

Abstract
Background
Leber's hereditary optic neuropathy (LHON) is a condition characterised by a rapid bilateral central vision loss due to death of the retinal ganglion cells, leading to visual impairment commonly occurring during young adulthood. The disease manifests itself more in male patients than female patients. The mtDNA mutations m.11778G>A, m.3460G>A and m.14484T>C are by far more frequent in LHON than any other mutation. In this report, a multi-generational Arab family from Kuwait with 14 male members with LHON was investigated.
Methods
Complete mtDNA mutational analysis by direct Sanger's sequencing was carried out to detect pathogenic mutations, polymorphisms and haplogrouping.
Results
All maternally related subjects from this study who were examined expressed the L3 haplotype background, with two concurrent mtDNA mutations, 10609T>C and 10663T>C, that led to non-conservative amino acid changes of Ile47Thr and Val65Ala, respectively. The two variations were absent in 144 normal and ethnicity-matched controls.
Conclusions
The two identified mutations associated with LHON in this family may exert their pathogenicity through a cumulative or haplogroup effect. This is the first report of the presence of two concurrent mutations in the ND4L gene in individuals with LHON who carry the L3 haplogroup.
Keywords: LHON, Pedigree, ND4L, mutation, Kuwait
Introduction
Leber's hereditary optic neuropathy (LHON) is a condition characterised by a rapid bilateral central vision loss due to the death of the retinal ganglion cells, leading to visual impairment. The peak age of onset in LHON is between 15 and 30 years.1 The disease usually affects male patients, whereas female patients tend to be carriers with a male to female ratio greater than 4 to 1. However, the expression of LHON in female patients is no less severe compared with male patients.2 Environmental factors such as tobacco and alcohol use increase the risk of LHON in carriers of the mtDNA mutations.3 4
Mutational defects in mtDNA genes can lead to a wide range of organ disorders due to defective mitochondrial oxidative phosphorylation pathway. The mitochondrial oxidative phosphorylation machinery is structured from five enzymatic complexes (I–V). The first of these complexes, complex I, is composed of seven structural subunits that are encoded by seven mitochondrial genes (ND1–6 and ND4L). While pathogenic mutations in all seven genes have been reported to cause a wide variety of clinical phenotypes, mutations in four of these (ND1, ND4, ND4L and ND6) are responsible for almost 90–95% of all LHON cases.5 The three primary mtDNA mutations, m.11778G>A, m.3460G>A or m.14484T>C (in the genes encoding the mitochondrial complex I subunit including ND4, ND1 and ND6, respectively) are far more frequent in LHON than any other mutation, and account for more than 90% of LHON cases.6 The prevalence of these mutations varies worldwide, but m.11778G>A is the most common; this mutation accounts for 70% of all cases in northern Europeans.7 m.14484T>C is the most common among French Canadians and can be tracked to a founder effect,8 but this mutation is uncommon in the UK and in Scandinavia.7 However, 10% of individuals with LHON do not harbour one of the above three common mutations.9
mtDNA mutational analysis for patients with LHON in some instances has revealed the presence of additional rare but pathological mutations in ND1 and ND6 genes.10 Polymorphic nucleotide changes (secondary mutations) characteristic of Eurasian mtDNA haplogroup J and its subclasses have been indicated to play a synergistic role in penetrance of the primary mutations 11778G>A and 14484T>C.11 Other mtDNA analysis of patients with LHON resulted in discovering more mutations in ND1 (4171C>A, 3733G>A and m.3635G>A) and in ND6 (14482C>G/A, 14568C>T and 14495A>G).12 Further genetic testing for families with LHON who belong to the J haplogroup has shown the presence of ND4L 10663T>C mutation.13 More mutations like m.14498C>T in ND614 and m.4640C>A in ND215 were also described, suggesting a pathogenic role in LHON.
In this report we describe a multigenerational Arab family with 14 LHON-affected members who lack the primary mtDNA mutations but carry two uniquely concurrent mutations in the ND4L gene and express the L3 haplogroup.
Methods
Cases enrolment
Maternally related family members from Kuwait were investigated in this study. This study has adhered to the tenets of the Declaration of Helsinki, and was approved by our institutional Ethics Review Committee. Written informed consent was obtained from all 13 participants who enrolled in this study. For minors (age <18 years), parental consent was obtained.
Clinical assessment
Out of 14 LHON-affected family members (figure 1), six were enrolled and were available for neuro-ophthalmic assessment, including best corrected visual acuity, colour vision testing using the pseudoisochromatic colour plates, pupillary testing, dilated fundus examination, Humphrey automated visual field and spectral domain optical coherence tomography (OCT) of the retinal nerve fibre layer (RNFL) using a circular 3.4 mm (1024 A-scans and 6×6 mm) and a macular 3D scan 512×128 (128 horizontal scan lines comprised of 512 A-scans) for measuring macular thickness and volume (Topcon 3D OCT 2000). The age range of enrolled affected subjects was between 12 and 24 years. The cases comprised three kindreds: the first was composed of one male member (V-1); the second consisted of seven members, including the two parents, a sister and four brothers; the third comprised two parents and two male children. The proband (VI-9) belonged to the third kindred. For non-pathological mutations validation/exclusion, unrelated Kuwaiti adult controls (144 in total; 80 women and 64 men) were used as a normal mtDNA sequence referencing.
Figure 1 .
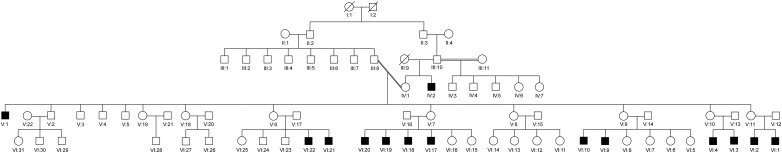
Pedigree for family structures and members’ IDs. Pedigree consisted of five generations of which 14 male members expressed Leber's hereditary optic neuropathy. All participants (V-1, V-2, V-7, V-9, V-14, V-16, VI-9, VI-10, VI-16, VI-17, VI-18, VI-19 and VI-20) received full mtDNA analysis.
Preparation of DNA samples
A 4 ml sample of blood from each patient was collected with informed consent which was approved by the Institutional Ethics Review Board. DNA was isolated using the standard procedure (PURAGENE) and resuspended in 300 µLs of TE buffer and then stored at −40°C until needed.
PCR amplification and sequencing analysis for LHON mutation screening
Full mtDNA sequence was carried out on all participating family members for haplogrouping and detection of LHON-causing mutations in ND1, ND4L, ND4 and ND6 genes. The primers’ used for sequencing were according to previous work by Rieder et al.16 A subset of these primer pairs with additional information is given in table 1. M13 oligo tags were added to the 5′ ends of all primers for robustness. Nucleotide coordinates are in accordance with the revised Cambridge reference sequence (rCRS) of human mtDNA.17 The resulting sequences were aligned against the reference sequence using the Applied Biosystems SeqScape sequence analysis software.
Table 1 .
List of overlapping primer pairs used for mtDNA sequencing
| Gene/region | Forward primer | Reverse primer | mtDNA region | PCR product size |
|---|---|---|---|---|
| ND1 | ND1F1 TACTTCACAAAGCGCCTTCC | ND1R1 ATGAAGAATAGGGCGAAGGG | 3150–3980 | 830bP |
| ND1F2 TGGCTCCTTTAACCTCTCCA | ND1R2 AAGGATTATGGATGCGGTTG | 3777–4660 | 883bP | |
| ND4L | ND4LF TCTCCATCTATTGATGAGGGTCT | ND4LR AATTAGGCTGTGGGTGGTTG | 9967–10 859 | 892bP |
| ND4 | ND4F1 GCCATACTAGTCTTTGCCGC | ND4R1 TTGAGAATGAGTGTGAGGCG | 10 653–11 511 | 858bP |
| ND4F2 TCACTCTCACTGCCCAAGAA | ND4R2 GGAGAATGGGGGATAGGTGT | 11 314–12 096 | 782bP | |
| ND4F3 TATCACTCTCCTACTTACAG | ND4R2 AGAAGGTTATAATTCCTACG | 11 929–12 793 | 864bP | |
| ND6 | ND6F GCATAATTAAACTTTACTTC | ND6R AGAATATTGAGGCGCCATTG | 14 081–15 017 | 936bP |
| D-loop | HSV I F1 TCATTGGACAAGTAGCATCC | HSV I R1 GAGTGGTTAATAGGGTGATAG | 15 792–16 400& 1–31 | 809bP |
| HSV II F2 GAGTGGTTAATAGGGTGATAG | HSV II R2 AGGCTAAGCGTTTTGAGCTG | 10–794 | 784bP | |
| M13 5′ Tag | M13F GTAAAACGACGGCCAGT | M13R CAGGAAACAGCTATGACC | – | – |
Six primer pairs were used to sequence ND1, ND4, ND4L and ND6 genes; two primer pairs were also used to sequence the hypervariable region of the mtDNA. M13 generic tag sequences were engineered into the 5′ end of each primer for sequencing reactions robustness.
Results
From the 14 LHON-affected male subjects in the studied pedigree, six had received complete ophthalmic assessment. The clinical presentation of two male subjects is summarised below.
Proband (VI-9): This 15-year-old male patient presented with sudden decrease in vision in the right eye. His brother had the same condition at 8 years of age and he was suspected of having LHON. There was also family history in his maternal uncle and cousins. Visual acuity was 20/400 in the right eye and 20/30 in the left eye. He recognised one of eight pseudoisochromatic colour plates in the right eye and eight of eight in the left eye. Pupillary testing did not reveal a relative afferent pupillary defect. Fundus examination shows a bilateral mild disc oedema (figure 2A). OCT showed thickening of the RNFL in both eyes (figure 2B) and OCT of the macula showed reduced macular thickness in a peri-central pattern in both eyes (figure 2C). The patient was started on coenzyme q10 600 mg/day. At 8-month follow-up, his visual acuity was 20/400 in the right and 20/25 in the left eye, and fundus examination showed temporal pallor of both optic discs. His OCT showed reduced RNFL thickness (figure 2D) and macular thickness in the right more than the left eye (figure 2E).
Figure 2 .

(A) Fundus photograph of proband VI-9 showing mild bilateral swelling of optic nerves. (B) Optical coherence tomography (OCT) of the retinal nerve fibre layer showing mild thickening of the inferior and temporal quadrants with resolution and subsequent thinning 8 months later (D). (C) OCT of the macula showing reduced macular thickness in both eyes and further thinning 8 months later (E). OD, right eye; OS, left eye.
Case 2 (VI-19): This 12-year-old male patient complained of being ‘unable to see at far’ and this started 3 months previously but remained stable since then. He also has a maternal uncle diagnosed with a similar condition. On examination his visual acuity was 20/30 in both eyes and he recognised only one of eight pseudoisochromatic colour plates in the right and two of eight in the left eye. His pupillary responses were brisk in both eyes and dilated fundus examination showed bilateral optic nerve head pallor (figure 3A). Humphrey visual fields (10-2) showed central visual field defects. OCT showed reduced RNFL thickness and macular thickness, and volume and follow-up examination 6 months later showed no significant reduction in the central macular thickness and volume and stable RNFL thickness (figure 3B–E).
Figure 3 .

(A) Fundus photograph of proband VI-19 showing optic nerve head pallor in both eyes. (B) Optical coherence tomography (OCT) of the retinal nerve fibre layer (RNFL) shows thinning in the temporal quadrant and repeat OCT 6 months later (3D) shows further thinning of the RNFL extending into the superior and inferior quadrants. (C) OCT of the macula shows thinning and a repeat OCT 6 months later shows progressive thinning as well (E). Four other family participants (VI-10, VI-17, VI-20 and V-1) received full clinical assessment and showed similar features (data not shown). OD, right eye; OS, left eye.
In comparing all six clinical cases we examined, various stages of the disease were observed (table 2). Cases VI-9, VI-19 and VI-20 presented with recent onset of visual loss (within 3 months). Cases VI-10, VI-17, VI-20 and V-1 had remote sudden onset of visual loss.
Table 2.
Clinical characteristics of the six patients who received full ophthalmic assessment
| Case no | Age | Visual acuity | Colour vision | Onset | Optic disc appearance | RNFL (μm) OD | RNFL (μm) OS | Macular thickness (μm) OD | Macular thickness (μm) OS | Macular volume (μm) OD | Macular volume (μm) OS | Treatment | Smoker? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VI-20 | 12 | 20/25 OU | 1/8 OU | 3 months subacute | Pallor | 89 | 86 | 250.2 | 245.4 | 7.07 | 6.94 | Coeq10 600 mg |
– |
| VI-19 | 14 | 20/400 OU | 1/8 OU | Acute | Disc oedema | 109 | 96 | 242 | 235.6 | 6.84 | 6.66 | Coeq10 600 mg |
– |
| VI-17 | 22 | CF OU | 0/8 | Acute (5 years ago) | Pallor | 56 | 49 | 217.8 | 221.8 | 6.16 | 6.27 | Coeq10 600 mg |
1 pack/day |
| VI-10 | 18 | 20/40 OD 20/25 OS |
1/8 OU | Acute (4 years ago) | Pallor | 72 | 73 | 229.2 | 230.7 | 6.48 | 6.52 | None | – |
| V-1 | 24 | 20/40 OU | 1/8 OU | Acute (4 years ago) | Disc oedema then pallor | 56 | 59 | 233.2 | 229.5 | 6.59 | 6.49 | None | – |
| VI-9 | 15 | 20/400 OD 20/30 OS |
1/8 OD 8/8 OS |
Acute (2 weeks) | Disc oedema OD >OS | 126 | 130 | 253.5 | 267 | 7.17 | 7.55 | Coeq10 600 mg |
Coeq10, coenzyme q10; OD, right eye; OS, left eye; OU, both eyes; RNFL, retinal nerve layer thickness.
Mutational analysis of mtDNA
Initially, sequence analysis of all four mitochondrial genes (ND1, ND4, ND4L and ND6) that are known to cause LHON when defective was carried out for all participants. Care was given during analysis to capture the existence of multiple variants. Three kindreds (composed of 13 individuals) from the family pedigree were enrolled and analysed. For a comprehensive analysis and haplotyping, full mtDNA (including HSV I and II) sequence was also pursued for all participants (V-1, V-2, V-7, V-9, V-14, V-16, VI-9, VI-10, VI-16, VI-17, VI-18, VI-19, VI-20).
Relative to the Cambridge sequence,17 52m tDNA homoplasmic variations could be detected (data available in online supplementary table S1). Among these, two variants mapped to the ND4L gene including 10609T>C and 10663T>C, which were coinherited by all examined maternally related descendent male and female participants. The unrelated paternal participants V-14 and V-16 did not carry these mutations. The consequence of these two mtDNA mutations is change in amino acids encoding the ND4L protein at codon 47 and codon 65, from isoleucine to threonine (Ile47Thr) and valine to alanine (Val65Ala), respectively. The clinical expression of LHON in selected cases from the pedigree was given in the clinical description above. To rule out whether the mutations are not just benign polymorphisms, 144 normal and ethnicity matched male and female controls were screened by sequencing for comparison. While 10663T>C was absent in all 144 controls, 10609T>C was only observed in 2/144, with a frequency of 0.01%.
The entire mtDNA genome sequences of all participants were also analysed for haplogrouping by utilising the haplotyping HmtDB database (http://www.hmtdb.uniba.it:8080/hmdb). All participants except V-14 and V-16 pointed to haplogroup L3'4 with 100% clustering, which further maps as a unique haplotype within the HmtDB database. However, both fathers (they are cousins) were classified into haplogroup HV with 100% clustering, and this haplogroup is distinguished as a unique haplotype within the HmtDB database. Haplogroup L3'4 has its ancestry in L3, and is believed to have arisen in Eastern Africa between 84 000 and 104 000 years ago. Immigrants carried this haplogroup across the Red Sea to the Arabian Peninsula.18 Haplogroup HV, however, is a west Eurasian haplogroup. It is also found to a much lesser extent in parts of East Africa, mainly in the population of Sudanese Arabs, where the frequency of Eurasian ancestry is marked at 22.5%.19
Discussion
We have identified in this study a unique double-mutant ND4L in an extended Arab pedigree from Kuwait. We detected two concurrent mutations 10609T>C and 10663T>C in the fifth and sixth generation participants. The mutations were coinherited by all maternally related descendants and were absent in the unrelated paternal samples (V-14 and V-16). These two neighbouring mutations led to non-conservative amino acid change in the encoded ND4L subunit protein at codon 47 and codon 65, from isoleucine to threonine (Ile47Thr) and valine to alanine (Val65Ala), respectively. We could not find in previous reports similar results of the dual mutant ND4L gene in association with LHON.
The mutations could affect oxidase complex formation resulting in loss (or suboptimal) complex function. Gender expression of LHON in this pedigree was consistent with the maternal transmission model and male affection. In general, male and female individuals carrying any of the three primary mtDNA mutations in a homoplasmic state are at an approximate 50% and 10% lifetime risk of developing visual loss, respectively.8 20–21 In addition to gender and age determinants of LHON expression, the disease variability is determined by interplay between mtDNA defects, haplogroup and the environmental effects.1
From the 52 mutations detected in our study, only 4541G>A was found to be heteroplasmic and the rest of the mutations were in a homoplasmic state (see online supplementary table 1). This overwhelming homoplasmy may have contributed to the penetrance of the mutant ND4L gene. The single observed heteroplasmy was seen in only three individuals (V2, V-7 and VI-19), and that did not provide a protective effect against LHON. Heteroplasmy tends to drift toward homoplasmy over time,22 and perhaps this was the case in our studied family in this report.
Another interesting feature was from fourth-generation siblings; only one man (V-1) at age 35 years has so far expressed a mild LHON phenotype. He carries the double-mutant ND4L gene. His brother (V-2), age 33 years, also showed the double-mutant ND4L, but interestingly so far has not developed LHON.
The early onset manifestation of LHON in the fifth generation compared with the older generations may be due to unidentifiable non-genetic (environmental) factors. One such factor could be exposure to tobacco smoking (VI-17 from third kindred is a smoker). In one large multicentre epidemiological study, smoking had a strong and consistent association with visual loss, independent of gender and alcohol intake. In that study, men who smoked and carried any of the three primary LHON mutations had clinical penetrance of 93%, although the authors did not control for pedigree.3 Another hypothesis to explain disparity in disease manifestation could be toxigenic environment factors and psychological stress; the fifth generation was born in the post-Gulf War era when environmental pollution resulting from the fire in oil wells was linked to many health problems.23 There were no other rational or possible factors that could trigger the onset of LHON in this studied sixth (VI) generation, including nutritional limitations, antimicrobial drugs and acute illness.
In a recent meta-analysis of 159 European LHON pedigrees, it was suggested the mtDNA haplogroup does impact on the visual loss risk as underlined by the three LHON-causing primary mutations.24 There are conflicting reports on vision loss risk due to a combination of primary mutations with a particular haplogroup.25 Other reports suggested a synergistic and deleterious effect between LHON-causing mutations when associated with haplogroup J lineage.26 We performed phylogenetic analysis for haplogrouping of all participants. Two haplogroups (L3'4 and HV) could be distinguished. All except the two parents (V-14 and V-16) belonged to the haplogroup L3'4 which has its ancestry in L3 and is believed to have arisen in Eastern Africa. Immigrants have carried this haplogroup across the Red Sea to the Arabian Peninsula.18 However, haplogroup HV (seen only in V-14 and V-16 parents) is west Eurasian, and can be found to a much lesser extent in parts of East Africa, mainly in the population of Sudanese Arabs, where the frequency of Eurasian ancestry is marked at 22.5%.19 At present, no other reports have described the expression of LHON in an L3 haplogroup, especially in association with two concurrent mutations discovered here.
In summary, we have identified in this report a unique Arab family of haplogroup L3, with LHON phenotype that appears to have arisen through a maternal transmission mode, with two concurrent mtDNA mutations, 10609T>C and 10663T>C. We are unaware of a previous report of a dual-mutant ND4L gene in association with LHON. These mutations may potentially be considered as primary LHON mutations, in the L3 haplotype background, and they are distinguished from the other three well established primary mutations that usually cause LHON to manifest in subjects who are carrying the haplogroup J lineage.
Supplementary Material
Acknowledgments
The authors would like to thank Professor Alfredo Sadun and Dr Khaled Abu-Amero for their critical manuscript review and suggestions. We thank Mrs Daisy Thomas for supporting participants’ recruitment and Ms Hunouf Mahmoud for the pilot sequencing work. The authors also want to thank the tissue bank for their support of DNA extraction form participants’ blood samples, the Ethical Committee and the Scientific Advisory Board in Dasman Diabetes Institute for approving the study. The authors would also like to thank the Senior Management at Dasman Institute and the Kuwait Foundation of Advancement of Science (KFAS) for their support and funding.
Footnotes
Contributors: RB carried out clinical assessments and wrote clinical summaries. MM carried out sequencing work. GA carried out sequencing primers selection and optimisation. KB codirected the research and study design. OAA designed and directed the research and developed the manuscript.
Funding: Funding in full was through the Kuwait Foundation for the advancement of Science.
Competing interests: None.
Patient consent: Obtained.
Ethics approval: Dasman Diabetes Institute Ethics Review Board.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Yu-Wai-Man P, Griffiths PG, Hudson G, et al. Inherited mitochondrial optic neuropathies. J Med Genet 2009;46:145–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newman NJ, Wallace DC. Mitochondria and Leber's hereditary optic neuropathy. Am J Ophthalmol 1990;109:726–30 [DOI] [PubMed] [Google Scholar]
- 3.Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene–environment interactions in Leber hereditary optic neuropathy. Brain 2009;132(Pt 9):2317–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Z, Harrison CM, Chuang GC, et al. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat Res 2007;621:61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirches E. LHON: mitochondrial mutations and more. Curr Genomics 2011;12:44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 2004;23:53–89 [DOI] [PubMed] [Google Scholar]
- 7.Mackey DA, Oostra RJ, Rosenberg T, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet 1996;59:481–5 [PMC free article] [PubMed] [Google Scholar]
- 8.Macmillan C, Kirkham T, Fu K, et al. Pedigree analysis of French Canadian families with T14484C Leber's hereditary optic neuropathy. Neurology 1998;50:417–22 [DOI] [PubMed] [Google Scholar]
- 9.Taylor RW, Jobling MS, Turnbull DM, et al. Frequency of rare mitochondrial DNA mutations in patients with suspected Leber's hereditary optic neuropathy. J Med Genet 2003;40:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valentino ML, Barboni P, Ghelli A, et al. The ND1 gene of complex I is a mutational hot spot for Leber's hereditary optic neuropathy. Ann Neurol 2004;56:631–41 [DOI] [PubMed] [Google Scholar]
- 11.Carelli V, Achilli A, Valentino ML, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet 2006;78:564–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Achilli A, Iommarini L, Olivieri A, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in Leber's hereditary optic neuropathy. PLoS ONE 2012;7:e42242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abu-Amero KK, Bosley TM. Mitochondrial abnormalities in patients with LHON-like optic neuropathies. Invest Ophthalmol Vis Sci 2006;47:4211–20 [DOI] [PubMed] [Google Scholar]
- 14.Wissinger B, Besch D, Baumann B, et al. Mutation analysis of the ND6 gene in patients with Lebers hereditary optic neuropathy. Biochem Biophys Res Commun 1997;234:511–15 [DOI] [PubMed] [Google Scholar]
- 15.Brown MD, Zhadanov S, Allen JC, et al. Novel mtDNA mutations and oxidative phosphorylation dysfunction in Russian LHON families. Hum Genet 2001;109:33–9 [DOI] [PubMed] [Google Scholar]
- 16.Rieder MJ, Taylor SL, Tobe VO, et al. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res 1998;26:967–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews RM, Kubacka I, Chinnery PF, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999;23:147. [DOI] [PubMed] [Google Scholar]
- 18.Gonder MK, Mortensen HM, Reed FA, et al. Whole-mtDNA genome sequence analysis of ancient African lineages. Mol Biol Evol 2007;24:757–68 [DOI] [PubMed] [Google Scholar]
- 19.Afonso C, Alshamali F, Pereira JB, et al. MtDNA diversity in Sudan (East Africa). Forensic Science International: Genetics Supplement Series 2008;1:257–8 [Google Scholar]
- 20.Harding AE, Sweeney MG, Govan GG, et al. Pedigree analysis in Leber hereditary optic neuropathy families with a pathogenic mtDNA mutation. Am J Hum Genet 1995;57:77–86 [PMC free article] [PubMed] [Google Scholar]
- 21.Man PY, Griffiths PG, Brown DT, et al. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet 2003;72:333–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace DC. Mitotic segregation of mitochondrial DNAs in human cell hybrids and expression of chloramphenicol resistance. Somat Cell Mol Genet 1986;12:41–9 [DOI] [PubMed] [Google Scholar]
- 23.Petruccelli BP, Goldenbaum M, Scott B, et al. Health effects of the 1991 Kuwait oil fires: a survey of US army troops. J Occup Environ Med 1999;41:433–9 [DOI] [PubMed] [Google Scholar]
- 24.Hudson G, Carelli V, Spruijt L, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet 2007;81:228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tharaphan P, Chuenkongkaew WL, Luangtrakool K, et al. Mitochondrial DNA haplogroup distribution in pedigrees of Southeast Asian G11778A Leber hereditary optic neuropathy. J Neuroophthalmol 2006;26:264–7 [DOI] [PubMed] [Google Scholar]
- 26.Brown MD, Starikovskaya E, Derbeneva O, et al. The role of mtDNA background in disease expression: a new primary LHON mutation associated with Western Eurasian haplogroup. J Hum Genet 2002;110:130–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.