supplemental Table S1).
Figure 1.

Spontaneous mutagenesis in TLS-deficient strains. (A) Generation of mutation accumulation (MA) lines. For each genotype, multiple populations were started by cloning out single worms from a single hermaphrodite P0. Cultures were propagated by transferring animals to new plates each generation. At generation Fn, a single animal was grown to a full population, from which genomic DNA was isolated and subjected to whole-genome sequencing on an Illumina HiSeq. (B) Substitution and microsatellite mutation rates for the indicated genotypes. Mutation rates are expressed as the number of mutations per generation divided by the fraction of the genome covered >3 times in all samples. (C) Rates of structural variations for the indicated genotypes. (D) Size distribution of deletions in the different mutant backgrounds. The median sizes are indicated by the gray horizontal lines.
Although pol eta and pol kappa have reduced accuracy while replicating from undamaged as well as damaged DNA templates (Matsuda et al. 2000; Ohashi et al. 2000), we found that these proteins hardly contribute to base substitution processes or microsatellite instability under normal growth conditions. No significant elevation in the substitution or microsatellite mutation rates were found in polh-1polk-1 worms compared to wild-type controls (Fig. 1B), which shows that another class of genetic changes must be responsible for the observed mutator phenotype. To detect other structural variations, we used Pindel software, developed to identify deletions and/or insertions in whole-genome sequencing data (Ye et al. 2009). Strikingly, a unique class of deletions emerged in polh-1 and polh-1polk-1 mutants which were not associated with repetitive loci, or with sequences able to adopt a stable secondary structure (e.g., G4 DNA) or with any other obvious genomic trait. Instead, these deletions occurred at seemingly random locations throughout the genome (Fig. 1C; Supplemental Fig. S2). The vast majority of deletions ranged between 50 and 200 bp in size, with just a few exceptions being larger or smaller (Fig. 1D). The median size of 107 bp was similar for deletions derived from either polh-1 or polh-1polk-1-mutant animals (Fig. 1D). Control wild-type and msh-6 samples did not display any mutations from this class. Deletions occurred in polh-1 single mutants with a rate of ∼0.4 per animal generation, which translates to an average of ∼0.03 deletion per genome per cell division. polk-1 single mutants hardly suffered from deletions; however, polh-1polk-1 double mutants had fivefold increased rates of deletion induction compared to polh-1 single mutant animals, implying that C. elegans pol eta and pol kappa function redundantly on a subset of endogenous lesions.
DSB induction in TLS deficient mutants
To further investigate the origin of the high number of deletions in polh-1polk-1 deficient strains, we looked for manifestations of genomic instability in germ cells of these animals. We observed a mild but statistically significant increase in the number of foci of the DSB marker RAD-51 in proliferating germ cells of polh-1polk-1 mutant animals (Supplemental Fig. S3A,B). Elevated levels of DSBs, are also suggested by the spontaneous emergence of dominant him mutants in polh-1polk-1 mutant populations (Supplemental Fig. S1). This phenotype, which is defined by dominant inheritance of an increased number of males (XO) in predominantly hermaphroditic (XX) populations, has previously been found upon exposure to gamma irradiation and in mutants with enhanced telomere shortening, where it proved to result from X/autosome translocations (Herman et al. 1982; Meier et al. 2009). Despite these manifestations of enhanced replication stress in polh-1polk-1 mutants, the levels of DSBs were insufficient to activate the two DNA damage checkpoints that operate in the C. elegans germline: cell cycle arrest and apoptosis (Gartner et al. 2000). We found neither a reduction in germ cell proliferation nor an increase of apoptotic bodies in polh-1polk-1 mutant germlines, suggesting that TLS-compromised germ cells proliferate in the presence of elevated levels of DSBs, with genomic deletions as a consequence (Supplemental Fig. S3C–E).
Footprints of error-prone DSB repair
To obtain mechanistic insight on the biology of deletion formation, we performed a detailed analysis on the sequence context of 141 polh-1polk-1-derived deletions (Supplemental Data File). Although the majority had simple deletion junctions (without inserts), ∼25% of the footprints showed insertions of short sequence stretches (Fig. 2A). Cases with inserts sufficiently long to faithfully trace their origin revealed that the inserted stretch, or part of it, is identical to sequences flanking the deletion (Fig. 2B,C). This finding strongly suggests that DNA close to the break site was used as a template for de novo synthesis before both DNA ends were joined.
Figure 2.
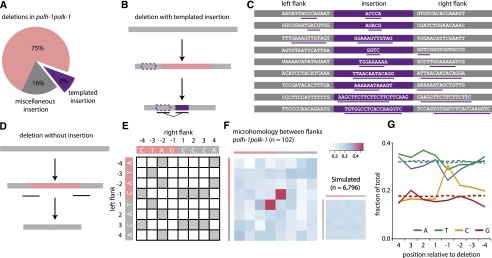
Deletion footprints in TLS mutants indicate a priming-based end joining mechanism. (A) Distribution of deletion footprints in polh-1polk-1 mutants. (B) Schematic illustration of a deletion associated with a templated insertion. Deleted sequence in pink; newly inserted sequence in purple and its template boxed; nonaltered DNA in gray. (C) Sequence context of deletions with templated insertions derived from polh-1polk-1 animals. Matching sequences are underlined. (D) Schematic illustration of a deletion not accompanied by insertions. Deleted sequence in pink; nonaltered DNA in gray. The eight-nucleotide window, capturing neighboring flanking and deleted sequences and used for the generation of the heat maps, is underlined. (E) The strategy to score junction homology: For each simple deletion, matching bases between the 5′ and 3′ junction were scored 1, nonmatching bases were scored 0, thus creating one map per deletion. (F) A heat map representing the sum of all individual deletion maps derived from polh-1polk-1 animals (n = 102). A heat map for a simulated set of deletions (n = 6796) with random distribution is displayed on the right. (G) Base composition at the 5′ and 3′ junctions. The flanking sequences have positive numbers, the deleted sequences have negative; −1 being the first nucleotide within the deletion. Dotted lines indicate the relative abundance of a particular base for a simulated set of deletions (n = 6796).
A DSB repair mechanism involving DNA synthesis is also suggested by the notion of a “priming” nucleotide in >80% of all deletions: 83 of the 102 deletions without inserts contain at the junction at least one nucleotide that could have originated from either flank; in 51 cases, this is restricted to a single nucleotide. To systematically assess the significance of this observation, we constructed deletion junction heat maps, which reflect the level of (micro)homology between 5′ and 3′ junctions (Fig. 2D–F). We scored the degree of sequence identity in an 8-nt window, encompassing the four outermost nucleotides of the flanking sequence and the four nucleotides of the adjacent, but deleted, sequence. Indeed, compared to a randomly distributed simulated set, we found a very high similarity score for the nucleotide at the −1 position of the deletions and the +1 position of the opposing flanks (P = 7.3 × 10−17). Importantly, this profound degree of microhomology is restricted to only a single one—the terminal nucleotide.
Replication-blocking endogenous damage resides at guanines
We next investigated whether the deletion specifics would reveal the nature of the spontaneous damage underlying fork stalling and break formation in TLS-compromised animals using the following rationale. Deletions in TLS-deficient animals are likely brought about by an inability to incorporate a base across endogenous lesions. If the nascent strand, blocked at the site of base damage, defines one end of the deletion junction, then the −1 position of the corresponding junction will represent the nucleotide complementary to the damaged base: It is the first base not to be incorporated. To test this hypothesis, we plotted the base distribution for each position of the junction and indeed found it not to be random at the −1 position, but rather found it to be dominated by cytosines (Fig. 2G). This result strongly suggests that spontaneous base damage that requires pol eta and pol kappa to avoid DSB induction resides at guanines, which may point toward N2-dG and/or 8-oxo-dG adducted sites as a primary source of spontaneous mutagenesis.
Deletion formation is dependent on pol theta
The frequent occurrence of templated insertions at the deletion junctions suggests the involvement of a DNA polymerase to repair DSBs that are induced at replication-blocking dG bases. One candidate is the A-family DNA polymerase theta, which was previously implicated in the repair of interstrand crosslinks in various models and in the repair of transposition-induced DSBs in Drosophila (Shima et al. 2004; Muzzini et al. 2008; Yousefzadeh and Wood 2012). We recently identified a role for pol theta in preventing genomic instability at endogenous sequences that are able to fold into potentially replication-blocking G-quadruplex structures (Koole et al. 2014). To test a possible role for this protein in deletion formation at spontaneous damage, we generated animals defective for polh-1polk-1 and the C. elegans pol theta homolog polq-1. Strikingly, these animals are severely compromised in normal growth: Although polq-1 and polh-1polk-1 animals had nearly wild-type growth characteristics, polh-1polk-1polq-1 triple mutant animals had a high rate of reduced fertility, albeit in a stochastic fashion, ranging from complete sterility to brood sizes of 30% of wild-type levels (Fig. 3A). Associated with these fertility defects, we observed a profound increase in the number of RAD-51 foci in the proliferative zone of the germline as well as activation of the DNA damage checkpoint, suggesting increased DNA end-resection and DSB signaling (Fig. 3B,C; Supplemental Fig. S3E). From this, we conclude that when damage cannot be bypassed, pol theta action safeguards animal fertility by preventing undesired HR-related processing of replication-associated breaks, which trigger checkpoint activation and prohibit proliferation.
Figure 3.

Pol theta mediates end joining of breaks in pol eta- and pol kappa-deficient animals. (A) Fecundity of single, double, and triple knockout mutants of pol theta and TLS polymerases pol eta and pol kappa. (B) Quantification and (C) representative pictures of RAD-51 immunostainings on germlines of the indicated genotype. Scale bar, 10 µm. (D) Schematic representation of the unc-22 reporter gene and spontaneous deletions (in red) isolated from either polh-1polk-1 or polh-1polk-1polq-1 mutant animals. Three of five deletions extended beyond the borders of the unc-22 locus.
Because the notion of endogenous damage blocking the replication fork can only be inferred indirectly from our data, we tested whether a similar detrimental effect of knocking out pol theta is also observed on bona-fide fork-stalling lesions, such as UV-induced photoproducts. Indeed, mutating pol theta strongly sensitizes polh-1 mutant animals—but not wild-type animals—to UV exposure (Supplemental Fig. S3F). This further strengthens the conclusion that pol theta action minimizes the toxic effects of persistent replication-blocking DNA lesions that result from either an endogenous or exogenous source.
To study the role of pol theta in deletion formation on a molecular level, we assessed mutagenesis using an endogenous unc-22 reporter gene (Fig. 3D). We isolated spontaneous unc-22 mutants from polh-1polk-1 and polh-1polk-1polq-1 populations and determined their molecular nature using PCR and Sanger sequencing. In perfect agreement with our whole-genome sequencing data, all unc-22 mutations derived from polh-1polk-1 animals were 50- to 200-bp deletions characterized by single nucleotide homology and templated insertions (Fig. 3D; Supplemental Table S2). In sharp contrast, unc-22 mutants derived from polh-1polk-1polq-1 triple mutants, while being induced at comparable rates, were of a completely different size category. Here, deletions were typically larger than 5 kb, with some spanning >30 kb of genomic sequence, thus amplifying the deleterious impact of replication-stalling lesions >100-fold (Fig. 3D; Supplemental Tables S2, S3). We conclude that a pol theta-mediated end joining mechanism is responsible for the generation of small-sized deletions induced by replication fork stalling endogenous lesions. In its absence, large stretches of DNA surrounding DSBs are resected, resulting in abundant RAD-51 filament formation, mitotic checkpoint activation, and excessive loss of DNA.
Pol theta in wild-type C. elegans strains
The notion that we have uncovered a role for pol theta in genome protection under TLS-deficient conditions raises the following question: Does pol theta-mediated repair also act when TLS is functional? Or, in other words, how relevant is this error-prone repair pathway for animal fitness? We hypothesized that the action of an error-prone repair mechanism with such a clear and distinct signature, i.e., a distinct size class, single nucleotide homology, and templated insertions, may leave its fingerprint in evolving genomes. For this reason, we compared the genomes of different natural isolates of C. elegans to identify structural variations and defined their characteristics (Fig. 4). The majority of deletions are of small size—60% being smaller than 10 bp—while the number of deletions decreases with increasing size in an exponential manner. However, we found deletions in the size range 50–200 bp much more abundantly present than expected from this exponentially declining trend (Fig. 4B). Moreover, deletions in this size range bear the pol theta signature: templated insertions and a strong overrepresentation (>80%) of having at least one nucleotide homology (Fig. 4C), which supports a role for pol theta in genome change during nonchallenged growth. Unexpectedly, we observed templated insertions (2%) also in the small size range of deletions, and found also this class to be dominated by ≥1 nucleotide homology at the junction (Fig. 4C,D), hinting to a much broader involvement of pol theta in genomic change, not being restricted to the creation of 50- to 200-bp deletions.
Figure 4.
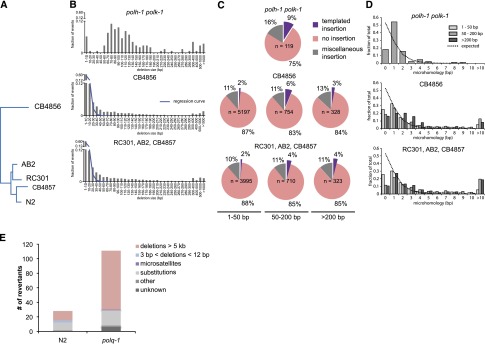
Signature of pol theta-mediated end joining in natural isolates of C. elegans. (A) Phylogenetic tree diagram of the different isolates of C. elegans used in this study. (B) Size distribution of deletions of evolutionary distinct C. elegans species compared to size distribution of polh-1polk-1-derived deletions. An exponential regression curve describes the size distribution of deletions in both natural isolates up to 20 bp; deletions up to 200 bp are overrepresented. (C) Deletions in natural isolates, especially in size class 50–200 bp, show templated insertions analogously to deletion footprints in polh-1polk-1 animals. (D) Microhomology for deletions in natural isolates as compared to deletions in polh-1polk-1 animals. (E) unc-93 mutagenesis in polq-1 worms and wild-type controls.
To further investigate the potential contribution of pol theta in spontaneous mutation induction under nonchallenged growth conditions, we used a forward mutagenesis assay that is based on the uncoordinated movement of animals carrying a dominant mutation (e1500) in the UNC-93 protein that affects muscle contraction (De Stasio et al. 1997; Greenwald and Horvitz 2003). This phenotype is suppressed by complete loss of function of unc-93, or by loss of one of several extragenic suppressor genes (e.g., sup-9, sup-10). We propagated populations of wild-type and polq-1 mutant animals from which we isolated and molecularly characterized revertant animals that had normal movement. Strikingly, the total number of revertants was increased fourfold in polq-1 mutants (Fig. 4E; Supplemental Tables S4, S5), demonstrating that pol theta action also prevents mutation induction in wild-type animals during normal growth. The increased mutagenesis in polq-1 is mainly attributed to a selective increase in large chromosomal deletions, similar to those previously identified in unc-22 in polh-1polk-1polq-1 deficient strains (Supplemental Fig. S5). Interestingly, we observed that one mutation class, i.e., small deletions ≥3 bp in size, was completely absent in animals polq-1 (3/28 in wild types versus 0/111 in polq-1 mutants), arguing, together with the identification of the pol theta signature carrying small-sized deletions in the genomes of natural isolates, that pol theta protect cells against arrest and the genome against large chromosomal DNA loss, but at the price of small deletions.
Discussion
TLS polymerases eta and kappa operate on endogenous lesions in an error-free manner
Our data present the first evaluation of the contribution of two main members from the Y family polymerases eta and kappa to the stability of an animal’s entire genome under unchallenged conditions. We show that these TLS polymerases prevent the induction of spontaneous deletions. Although in vitro studies demonstrated the reduced accuracy of pol eta and pol kappa while replicating from undamaged and damaged DNA templates (Johnson et al. 1999; Masutani et al. 1999a; Haracska et al. 2000; Matsuda et al. 2000; Ohashi et al. 2000; Fischhaber et al. 2002; Kusumoto et al. 2002), our in vivo data show that the biologically desired bypass action of pol eta and pol kappa is largely error-free: Their joined action prevents ∼2 deletions per animal generation without significantly affecting the overall substitution rate (Fig. 1B).
Deletions were found in animals deficient for pol eta but not in pol kappa mutant strains. Pol kappa nevertheless can act on spontaneous damage because a greatly increased number of deletions result from the combined absence of both pol eta and pol kappa. This outcome suggests that the two Y-family members function redundantly on a subset of endogenous lesions, a conclusion that is further supported by a similar genetic interaction for sensitivity toward the guanine alkylator MMS. Also for this exogenous source of DNA damage, animals deficient for both pol eta and pol kappa are profoundly more sensitive than animals deficient for only pol eta, whereas pol kappa disruption by itself only very mildly increases the sensitivity of wild-type worms. (Roerink et al. 2012). Under nonchallenged conditions, we found deletion junctions to preferentially result from replication fork stalling at dG residues (Fig. 2G), which may point toward N2-dG and/or 8-oxo-dG adducted sites as a primary source of spontaneous mutagenesis, because bypass activities of pol eta and pol kappa have been reported for these lesions (Haracska et al. 2000; Avkin et al. 2004).
An error-prone pol theta-mediated mechanism for repair of replication-associated DSBs
The footprints of the deletions that are suppressed by TLS polymerases fit best with a model in which one end of a DSB, induced at replication-blocking lesions, is extended using the other end as a template with just a single base-paired nucleotide as a primer (explaining both single nucleotide homology and templated insertions). In this model, templated inserts can be explained as the result of iterative rounds of annealing and extension (Fig. 5). The close proximity of insertions to their template also suggests that the extendable end of the DSB is not subjected to extensive trimming and suggests that DNA close to the break site was used as a template for de novo synthesis before both DNA ends were joined. A “priming” nucleotide in >80% of all deletions further strengthened our hypothesized model of a DSB repair mechanism involving DNA synthesis. Further support is provided by the identification of a polymerase, the A-family polymerase pol theta, which we found to be essential for the formation of small-sized deletions. The molecular function of this protein in previously identified phenotypes, such as sensitivity toward crosslinking agents and radiation, as well as spontaneous genome instability in mice, was largely unknown (Shima et al. 2004; Muzzini et al. 2008; Yousefzadeh and Wood 2012). We now show that a pol theta-dependent repair route provides cells with the ability to repair replication-associated breaks; we propose to refer to this pathway as TMEJ, for pol theta-mediated end joining, to set it apart from canonical NHEJ. We hypothesize that TMEJ may be specifically important in cases in which the sister chromatid cannot be used as a DSB repair template for, e.g., homologous recombination, because that template still contains the original replication-blocking lesion (Fig. 5). In favor of a role for TMEJ in preventing futile HR, we observed abundant RAD-51 filament formation, mitotic checkpoint activation, and excessive loss of DNA in the absence of pol theta. When damage cannot be bypassed, pol theta action safeguards animal fertility by preventing undesired HR-related processing of replication-associated breaks, which would trigger checkpoint activation and prohibit proliferation.
Figure 5.

A tentative model for TMEJ of breaks induced at replication fork barriers. DNA lesions from endogenous sources—with increased frequency in the absence of functional TLS—causes replication fork blocks, leading to double-stranded breaks. The broken ends are repaired by pol theta-mediated end joining (TMEJ), which is stimulated by minimal priming of 1 base pair, explaining deletions with single nucleotide homology (left). Iterative cycles of priming, extending, and dissociation will result in deletions with templated insertions (right). In pol theta-deficient cells, DNA breaks resulting from replication fork stalling are differently processed, eventually leading to larger size deletions.
Our model for TMEJ is conceptually different from the models that have previously been proposed to explain copy number variations and complex rearrangements in tumors and congenital disorders: microhomology-mediated break-induced replication (MMBIR), and Fork stalling and Template switching (FoSTeS) (Zhang et al. 2001; Lee et al. 2007; Hastings et al. 2009). The genome rearrangements explained by these models are also characterized by the presence of limited sequence homology at the rearranged DNA junctions. However, both these models invoke the invasion of a 3′ single-strand end, either resulting from DNA breaks (MMBIR) or stalled forks (FoSTeS) into the sister molecule or into another replication fork that is in 3D physical proximity, to ensure ongoing DNA replication. Our data on deletion junctions that result from blocked replication either at endogenous lesions (this manuscript), or secondary structures such as G4 DNA (Koole et al. 2014), favor an end joining mechanism based on the presence of two-ended double-strand breaks, which may be the result of replication of gapped DNA intermediates that form at persistent replication fork blocks (Fig. 5), as opposed to restarting replication of a one-ended break. The observation that Mus308, the Drosophila ortholog of pol theta, can act on dsDNA breaks resulting from P-element excision, is also in concert with an end joining mechanism.
Another difference between TMEJ and MMBIR/FoSTeS relates to size; whereas TMEJ deletions are typically 50–200 bp, the rearrangements that are explained by MMBIR/FoSTeS models span kilobases. Nevertheless, all models evoke the presence of flexible primer-template intermediates that can be extended in recurrent cycles and imply DNA polymerase action. Important in that respect is the recent observation that MMBIR-type rearrangement in mammalian cells can be induced by replication stress and depends at least in part on the Pol delta subunit PolD4 (Costantino et al. 2014).
Interestingly, although the vast majority of genomic rearrangements that we observed in TLS-compromised animals are 50–200 deletions, we nevertheless found a very small number of more complex rearrangements (Supplemental Data File). These events may, because of their complexity and size, more closely resemble the complex rearrangement found in mammalian cells; however, their number was too limited to allow systematic analyses, and none were found in any of our other less sensitive phenotype-based assays, thus precluding genetic analysis at this stage.
TMEJ footprints in evolving genomes
The observation that pol theta also suppresses mutagenesis in wild-type animals, together with the notion that the signature of TMEJ is apparent in the genomes of natural isolates of C. elegans, argues for a prominent role of this error-prone pathway to protect genomes against large chromosomal rearrangements. This role seems not to be restricted to replication fork stalls. Although the class of 50- to 200-bp deletions that is seen in TLS-deficient animals is overrepresented in genomes of natural isolates, the predominant fraction of deletions are smaller in size. Still, these smaller-sized deletions bear a TMEJ signature in that they are characterized by single-nucleotide homology and frequently are associated with templated insertions. A broader role for TMEJ, thus being responsible for many types of structural variations, is also supported by the unc-93 forward mutagenesis assay, in which small deletions (3–12 bp) were exclusively found in pol theta-proficient strains.
Although mutagenic processes are drivers of evolution, they also fuel malignant transformation of cells. It is a current challenge to recognize specific classes of mutations in cancer genomes and attribute these either to underlying sources of DNA damage or to error-prone repair mechanisms. Identifying mutational signatures typifying specific repair processes is pivotal to this ambition. Templated insertions and the use of minimal homology—two characteristics of TMEJ—have frequently been observed in higher-order eukaryotes and in cancer tissues (Chen et al. 2010; Nik-Zainal et al. 2012; Carvalho et al. 2013), and have been ascribed to either classical nonhomologous end joining or the molecularly ill-defined process of microhomology-mediated end joining (Honma et al. 2007; Kloosterman et al. 2012). Here, we describe a mechanistic alternative for repair of DSBs induced at stalled forks, which leaves a distinct and well-defined footprint in evolving genomes.
Methods
C. elegans genetics
All strains were cultured according to standard methods (Brenner 1974). Wild-type N2 (Bristol) worms were used in all control experiments. Alleles used in this study are: polh-1 (lf31); polh-1 (ok3317); polk-1 (lf29); polq-1(tm2026); msh-6(pk2504); bcIs39[P(lim-7)ced-1∷GFP + lin-15(+)]); unc-93(e1500). All mutant strains were backcrossed six times before performing experiments.
Whole-genome sequencing of MA lines
Mutation accumulation (MA) lines were generated by cloning out F1 animals from one hermaphrodite. Each generation, about five worms, were transferred to new plates. MA lines were maintained for 60 generations or until severe growth defects developed. Single animals were then cloned out and propagated to obtain full plates for DNA isolation. Worms were washed off with M9 and incubated for 1 h at room temperature while shaking in order to remove bacteria from the animal’s intestine. After two washes, worm pellets were lysed for 2 h at 65°C with SDS containing lysis buffer. Genomic DNA was purified by using a DNeasy kit (Qiagen). Paired-end (PE) libraries for whole-genome sequencing (HiSeq 2000 Illumina) were constructed from genomic DNA according to manufacturers’ protocols with some adaptations. Shortly, 5 μg DNA was sheared using a Covaris S220 ultrasonicator, followed by DNA end-repair, formation of 3′A overhangs using Klenow and ligation to Illumina PE adapters. Adapter-ligated products were purified on QIAquick spin columns (Qiagen) and PCR-amplified using Phusion DNA polymerase and barcoded Illumina PE primers for 10 cycles. PCR products of the 300–400 bp size range were selected on a 2% ultrapure agarose gel and purified on QIAquick spin columns. DNA quality was assessed and quantified using an Agilent DNA 1000 assay. Four to five barcoded libraries were pooled in one lane for sequencing on a HiSeq.
Bioinformatic analysis
Image analysis, basecalling, and error calibration were performed using standard Illumina software. For the analysis of the natural isolates, paired-end whole-genome sequence data were downloaded from the NCBI Sequence Read Archive (SRP011413) (Grishkevich et al. 2012), and sequence reads were mapped to the C. elegans reference genome (WormBase release 225) by BWA (Li and Durbin 2009). SAMtools was used for SNP and indel calling, with BAQ calculation turned off (Li et al. 2009). All nonunique SNPs and indels are considered to be preexisting and were filtered out using custom Perl scripts. To identify microsatellite mutations and deletions, we used Pindel, developed by Ye et al. (2009). A more detailed description of the bioinformatic procedures can be found in the Supplemental Material.
Microscopy
To study RAD-51 foci formation, germlines were dissected, freeze cracked, and subsequently washed with 1% Triton and methanol (−20°C). RAD-51 was visualized by using an anti-RAD-51 rabbit monoclonal antibody and an Alexa488-labeled goat-anti-rabbit secondary antibody (Molecular Probes, Inc.), combined with 10 μg/mL DAPI. Dissected worms and eggs were mounted using Vectashield. Apoptosis was monitored using a lim-7 driven CED-1∷GFP fusion, which visualizes sheath cells surrounding apoptotic germ cells. All microscopy was performed with a Leica DM6000 microscope.
UV sensitivity assay
To assess the sensitivity of germ cells to UV-exposure, young adults were exposed to various doses of UV light and subsequently allowed to lay eggs for 48 h; 24 h later, the number of nonhatched eggs and surviving progeny were determined.
Unc-22 mutagenesis assay
To identify spontaneous mutations in the unc-22 muscle gene, we started 50 populations by transferring a single animal to 9-cm plates seeded with OP50. In the case of the synthetically sick polh-1polk-1polq-1 mutant, we started 200 populations with five worms per plate. Animals were washed off with 2 mM levamisole and transferred to six-well plates to facilitate scoring of unc-22 mutants, which are insensitive to the hypercontracting effects of the drug levamisole. Independent unc-22 mutant animals were isolated. Genomic DNA was isolated from homozygous animals for subsequent PCR and sequence analysis.
Unc-93 (e1500) mutagenesis assay
To generate a complete spectrum of spontaneous mutations, we used a mutagenesis assay based on reversion of the so-called “rubber band” phenotype, caused by a dominant mutation in the muscle gene unc-93 (De Stasio et al. 1997; Greenwald and Horvitz 2003). Reversion of the unc-93(e1500) phenotype is caused by homozygous loss of unc-93 or one of the suppressor genes sup-9, sup-10, sup-11, and sup-18. Polq-1(tm2026) unc-93(e1500) and unc-93(e1500) animals were singled to 2 × 400 6-cm plates. These plates were grown until starvation, and equal fractions (chunks of 2 × 2 cm) were then transferred to 9-cm plates. Before these plates were fully grown, they were inspected for wild-type moving animals. From each starting culture, only one revertant animal was isolated to ensure independent events.
Large chromosomal deletions in unc-93, sup-9, and sup-10 were identified by PCR amplification of exonic regions and two regions 5-kb upstream of and downstream from the respective genes. Smaller genetic changes and substitutions were first classified into events in either the unc-93 gene or in one of the suppressor genes by their ability to complement a known unc-93 deletion allele. All unc-93 exons were sequenced in revertant animals that failed to complement unc-93, whereas all exons of sup-9 and sup-10 were sequenced in revertants that complemented unc-93. Sup-11 or sup-18 could not be subjected to molecular analysis due to lack of sequence data. Revertants that complemented unc-93 but that did not have a detectable mutation of sup-9 or sup-10 were classified as “unknown.”
Data access
The sequencing data have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP020555.
Acknowledgments
We thank the C. elegans Knockout Consortium, Shohei Mitani, and the C. elegans Genetics Center for providing strains. We thank Wouter Koole and Jane van Heteren for critical reading of the manuscript and Bennie Lemmens and Harry Vrieling for discussions. M.T. is supported by grants from the European Research Council (203379, DSBrepair) and ZonMW/NGI-Horizon, Zenith.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.170431.113.
References
- Avkin S, Goldsmith M, Velasco-Miguel S, Geacintov N, Friedberg EC, Livneh Z 2004. Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase κ. J Biol Chem 279: 53298–53305 [DOI] [PubMed] [Google Scholar]
- Brenner S 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR 2013. Replicative mechanisms for CNV formation are error prone. Nat Genet 45: 1319–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Férec C, Kehrer-Sawatzki H, Patrinos GP 2010. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol 20: 222–233 [DOI] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD 2014. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stasio E, Lephoto C, Azuma L, Holst C, Stanislaus D, Uttam J 1997. Characterization of revertants of unc-93(e1500) in Caenorhabditis elegans induced by N-ethyl-N-nitrosourea. Genetics 147: 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischhaber PL, Gerlach VL, Feaver WJ, Hatahet Z, Wallace SS, Friedberg EC 2002. Human DNA polymerase κ bypasses and extends beyond thymine glycols during translesion synthesis in vitro, preferentially incorporating correct nucleotides. J Biol Chem 277: 37604–37611 [DOI] [PubMed] [Google Scholar]
- Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO 2000. A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 5: 435–443 [DOI] [PubMed] [Google Scholar]
- Greenwald IS, Horvitz HR 2003. unc-93(e1500): a behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 96: 147–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishkevich V, Ben-Elazar S, Hashimshony T, Schott DH, Hunter CP, Yanai I 2012. A genomic bias for genotype-environment interactions in C. elegans. Mol Syst Biol 8: 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L, Yu SL, Johnson RE, Prakash L, Prakash S 2000. Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase η. Nat Genet 25: 458–461 [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman RK, Kari CK, Hartman PS 1982. Dominant X-chromosome nondisjunction mutants of Caenorhabditis elegans. Genetics 102: 379–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma M, Sakuraba M, Koizumi T, Takashima Y, Sakamoto H, Hayashi M 2007. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair (Amst) 6: 781–788 [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash S, Prakash L 1999. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Polη. Science 283: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Johnson RE, Yu SL, Prakash S, Prakash L 2007. A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol Cell Biol 27: 7198–7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, Ballarati L, Vergult S, Giardino D, Hansson K, et al. 2012. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell Rep 1: 648–655 [DOI] [PubMed] [Google Scholar]
- Knobel PA, Marti TM 2011. Translesion DNA synthesis in the context of cancer research. Cancer Cell Int 11: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole W, van Schendel R, Karambelas AE, van Heteren JT, Okihara KL, Tijsterman M 2014. A polymerase θ-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat Commun 5: 3216. [DOI] [PubMed] [Google Scholar]
- Kusumoto R, Masutani C, Iwai S, Hanaoka F 2002. Translesion synthesis by human DNA polymerase η across thymine glycol lesions. Biochemistry 41: 6090–6099 [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CMB, Lupski JR 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M 2008. The cellular, developmental and population-genetic determinants of mutation-rate evolution. Genetics 180: 933–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Araki M, Yamada A, Kusumoto R, Nogimori T, Maekawa T, Iwai S, Hanaoka F 1999a. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J 18: 3491–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F 1999b. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399: 700–704 [DOI] [PubMed] [Google Scholar]
- Matsuda T, Bebenek K, Masutani C, Hanaoka F, Kunkel TA 2000. Low fidelity DNA synthesis by human DNA polymerase-η. Nature 404: 1011–1013 [DOI] [PubMed] [Google Scholar]
- Meier B, Barber LJ, Liu Y, Shtessel L, Boulton SJ, Gartner A, Ahmed S 2009. The MRT-1 nuclease is required for DNA crosslink repair and telomerase activity in vivo in Caenorhabditis elegans. EMBO J 28: 3549–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F 2007. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7: 233–245 [DOI] [PubMed] [Google Scholar]
- Muzzini DM, Plevani P, Boulton SJ, Cassata G, Marini F 2008. Caenorhabditis elegans POLQ-1 and HEL-308 function in two distinct DNA interstrand cross-link repair pathways. DNA Repair (Amst) 7: 941–950 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi E, Bebenek K, Matsuda T, Feaver WJ, Gerlach VL, Friedberg EC, Ohmori H, Kunkel TA 2000. Fidelity and processivity of DNA synthesis by DNA polymerase κ, the product of the human DINB1 gene. J Biol Chem 275: 39678–39684 [DOI] [PubMed] [Google Scholar]
- Roerink SF, Koole W, Stapel LC, Romeijn RJ, Tijsterman M 2012. A broad requirement for TLS polymerases η and κ, and interacting sumoylation and nuclear pore proteins, in lesion bypass during C. elegans embryogenesis. PLoS Genet 8: e1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima N, Munroe RJ, Schimenti JC 2004. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol 24: 10381–10389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijsterman M, Pothof J, Plasterk RHA 2002. Frequent germline mutations and somatic repeat instability in DNA mismatch-repair-deficient Caenorhabditis elegans. Genetics 161: 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Wood RD 2012. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair (Amst) 12: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wu X, Yuan F, Xie Z, Wang Z 2001. Highly frequent frameshift DNA synthesis by human DNA polymerase μ. Mol Cell Biol 21: 7995–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]