

Abstract
Objective
This study investigated the capacity of genetic analysis of uveal melanoma samples to identify high-risk patients and discusses its clinical implications.
Methods
Patients with posterior uveal melanoma were prospectively enrolled. Tumour samples were derived from enucleated globe, fine-needle aspirates or endoresection. Chromosome 3 and 8 status was determined by array comparative genomic hybridisation (array-CGH). Patients were followed after treatment to detect metastasis.
Results
Four groups were classified by array-CGH. Patients were divided into disomy 3 and normal chromosome 8 (D3/8nl), disomy 3 and 8q gain (D3/8g), monosomy 3 and normal chromosome 8 (M3/8nl) and monosomy 3 and 8 or 8q gain (M3/8g). Median follow-up was 28 months (range: 1–147 months). At the end of the study, 128 patients (33.7%) had developed metastasis and 96 patients had died. Univariate Cox proportional hazard analysis showed that factors associated with metastasis included basal tumour diameter p=0.0007, tumour thickness p=0.01, mixed/epithelioid cell type p=0.0009 and genomic data p<0.0001. High-risk profile was more strongly associated with metastasis than the other prognostic factors p<0.001. Multivariate Cox modelling analysis showed that the status of chromosomes 3 and 8 were the only two variables that independently contributed to prognosis: monosomy 3 alone p=0.001 and monosomy 3 and 8q gain p<0.0001.
Conclusions
Array-CGH allowed identification of three prognostic groups with low, intermediate and high risk of developing metastasis. Array-CGH is a reliable and inexpensive method for uveal melanoma prognosis. This method is now currently used in France.
Keywords: Choroid, Pathology, Diagnostic tests/Investigation, Genetics
Introduction
Uveal melanoma is the most common ocular malignancy in Caucasian adults. Even when localised, this tumour metastasises in about 50% of cases, mostly to the liver.1 Progress in treatment of the primary tumour has allowed better eye conservation rates, but has had no impact on patient survival.2 As metastatic disease usually remains fatal regardless of the modality of conventional treatment used, physicians have recently considered systemic adjuvant therapy trials for high-risk patients,3 4 but which require accurate assessment of the patient's prognosis. Many anatomical, histological and, more recently, genomic prognostic features have been used to identify patients with a higher risk of recurrence: large basal diameter, ciliary body involvement, extrascleral extension, epithelioid cell histology, high mitotic rate, chromosome imbalances (monosomy 3, 8q gain) and class 2 gene expression profile (GEP).5–10 High-risk patients require closer follow-up to allow rapid initiation of local hepatic therapy that may prolong survival.11–14
In our institution, we routinely perform tumour analysis by genome-wide profiling associated with pathology. This article investigates the respective value of the various prognostic features assessed, their capacity to identify high-risk patients and discusses the clinical implications of these results.
Materials and methods
This study was approved by our institutional ethics committee. Written informed consent for the use of tissues and data for research was signed by each patient. The study complied with the principles of the Declaration of Helsinki.
Patients
All patients were referred to our institution. Clinical diagnosis of posterior uveal melanoma was based on the presence of typical clinical findings, thickness >2.5 mm, orange pigment, serous retinal detachment, mushroom shape, extrascleral extension and growth. Demographic data were collected, including patient age, gender and tumour thickness measured by ultrasound biomicroscopy; basal tumour diameter measured by both ultrasound and transillumination of the globe or fundus photographs; tumour location; the presence of ciliary body invasion evaluated by transillumination of the globe or histologically when available; the presence of clinical serous retinal detachment and the presence of extrascleral extension measured clinically or by ultrasound when macroscopic or by histology. Liver ultrasound was performed prior to treatment. Each case was staged according to the collaborative ocular melanoma study classification and the guidelines of the 7th edition of the American Joint Committee on Cancer Staging TNM (Tumour Node Metastasis) staging system.15 16 Local treatment consisted of enucleation for large tumours (basal diameter >16 mm and/or tumour thickness >10 mm) and proton beam radiotherapy or iodine 125 brachytherapy for small-to-medium-sized tumours. Tumour samples were obtained by enucleation or endoresection, or fine-needle aspiration (FNA) at the time of clip/plaque positioning.17 FNA was proposed when tumour thickness was >5 mm, and endoresection was indicated for tumour diameter <16 mm and tumour thickness >8 mm.17 Patients with a small choroidal melanoma <5 mm thick were excluded from the study. Histological examination was not available when tumour sampling was performed by FNA. Patients were reviewed every 6 months with complete eye examination, ultrasound biomicroscopy and liver ultrasonography or MRI. Suspicion of liver metastasis was confirmed by liver biopsy.
Genomic analysis
Tumour specimens were collected in Hanks’ balanced salt solution. Tumour fragments were snap frozen in liquid nitrogen, then stored at −80°C. Tumour cellularity was checked to be >60% on H&E-stained cryosections. FNAs were centrifuged and then frozen. Comparative genomic hybridisation on microarrays (array-CGH) was performed on two different platforms according to when the test was performed. Home-made bacterial artificial chromosome arrays, as described in Trolet et al8 were used until 2009. Subsequent analyses were performed on NimbleGen 4×72 K arrays (Roche NimbleGen, Madison, Wisconsin, USA). Genomic DNA was extracted with phenol/chloroform and Phase Lock Gel Light (Eppendorf, Hamburg, Germany) procedure and analysed versus normal reference DNA. For each sample, 700 ng of tumour DNA and reference DNA were labelled and cohybridised to the NimbleGen arrays according to the manufacturer's protocol. Arrays were washed and then scanned on a GenePix 4000B scanner using GenePix V.6.0 Software (Molecular Devices, Sunnyvale, California, USA). Raw data were normalised and processed using the NimbleScan V.2.5 Software (Roche NimbleGen). Data were then visualised with SignalMap V.1.9 (Roche NimbleGen) before analysis. Gains were defined as a log2ratio ≥0.58 and homozygous losses as a log2ratio ≤−0.60.18
On the basis of the work by Trolet et al8 showing that chromosome 3 and 8 status had the highest prognostic correlation, only these two chromosomes were considered in this study. Cases were interpreted by two of the authors (JC and MJR). Chromosome 3 was present either as two copies (disomy) or as a single copy (monosomy). Cases with partial loss of chromosome 3 were excluded. Chromosome 8 showed three possible types of gain: gain of the entire chromosome, gain of the entire long arm (8q) and gain of a distal part of the long arm (distal 8q). Four genomic types were subsequently defined taking into account the status of chromosomes 3 and 8: tumours with disomy 3 and normal dosage of chromosome 8 (named as ‘D3/8nl’), monosomy 3 and normal dosage of chromosome 8 (‘M3/8nl’), disomy 3 and any type of gain of chromosome 8 (‘D3/8g’) and monosomy 3 with any type of gain of chromosome 8 (‘M3/8g’). The different genomic profiles are shown in figure 1.
Figure 1.

Four examples of Nimblegen 4×72 K comparative genomic hybridisation-array (array-CGH) profiles. From upper to lower: profiles from D3/8nl, M3/8nl, D3/8nl and M3/8 g groups. In these array-CGH profiles, every point represents the mean level of the probes over 400 kilobases. Chromosome imbalances are presented in log2 ratio.
Statistical analysis
Clinical, pathological and genomic data at diagnosis and follow-up events (local and distant recurrences, second cancers, death from uveal melanoma or from any other cause) were prospectively collected. The French Death Registry was consulted for patients lost to follow-up. Metastases were proven by biopsy. Patients with metastatic disease were treated by an oncologist at our institution.
Statistical analysis was performed using R software V2.13 (http://www.r-project.org/). The metastasis-free interval (MFI) was defined as the interval between the date of diagnosis of primary uveal melanoma and the date of distant metastasis. Survival distributions were estimated by the Kaplan–Meier method and compared using the log-rank test. Differences were considered to be significant when the log-rank p value was<0.05. In order to identify variables associated with MFI, a Co x regression analysis of candidate prognostic factors was performed using a forward stepwise selection procedure at the p=0.10 level of significance. The added value of each variable to the Cox model was determined using a likelihood ratio test. Genomic factors were coded by three dummy variables, with the reference group corresponding to D3/8nl, to calculate a HR for each group in comparison with this reference group.
Results
The study started in February 1996 and ended in May 2012. Of the 361 patients with tumour samples prospectively included, 338 were included in the present analysis. Twenty-three patients lost to follow-up 3 months after radiotherapy (three patients) or with an insufficient sample for analysis (18 patients) were excluded. Median follow-up was 28 months (range: 1–147 months).
Demographic data and tumour characteristics of the 338 patients are summarised in table 1. Genomic analysis by array-CGH was performed on 338 patients.
Table 1.
Demographic and baseline disease characteristics of the patients
| Age (years) n=338 | Median: 59 Range: 17–92 |
| Gender | Women: 136 Men: 202 |
| Tumour diameter (mm) | Median: 15.7 Range: 7–25 |
| Tumour thickness (mm) | Median: 10.4 Range: 2.7–17 |
| Retinal detachment | Present: 228 Absent: 110 |
| Ciliary body involvement | Present: 106 Absent: 232 |
| Extrascleral extension (macroscopic or microscopic) | Present: 25 Absent: 313 |
| TNM classification (7th TNM) | |
| T1 | 2/338 0.6% |
| T2 | 30/338 9.4% |
| T3 | 155/338 48% |
| T4 | 134/338 42% |
| Samples n=338 | Enucleation: 219 Endoresection: 49 FNA: 70 |
| Histology: mixed or epithelioid cytology n=206 | Yes: 138 No: 65 Indeterminate: 3 |
FNA, fine-needle aspiration; TNM, tumour node metastasis.
Patients were classified into the four prognostic groups defined above on the basis of their genomic profile. Ninety-one patients were classified in the D3/8nl group, 47 in the D3/8g group, 41 in the M3/8nl group and 159 in the M3/8g group (table 2).
Table 2.
Univariate analysis of risk factors for metastasis
| Prognostic factors | N=338 | Metastatic events | Two-year survival rate ± SD | HR | p Value |
|---|---|---|---|---|---|
| Age at uveal melanoma diagnosis | |||||
| <60 years | 177 | 52 | 71.9±4 | 1 | 0.02 |
| ≥60 years | 161 | 66 | 56.8±4.5 | 1.50 (1.05–2.17) | |
| Gender | |||||
| Men | 202 | 64 | 66.5±3.8 | 1 | 0.168 NS |
| Women | 136 | 54 | 61.8± 4.8 | 1. 26 (0.88–1.08) | |
| Ocular tumour diameter | |||||
| ≤15 mm | 134 | 32 | 78.3±4.2 | 1 | 0.0007 |
| >15 mm (md=17) | 187 | 78 | 56.3±4.2 | 2.0 (1.33–3.0) | |
| Ocular tumour thickness | |||||
| ≤10 mm | 159 | 42 | 72.5±4 | 1 | 0.01 |
| >10 mm (md=1) | 178 | 76 | 57.4±4 | 1.6 (1.1–2.3) | |
| Ocular tumour location | |||||
| Anterior to equator | 19 | 7 | 45.0±14 | 1 | 0.411 NS |
| On equator | 234 | 85 | 63.7±3.6 | 1.2 (0.38–18) | |
| Posterior to equator (md=4) | 81 | 24 | 73.0± 6 | 1.6 (0.26–1.44) | |
| Retinal detachment | |||||
| No | 94 | 31 | 66.6±6 | 1 |
0.343 NS |
| Yes (md=16) | 228 | 77 | 65.4±3 | 1.22 (0.80–1.8) | |
| Histological type | |||||
| Spindle | 78 | 22 | 76.0±5.3 | 1 | 0.0009 |
| Epithelioid—mixed (md=91) | 169 | 78 | 54.7±4.4 | 2.2 (1.3–3.5) | |
| Genomic data | |||||
| D3/8nl | 91 | 2 | 100±0 | 1 | <0.0001 |
| D3/8g | 47 | 8 | 85.0±6 | 8.1 (01.7–38.2) | |
| M3/8nl | 41 | 8 | 82.1±6 | 9.3 (01.9–44.2) | |
| M3/8g | 159 | 100 | 37.1±4 | 46.6 (11.4–189.4) | |
Metastasis rate was estimated by the Kaplan–Meier method and the HR was determined by the Cox model (md, missing data).
Variables identified by univariate analysis were genomic classification, histology, tumour diameter, tumour thickness and age. Genomic classification was the only significant factor that independently contributed to prognosis in multivariate analysis (table 3).
Table 3.
HR for metastasis according to genomic group.
| HR | 95% CI | p Value | |
|---|---|---|---|
| Genomic groups | |||
| D3/8nl | 1 | (1.5 to 34.6) | 0.01 |
| D3/8g | 7.3 | (2.4 to 50.0) | 0.002 |
| M3/8nl | 10.6 | (10.4 to 174.3) | <0.0001 |
| M3/8g | 42.6 | ||
HR was estimated by Cox model.
The 2-year MFI was excellent with 100% patients free of metastasis in the D3/8nl group. The D3/8g and M3/8nl groups appeared to have an intermediate prognosis with a 2-year MFI of 85.0% and 82.1%, respectively. Finally, patients in the M3/8g group had a poor prognosis with a 2-year MFI of 37.1% (figure 2).
Figure 2.
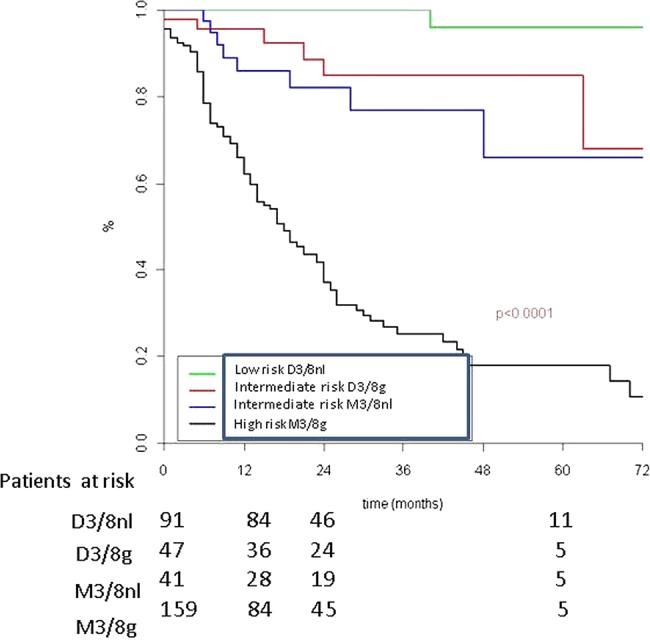
Metastatic-free interval according to genomic results: M3/8nl (disomy 3, normal ch 8); D3/8g (disomy 3 ch 8 gain); M3/8nl (monosomy 3 normal ch.8) and M3/8g (monosomy 3 and ch 8 gain).
Compared with the D3/8nl group, the HR of developing a metastasis was similar for patients with monosomy 3 alone (HR=7.3; p=0.01) and for those with gain of chromosome 8 alone (HR=10.6; p=0.002). HR was 42.6 for patients with monosomy 3 and gain of chromosome 8 (p<0.0001). In contrast, the HR for metastasis was 1.4 when basal tumour diameter was greater than 15 mm (p=0.07) (table 3).
Three genomic prognostic groups were able to be identified on the basis of these data: a low-risk group corresponding to patients in the D3/8nl genomic group, an intermediate-risk group corresponding to patients with a tumour presenting either monosomy 3 (M3/8nl) or gain of chromosome 8 (D3/8g) and a high-risk group corresponding to patients in the M3/8g group.
Two (0.6%) of the 338 patients developed metastases in the low-risk genomic group, 16 (4.7%) of the 338 patients developed metastases in the intermediate-risk group and 100 (29.6%) of the 338 patients developed metastases in the high-risk group.
These two patients had extrascleral extension at diagnosis. The first patient was enucleated for a tumour with a basal diameter of 15.8 mm, tumour thickness of 12.6 mm, retinal detachment and ciliary body invasion. Histological examination demonstrated a mixed cell type uveal melanoma with 2 mm thick extrascleral extension. The second patient was also enucleated for a large tumour with a basal diameter of 19 mm, tumour thickness of 12.4 mm, retinal detachment and ciliary body invasion. Histological examination demonstrated spindle cell melanoma associated with 5 mm thick extrascleral extension.
Discussion
As in most centres, the prognosis of uveal melanoma in our institution is classically defined according to clinical, histological and genomic criteria. The present study shows that genomic characteristics are the main prognostic factor ahead of clinical and histological features. Three prognostic groups can be very simply defined on the basis of the presence of monosomy 3 and chromosome 8 gain (either whole chromosome 8 gain, 8q gain or distal 8q gain). The 2-year MFI was 100% for patients with a tumour presenting no imbalances of chromosomes 3 or 8, about 82.1–85.0% for tumours presenting an imbalance of one of these two chromosomes (either one of the two classes of imbalances) and 37.1% for tumours with monosomy 3 and any type of gain of chromosome 8.
As most tumours in this series were stage T3/T4, our cohort is probably biased and does not reflect the usual population of uveal melanoma patients. However, large uveal melanomas are at higher risk of developing metastasis than small tumours. Furthermore, these large tumours require more accurate classification than uveal melanomas in general, based on clinical or histological findings. However, the study of the genomic profiles of small tumours needs to be improved, which could be facilitated by improvement of our FNA techniques in order to include tumours less than 5 mm thick. To achieve reliable results, array-CGH analysis requires a minimum of 500 ng of tumour DNA, but FNAs from small tumours were sometimes associated with small quantities of DNA. Until new, more sensitive technologies become available, array-CGH after FNA in small tumours will continue to be associated with a significant number of technical failures.
Array-CGH assesses only chromosome copy number representing some method limits. A small proportion of disomy 3 are isodysomic, in which one copy of chromosome 3 is lost while the other one is duplicated, leading to a loss of heterozygosity.19 20 Single nucleotide polymorphism array gives the allelic status of chromosomes and their copy number. However, this array is expensive and was used as control in selected cases.
Array-CGH is now a simple genomic technology used in other tumour types, but requires a genetics laboratory with appropriate equipment, such as a array-CGH scanner and a rigorous sample circuit. Although arrays and reagents are now relatively inexpensive, these installations require a significant initial financial investment.
Other teams have tried to establish prognostic groups according to DNA and RNA alterations in uveal melanoma. Some studies have assessed the value of chromosome 3 status by fluorescence in situ hybridisation (FISH).19 Damato et al, based on a study of 356 patients, showed that monosomy 3 evaluated by FISH was strongly associated with metastatic deaths.20 However, they described 12 patients without monosomy 3 determined by FISH who died from metastases. These cases may have been wrongly classified as low risk because of failure of the FISH method or because chromosome 8 imbalances were not investigated. This team is now using multiplex ligation-dependent probe amplification (MLPA) assays to characterise frequent genomic imbalances in uveal melanoma in chromosomes 1p, 3, 6 and 8. They have recently published a large series of 452 patients showing an increased mortality when loss of chromosome 3 is associated with gain of 8q versus loss of chromosome 3 alone.9 MLPA is an interesting technology because of its high sensitivity (interpretable results with less DNA), reproducibility of the results and its low cost. However, MLPA is limited by its partial view of genomic imbalances, as MLPA is less sensitive as it only explores a few genomic positions and may therefore fail to distinguish between partial and total alteration of an entire arm or chromosome. This study also reported discordant results to those obtained in our study. In this series, patients with tumours presenting 8q gain without chromosome 3 monosomy were at very low risk of metastatic recurrence. Further studies with new technologies are required to elucidate these discrepancies.
Another way to estimate the risk of recurrence is to study GEPs, as described by Onken et al in their prospective validation.10 Their array explores the expression of 12 genes, five of which (EIF1B, FXR1, LMCD1, ROBO1, SATB1) are situated on chromosome 3. Down-expression of these genes, possibly due to monosomy 3, is associated with poor prognosis. This array separates uveal melanoma into two subsets either low metastatic risk, called ‘class 1’ tumours (1% of metastatic recurrence) or high metastatic risk, called ‘class 2’ tumours (26% of metastasis).10 These authors considered that loss of chromosome 3 was insufficient to efficiently discriminate high-risk patients from low-risk patients. However, they compared GEP with chromosome 3 FISH rather than a more global analysis evaluating chromosome 8 imbalances such as that provided by MLPA or array-CGH.21 Although the GEP classification is the first method to meet the National Comprehensive Cancer Network ‘1A’ level of evidence for a biomarker in uveal melanoma and the first method to be validated in a prospective series, this method, nevertheless, remains expensive and inaccessible for many countries. Our classification system is efficient and less expensive than the GEP classification. Our classification also introduces an intermediate-risk group so that our low-risk group really has a very low risk of metastasis and our high-risk group really has a very high risk of metastasis. Recent updates show that Decision-Dx, the GEP provider, has introduced a third, intermediate-risk group. To our knowledge, no international publication has accompanied this announcement. Knowledge of the GEP and functional analysis of the genes involved in the metastatic process remains essential to understand carcinogenesis and to develop targeted therapies, but these procedures may be reserved for research programmes.22 23
Local treatment of uveal melanoma has now been clearly defined,24–26 but little progress has been made in terms of survival.27 The next challenge is to increase overall survival of patients with localised disease. To achieve this objective, trials of adjuvant therapy must be conducted in high-risk patients. For example, a recent randomised trial, initiated in 2009, is comparing adjuvant fotemustine versus close follow-up only in high-risk patients as defined on clinical and genomic criteria (ie, diameter >15 mm with retinal detachment or diameter >18 mm or monosomy 3 with or without gain of 8q; EudraCT No.: 2008-005691-27).
In future, the prognosis of patients with uveal melanoma will probably use new genomic technologies such as next-generation sequencing not only to refine prognosis by looking for additional mutations in BAP1, GNAQ, GNA11,28 SF3B129 or loss of heterozygosity in the primary tumour but also to follow circulating tumour DNA.30 31
Conclusion
Array-CGH is a robust and affordable method for uveal melanoma prognostic assessment. This method is currently used in France.
Footnotes
Contributors: NC designed the study, collected data, analysed the data and prepared the final draft of the manuscript. MJR contributed to data collection and analysis, and reviewed and approved the final manuscript. CP and BA analysed data, performed statistical analysis, revised the manuscript and approved the final draft. CL-G, LL-LR, RD, XS and SP-N contributed to data collection, and revised and approved the final manuscript. LD and JC contributed equally to the work of conception and design, revised the manuscript, supervised data analysis and approved the final draft.
Funding: This research was supported by the Institut Curie Research and Development fund.
Competing interests: None.
Patient consent: Obtained.
Ethics approval: Institut Curie ethics committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Genetic profile of the patients are available at Department of Transfert, Institut Curie.
References
- 1.Kujalä E, Makitie T, Kivelä T. Very long-term prognosis of patients with malignant uveal melanoma. Invest Ophthalmol Vis Sci 2003;44:4651–9 [DOI] [PubMed] [Google Scholar]
- 2.Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology 2012;118:1881–5 [DOI] [PubMed] [Google Scholar]
- 3.Lane AM, Egan KM, Harmon D, et al. Adjuvant interferon therapy for patients with uveal melanoma at high risk of metastasis. Ophthalmology 2009;116:2206–12 [DOI] [PubMed] [Google Scholar]
- 4.Voelter V, Schalenbourg A, Pampallona S, et al. Adjuvant intra-arterial hepatic fotemustine for high-risk uveal melanoma patients. Melanoma Res 2008;18:220–4 [DOI] [PubMed] [Google Scholar]
- 5.Gamel JW, McCurdy JB, McLean IW. A comparison of prognostic covariates for uveal melanoma. Invest Ophthalmol Vis Sci 1992;33:1919–22 [PubMed] [Google Scholar]
- 6.Hughes S, Damato BE, Giddings I, et al. Microarray comparative genomic hybridisation analysis of intraocular uveal melanomas identifies distinctive imbalances associated with loss of chromosome 3. Br J Cancer 2005;93:1191–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehlers JP, Worley L, Onken MD, et al. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin Cancer Res 2008;14:115–22 [DOI] [PubMed] [Google Scholar]
- 8.Trolet J, Hupe P, Huon I, et al. Genomic profiling and identification of high-risk uveal melanoma by array CGH analysis of primary tumors and liver metastases. Invest Ophthalmol Vis Sci 2009;50:2572–80 [DOI] [PubMed] [Google Scholar]
- 9.Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res 2010;16:6083–92 [DOI] [PubMed] [Google Scholar]
- 10.Onken MD, Worley LA, Tuscan MD, et al. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol diag 2010;12:461–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mariani P, Piperno-Neumann S, Servois V, et al. Surgical management of liver metastases from uveal melanoma: 16 years’ experience at the Institut Curie. Eur J Surg Oncol 2009;35:1192–7 [DOI] [PubMed] [Google Scholar]
- 12.Leyvraz S, Keilholz U. Ocular melanoma: what's new? Curr Op Oncol 2010;24:162–9 [DOI] [PubMed] [Google Scholar]
- 13.Mariani P, Servois V, Piperno-Neumann S. Therapeutic options in metastatic uveal melanoma. Dev Ophthalmol 2012;49:166–81 [DOI] [PubMed] [Google Scholar]
- 14.Frenkel S, Nir I, Hendler K, et al. Long-term survival of uveal melanoma patients after surgery for liver metastases. Br J Ophthalmol 2009;93:1042–6 [DOI] [PubMed] [Google Scholar]
- 15.Straatsma BR, Fine SL, Earle JD, et al. Enucleation versus plaque irradiation for choroidal melanoma. Ophthalmology 1988;95:1000–4 [DOI] [PubMed] [Google Scholar]
- 16.Kivelä T, Kujalä E. Prognostication in eye cancer: the latest tumor, node, metastasis classification and beyond. Eye 2013;27:243–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassoux N, Cayette S, Plancher C, et al. CHOROIDAL MELANOMA: Does endoresection prevent neovascular glaucoma in patient treated with proton beam irradiation? Retina 2013;33:1441–7 [DOI] [PubMed] [Google Scholar]
- 18.La Rosa P, Viara E, Hupe P, et al. VAMP: visualization and analysis of array-CGH, transcriptome and other molecular profiles. Bioinformatics 2006;22:2066–73 [DOI] [PubMed] [Google Scholar]
- 19.Onken MD, Worley LA, Char DH, et al. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology 2012;119:1596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harbour JW. The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res 2012;25:171–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shields CL, Ganguly A, Bianciotto CG, et al. Prognosis of uveal melanoma in 500 cases using genetic testing of fine-needle aspiration biopsy specimens. Ophthalmology 2011;118:396–401 [DOI] [PubMed] [Google Scholar]
- 22.Damato B, Duke C, Coupland SE, et al. Cytogenetics of uveal melanoma: a 7-year clinical experience. Ophthalmology 2007;114:1925–31 [DOI] [PubMed] [Google Scholar]
- 23.Woodman SE. Metastatic uveal melanoma: biology and emerging treatments. Cancer Res 2012;18:148–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel M, Smyth E, Chapman PB, et al. Therapeutic implications of the emerging molecular biology of uveal melanoma. Clin cancer res 2011;(8):2087–100 [DOI] [PubMed] [Google Scholar]
- 25.Hawkins BS. Collaborative ocular melanoma study randomized trial of I-125 brachytherapy. Clin trials 2011;8:661–73 [DOI] [PubMed] [Google Scholar]
- 26.Dendale R, Lumbroso-Le Rouic L, Noel G, et al. Proton beam radiotherapy for uveal melanoma: results of Curie Institut-Orsay proton therapy center (ICPO). Int J Radiat Oncol Biol Phys 2006;65:780–7 [DOI] [PubMed] [Google Scholar]
- 27.Egan KM, Ryan LM, Gragoudas ES. Survival implications of enucleation after definitive radiotherapy for choroidal melanoma: an example of regression on time-dependent covariates. Arch Ophthalmol 1998;116:366–70 [DOI] [PubMed] [Google Scholar]
- 28.Damato B, Lecuona K. Conservation of eyes with choroidal melanoma by a multimodality approach to treatment: an audit of 1632 patients. Ophthalmology 2004;111:977–83 [DOI] [PubMed] [Google Scholar]
- 29.Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010;330:1410–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harbour JW, Roberson ED, Anbunathan H, et al. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 2013;45:133–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madic J, Piperno-Neumann S, Servois V, et al. Pyrophosphorolysis-activated polymerization detects circulating tumor DNA in metastatic uveal melanoma. Clin Cancer Res 2012;18:3934–41 [DOI] [PubMed] [Google Scholar]