

Abstract
Hepatitis C virus (HCV) is a single-stranded, positive-sense RNA virus of clinical importance. The virus establishes a chronic infection and can progress from chronic hepatitis, steatosis to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). The mechanisms of viral persistence and pathogenesis are poorly understood. Recently the unfolded protein response (UPR), a cellular homeostatic response to endoplasmic reticulum (ER) stress, has emerged to be a major contributing factor in many human diseases. It is also evident that viruses interact with the host UPR in many different ways and the outcome could be pro-viral, anti-viral or pathogenic, depending on the particular type of infection. Here we present evidence for the elicitation of chronic ER stress in HCV infection. We analyze the UPR signaling pathways involved in HCV infection, the various levels of UPR regulation by different viral proteins and finally, we propose several mechanisms by which the virus provokes the UPR.
Keywords: hepatitis C virus, unfolded protein response, endoplasmic reticulum stress, hepacivirus, virus-host interaction
Introduction
Hepatitis C virus (HCV) infection produces a clinically important disease affecting around 3% of the world population (Thomas, 2013). The disease usually manifests itself as chronic hepatitis, which can progress into fibrosis, cirrhosis and eventually into hepatocellular carcinoma (HCC). How the virus establishes persistence and causes diseases is still far from clear. Understanding how the virus interacts with the host is key to answer these questions. Recently the unfolded protein response (UPR), a host homeostatic response to endoplasmic reticulum (ER) stress, has emerged to be a novel mechanism involved in a number of human diseases including that caused by virus infections (Dimcheff et al., 2003; Favreau et al., 2009; Wang and Kaufman, 2012). UPR has also been frequently manipulated by a number of viruses to aid in infection and to attenuate anti-viral response (Isler et al., 2005; Smith et al., 2006; Yu et al., 2006; Liu et al., 2009; Jheng et al., 2010; Ambrose and Mackenzie, 2011; Pena and Harris, 2011; Burnett et al., 2012; Galindo et al., 2012; Qian et al., 2012; Rathore et al., 2013; Stahl et al., 2013). On the other hand, UPR is recruited by the host anti-viral machinery to help eliminating virus infection (Clavarino et al., 2012). Thus UPR is far from being a homeostatic response in terms of virus infection. For viruses that establish persistent infection it is even more important to be able to adapt to chronic ER stress, otherwise diseases will ensue. It is therefore important to understand how HCV interacts with the host UPR, taking into consideration the genotypes and the various experimental systems used to dissect this virus-host interaction.
Hepatitis C virus
Life cycle
HCV is a single-stranded, positive-sense RNA Hepacivirus (a genus of the family Flaviviridae) with a 9.6 kb genome (Figure 1) (Scheel and Rice, 2013; Simmonds, 2013). The virus particles are unusual in that they are associated with low-density lipoproteins (LDLs) and very low-density lipoproteins (VLDLs) to form the lipoviroparticles (Andre et al., 2002). The virus enters host cells (hepatocytes) by initial binding to low-affinity receptors the LDL receptor and glycosaminoglycans on the heparan sulphate proteoglycans, followed by binding to scavenger receptor class B member 1 and stepwise translocation to post-binding co-receptors the tetraspanin CD81 and tight junction proteins claudin 1 and occludin (Pileri et al., 1998; Agnello et al., 1999; Scarselli et al., 2002; Evans et al., 2007; Ploss et al., 2009; Jiang et al., 2012; Lindenbach and Rice, 2013). Interaction between CD81 and claudin 1 facilitates viral uptake by clathrin-mediated endocytosis (Meertens et al., 2006; Farquhar et al., 2012). Exposure to low pH in the endosome triggers membrane fusion and release of the RNA genome into the cytoplasm (Lavillette et al., 2006). Translation of the RNA genome into a single polypeptide is mediated from an internal ribosome entry site element at the 5' untranslated region (Tsukiyama-Kohara et al., 1992). The polypeptide is then cleaved by the host signal peptidase and signal peptide peptidase and viral autoprotease NS2-3 and serine protease NS3/NS4A co-factor into the structural proteins core, envelopes E1 and E2, and non-structural (NS) proteins p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Hijikata et al., 1991b; Grakoui et al., 1993a; Failla et al., 1994; Lin et al., 1994; Santolini et al., 1994; McLauchlan et al., 2002; Schregel et al., 2009; Scheel and Rice, 2013). Replication takes place in a membranous web, which is a re-organized intracellular membrane structure consisting of single, double, and multiple membrane vesicles (Behrens et al., 1996; Romero-Brey et al., 2012; Bartenschlager et al., 2013; Paul et al., 2013). Formation of the membranous web is mainly induced by NS4B and NS5A (Romero-Brey et al., 2012). Replication is catalyzed by an RNA-dependent RNA polymerase, NS5B, via a negative-sense RNA intermediate, and assisted by the helicase activity of NS3 and host factors cyclophilin A and miR-122 (Behrens et al., 1996; Banerjee and Dasgupta, 2001; Jopling et al., 2005; Kaul et al., 2009). Virion assembly is initiated on core-coated lipid droplets followed by budding into the ER, where the two envelope glycoproteins, E1 and E2, form non-covalently-bonded heterodimers and disulphide-bonded aggregates (Dubuisson et al., 1994; Deleersnyder et al., 1997; Lindenbach and Rice, 2013). Virus particles are released via trafficking through the secretory pathway, where the envelope proteins undergo further glycan modifications and structural re-arrangement into higher ordered oligomeric aggregates (Vieyres et al., 2010, 2014). The viroporin, p7, forms ion channels to equilibrate pH gradients during trafficking through the secretory pathway to protect the virion (Wozniak et al., 2010). Assembly, budding and egress are tightly coupled to host lipoprotein synthesis (Lindenbach and Rice, 2013).
Figure 1.

Hepatitis C virus life cycle. Hepatitis C virus enters cells by stepwise binding through host receptors low-density lipoprotein receptor (LDLR), glycosaminoglycans (GAGs), scavenger receptor class B member 1 (SRB1), CD81, and the tight junction proteins claudin 1 and occludin. Interaction between CD81 and claudin 1 facilitates viral uptake by clathrin-mediated endocytosis. Endosomal low pH triggers membrane fusion and release of genome into the cytoplasm. The positive-sense (+), single-stranded RNA is translated by an internal ribosome entry site (IRES) element at its 5' untranslated region (UTR) into a single polypeptide, which is then cleaved into the core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B by host signal peptidase (purple scissor) and signal peptide peptidase (pink scissor) and viral autoprotease (NS2-3) (fluorescent green scissor) and serine protease (NS3-NS4A) (dark green scissor). Replication is catalyzed by the RNA-dependent RNA polymerase NS5B, assisted by the helicase activity of NS3, via a negative-sense (−) intermediate RNA (red arrows). Replication takes place in the membranous web, which consists of single, double and multiple membrane vesicles. Formation of the membranous web is induced by NS4B and NS5A. Assembly of virion is initiated on core-coated lipid droplets (LD) followed by budding into the endoplasmic reticulum (ER), where it is coated by the ER-resident envelope proteins E1 and E2. Egress follows the secretory pathway to release the virion to extracellular space. The viroporin, p7, forms ion channels to equilibrate pH gradients during trafficking through the secretory pathway to protect the virion.
Genotypes
HCV is classified into 7 genotypes (with >30% sequence variation), sub-divided into sub-types a, b, c, etc. (with 20–25% sequence variation) and then strains/isolates (Simmonds et al., 1994, 2005; Kuiken and Simmonds, 2009). Genotype is a major determining factor in responsiveness to interferon (IFN) treatment and in disease progression in hepatitis C patients (Chayama and Hayes, 2011; Ripoli and Pazienza, 2011). Infection with genotype 1 is more resistant to IFN treatment and presents a more aggressive disease course with the chance of progression into HCC significantly higher. Moreover, HCV in infected patients exists as a population of quasispecies/intrahost variants (Martell et al., 1992; Simmonds et al., 1994, 2005; Holmes, 2010; Domingo et al., 2012). It is anticipated that virus-host interaction is determined at genotypic, sub-genotypic, strain/isolate and even quasispecies/intrahost variants levels. Therefore in this review, we will refer to the genotypes, sub-types and strains/isolates used in various studies.
HCV experimental systems
When HCV was discovered in 1989 as the causative agent of post-transfusional non-A, non-B hepatitis, study on the virus was limited to the use of in vitro cell-free systems and cell culture expression systems employing transient transfection or viral vectors (Choo et al., 1989; Hijikata et al., 1991b, 1993; Grakoui et al., 1993b). Nevertheless, much has been known about the genomic structure and viral protein functions. In vivo study was made possible by the successful infection of chimpanzees by intrahepatic inoculation of the RNA transcript (Kolykhalov et al., 1997). However, the use of chimpanzees is limited and restricted (Mailly et al., 2013). Small animal models have become available by the creation of transgenic mice expressing viral proteins in their livers and chimeric mice with humanized livers (Moriya et al., 1998; Mercer et al., 2001; Dorner et al., 2011). It was not until 1999 when a selectable sub-genomic replicon (SGR) of genotype 1b Con1 isolate was successfully established which allowed the study of the intracellular steps of the virus life cycle (Figure 2A) (Lohmann et al., 1999). Since then some other SGR and genomic replicons have been established (Figure 2B) (Ikeda et al., 2002; Blight et al., 2003; Kato et al., 2003). A pseudotyped virus containing HCV envelope proteins in a retrovirus or lentivirus genomic backbone (HCVpp) was also established to facilitate the study of virus entry (Bartosch et al., 2003). The breakthrough came in 2005 when a cell-cultured infectious system (HCVcc) was established from a wild type genotype 2a JFH1 strain fulminant hepatitis C patient, coupled with derivation of cell lines (Huh7.5, Huh7.5.1) from the parental Huh7 with improved infectivity (Figure 2C) (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005). Chimeric viruses were then created by fusing core-NS2 from other genotypes or sub-types to the NS3-5B backbone of JFH1, allowing partial studies of other genotypes (Figure 2D) (Gottwein et al., 2007, 2009; Jensen et al., 2008; Scheel et al., 2008; Li et al., 2011). Currently there has been some success in establishing HCVcc from other genotypes but they all require adaptive mutations, thus do not represent the wild type repertoires (Yi et al., 2006; Date et al., 2012; Li et al., 2012a,b; Ramirez et al., 2014). With the availability of so many systems, therefore in this review, we will refer to the systems and cell lines used in various studies.
Figure 2.
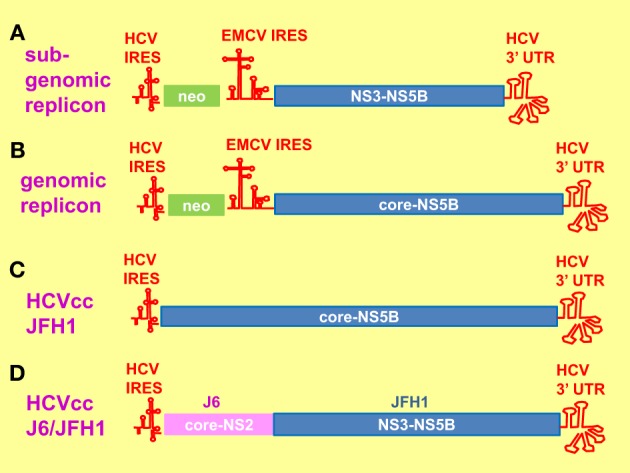
Hepatitis C virus replication systems. (A) Sub-genomic replicon (SGR) consists of a bicistronic mRNA. The 5' neomycin (neo) mRNA is translated by the hepatitis C virus (HCV) internal ribosome entry site (IRES) element whereas the 3' mRNA encoding HCV NS3-NS5B plus the 3' untranslated region (UTR) is translated by the encephalomyocarditis virus (EMCV) IRES element. Cell lines harboring the SGR were established by neomycin selection. (B) The genomic replicon is similar to that of SGR, apart from that the 3' mRNA encodes core-NS5B plus 3' UTR. (C) The HCV cell-cultured infectious system (HCVcc) consists of the entire genomic RNA from JFH1. (D) The chimeric J6/JFH1 is created by fusing the core-NS2 from J6 to NS3-NS5B plus 3' UTR from JFH1.
Unfolded protein response
UPR is a cellular adaptive response for restoring ER homeostasis in response to ER stress (Figure 3) (Walter and Ron, 2011). UPR transduces into a programme of cellular transcriptional and translational responses culminating in upregulation of the molecular chaperone the immunoglobulin heavy-chain binding protein (BiP) to promote protein folding, global inhibition in protein synthesis to reduce protein load and potentiation of ER-associated degradation (ERAD) to eliminate unfolded/malfolded proteins from the ER (Travers et al., 2000; Walter and Ron, 2011).
Figure 3.

Unfolded protein response. Mammalian unfolded protein response (UPR) is a tripartite response involving three proximal sensors: activating factor (ATF) 6, RNA-dependent protein kinase-like ER-resident kinase (PERK) and inositol-requiring enzyme 1 (IRE1). Left: ATF6 is sequestered in an inactive state by the molecular chaperone the immunoglobulin heavy-chain binding protein (BiP). Unfolded/malfolded proteins “distract” BiP from ATF6. ATF6 de-oligomerizes and migrates to the Golgi, where the monomer is cleaved by site-1 and site-2 proteases (red scissor) into an active transcription factor. The truncated N-terminal ATF6 is translocated to the nucleus where it transactivates UPR genes harboring an ERSE in their promoters e.g., BiP, glucose-regulated protein 94 (GRP94), P58IPK. Middle: PERK is sequestered in an inactive state by BiP. Unfolded/malfolded proteins “distract” BiP from PERK, allowing its oligomerization and auto-phosphorylation (red asterisk). The activated PERK then phosphorylates its substrate, the alpha subunit of the eukaryotic initiation factor 2 (eIF2α) (red asterisk) to inhibit global protein synthesis. Paradoxically, translation of ATF4 is upregulated to drive transcription of UPR genes with an ATF4 element in their promoters e.g., the CCAAT/enhancer-binding protein-homologous protein (CHOP). CHOP is a pro-apoptotic transcription factor, as it transactivates a number of apoptotic genes and downregulates the anti-apoptotic Bcl-2. ATF4 co-operates with CHOP to transactivate ATF3 and the growth arrest and DNA damage-inducible protein 34 (GADD34). GADD34 is the regulatory subunit of the protein phosphatase 1 (PP1). It recruits PP1 to dephosphorylate eIF2α (red blunt arrow), thus establishing a negative feedback loop. Right: Analogous to yeast, it is thought that IRE1 is activated by direct binding of unfolded/malfolded proteins to its luminal domain and BiP plays a regulatory role. IRE1 possesses endoribonuclease and kinase activity. The endoribonuclease activity mediates unconventional splicing of XBP1 (purple scissor) (usXBP1, unspliced XBP1 mRNA; sXBP1, spliced XBP1 mRNA). The sXBP1 mRNA is translated into an active transcription factor sXBP1 to transactivate genes with ERSE or UPRE in their promoters. XBP1 upregulation of UPR genes such as BiP and ERAD genes such as EDEM and ERdj4 provides a link between UPR and ERAD. XBP1 provides a link between the IRE1 and PERK pathways by upregulating P58IPK, an inhibitor of PERK. XBP1 also orchestrates lipogenesis and ER expansion. The other endoribonuclease activity of IRE1 cleaves the ribosomal RNA (rRNA) (purple scissor) and mediates regulated IRE1-dependent decay (RIDD) to cleave a subset of mRNAs (purple scissor) to inhibit protein synthesis. The kinase activity of IRE1 plays a role in cell death/survival. Phosphorylated IRE1 (red asterisk) recruits the adaptor protein tumor necrosis factor receptor-associated factor 2 (TRAF2) to activate a cascade of phosphorylation culminating in pro-apoptotic Jun amino-terminal kinase (JNK) (red asterisk) and pro-survival c-Jun (red asterisk). Red asterisk, activation by phosphorylation.
BiP has been attributed a pivotal role as the master negative regulator of UPR by binding to and repressing the activities of the three proximal UPR sensors: activating transcription factor (ATF) 6, RNA-dependent protein kinase-like ER-resident kinase (PERK), and inositol-requiring enzyme 1 (IRE1) (Bertolotti et al., 2000; Shen et al., 2002a). Accumulation of unfolded/malfolded proteins “ distract” BiP from binding to the UPR sensors. ATF6 de-oligomerizes and migrates to the Golgi where it is cleaved sequentially by site-1 protease and site-2 protease to release an active transcription factor into the nucleus where it transactivates UPR genes harboring an ER-stress element (ERSE) in their promoters e.g., BiP, glucose-regulated protein 94 (GRP94) and P58IPK (Yoshida et al., 1998; Shen et al., 2002a; Nadanaka et al., 2007). P58IPK is an inhibitor of PERK, thus linking the ATF6 pathway to the PERK pathway (Van Huizen et al., 2003).
PERK is an ER stress kinase, activated by dimerisation and autophosphorylation (Harding et al., 1999; Bertolotti et al., 2000). PERK specifically phosphorylates the alpha subunit of the eukaryotic translation initiation factor 2 (eIF2α) causing global inhibition of protein synthesis but paradoxically enhances translation of the transcription factor ATF4 (Harding et al., 2000). ATF4 transactivates UPR genes with an ATF4 element in their promoters e.g., the CCAAT/enhancer-binding protein-homologous protein (CHOP). CHOP is a pro-apoptotic transcription factor owing to its ability to transactivate a number of apoptotic genes and downregulate the anti-apoptotic Bcl-2 (McCullough et al., 2001; Tabas and Ron, 2011). ATF4 and CHOP co-operate to transactivate downstream effectors e.g., ATF3, growth arrest and DNA damage-inducible protein 34 (GADD34) (Han et al., 2013). GADD34 promotes translational recovery by recruiting protein phosphatase 1 to dephosphorylate eIF2α, thus establishing a negative feedback loop (Connor et al., 2001; Novoa et al., 2001, 2003).
IRE1 is a kinase/endoribonuclease activated by self-oligomerisation and autophosphorylation (Sidrauski and Walter, 1997; Bertolotti et al., 2000; Shen et al., 2002a). The IRE1 pathway is an ancient pathway shared with yeast (Tirasophon et al., 1998; Hollien, 2013). In yeast, there is evidence to suggest that IRE1 is activated by direct binding of unfolded protein ligands to its luminal domain whereas BiP plays a regulatory role by maintaining IRE1 oligomeric equilibrium (Credle et al., 2005; Gardner and Walter, 2011; Gardner et al., 2013). The endoribonuclease activity of IRE1 mediates unconventional splicing of the X-box binding protein 1 (XBP1) for its productive translation into an active, multi-functional transcription factor, the spliced XBP1 (sXBP1) (Calfon et al., 2002). sXBP1 transactivates ERSE in the promoters of UPR genes and the mammalian UPR element (UPRE) in the promoters of ERAD genes, thus providing a link between UPR and ERAD (Yoshida et al., 2003). Indeed, UPRE-mediated transcriptional induction of the ER degradation-enhancing α-mannosidase-like protein (EDEM) is directly involved in the recognition of malfolded proteins for degradation. Another protein ERdj4 transactivated by sXBP1 also participates in ERAD (Shen et al., 2002b; Lee et al., 2003; Lai et al., 2012). Similar to that of ATF6, XBP1 also links the IRE1 pathway to the PERK pathway by upregulating the inhibitor of PERK, P58IPK, to aid in translational recovery (Yan et al., 2002; Lee et al., 2003; Van Huizen et al., 2003). XBP1 also assumes additional function in the regulation of lipogenesis and ER expansion (Lee et al., 2008; Glimcher and Lee, 2009; Brewer and Jackowski, 2012). The endoribonuclease activity of IRE1 also participates in translational repression by cleavage of the 28S ribosomal RNA and a subset of mRNAs via regulated IRE1-dependent decay (Iwawaki et al., 2001; Hollien et al., 2009). On the other hand, the kinase activity of IRE1 regulates cell death/survival (Urano et al., 2000; Tabas and Ron, 2011). Phosphorylated IRE1 associates with the adaptor protein tumor necrosis factor receptor-associated factor 2 to initiate a cascade of phosphorylation culminating in that of the pro-apoptotic Jun amino-terminal kinase (JNK) and pro-survival c-Jun (Darling and Cook, 2014).
Evidence of UPR in hepatitis C
There is as yet no consistent clinical data to support or refute the presence of ER stress in hepatitis C patients (Asselah et al., 2010; McPherson et al., 2011). Comparison between HCV-positive and -negative liver biopsy using real-time RT-PCR did not reveal any significant variation in the mRNA levels of GRP94, sXBP1 and EDEM (McPherson et al., 2011). Immunohistochemistry also did not detect any overall difference in the intensity of BiP between chronic hepatitis C and non-diseased livers, however, the staining was variable and one HCV sample showed a very high level of BiP. This may be explained by HCV being a focal infection, infecting only 7–20% of the liver (Liang et al., 2009; Stiffler et al., 2009). As a result, random sampling may not be able to detect a significant change in the mRNA/protein level in an area of mixed infected- and uninfected-hepatocytes. Indeed, using electron microscopy, dilated and disorganized ER indicative of ER stress was observed in hepatocytes from liver biopsy of mild chronic hepatitis C patients (Asselah et al., 2010). Evidence of ER stress in these liver samples was further confirmed using Western blotting which showed marked elevation in the levels of the proximal sensors ATF6α, ATF6β, sXBP1, and phosphorylated PERK and select subsets of downstream effectors BiP, phospho-eIF2α, ATF4, and EDEM. A study on a cohort of HCV HCC patients also demonstrated increased UPR markers of sXBP1, BiP, and ATF6 in liver biopsy by using immunohistochemistry and Western blotting (Shuda et al., 2003). It is therefore essential that concrete clinical evidence should await the use of more sensitive methods to detect, at single cells level, co-localization of ER stress markers in infected cells as compared to neighboring uninfected cells. Nevertheless, there is overwhelming evidence from in vivo and in vitro experiments to suggest that the ER stress response plays an important role in the life cycle of HCV (Liberman et al., 1999; Tardif et al., 2002, 2004; Benali-Furet et al., 2005; Chan and Egan, 2005, 2009; Ciccaglione et al., 2005, 2007; Zheng et al., 2005; Tumurbaatar et al., 2007; Sekine-Osajima et al., 2008; Joyce et al., 2009; Li et al., 2009; Mishima et al., 2010; Von Dem Bussche et al., 2010; Funaoka et al., 2011; Merquiol et al., 2011; Shinohara et al., 2013). Importantly, by using immunohistochemistry and confocal microscopy, increased level of the UPR marker, BiP, was found to co-localize with HCV-infected hepatocytes in SCID/Alb/uPA mice (chimeric mice with humanized livers) infected with genotype 1a H77 or intrahepatically inoculated with H77 RNA (Joyce et al., 2009). Infection of humanized mice with another genotype (2a) JFH1 strain also resulted in increased levels of BiP and CHOP in the livers (Mishima et al., 2010). Further in vivo evidence of ER stress was obtained in transgenic mice stably expressing the entire open reading frame, the core protein or inducibly expressing C-E1-E2-p7 in the livers (Benali-Furet et al., 2005; Tumurbaatar et al., 2007; Merquiol et al., 2011). Modulation of the UPR was widely observed in tissue-cultured hepatocytes infected with HCV; in cells harboring the HCV genomic replicon and SGR and in cells ectopically expressing individual viral proteins (Liberman et al., 1999; Tardif et al., 2002, 2004; Benali-Furet et al., 2005; Chan and Egan, 2005, 2009; Ciccaglione et al., 2005, 2007; Zheng et al., 2005; Sekine-Osajima et al., 2008; Li et al., 2009; Von Dem Bussche et al., 2010; Funaoka et al., 2011; Shinohara et al., 2013).
UPR signaling in hepatitis C
Some viruses can selectively activate or suppress one or more of the UPR tripartite pathways to facilitate their own replication (Isler et al., 2005; Smith et al., 2006; Yu et al., 2006; Jheng et al., 2010; Ambrose and Mackenzie, 2011; Pena and Harris, 2011; Burnett et al., 2012; Galindo et al., 2012; Qian et al., 2012; Rathore et al., 2013; Stahl et al., 2013). It is apparent that HCV infection activates all three proximal sensors (Ke and Chen, 2011; Merquiol et al., 2011). Infection of the hepatocyte sub-line Huh7.5.1 with JFH1 (2a) induced an acute ER stress peaking at 2–5 days post-infection (dpi), concomitant with phosphorylation of IRE1, eIF2α, and JNK, XBP1 splicing, ATF6 cleavage and upregulation of GADD34, ERdj4, P58IPK, ATF3, ATF4, and CHOP (Merquiol et al., 2011). It then subsided into a chronic and milder ER stress response persisting up to 14 dpi, with elevated mRNA levels of CHOP, ATF3, sXBP1, and P58IPK and increased level of phospho-eIF2α. ER stress response is not restricted to the sub-line Huh7.5.1, as infection of the parental lines Huh7 or Huh7.5 with JFH1 (2a) also induced ER stress (Ke and Chen, 2011). Infection of Huh7 with JFH1 provoked an acute ER stress response concomitant with ATF6 cleavage, XBP1 splicing and PERK phosphorylation at 6–9 dpi followed by a chronic and milder ER stress with a diminished CHOP level at 15–22 dpi. Similarly, infection of Huh7.5 with JFH1 (2a) has been shown to transactivate the Bip, CHOP, and ATF6 promoters (Von Dem Bussche et al., 2010).
Currently in vitro infection study with wild type genotype is only achievable with the strain JFH1 and yet JFH1 was isolated from a patient with fulminant hepatitis-a rare manifestation of HCV diseases (Wakita et al., 2005; Lohmann and Bartenschlager, 2014). It is therefore important that studies should be extended to other genotypes before it can be generalized that ER stress is a common phenomenon of chronic hepatitis C. Chimeric HCV has been created by fusing the structural proteins from all seven genotypes with the NS proteins of JFH1, which should at least allow us to study the role of genotypic structural proteins in UPR (Gottwein et al., 2007, 2009; Jensen et al., 2008; Scheel et al., 2008; Li et al., 2011). An intragenotypic chimera J6/JFH1 has already been shown to be capable of eliciting the UPR, as evident by the increased levels of CHOP and sXBP1 at 1–3 dpi (Mohl et al., 2012). Some success has been achieved to establish cell-cultured infectious systems for genotypes 1a (H77 and TN), 1b (NC1), 2a (J6), and 2b (J8, DH8, DH10) but they require a number of adaptive mutations (Yi et al., 2006; Date et al., 2012; Li et al., 2012a,b; Ramirez et al., 2014). At the moment, studies with wild type genotypes other than JFH1 still rely on the use of genomic replicons (Benali-Furet et al., 2005; Shinohara et al., 2013). Similar to that in JFH1-infected Huh7, all three pathways have been activated in Huh7 cells harboring a genomic replicon of genotype 1b O strain, as indicated by the phosphorylation of eIF2α, XBP1 splicing and increased levels of IRE1, phospho-JNK, and phospho-c-Jun (Shinohara et al., 2013).
Which viral proteins mediate UPR?
A number of steps in the virus life cycle are potential trigger of ER stress e.g., the maturation of the viral envelope glycoproteins in the ER, formation of replication complex on the ER, virus assembly, and budding of virus particles into the ER (Scheel and Rice, 2013).
Envelope proteins
SGR (devoid of C-E1-E2-p7-NS2) is generally less capable (or incapable) of triggering the UPR than its full-length counterpart, suggesting that the main contributors to ER stress lie within the structural-NS2 region (Von Dem Bussche et al., 2010; Mohl et al., 2012). A genomic replicon devoid of the envelope proteins E1 and E2 failed to elicit UPR in transfected Huh7 cells, implicating a pivotal role of the envelope proteins in the elicitation of ER stress (Mohl et al., 2012). This is consistent with the ER residence of the envelope proteins. Using transient transfection of envelope proteins to physiological levels, we have confirmed that the HCV envelope proteins are capable of inducing the UPR in hepatocytes HepG2 and Huh7 as well as non-hepatocyte HeLa (Chan and Egan, 2005, 2009).
Core
The core protein, which does not enter the ER lumen but is important in lipid droplet formation and virus assembly and budding, also elicits the UPR (McLauchlan et al., 2002; Benali-Furet et al., 2005; Funaoka et al., 2011; Scheel and Rice, 2013). Evidence of ER stress has been documented in tissue-cultured cells transfected with the HCV-core and in the livers of HCV-core transgenic mice (Benali-Furet et al., 2005). The significant role of the core protein can be illustrated by the considerable effects of mutating the core residues R70Q, R70H, L91M on the UPR in Huh7 cells infected or transfected with JFH1 (Funaoka et al., 2011).
NS2
NS2 does not enter the ER lumen despite being a transmembrane protein (Bartenschlager et al., 2013). Apart from harboring a protease, NS2 is important in organizing the virus assembly complex (Lindenbach and Rice, 2013). One study implicated a major role of NS2 in provoking the UPR based on the detection of increased BiP in Huh-7 cells transfected with core-E1-E2-p7-NS2 compared with that transfected with core-E1-E2-p7, however, expression of NS2 was barely detectable (Von Dem Bussche et al., 2010). Whereas the increases of BiP at the promoter and mRNA levels were very modest, it is not clear why the more pronounced increase of BiP protein level necessitated detection by immunoprecipitation-Western blotting rather than the more straightforward Western blotting. Ectopic expression of NS2 from genotype 1a in Huh7 cells resulted in eIF2α phosphorylation and modest increases of BiP, CHOP, and ATF6 at the promoter and mRNA levels together with a more pronounced increase in BiP protein level (again, detected by immunoprecipitation-Western blotting). In contrast, another hepatocyte cell line Hep3B stably expressing NS2 from genotype 1b (k isolate) failed to induce ATF6 cleavage (Li et al., 2009). Therefore, whether NS2 is the main contributor of ER stress, as claimed, still needs robust testing (Von Dem Bussche et al., 2010).
NS4B
Huh7 cells harboring SGR were capable of inducing ATF6 cleavage and XBP1 splicing but suppressing the downstream activation of UPRE and EDEM by sXBP1 (Tardif et al., 2002, 2004). This suppressive effect could be attributed to NS4B as ectopic expression of NS4B in Huh7 cells displayed a similar pattern of ATF6 cleavage and XBP1 splicing without downstream activation of EDEM (Zheng et al., 2005; Li et al., 2009). NS4B is important in membranous web/replication complex formation (Bartenschlager et al., 2013). Similar to NS2, it also does not enter the ER lumen despite being a transmembrane protein. The role of NS4B in UPR could be modulating. Indeed, ERAD activation, as demonstrated by XBP1 splicing and upregulation of EDEMs mRNAs, clearly exists during infection of Huh7.5.1 with JFH1 (Saeed et al., 2011). EDEM interaction with E1 and E2 resulted in ubiquitination of E2 and decrease in virus particle production. This is not ideal for the virus. Subsequently, NS4B may act to modulate the UPR by suppressing the ERAD to help damping down the inhibitory effect of EDEM in order to regulate and fine-tune virus particle production.
NS5A/5B
NS5A/5B are integral to viral replication (Scheel and Rice, 2013). Infection of humanized mice with NS5A/5B mutants of JFH1 led to increased expression of BiP and CHOP, suggesting a role of the NS5 proteins in ER stress although it is not clear whether they act directly or indirectly (Mishima et al., 2010). It is possible that the enhancing effect of the NS5 mutants on the UPR may be an indirect result of a higher replication rate of these mutants leading to increased production of the responsible proteins i.e., core, E1, E2. Whether the NS5 proteins directly induce the UPR still needs to be shown but Hep3B cells stably expressing NS5B genotype 1b (k isolate) failed to induce ATF6 cleavage (Li et al., 2009).
How do envelope proteins elicit UPR?
Enveloped viruses either bud through the plasma membrane or an intracellular compartment e.g., ER (Figure 4) (Garoff et al., 2004; Stertz et al., 2007; Murakami, 2012; Prange, 2012; Vieyres et al., 2014). In either case, the envelope proteins will be first targeted to the ER for post-translational modification and maturation. Many viral envelope proteins are significant inducers of UPR, whether they are ER-resident proteins (for viruses budding into the ER) or are just trafficking through the ER en route to the plasma membrane (for viruses budding through the plasma membrane) (Dimcheff et al., 2003, 2004; Wang et al., 2003, 2006; Hsieh et al., 2004; Liu et al., 2004, 2006; Nanua and Yoshimura, 2004; Qiang et al., 2004; Chua et al., 2005; Chan et al., 2006; Yoshimura and Luo, 2007; Yoshimura et al., 2008; Zhao and Yoshimura, 2008; Favreau et al., 2009; Portis et al., 2009; Barry et al., 2010; Dediego et al., 2011; Hung et al., 2011).
Figure 4.
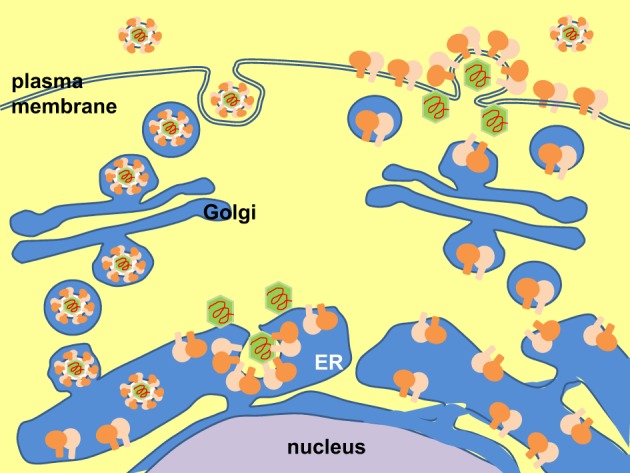
Two modes of virus budding. Left: Virus budding into the endoplasmic reticulum (ER). Virion assembles and buds into the ER, where it is coated by the ER-resident envelope proteins. Egress of virus particle follows the host secretory pathway and released into the extracellular space. Right: Virus budding from the plasma membrane. Envelope proteins are targeted to the ER and transported to the cell surface via the host secretory pathway. Virion assembles and buds through the plasma membrane.
Mutant envelope proteins as UPR inducers
Several mutated viral envelope proteins are significant UPR inducers and major determinants of virulence, in analogous to many human diseases which are caused by retention of mutated cellular proteins in the ER e.g., the genetic variant null Hong Kong of α1-antitrypsin and the ΔF508 cystic fibrosis transmembrane conductance regulator (Oda et al., 2003; Gnann et al., 2004). Retrovirus buds through the plasma membrane (Murakami, 2012). Virulent strains of retrovirus harbor mutations in the envelope proteins resulting in retention of inefficiently folded envelope proteins in the ER, leading to elicitation of the UPR which is a major determinant of neurovirulence (Dimcheff et al., 2003, 2004; Liu et al., 2004, 2006; Nanua and Yoshimura, 2004; Qiang et al., 2004; Yoshimura and Luo, 2007; Yoshimura et al., 2008; Zhao and Yoshimura, 2008; Portis et al., 2009). Hepatitis B virus buds into the ER-Golgi intermediate or other intracellular compartments, mutations in the large surface protein resulted in ER retention, provoking ER stress which is associated with hepatocarcinogenesis (Wang et al., 2003, 2006; Hsieh et al., 2004; Chua et al., 2005; Hung et al., 2011; Prange, 2012). Coronavirus also buds into the ER-Golgi intermediate compartment (Garoff et al., 1998; Stertz et al., 2007). Its spike protein is an UPR inducer (Chan et al., 2006; Versteeg et al., 2007; Siu et al., 2014). Mutations in the spike protein have been associated with persistence and translational attenuation and these mutations have also been found to enhance UPR, cytotoxicity and cell death and confer neurovirulence (Favreau et al., 2009).
Immature virion as UPR inducer
For viruses that bud into the ER it is not clear how the ER-residing envelope proteins will induce UPR. Many of these envelope proteins will undergo further processing and re-organization/conformational changes after incorporation into the immature virion and trafficking through the secretory pathway. In flavivirus, 60 trimeric prM/E assemble as immature virion in the ER (Pierson and Diamond, 2012). During transit through the Golgi E undergoes dramatic re-organization and collapses onto the virion surface whereas a cleavage site on prM is exposed for furin proteolysis. Flaviviruses are prolific inducers of UPR (Jordan et al., 2002; Su et al., 2002; Yu et al., 2006; Medigeshi et al., 2007; Umareddy et al., 2007; Ambrose and Mackenzie, 2011; Klomporn et al., 2011; Paradkar et al., 2011; Pena and Harris, 2011; Wu et al., 2011; Ambrose and Mackenzie, 2013; Blazquez et al., 2013; Yu et al., 2013; Bhattacharyya et al., 2014). UPR has been documented in infections of Dengue virus, West Nile virus, Japanese encephalitis virus, tick-borne encephalitis virus and Usutu virus. The HCV envelope proteins are synthesized as part of a single polypeptide (Grakoui et al., 1993b). After importing into the ER by signal peptides at their respective N-termini, they are cleaved into E1 and E2 by cellular signal peptidase (Hijikata et al., 1991a; Lin et al., 1994). Inside the ER, E1 and E2 form two types of complexes: non-covalently-bonded E1-E2 heterodimer and disulphide-bonded aggregates, neither is the mature form (Dubuisson et al., 1994; Dubuisson and Rice, 1996; Deleersnyder et al., 1997). It is not clear which of these forms of E1E2 is acquired by the virion when the virus buds into the ER as E1E2 undergo further conformational changes into aggregated oligomers when the virus particles transit through the secretory pathway (Vieyres et al., 2010, 2014). There has been evidence to suggest that HCV envelope proteins are major UPR inducers (Mohl et al., 2012). Alphavirus does not bud through the ER and yet its maturation resembles that of flavivirus in that the immature prE2/E1 trimer assembled in the ER undergoes furin cleavage in the Golgi into E3(=pr)/E2/E1 trimer (Garoff et al., 2004; Vaney et al., 2013). UPR has been documented in infections of Chikungunya virus and Sindbis virus (Joubert et al., 2012; Abraham et al., 2013; Rathore et al., 2013). The envelope proteins of Semliki Forest virus have been shown to be responsible for the induction of UPR (Barry et al., 2010). Therefore, we speculate that the immature ER form may be in itself a trigger of the UPR, irrespective of whether the virions bud through the ER or other sites.
How do HCV envelope proteins activate UPR?
E1 and E2 accumulate in the ER, placing them in proximity to interact with BiP (Choukhi et al., 1998). Folding of E1 and E2 into the non-covalently-bonded heterodimer utilizes the canonical chaperone calnexin and calreticulin whereas the E2 aggregates are bound by BiP (Figure 5) (Dubuisson and Rice, 1996; Choukhi et al., 1998). This may explain why E2 was able to elicit the UPR (Liberman et al., 1999). However, Bip binds to the E1 aggregates inefficiently or not at all, leading to the possibility that E1 may induce UPR by other means (Choukhi et al., 1998; Liberman et al., 1999; Merola et al., 2001).
Figure 5.

Proposed mechanisms of E1/E2 activation of UPR. E1 and E2 are targeted and mature in the endoplasmic reticulum (ER) to form non-covalently-bonded heterodimers and disulphide-bonded aggregates (pink arrows, 1). The E2 aggregates distract BiP from PERK (purple arrow), allowing PERK oligomerization and activation (brown arrows, 2). Retrotranslocated (3) and cytosolic E1/E2 from surplus protein synthesis (4) can also activate the unfolded protein response (UPR) by perturbation of proteasomal function (Ub, polyubiquitin). Conversely, cytosolic E1/E2 can bind to the cytoplasmic domain of PERK and inhibit its activation (red cross, 5). Direct binding of E1 and E2 aggregates to the luminal domain of IRE1 can also activate UPR (6). Red asterisk, activation by phosphorylation.
One plausible mechanism is that E1 (or even E2) induces UPR by impairing ERAD as ERAD and UPR exist in a regulatory loop (Travers et al., 2000). It is well known that cytosolic proteins such as the polyQ aggregates induce UPR by perturbation of proteasomal degradative function (Friedlander et al., 2000; Travers et al., 2000; Nishitoh et al., 2002). We have shown ERAD engagement in cells transfected with E1 and/or E2 by the demonstration of XBP1 splicing and UPRE induction in these cells (Chan and Egan, 2005). Although E1/E2 mature in the ER, it is possible that some of them have been directed to the cytoplasm during synthesis or as a result of retro-translocation from the ER. There is in vivo evidence of retrograde transport of E1 from the ER to the cytoplasm for proteasome degradation based on the detection of a deglycosylated-deamidated T-epitope from an HCV-infected chimpanzee (Selby et al., 1999). Cytosolic existence of E2 has been demonstrated in vector-expression system although it still yet has to show the cytosolic existence of E2 in infected cells (Pavio et al., 2002). By removing the signal peptides from E1 and E2 we re-directed expression of these proteins to the cytoplasm (Egan et al., 2013). These cytosolic-targeting E1/E2 did not induce UPR. Instead, they repressed tunicamycin-induced UPR possibly as a result of binding to the cytoplasmic domain of PERK and blocking its activation, suggesting that UPR induction by ERAD perturbation is unlikely (Pavio et al., 2003; Egan et al., 2013).
Another possibility is that E1 (or even E2) can trigger UPR by direct binding to one or more of the UPR sensors in the ER lumen. This is especially true when UPR triggered by many virus infections is often skewed suggesting a canonical tripartite-responsive BiP derepression mechanism may not be sufficient to explain these skewed UPR in cases of virus infections. Studies with yeast have shown that UPR can be triggered by direct binding of unfolded proteins to the luminal domain of IRE1 (Credle et al., 2005; Gardner and Walter, 2011; Gardner et al., 2013). The luminal domain of PERK bears secondary structure homology with that of IRE1, by extrapolation, direct binding of unfolded proteins to PERK can also be feasible (Gardner et al., 2013). Direct binding between the herpes simplex virus glycoprotein B and the luminal domain of PERK has been documented but in this case, binding results in repression rather than elicitation of the UPR (Mulvey et al., 2007). Toxic lipids are directly sensed by the transmembrane domains of IRE1 and PERK to provoke the UPR, further supporting the idea that mechanisms other than BiP derepression is possible (Volmer et al., 2013).
How do core and NS proteins elicit UPR?
During polyprotein processing, the signal peptide at the C terminus of the core protein directs the translocation of E1 into the ER, after that the signal peptidase will cleave at the C-terminal end of the core protein at amino acid (aa) residue 191 (Santolini et al., 1994). This intermediate core protein is anchored onto the cytosolic side of the ER membrane by a membrane anchor. Maturation of the core protein involves another intramembrane cleavage event at aa173–182 by signal peptide peptidase in the ER membrane (Okamoto et al., 2008a; Pene et al., 2009). The exact C terminus has not been determined but a minimum of 177 aa residues seems to be required for productive virus production (Kopp et al., 2010). The mature core protein is then released from the ER to traffic to lipid droplets to orchestrate virus assembly (McLauchlan et al., 2002). It appears that the core protein is never directed inside the ER to be able to interact with BiP to trigger the canonical UPR signaling. This is also true for the NS proteins which do not appear to enter the ER lumen even though NS2 and NS4B are transmembrane proteins (Romero-Brey et al., 2012). The question remains how then can their cytosolic presence elicit the UPR?
Proteasomal perturbation
Perturbation of proteasomal activity is one possibility (Friedlander et al., 2000; Travers et al., 2000; Nishitoh et al., 2002). The core, NS2 and NS5B proteins have been shown to interact with the proteasomal pathways (Figure 6) (Gao et al., 2003; Moriishi et al., 2003; Franck et al., 2005; Shirakura et al., 2007; Suzuki et al., 2009). Moreover, interaction of the core protein with the proteasome activator PA28γ is responsible for the pathogenesis of steatosis, HCC and other liver pathology in core-transgenic mice and virus propagation in JFH1-infected Huh7 cells (Moriishi et al., 2007, 2010; Tripathi et al., 2012).
Figure 6.

Viral proteins perturb proteasomal function to elicit UPR. Immature core protein (C) attaches to the cytosolic side of the endoplasmic reticulum (ER) membrane by a membrane anchor. Cleavage of the membrane anchor (pink scissor) releases mature core to the cytoplasm where it can be polyubiquitinated (Ub) and degraded by the cytosolic 26S proteasome. The mature core protein can also be imported into the nucleus by association with the proteasome activator PA28γ and importin (IMP) (purple arrow), where it is degraded by the 20S proteasome independent of ubiquitin. Also shown is the polyubiquitination and degradation of non-structural protein 2 (NS2) and NS5B by the 26S proteasome. Perturbation of proteasomal function elicits the unfolded protein response (UPR).
Perturbation of membranous web protein chaperone activity
The cytosolic chaperone heat shock protein 90 (HSP90) promotes HCV replication by facilitating host and viral protein folding in the replication complex of the membranous web (Figure 7) (Taguwa et al., 2009). Inhibition of HSP90 activity reduces protein folding, accelerates proteasome degradation and induces the UPR. HSP90 is recruited into the replication complex by means of interaction between its co-chaperones FK506-binding protein 8 (FKBP8) and the human butyrate-induced transcript 1 (hB-ind1) and NS5A (Okamoto et al., 2006; Taguwa et al., 2008). It is therefore possible that any changes in HSP90-FKBP8/hB-ind1-NS5A interaction can disrupt the chaperone activity of HSP90 leading to UPR. Indeed, interaction of NS5A with FKBP8 has already been implicated in pathogenesis via activation of mammalian target of rapamycin anti-apoptotic function (Peng et al., 2010). In contrast, a JFH1 NS5A/5B multiple mutants exhibited a higher replication rate and yet provoking a stronger UPR in humanized mice (Mishima et al., 2010). However, the sample size is small (only one mouse from each of the test and control groups was dissected for the UPR markers). Moreover, the mutations are not likely to be involved in co-chaperone binding, suggesting another mechanism of UPR regulation. Previously, it has been found that mutation of a single amino acid V/I121A in NS5A is sufficient to abolish its interaction with FKBP8 impairing virus replication (Okamoto et al., 2008b). It would be interesting to see whether this mutation will provoke a stronger UPR.
Figure 7.

Perturbation of replicase chaperone activity results in UPR. An enlarged view of a membrane vesicle (enclosed by a red square) within the membranous web. Heat shock protein 90 (HSP90) chaperones folding of viral non-structural (NS) 3-NS5B proteins and host protein cyclophilin A (CYPA) etc. in the replication complex to facilitate viral replication. HSP90 forms a complex with NS5A via interactions with its co-chaperone FK506-binding protein 8 (FKBP8) and the human butyrate-induced transcript 1 (hB-ind1). Disruption of co-chaperone-NS5A interaction (red serrated arrows) interrupts HSP90 chaperone activity, resulting in increased degradation of polyubiquitinated (Ub) proteins and perturbation of proteasomal function, leading to the unfolded protein response (UPR) (purple arrows).
Direct binding to cytosolic domains of UPR sensors
Another possibility for cytosolic proteins to trigger UPR is by direct binding to the cytoplasmic domain of the UPR sensors. Currently there is no evidence for interaction of the core protein with any of the cytosolic domains of the UPR sensors. NS4B, however, interacts with the bZIP motif of ATF6β via a predicted bZIP motif in its N-terminal cytoplasmic domain although there is as yet no functional analysis of whether this interaction leads to induction or suppression of ATF6β activity (Figure 8) (Tong et al., 2002; Welsch et al., 2007). On the other hand, NS4B also interacts with ATF6α, despite to a lesser extent, and it is plausible that this interaction signals ATF6α cleavage as observed in hepatocytes and non-hepatocytes expressing NS4B (Tardif et al., 2002; Tong et al., 2002; Zheng et al., 2005; Li et al., 2009). However, expression of NS4B alone also induced XBP1 splicing, suggesting that at least one other mechanism is operating to induce the UPR (Li et al., 2009).
Figure 8.
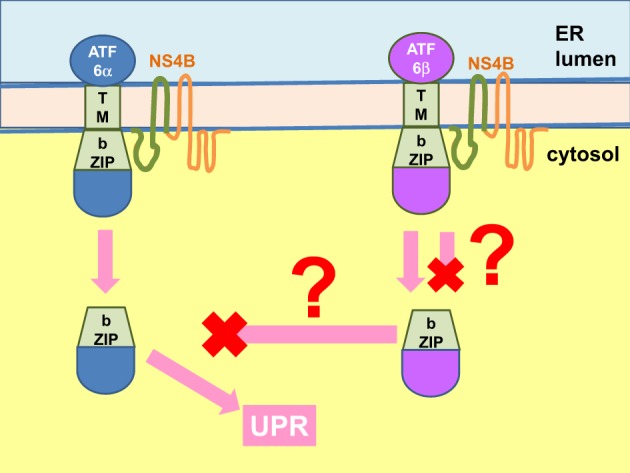
NS4B interacts with ATF6 to modulate UPR. The hepatitis C virus (HCV) non-structural (NS) 4B protein binds to the b-ZIP and transmembrane (TM) domains of the activating factor 6 (ATF6) α and β (binding domains shown in green for both NS4B and ATF6). Binding of ATF6α likely triggers its Golgi translocation and cleavage into an active transcription factor to mediate the unfolded protein response (UPR). The fate of NS4B binding to ATF6β is unknown. It can either trigger or inhibit ATF6β Golgi translocation and activation. The end result could be modulation of the UPR but whether ATF6β is an inhibitor of ATF6α is controversial. ER, endoplasmic reticulum.
Lipid perturbation
UPR regulates lipogenesis and ER membrane expansion (Lee et al., 2008; Glimcher and Lee, 2009; Brewer and Jackowski, 2012). On the contrary, lipid perturbation is one of the triggers of UPR (Volmer et al., 2013). HCV virus particle formation is intimately coupled to the host lipogenesis (Figure 9). The core protein targets to the lipid droplets which is the initial site of virus assembly (McLauchlan et al., 2002; Lindenbach and Rice, 2013). Virus infection also induces massive intracellular membrane re-organization to form the membranous web as the site of virus replication (Behrens et al., 1996; Romero-Brey et al., 2012; Bartenschlager et al., 2013; Paul et al., 2013). Budding of virion into the ER and egress through the secretory is tightly linked to lipoprotein synthesis (Lindenbach and Rice, 2013). Finally, the virus particle associates with lipoproteins to form lipoviroparticle which is essential for virus infectivity (Andre et al., 2002; Felmlee et al., 2013). Therefore, throughout the life cycle of the virus, there is a constant need for lipids and lipoproteins. Transactivation of the sterol regulatory element binding proteins, the master regulator of lipogenesis, has been observed in Huh7 cells infected with JFH1 (2a) or harboring a SGR and in cells ectopically expressing the core protein or NS4B (Waris et al., 2007; Rahman et al., 2009). It is not difficult to imagine that this can easily lead to perturbation of lipid homeostasis and trigger the UPR. Indeed, hepatitis C patients exhibit many lipid and lipoprotein metabolism disorders such as hepatic steatosis (fatty liver), hypobetalipoproteinaemia, and hypocholesterolemia (Serfaty et al., 2001; Colloredo et al., 2004; Felmlee et al., 2013). Experimentally, both exogenous and endogenous sources of fatty acids were capable of inducing ER stress in Huh7 cells infected with JFH1 or harboring a SGR (Rahman et al., 2009; Gunduz et al., 2012).
Figure 9.

Lipid perturbation elicits unfolded protein response. The hepatitis C virus (HCV) core protein (C) plays a part in lipid droplet (LD) formation (green arrow), and also coats the lipid droplet to initiate virion assembly and budding into the envelope proteins E1- and E2-coated endoplasmic reticulum (ER). Maturation and egress of the virion is tightly coupled to host very low density lipoprotein (VLDL) synthesis (A, apolipoprotein A; C, apolipoprotein C; E, apolipoprotein E), resulting in the formation of a lipoviroparticle (LVP). The non-structural (NS) 4B and NS5A proteins induce formation of the membranous web (brown arrow). The core and NS4B proteins have been shown to transactivate the sterol regulatory element binding protein (SREBP), the master regulator of lipogenesis (purple arrow). Perturbation in any of these lipid synthesis pathways will easily signal to elicit the unfolded protein response (UPR) (pink arrow). Putative lipid metabolic steps vulnerable to perturbation are marked with red crosses.
Concluding remarks
Despite overwhelming evidence from in vivo (transgenic and humanized mice) and in vitro studies to indicate that HCV infection causes ER stress and induces the UPR, we still need to confirm the presence of ER stress in hepatitis C patients by conducting clinical studies at single cells level (Liberman et al., 1999; Tardif et al., 2002, 2004; Benali-Furet et al., 2005; Chan and Egan, 2005, 2009; Ciccaglione et al., 2005, 2007; Zheng et al., 2005; Tumurbaatar et al., 2007; Sekine-Osajima et al., 2008; Joyce et al., 2009; Li et al., 2009; Mishima et al., 2010; Von Dem Bussche et al., 2010; Funaoka et al., 2011; Merquiol et al., 2011; Shinohara et al., 2013). Clinical data will need to be further corroborated and elaborated using well-controlled experiments. Genotype is a major determinant of IFN responsiveness and disease progression, therefore, it is important that studies should be extended to other genotypes before it can be generalized that ER stress is a common phenomenon of chronic hepatitis C (Chayama and Hayes, 2011; Ripoli and Pazienza, 2011).
A number of steps in the virus life cycle are potential trigger of ER stress e.g., the maturation of the viral envelope glycoproteins in the ER, formation of replication complex on the ER and virus assembly and budding of virus particles into the ER (Scheel and Rice, 2013). Current evidence suggests a major role of the structural proteins, with the NS proteins playing a modulating role (Tardif et al., 2002, 2004; Mishima et al., 2010; Von Dem Bussche et al., 2010; Funaoka et al., 2011; Mohl et al., 2012). More work still needs to be done to decipher the mechanisms of UPR induction and the answer will lead to a better understanding of virus-host interaction and may uncover novel mechanisms of UPR sensing in general.
Author contributions
Shiu-Wan Chan conceived, designed, and wrote the review article.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Work quoted from my laboratory was supported by a grant from the Medical Research Council, United Kingdom.
References
- Abraham R., Mudaliar P., Padmanabhan A., Sreekumar E. (2013). Induction of cytopathogenicity in human glioblastoma cells by chikungunya virus. PLoS ONE 8:e75854 10.1371/journal.pone.0075854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnello V., Abel G., Elfahal M., Knight G. B., Zhang Q. X. (1999). Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 96, 12766–12771 10.1073/pnas.96.22.12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose R. L., Mackenzie J. M. (2011). West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 85, 2723–2732 10.1128/JVI.02050-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose R. L., Mackenzie J. M. (2013). ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 87, 2206–2214 10.1128/JVI.02097-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre P., Komurian-Pradel F., Deforges S., Perret M., Berland J. L., Sodoyer M., et al. (2002). Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 76, 6919–6928 10.1128/JVI.76.14.6919-6928.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselah T., Bieche I., Mansouri A., Laurendeau I., Cazals-Hatem D., Feldmann G., et al. (2010). In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 221, 264–274 10.1002/path.2703 [DOI] [PubMed] [Google Scholar]
- Banerjee R., Dasgupta A. (2001). Specific interaction of hepatitis C virus protease/helicase NS3 with the 3'-terminal sequences of viral positive- and negative-strand RNA. J. Virol. 75, 1708–1721 10.1128/JVI.75.4.1708-1721.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry G., Fragkoudis R., Ferguson M. C., Lulla A., Merits A., Kohl A., et al. (2010). Semliki forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J. Virol. 84, 7369–7377 10.1128/JVI.02310-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R., Lohmann V., Penin F. (2013). The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 11, 482–496 10.1038/nrmicro3046 [DOI] [PubMed] [Google Scholar]
- Bartosch B., Dubuisson J., Cosset F. L. (2003). Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197, 633–642 10.1084/jem.20021756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens S. E., Tomei L., De Francesco R. (1996). Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 15, 12–22 [PMC free article] [PubMed] [Google Scholar]
- Benali-Furet N. L., Chami M., Houel L., De Giorgi F., Vernejoul F., Lagorce D., et al. (2005). Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24, 4921–4933 10.1038/sj.onc.1208673 [DOI] [PubMed] [Google Scholar]
- Bertolotti A., Zhang Y. H., Hendershot L. M., Harding H. P., Ron D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 10.1038/35014014 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S., Sen U., Vrati S. (2014). Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J. Gen. Virol. 95, 71–79 10.1099/vir.0.057265-0 [DOI] [PubMed] [Google Scholar]
- Blazquez A. B., Escribano-Romero E., Merino-Ramos T., Saiz J. C., Martin-Acebes M. A. (2013). Infection with Usutu virus induces an autophagic response in mammalian cells. PLoS Negl. Trop. Dis. 7:e2509 10.1371/journal.pntd.0002509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight K. J., McKeating J. A., Marcotrigiano J., Rice C. M. (2003). Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77, 3181–3190 10.1128/JVI.77.5.3181-3190.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer J. W., Jackowski S. (2012). UPR-mediated membrane biogenesis in B cells. Biochem. Res. Int. 2012:738471 10.1155/2012/738471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett H. F., Audas T. E., Liang G., Lu R. R. (2012). Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 17, 473–483 10.1007/s12192-012-0324-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M., Zeng H. Q., Urano F., Till J. H., Hubbard S. R., Harding H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- Chan C. P., Siu K. L., Chin K. T., Yuen K. Y., Zheng B., Jin D. Y. (2006). Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 80, 9279–9287 10.1128/JVI.00659-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S.-W., Egan P. A. (2005). Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 19, 1510–1512 10.1096/fj.04-3455fje [DOI] [PubMed] [Google Scholar]
- Chan S.-W., Egan P. A. (2009). Effects of hepatitis C virus envelope glycoprotein unfolded protein response activation on translation and transcription. Arch. Virol. 154, 1631–1640 10.1007/s00705-009-0495-5 [DOI] [PubMed] [Google Scholar]
- Chayama K., Hayes C. N. (2011). Hepatitis C virus: How genetic variability affects pathobiology of disease. J. Gastroenterol. Hepatol. 26 Suppl. 1, 83–95 10.1111/j.1440-1746.2010.06550.x [DOI] [PubMed] [Google Scholar]
- Choo Q. L., Kuo G., Weiner A. J., Overby L. R., Bradley D. W., Houghton M. (1989). Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral-hepatitis genome. Science 244, 359–362 10.1126/science.2523562 [DOI] [PubMed] [Google Scholar]
- Choukhi A., Ung S., Wychowski C., Dubuisson J. (1998). Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J. Virol. 72, 3851–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua P. K., Wang R. Y., Lin M. H., Masuda T., Suk F. M., Shih C. (2005). Reduced secretion of virions and hepatitis B virus (HBV) surface antigen of a naturally occurring HBV variant correlates with the accumulation of the small S envelope protein in the endoplasmic reticulum and Golgi apparatus. J. Virol. 79, 13483–13496 10.1128/JVI.79.21.13483-13496.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccaglione A. R., Costantino A., Tritarelli E., Marcantonio C., Equestre M., Marziliano N., et al. (2005). Activation of endoplasmic reticulum stress response by hepatitis C virus proteins. Arch. Virol. 150, 1339–1356 10.1007/s00705-004-0487-4 [DOI] [PubMed] [Google Scholar]
- Ciccaglione A. R., Marcantonio C., Tritarelli E., Equestre M., Vendittelli F., Costantino A., et al. (2007). Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus Res. 126, 128–138 10.1016/j.virusres.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Clavarino G., Claudio N., Couderc T., Dalet A., Judith D., Camosseto V., et al. (2012). Induction of GADD34 is necessary for dsRNA-dependent interferon-beta production and participates in the control of Chikungunya virus infection. PLoS Pathog. 8:e1002708 10.1371/journal.ppat.1002708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colloredo G., Sonzogni A., Rubbia-Brandt L., Negro F. (2004). Hepatitis C virus genotype 1 associated with massive steatosis of the liver and hypo-beta-lipoproteinemia. J. Hepatol. 40, 562–563 10.1016/j.jhep.2003.11.027 [DOI] [PubMed] [Google Scholar]
- Connor J. H., Weiser D. C., Li S., Hallenbeck J. M., Shenolikar S. (2001). Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell. Biol. 21, 6841–6850 10.1128/MCB.21.20.6841-6850.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle J. J., Finer-Moore J. S., Papa F. R., Stroud R. M., Walter P. (2005). On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 102, 18773–18784 10.1073/pnas.0509487102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling N. J., Cook S. J. (2014). The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta. [Epub ahead of print]. 10.1016/j.bbamcr.2014.01.009 [DOI] [PubMed] [Google Scholar]
- Date T., Morikawa K., Tanaka Y., Tanaka-Kaneko K., Sata T., Mizokami M., et al. (2012). Replication and infectivity of a novel genotype 1b hepatitis C virus clone. Microbiol. Immunol. 56, 308–317 10.1111/j.1348-0421.2012.00437.x [DOI] [PubMed] [Google Scholar]
- Dediego M. L., Nieto-Torres J. L., Jimenez-Guardeno J. M., Regla-Nava J. A., Alvarez E., Oliveros J. C., et al. (2011). Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 7:e1002315 10.1371/journal.ppat.1002315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleersnyder V., Pillez A., Wychowski C., Blight K., Xu J., Hahn Y. S., et al. (1997). Formation of native hepatitis C virus glycoprotein complexes. J. Virol. 71, 697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimcheff D. E., Askovic S., Baker A. H., Johnson-Fowler C., Portis J. L. (2003). Endoplasmic reticulum stress is a determinant of retrovirus-induced spongiform neurodegeneration. J. Virol. 77, 12617–12629 10.1128/JVI.77.23.12617-12629.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimcheff D. E., Faasse M. A., McAtee F. J., Portis J. L. (2004). Endoplasmic reticulum (ER) stress induced by a neurovirulent mouse retrovirus is associated with prolonged BiP binding and retention of a viral protein in the ER. J. Biol. Chem. 279, 33782–33790 10.1074/jbc.M403304200 [DOI] [PubMed] [Google Scholar]
- Domingo E., Sheldon J., Perales C. (2012). Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76, 159–216 10.1128/mmbr.05023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner M., Horwitz J. A., Robbins J. B., Barry W. T., Feng Q., Mu K., et al. (2011). A genetically humanized mouse model for hepatitis C virus infection. Nature 474, 208–211 10.1038/nature10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson J., Hsu H. H., Cheung R. C., Greenberg H. B., Russell D. G., Rice C. M. (1994). Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68, 6147–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson J., Rice C. M. (1996). Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 70, 778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan P. A., Sobkowiak M., Chan S. W. (2013). Hepatitis C virus envelope protein E1 binds PERK and represses the unfolded protein response. Open Virol. J. 7, 37–40 10.2174/1874357901307010037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. J., Von Hahn T., Tscherne D. M., Syder A. J., Panis M., Wolk B., et al. (2007). Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- Failla C., Tomei L., De Francesco R. (1994). Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 68, 3753–3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar M. J., Hu K., Harris H. J., Davis C., Brimacombe C. L., Fletcher S. J., et al. (2012). Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 86, 4305–4316 10.1128/JVI.06996-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favreau D. J., Desforges M., St-Jean J. R., Talbot P. J. (2009). A human coronavirus OC43 variant harboring persistence-associated mutations in the S glycoprotein differentially induces the unfolded protein response in human neurons as compared to wild-type virus. Virology 395, 255–267 10.1016/j.virol.2009.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmlee D. J., Hafirassou M. L., Lefevre M., Baumert T. F., Schuster C. (2013). Hepatitis C virus, cholesterol and lipoproteins–impact for the viral life cycle and pathogenesis of liver disease. Viruses 5, 1292–1324 10.3390/v5051292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franck N., Le Seyec J., Guguen-Guillouzo C., Erdtmann L. (2005). Hepatitis C virus NS2 protein is phosphorylated by the protein kinase CK2 and targeted for degradation to the proteasome. J. Virol. 79, 2700–2708 10.1128/JVI.79.5.2700-2708.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R., Jarosch E., Urban J., Volkwein C., Sommer T. (2000). A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2, 379–384 10.1038/35017001 [DOI] [PubMed] [Google Scholar]
- Funaoka Y., Sakamoto N., Suda G., Itsui Y., Nakagawa M., Kakinuma S., et al. (2011). Analysis of interferon signaling by infectious hepatitis C virus clones with substitutions of core amino acids 70 and 91. J. Virol. 85, 5986–5994 10.1128/JVI.02583-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo I., Hernaez B., Munoz-Moreno R., Cuesta-Geijo M. A., Dalmau-Mena I., Alonso C. (2012). The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 3, e341 10.1038/cddis.2012.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L., Tu H., Shi S. T., Lee K. J., Asanaka M., Hwang S. B., et al. (2003). Interaction with a ubiquitin-like protein enhances the ubiquitination and degradation of hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77, 4149–4159 10.1128/JVI.77.7.4149-4159.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M., Pincus D., Gotthardt K., Gallagher C. M., Walter P. (2013). Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5:a013169 10.1101/cshperspect.a013169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner B. M., Walter P. (2011). Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894 10.1126/science.1209126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff H., Hewson R., Opstelten D. J. (1998). Virus maturation by budding. Microbiol. Mol. Biol. Rev. 62, 1171–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garoff H., Sjoberg M., Cheng R. H. (2004). Budding of alphaviruses. Virus Res. 106, 103–116 10.1016/j.virusres.2004.08.008 [DOI] [PubMed] [Google Scholar]
- Glimcher L. H., Lee A. H. (2009). From sugar to fat: How the transcription factor XBP1 regulates hepatic lipogenesis. Ann. N.Y. Acad. Sci. 1173 Suppl. 1, E2–E9 10.1111/j.1749-6632.2009.04956.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnann A., Riordan J. R., Wolf D. H. (2004). Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol. Biol. Cell 15, 4125–4135 10.1091/mbc.E04-01-0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein J. M., Scheel T. K., Hoegh A. M., Lademann J. B., Eugen-Olsen J., Lisby G., et al. (2007). Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 133, 1614–1626 10.1053/j.gastro.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Gottwein J. M., Scheel T. K., Jensen T. B., Lademann J. B., Prentoe J. C., Knudsen M. L., et al. (2009). Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49, 364–377 10.1002/hep.22673 [DOI] [PubMed] [Google Scholar]
- Grakoui A., McCourt D. W., Wychowski C., Feinstone S. M., Rice C. M. (1993a). Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 67, 2832–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A., Wychowski C., Lin C., Feinstone S. M., Rice C. M. (1993b). Expression and identification of hepatitis C virus polyprotein cleavage products. J. Virol. 67, 1385–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz F., Aboulnasr F. M., Chandra P. K., Hazari S., Poat B., Baker D. P., et al. (2012). Free fatty acids induce ER stress and block antiviral activity of interferon alpha against hepatitis C virus in cell culture. Virol. J. 9, 143 10.1186/1743-422X-9-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Back S. H., Hur J., Lin Y. H., Gildersleeve R., Shan J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490 10.1038/ncb2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H. P., Novoa I., Zhang Y. H., Zeng H. Q., Wek R., Schapira M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y. H., Ron D. (1999). Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature 397, 271–274 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- Hijikata M., Kato N., Ootsuyama Y., Nakagawa M., Ohkoshi S., Shimotohno K. (1991a). Hypervariable regions in the putative glycoprotein of hepatitis-c virus. Biochem. Biophys. Res. Commun. 175, 220–228 10.1016/S0006-291X(05)81223-9 [DOI] [PubMed] [Google Scholar]
- Hijikata M., Kato N., Ootsuyama Y., Nakagawa M., Shimotohno K. (1991b). Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. U.S.A. 88, 5547–5551 10.1073/pnas.88.13.5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata M., Mizushima H., Tanji Y., Komoda Y., Hirowatari Y., Akagi T., et al. (1993). Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 90, 10773–10777 10.1073/pnas.90.22.10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J. (2013). Evolution of the unfolded protein response. Biochim. Biophys. Acta 1833, 2458–2463 10.1016/j.bbamcr.2013.01.016 [DOI] [PubMed] [Google Scholar]
- Hollien J., Lin J. H., Li H., Stevens N., Walter P., Weissman J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331 10.1083/jcb.200903014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C. (2010). Does hepatitis C virus really form quasispecies? Infect. Genet. Evol. 10, 431–432 10.1016/j.meegid.2010.03.006 [DOI] [PubMed] [Google Scholar]
- Hsieh Y. H., Su I. J., Wang H. C., Chang W. W., Lei H. Y., Lai M. D., et al. (2004). Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 25, 2023–2032 10.1093/carcin/bgh207 [DOI] [PubMed] [Google Scholar]
- Hung J. H., Teng Y. N., Wang L. H., Su I. J., Wang C. C., Huang W., et al. (2011). Induction of Bcl-2 expression by hepatitis B virus pre-S2 mutant large surface protein resistance to 5-fluorouracil treatment in Huh-7 cells. PLoS ONE 6:e28977 10.1371/journal.pone.0028977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M., Yi M. K., Li K., Lemon S. A. (2002). Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 76, 2997–3006 10.1128/JVI.76.6.2997-3006.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isler J. A., Skalet A. H., Alwine J. C. (2005). Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79, 6890–6899 10.1128/JVI.79.11.6890-6899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T., Hosoda A., Okuda T., Kamigori Y., Nomura-Furuwatari C., Kimata Y., et al. (2001). Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 3, 158–164 10.1038/35055065 [DOI] [PubMed] [Google Scholar]
- Jensen T. B., Gottwein J. M., Scheel T. K., Hoegh A. M., Eugen-Olsen J., Bukh J. (2008). Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 198, 1756–1765 10.1086/593021 [DOI] [PubMed] [Google Scholar]
- Jheng J. R., Lau K. S., Tang W. F., Wu M. S., Horng J. T. (2010). Endoplasmic reticulum stress is induced and modulated by enterovirus 71. Cell. Microbiol. 12, 796–813 10.1111/j.1462-5822.2010.01434.x [DOI] [PubMed] [Google Scholar]
- Jiang J., Cun W., Wu X., Shi Q., Tang H., Luo G. (2012). Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 86, 7256–7267 10.1128/JVI.07222-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling C. L., Yi M., Lancaster A. M., Lemon S. M., Sarnow P. (2005). Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309, 1577–1581 10.1126/science.1113329 [DOI] [PubMed] [Google Scholar]
- Jordan R., Wang L. J., Graczyk T. M., Block T. M., Romano P. R. (2002). Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress- mediated apoptosis of MDBK cells. J. Virol. 76, 9588–9599 10.1128/JVI.76.19.9588-9599.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert P. E., Werneke S. W., De La Calle C., Guivel-Benhassine F., Giodini A., Peduto L., et al. (2012). Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 209, 1029–1047 10.1084/jem.20110996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce M. A., Walters K.-A., Lamb S.-E., Yeh M. M., Zhu L.-F., Kneteman N., et al. (2009). HCV Induces Oxidative and ER Stress, and Sensitizes Infected Cells to Apoptosis in SCID/Alb-uPA Mice. PLoS Pathog. 5:e1000291 10.1371/journal.ppat.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T., Date T., Miyamoto M., Furusaka A., Tokushige K., Mizokami M., et al. (2003). Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125, 1808–1817 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- Kaul A., Stauffer S., Berger C., Pertel T., Schmitt J., Kallis S., et al. (2009). Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546 10.1371/annotation/29dffb2a-5d37-4196-b166-b0b15bc2f3fd [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke P. Y., Chen S. S. (2011). Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 121, 37–56 10.1172/JCI41474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klomporn P., Panyasrivanit M., Wikan N., Smith D. R. (2011). Dengue infection of monocytic cells activates ER stress pathways, but apoptosis is induced through both extrinsic and intrinsic pathways. Virology 409, 189–197 10.1016/j.virol.2010.10.010 [DOI] [PubMed] [Google Scholar]
- Kolykhalov A. A., Agapov E. V., Blight K. J., Mihalik K., Feinstone S. M., Rice C. M. (1997). Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277, 570–574 10.1126/science.277.5325.570 [DOI] [PubMed] [Google Scholar]
- Kopp M., Murray C. L., Jones C. T., Rice C. M. (2010). Genetic analysis of the carboxy-terminal region of the hepatitis C virus core protein. J. Virol. 84, 1666–1673 10.1128/JVI.02043-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiken C., Simmonds P. (2009). Nomenclature and numbering of the Hepatitis C virus, in Methods in Molecular Biology, ed Tang H. (New York, NY: Humana Press; ), 33–53 [DOI] [PubMed] [Google Scholar]
- Lai C. W., Otero J. H., Hendershot L. M., Snapp E. (2012). ERdj4 protein is a soluble endoplasmic reticulum (ER) DnaJ family protein that interacts with ER-associated degradation machinery. J. Biol. Chem. 287, 7969–7978 10.1074/jbc.M111.311290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillette D., Bartosch B., Nourrisson D., Verney G., Cosset F. L., Penin F., et al. (2006). Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J. Biol. Chem. 281, 3909–3917 10.1074/jbc.M509747200 [DOI] [PubMed] [Google Scholar]
- Lee A. H., Iwakoshi N. N., Glimcher L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 10.1128/MCB.23.21.7448-7459.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. H., Scapa E. F., Cohen D. E., Glimcher L. H. (2008). Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496 10.1126/science.1158042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Ye L., Yu X., Xu B., Li K., Zhu X., et al. (2009). Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappa B activation. Virology 391, 257–264 10.1016/j.virol.2009.06.039 [DOI] [PubMed] [Google Scholar]
- Li Y. P., Gottwein J. M., Scheel T. K., Jensen T. B., Bukh J. (2011). MicroRNA-122 antagonism against hepatitis C virus genotypes 1-6 and reduced efficacy by host RNA insertion or mutations in the HCV 5' UTR. Proc. Natl. Acad. Sci. U.S.A. 108, 4991–4996 10.1073/pnas.1016606108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. P., Ramirez S., Gottwein J. M., Scheel T. K., Mikkelsen L., Purcell R. H., et al. (2012a). Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc. Natl. Acad. Sci. U.S.A. 109, E1101–E1110 10.1073/pnas.1203829109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. P., Ramirez S., Jensen S. B., Purcell R. H., Gottwein J. M., Bukh J. (2012b). Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc. Natl. Acad. Sci. U.S.A. 109, 19757–19762 10.1073/pnas.1218260109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Shilagard T., Xiao S. Y., Snyder N., Lau D., Cicalese L., et al. (2009). Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology 137, 1448–1458 10.1053/j.gastro.2009.07.050 [DOI] [PubMed] [Google Scholar]
- Liberman E., Fong Y. L., Selby M. J., Choo Q. L., Cousens L., Houghton M., et al. (1999). Activation of the grp78 and grp94 promoters by hepatitis C virus E2 envelope protein. J. Virol. 73, 3718–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Lindenbach B. D., Pragai B. M., McCourt D. W., Rice C. M. (1994). Processing in the hepatitis-c virus e2-ns2 region - identification of p7 and 2 distinct e2-specific products with different c-termini. J. Virol. 68, 5063–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach B. D., Evans M. J., Syder A. J., Wolk B., Tellinghuisen T. L., Liu C. C., et al. (2005). Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- Lindenbach B. D., Rice C. M. (2013). The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 11, 688–700 10.1038/nrmicro3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Huangfu W. C., Kumar K. G., Qian J., Casey J. P., Hamanaka R. B., et al. (2009). Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5, 72–83 10.1016/j.chom.2008.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Kuang X., Kim H. T., Stoica G., Qiang W., Scofield V. L., et al. (2004). Possible involvement of both endoplasmic reticulum- and mitochondria-dependent pathways in MoMuLV-ts1-induced apoptosis in astrocytes. J. Neurovirol. 10, 189–198 10.1080/13550280490448043 [DOI] [PubMed] [Google Scholar]
- Liu N., Scofield V. L., Qiang W., Yan M., Kuang X., Wong P. K. (2006). Interaction between endoplasmic reticulum stress and caspase 8 activation in retrovirus MoMuLV-ts1-infected astrocytes. Virology 348, 398–405 10.1016/j.virol.2006.01.002 [DOI] [PubMed] [Google Scholar]
- Lohmann V., Bartenschlager R. (2014). On the history of hepatitis C virus cell culture systems. J. Med. Chem. 57, 1627–1642 10.1021/jm401401n [DOI] [PubMed] [Google Scholar]
- Lohmann V., Korner F., Koch J. O., Herian U., Theilmann L., Bartenschlager R. (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 10.1126/science.285.5424.110 [DOI] [PubMed] [Google Scholar]
- Mailly L., Robinet E., Meuleman P., Baumert T. F., Zeisel M. B. (2013). Hepatitis C virus infection and related liver disease: the quest for the best animal model. Front. Microbiol. 4:213 10.3389/fmicb.2013.00212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell M., Esteban J. I., Quer J., Genesca J., Weiner A., Esteban R., et al. (1992). Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol. 66, 3225–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough K. D., Martindale J. L., Klotz L. O., Aw T. Y., Holbrook N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259 10.1128/MCB.21.4.1249-1259.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLauchlan J., Lemberg M. K., Hope G., Martoglio B. (2002). Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 21, 3980–3988 10.1093/emboj/cdf414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson S., Powell E. E., Barrie H. D., Clouston A. D., McGuckin M., Jonsson J. R. (2011). No evidence of the unfolded protein response in patients with chronic hepatitis C virus infection. J. Gastroenterol. Hepatol. 26, 319–327 10.1111/j.1440-1746.2010.06368.x [DOI] [PubMed] [Google Scholar]
- Medigeshi G. R., Lancaster A. M., Hirsch A. J., Briese T., Lipkin W. I., Defilippis V., et al. (2007). West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81, 10849–10860 10.1128/JVI.01151-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meertens L., Bertaux C., Dragic T. (2006). Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 80, 11571–11578 10.1128/JVI.01717-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer D. F., Schiller D. E., Elliott J. F., Douglas D. N., Hao C., Rinfret A., et al. (2001). Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7, 927–933 10.1038/90968 [DOI] [PubMed] [Google Scholar]
- Merola M., Brazzoli M., Cocchiarella F., Heile J. M., Helenius A., Weiner A. J., et al. (2001). Folding of hepatitis C virus E1 glycoprotein in a cell-free system. J. Virol. 75, 11205–11217 10.1128/JVI.75.22.11205-11217.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merquiol E., Uzi D., Mueller T., Goldenberg D., Nahmias Y., Xavier R. J., et al. (2011). HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS ONE 6:e24660 10.1371/journal.pone.0024660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima K., Sakamoto N., Sekine-Osajima Y., Nakagawa M., Itsui Y., Azuma S., et al. (2010). Cell culture and in vivo analyses of cytopathic hepatitis C virus mutants. Virology 405, 361–369 10.1016/j.virol.2010.06.020 [DOI] [PubMed] [Google Scholar]
- Mohl B. P., Tedbury P. R., Griffin S., Harris M. (2012). Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 86, 10724–10732 10.1128/JVI.01667-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriishi K., Mochizuki R., Moriya K., Miyamoto H., Mori Y., Abe T., et al. (2007). Critical role of PA28gamma in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 1661–1666 10.1073/pnas.0607312104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriishi K., Okabayashi T., Nakai K., Moriya K., Koike K., Murata S., et al. (2003). Proteasome activator PA28gamma-dependent nuclear retention and degradation of hepatitis C virus core protein. J. Virol. 77, 10237–10249 10.1128/JVI.77.19.10237-10249.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriishi K., Shoji I., Mori Y., Suzuki R., Suzuki T., Kataoka C., et al. (2010). Involvement of PA28gamma in the propagation of hepatitis C virus. Hepatology 52, 411–420 10.1002/hep.23680 [DOI] [PubMed] [Google Scholar]
- Moriya K., Fujie H., Shintani Y., Yotsuyanagi H., Tsutsumi T., Ishibashi K., et al. (1998). The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 4, 1065–1067 10.1038/2053 [DOI] [PubMed] [Google Scholar]
- Mulvey M., Arias C., Mohr I. (2007). Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J. Virol. 81, 3377–3390 10.1128/JVI.02191-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T. (2012). Retroviral env glycoprotein trafficking and incorporation into virions. Mol. Biol. Int. 2012:682850 10.1155/2012/682850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadanaka S., Okada T., Yoshida H., Mori K. (2007). Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 27, 1027–1043 10.1128/MCB.00408-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanua S., Yoshimura F. K. (2004). Mink epithelial cell killing by pathogenic murine leukemia viruses involves endoplasmic reticulum stress. J. Virol. 78, 12071–12074 10.1128/JVI.78.21.12071-12074.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., et al. (2002). ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355 10.1101/gad.992302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I., Zeng H. Q., Harding H. P., Ron D. (2001). Feedback inhibition of the unfolded protein response by GADD34- mediated dephosphorylation of eIF2 alpha. J. Cell Biol. 153, 1011–1022 10.1083/jcb.153.5.1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I., Zhang Y., Zeng H., Jungreis R., Harding H. P., Ron D. (2003). Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22, 1180–1187 10.1093/emboj/cdg112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y., Hosokawa N., Wada I., Nagata K. (2003). EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 10.1126/science.1079181 [DOI] [PubMed] [Google Scholar]
- Okamoto K., Mori Y., Komoda Y., Okamoto T., Okochi M., Takeda M., et al. (2008a). Intramembrane processing by signal peptide peptidase regulates the membrane localization of hepatitis C virus core protein and viral propagation. J. Virol. 82, 8349–8361 10.1128/JVI.00306-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T., Nishimura Y., Ichimura T., Suzuki K., Miyamura T., Suzuki T., et al. (2006). Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 25, 5015–5025 10.1038/sj.emboj.7601367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T., Omori H., Kaname Y., Abe T., Nishimura Y., Suzuki T., et al. (2008b). A single-amino-acid mutation in hepatitis C virus NS5A disrupting FKBP8 interaction impairs viral replication. J. Virol. 82, 3480–3489 10.1128/JVI.02253-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradkar P. N., Ooi E. E., Hanson B. J., Gubler D. J., Vasudevan S. G. (2011). Unfolded protein response (UPR) gene expression during antibody-dependent enhanced infection of cultured monocytes correlates with dengue disease severity. Biosci. Rep. 31, 221–230 10.1042/BSR20100078 [DOI] [PubMed] [Google Scholar]
- Paul D., Hoppe S., Saher G., Krijnse-Locker J., Bartenschlager R. (2013). Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 87, 10612–10627 10.1128/JVI.01370-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavio N., Romano P. R., Graczyk T. M., Feinstone S. M., Taylor D. R. (2003). Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 77, 3578–3585 10.1128/JVI.77.6.3578-3585.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavio N., Taylor D. R., Lai M. M. C. (2002). Detection of a novel unglycosylated form of hepatitis C virus E2 envelope protein that is located in the cytosol and interacts with PKR. J. Virol. 76, 1265–1272 10.1128/JVI.76.3.1265-1272.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena J., Harris E. (2011). Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 286, 14226–14236 10.1074/jbc.M111.222703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pene V., Hernandez C., Vauloup-Fellous C., Garaud-Aunis J., Rosenberg A. R. (2009). Sequential processing of hepatitis C virus core protein by host cell signal peptidase and signal peptide peptidase: a reassessment. J. Viral Hepat. 16, 705–715 10.1111/j.1365-2893.2009.01118.x [DOI] [PubMed] [Google Scholar]
- Peng L., Liang D., Tong W., Li J., Yuan Z. (2010). Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J. Biol. Chem. 285, 20870–20881 10.1074/jbc.M110.112045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson T. C., Diamond M. S. (2012). Degrees of maturity: the complex structure and biology of flaviviruses. Curr. Opin. Virol. 2, 168–175 10.1016/j.coviro.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., et al. (1998). Binding of hepatitis C virus to CD81. Science 282, 938–941 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- Ploss A., Evans M. J., Gaysinskaya V. A., Panis M., You H., De Jong Y. P., et al. (2009). Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portis J. L., Askovich P., Austin J., Gutierrez-Cotto Y., McAtee F. J. (2009). The degree of folding instability of the envelope protein of a neurovirulent murine retrovirus correlates with the severity of the neurological disease. J. Virol. 83, 6079–6086 10.1128/JVI.02647-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange R. (2012). Host factors involved in hepatitis B virus maturation, assembly, and egress. Med. Microbiol. Immunol. 201, 449–461 10.1007/s00430-012-0267-9 [DOI] [PubMed] [Google Scholar]
- Qian Z., Xuan B., Chapa T. J., Gualberto N., Yu D. (2012). Murine cytomegalovirus targets transcription factor ATF4 to exploit the unfolded-protein response. J. Virol. 86, 6712–6723 10.1128/JVI.00200-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W., Cahill J. M., Liu J., Kuang X., Liu N., Scofield V. L., et al. (2004). Activation of transcription factor Nrf-2 and its downstream targets in response to moloney murine leukemia virus ts1-induced thiol depletion and oxidative stress in astrocytes. J. Virol. 78, 11926–11938 10.1128/JVI.78.21.11926-11938.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S. M., Qadri I., Janssen R. C., Friedman J. E. (2009). Fenofibrate and PBA prevent fatty acid-induced loss of adiponectin receptor and pAMPK in human hepatoma cells and in hepatitis C virus-induced steatosis. J. Lipid Res. 50, 2193–2202 10.1194/jlr.M800633-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S., Li Y. P., Jensen S. B., Pedersen J., Gottwein J. M., Bukh J. (2014). Highly efficient infectious cell culture of three hepatitis C virus genotype 2b strains and sensitivity to lead protease, nonstructural protein 5A, and polymerase inhibitors. Hepatology 59, 395–407 10.1002/hep.26660 [DOI] [PubMed] [Google Scholar]
- Rathore A. P., Ng M. L., Vasudevan S. G. (2013). Differential unfolded protein response during Chikungunya and Sindbis virus infection: CHIKV nsP4 suppresses eIF2alpha phosphorylation. Virol. J. 10, 36 10.1186/1743-422X-10-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoli M., Pazienza V. (2011). Impact of HCV genetic differences on pathobiology of disease. Expert Rev. Anti Infect. Ther. 9, 747–759 10.1586/eri.11.94 [DOI] [PubMed] [Google Scholar]
- Romero-Brey I., Merz A., Chiramel A., Lee J. Y., Chlanda P., Haselman U., et al. (2012). Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8:e1003056 10.1371/journal.ppat.1003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M., Suzuki R., Watanabe N., Masaki T., Tomonaga M., Muhammad A., et al. (2011). Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 286, 37264–37273 10.1074/jbc.M111.259085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santolini E., Migliaccio G., La Monica N. (1994). Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 68, 3631–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarselli E., Ansuini H., Cerino R., Roccasecca R. M., Acali S., Filocamo G., et al. (2002). The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel T. K., Gottwein J. M., Jensen T. B., Prentoe J. C., Hoegh A. M., Alter H. J., et al. (2008). Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. U.S.A. 105, 997–1002 10.1073/pnas.0711044105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel T. K. H., Rice C. M. (2013). Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849 10.1038/nm.3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schregel V., Jacobi S., Penin F., Tautz N. (2009). Hepatitis C virus NS2 is a protease stimulated by cofactor domains in NS3. Proc. Natl. Acad. Sci. U.S.A. 106, 5342–5347 10.1073/pnas.0810950106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine-Osajima Y., Sakamoto N., Mishima K., Nakagawa M., Itsui Y., Tasaka M., et al. (2008). Development of plaque assays for hepatitis C virus-JFH1 strain and isolation of mutants with enhanced cytopathogenicity and replication capacity. Virology 371, 71–85 10.1016/j.virol.2007.09.019 [DOI] [PubMed] [Google Scholar]
- Selby M., Erickson A., Dong C., Cooper S., Parham P., Houghton M., et al. (1999). Hepatitis C virus envelope glycoprotein E1 originates in the endoplasmic reticulum and requires cytoplasmic processing for presentation by class I MHC molecules. J. Immunol. 162, 669–676 [PubMed] [Google Scholar]
- Serfaty L., Andreani T., Giral P., Carbonell N., Chazouilleres O., Poupon R. (2001). Hepatitis C virus induced hypobetalipoproteinemia: a possible mechanism for steatosis in chronic hepatitis C. J. Hepatol. 34, 428–434 10.1016/S0168-8278(00)00036-2 [DOI] [PubMed] [Google Scholar]
- Shen J. S., Chen X., Hendershot L., Prywes R. (2002a). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 3, 99–111 10.1016/S1534-5807(02)00203-4 [DOI] [PubMed] [Google Scholar]
- Shen Y., Meunier L., Hendershot L. M. (2002b). Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem. 277, 15947–15956 10.1074/jbc.M112214200 [DOI] [PubMed] [Google Scholar]
- Shinohara Y., Imajo K., Yoneda M., Tomeno W., Ogawa Y., Kirikoshi H., et al. (2013). Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochem. Biophys. Res. Commun. 432, 326–332 10.1016/j.bbrc.2013.01.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakura M., Murakami K., Ichimura T., Suzuki R., Shimoji T., Fukuda K., et al. (2007). E6AP ubiquitin ligase mediates ubiquitylation and degradation of hepatitis C virus core protein. J. Virol. 81, 1174–1185 10.1128/JVI.01684-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuda M., Kondoh N., Imazeki N., Tanaka K., Okada T., Mori K., et al. (2003). Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 38, 605–614 10.1016/S0168-8278(03)00029-1 [DOI] [PubMed] [Google Scholar]
- Sidrauski C., Walter P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 10.1016/S0092-8674(00)80369-4 [DOI] [PubMed] [Google Scholar]
- Simmonds P. (2013). The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 369, 1–15 10.1007/978-3-642-27340-7_1 [DOI] [PubMed] [Google Scholar]
- Simmonds P., Alberti A., Alter H. J., Bonino F., Bradley D. W., Brechot C., et al. (1994). A Proposed System For the Nomenclature Of Hepatitis-C Viral Genotypes. Hepatology 19, 1321–1324 10.1002/hep.1840190538 [DOI] [PubMed] [Google Scholar]
- Simmonds P., Bukh J., Combet C., Deleage G., Enomoto N., Feinstone S., et al. (2005). Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42, 962–973 10.1002/hep.20819 [DOI] [PubMed] [Google Scholar]
- Siu K. L., Chan C. P., Kok K. H., Woo P. C., Jin D. Y. (2014). Comparative analysis of the activation of unfolded protein response by spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus HKU1. Cell Biosci. 4:3 10.1186/2045-3701-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. A., Schmechel S. C., Raghavan A., Abelson M., Reilly C., Katze M. G., et al. (2006). Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80, 2019–2033 10.1128/JVI.80.4.2019-2033.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl S., Burkhart J. M., Hinte F., Tirosh B., Mohr H., Zahedi R. P., et al. (2013). Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 9:e1003544 10.1371/journal.ppat.1003544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stertz S., Reichelt M., Spiegel M., Kuri T., Martinez-Sobrido L., Garcia-Sastre A., et al. (2007). The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361, 304–315 10.1016/j.virol.2006.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiffler J. D., Nguyen M., Sohn J. A., Liu C., Kaplan D., Seeger C. (2009). Focal distribution of hepatitis C virus RNA in infected livers. PLoS ONE 4:e6661 10.1371/journal.pone.0006661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H. L., Liao C. L., Lin Y. L. (2002). Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 76, 4162–4171 10.1128/JVI.76.9.4162-4171.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R., Moriishi K., Fukuda K., Shirakura M., Ishii K., Shoji I., et al. (2009). Proteasomal turnover of hepatitis C virus core protein is regulated by two distinct mechanisms: a ubiquitin-dependent mechanism and a ubiquitin-independent but PA28gamma-dependent mechanism. J. Virol. 83, 2389–2392 10.1128/JVI.01690-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I., Ron D. (2011). Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 10.1038/ncb0311-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguwa S., Kambara H., Omori H., Tani H., Abe T., Mori Y., et al. (2009). Cochaperone activity of human butyrate-induced transcript 1 facilitates hepatitis C virus replication through an Hsp90-dependent pathway. J. Virol. 83, 10427–10436 10.1128/JVI.01035-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguwa S., Okamoto T., Abe T., Mori Y., Suzuki T., Moriishi K., et al. (2008). Human butyrate-induced transcript 1 interacts with hepatitis C virus NS5A and regulates viral replication. J. Virol. 82, 2631–2641 10.1128/JVI.02153-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif K. D., Mori K., Kaufman R. J., Siddiqui A. (2004). Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164 10.1074/jbc.M312144200 [DOI] [PubMed] [Google Scholar]
- Tardif K. D., Mori K., Siddiqui A. (2002). Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 76, 7453–7459 10.1128/JVI.76.15.7453-7459.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. L. (2013). Global control of hepatitis C: where challenge meets opportunity. Nat. Med. 19, 850–858 10.1038/nm.3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W., Welihinda A. A., Kaufman R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 10.1101/gad.12.12.1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W. Y., Nagano-Fujii M., Hidajat R., Deng L., Takigawa Y., Hotta H. (2002). Physical interaction between hepatitis C virus NS4B protein and CREB-RP/ATF6beta. Biochem. Biophys. Res. Commun. 299, 366–372 10.1016/S0006-291X(02)02638-4 [DOI] [PubMed] [Google Scholar]
- Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., Walter P. (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER- associated degradation. Cell 101, 249–258 10.1016/S0092-8674(00)80835-1 [DOI] [PubMed] [Google Scholar]
- Tripathi L. P., Kambara H., Moriishi K., Morita E., Abe T., Mori Y., et al. (2012). Proteomic analysis of hepatitis C virus (HCV) core protein transfection and host regulator PA28gamma knockout in HCV pathogenesis: a network-based study. J. Proteome Res. 11, 3664–3679 10.1021/pr300121a [DOI] [PubMed] [Google Scholar]
- Tsukiyama-Kohara K., Iizuka N., Kohara M., Nomoto A. (1992). Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 66, 1476–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumurbaatar B., Sun Y., Chan T., Sun J. (2007). Cre-estrogen receptor-mediated hepatitis C virus structural protein expression in mice. J. Virol. Methods 146, 5–13 10.1016/j.jviromet.2007.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umareddy I., Pluquet O., Wang Q. Y., Vasudevan S. G., Chevet E., Gu F. (2007). Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol. J. 4:91 10.1186/1743-422X-4-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F., Wang X. Z., Bertolotti A., Zhang Y. H., Chung P., Harding H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- Vaney M. C., Duquerroy S., Rey F. A. (2013). Alphavirus structure: activation for entry at the target cell surface. Curr. Opin. Virol. 3, 151–158 10.1016/j.coviro.2013.04.003 [DOI] [PubMed] [Google Scholar]
- Van Huizen R., Martindale J. L., Gorospe M., Holbrook N. J. (2003). P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 278, 15558–15564 10.1074/jbc.M212074200 [DOI] [PubMed] [Google Scholar]
- Versteeg G. A., Van De Nes P. S., Bredenbeek P. J., Spaan W. J. (2007). The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J. Virol. 81, 10981–10990 10.1128/JVI.01033-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieyres G., Dubuisson J., Pietschmann T. (2014). Incorporation of hepatitis C virus e1 and e2 glycoproteins: the keystones on a peculiar virion. Viruses 6, 1149–1187 10.3390/v6031149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieyres G., Thomas X., Descamps V., Duverlie G., Patel A. H., Dubuisson J. (2010). Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 84, 10159–10168 10.1128/JVI.01180-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmer R., Van Der Ploeg K., Ron D. (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 110, 4628–4633 10.1073/pnas.1217611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dem Bussche A., Machida R., Li K., Loevinsohn G., Khander A., Wang J., et al. (2010). Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 53, 797–804 10.1016/j.jhep.2010.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z. J., et al. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wang H. C., Huang W., Lai M. D., Su I. J. (2006). Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 97, 683–688 10.1111/j.1349-7006.2006.00235.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. C., Wu H. C., Chen C. F., Fausto N., Lei H. Y., Su I. J. (2003). Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 163, 2441–2449 10.1016/S0002-9440(10)63599-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Kaufman R. J. (2012). The impact of the unfolded protein response on human disease. J. Cell Biol. 197, 857–867 10.1083/jcb.201110131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waris G., Felmlee D. J., Negro F., Siddiqui A. (2007). Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 81, 8122–8130 10.1128/JVI.00125-07 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Welsch C., Albrecht M., Maydt J., Herrmann E., Welker M. W., Sarrazin C., et al. (2007). Structural and functional comparison of the non-structural protein 4B in flaviviridae. J. Mol. Graph. Model. 26, 546–557 10.1016/j.jmgm.2007.03.012 [DOI] [PubMed] [Google Scholar]
- Wozniak A. L., Griffin S., Rowlands D., Harris M., Yi M., Lemon S. M., et al. (2010). Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 6:e1001087 10.1371/journal.ppat.1001087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y. P., Chang C. M., Hung C. Y., Tsai M. C., Schuyler S. C., Wang R. Y. (2011). Japanese encephalitis virus co-opts the ER-stress response protein GRP78 for viral infectivity. Virol. J. 8:128 10.1186/1743-422X-8-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W., Frank C. L., Korth M. J., Sopher B. L., Novoa I., Ron D., et al. (2002). Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. U.S.A. 99, 15920–15925 10.1073/pnas.252341799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M., Villanueva R. A., Thomas D. L., Wakita T., Lemon S. M. (2006). Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U.S.A. 103, 2310–2315 10.1073/pnas.0510727103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Haze K., Yanagi H., Yura T., Mori K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins - Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749 10.1074/jbc.273.50.33741 [DOI] [PubMed] [Google Scholar]
- Yoshida H., Matsui T., Hosokawa N., Kaufman R. J., Nagata K., Mori K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271 10.1016/S1534-5807(03)00022-4 [DOI] [PubMed] [Google Scholar]
- Yoshimura F. K., Luo X. (2007). Induction of endoplasmic reticulum stress in thymic lymphocytes by the envelope precursor polyprotein of a murine leukemia virus during the preleukemic period. J. Virol. 81, 4374–4377 10.1128/JVI.02292-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura F. K., Luo X., Zhao X., Gerard H. C., Hudson A. P. (2008). Up-regulation of a cellular protein at the translational level by a retrovirus. Proc. Natl. Acad. Sci. U.S.A. 105, 5543–5548 10.1073/pnas.0710526105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C., Achazi K., Niedrig M. (2013). Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Res. 178, 471–477 10.1016/j.virusres.2013.10.012 [DOI] [PubMed] [Google Scholar]
- Yu C. Y., Hsu Y. W., Liao C. L., Lin Y. L. (2006). Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 80, 11868–11880 10.1128/JVI.00879-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Yoshimura F. K. (2008). Expression of murine leukemia virus envelope protein is sufficient for the induction of apoptosis. J. Virol. 82, 2586–2589 10.1128/JVI.02291-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Gao B., Ye L., Kong L. B., Jing W., Yang X. J., et al. (2005). Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 43, 529–536 [PubMed] [Google Scholar]
- Zhong J., Gastaminza P., Cheng G. F., Kapadia S., Kato T., Burton D. R., et al. (2005). Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U.S.A. 102, 9294–9299 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]