

Abstract
Blockade of the vasopressin-2 receptor (V2R) in the kidney has recently emerged as a promising therapeutic strategy in autosomal dominant polycystic kidney disease. The pathophysiologic basis of V2R-dependent cyst proliferation and disease progression, however, is not fully understood. Recent evidence suggests that polycystic kidney disease is characterized by defects in urinary concentrating mechanisms and subsequent deregulation of vasopressin excretion by the neurohypophysis. On the cellular level, several recent studies revealed unexpected crosstalk of signaling pathways downstream of V2R activation in the kidney epithelium. This review summarizes some of the unexpected roles of V2R signaling and suggests that vasopressin signaling itself may contribute crucially to loss of polarity and enhanced proliferation in cystic kidney epithelium.
Keywords: ADPKD, vasopressin, Cell & Transport Physiology, Cell Signaling, collecting ducts
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common hereditary diseases of the kidney and a leading cause of ESRD. The past two decades have witnessed breakthrough discoveries in ADPKD research and provided a new and profound insight into the pathogenesis of this important chronic disorder. However, therapeutic strategies that halt or slow down disease progression are still lacking. Previously, pharmacological antagonism of the vasopressin-2 receptor (V2R) emerged as the most promising way to treat ADPKD.1,2 An earlier landmark study by Gattone et al.3 showed that V2R blockade was effective to reduce volume and cyst growth in polycystic kidneys in various polycystic kidney rodent models. These results prompted a number of pilot studies using both water loading and pharmacological inhibition of V2R in ADPKD patients1,4 and initiated a controlled, randomized trial testing tolvaptan, a competitive antagonist of V2R, for the treatment of ADPKD (TEMPO 3:4 trial). This large randomized, controlled trial confirmed earlier experimental studies and showed that tolvaptan can slow down disease progression in humans.5 Thus, the blockade of V2R is emerging as one of the few treatment options for ADPKD after inconclusive results obtained in studies using mammalian target of rapamycin inhibitors.6,7 It has to be stressed that, although the results of the TEMPO trial are highly encouraging, tolvaptan cannot be considered as an option for all patients, and it is not the ultimate solution for the treatment of ADPKD. First, the treatment comes at the price of considerable polyuria and polydipsia, which are not unexpected from a drug that blocks vasopressin action. In fact, about 20% of the patients enrolled in the TEMPO trial actually stopped the treatment because of the limiting side effects. Second, it is not clear how long the treatment should be prescribed and which subgroups of patients would benefit the most. In the landmark TEMPO trial, only patients with CKD stages 1–3 were included in the study. Most, if not all, of the patients had severe disease with grossly enlarged kidneys and considerable hypertension. It is important to stress that eGFR may not provide an ideal measure to monitor severity in the early years of presentation, because ADPKD patients develop an early hyperfiltration status mimicking near-normal eGFR; additionally, much of renal function was lost.
The effect of tolvaptan on eGFR was quite moderate. However, the reduced decrease of the eGFR was highly significant compared with the placebo group, although high- (i.e., with a high decrease of the GFR) and low-risk patients were lumped together in the analysis. Moreover, the blockade of V2R reduced the rate of kidney volume increase by approximately 50% in subjects with high and low GFRs. Of note is that cyst growth was only prevented in the kidney and not the liver, indicating the V2R-specific effect of the drug. Thus, it is expected that a better understanding of the V2R signaling pathways in cyst-lining epithelium may well provide additional innovative treatment options that may ultimately come to the clinic.
The effect of vasopressin V2R signaling on the tubular system in healthy subjects and ADPKD patients is still not entirely understood. Recent studies using system-wide perturbation, classic physiologic approaches, and state of the art molecular biology have provided important insight into unanticipated signaling pathways perturbed by V2R activation. Some of these signaling pathways expand the picture of the classic role of vasopressin mediating tubular water reabsorption through cAMP levels; in fact, vasopressin may regulate by far more general cellular functions (Figure 1). These findings may open avenues for future translational research that is guided by clinical observations and respects findings from basic science.
Figure 1.

A simplified view of V2R-dependent signaling in collecting duct cells revealed by phosphoproteomic and protomic approaches in various vasopressin-responsive tissues in vivo, ex vivo, and in vitro. The studies show alterations in cytoskeletal protein phosphorylation (e.g., Myh9 and Stmn). In addition, proteins involved in the maintenance of polarity and proliferative tone are significantly affected on functional phosphorylation sites. AC, adenylate cyclase; Atf2, activating transcription factor 2; Bad, Bcl2-agonist of cell death; CaM, calmodulin; CaMK-II, calmodulin-dependent kinase 2; cJun, proto-oncogene c-Jun; Ctnnb1, β-catenin; JNK, c-Jun N-terminal kinase; MLCK, myosin light chain kinase; Myh, myosin heavy chain; Ryr, ryanoidin receptor; Stmn, Stathmin; Yap1, yorkie homolog protein 1.
Pathophysiology of Osmoregulation in ADPKD
The altered regulation of vasopressin serum levels in ADPKD was reanalyzed in a number of recent studies.8–11 One key advance in the past 5 years was the development of more robust and reliable assays to measure and estimate concentration of vasopressin in the serum.12 Copeptin, a 39-amino acid glycopeptide comprising the C terminus of the vasopressin precursor (preprovasopressin), is cosecreted with vasopressin by the neurohypophysis, which is somewhat comparable with cosecretion of the C peptide and insulin by the pancreas.12,13 The copeptin assay circumvents shortcomings of vasopressin assays, such as low stability in serum, and shows significant correlation with vasopressin in a variety of physiologic and pathologic conditions, although its pharmacokinetics are dependent on the GFR. Indeed, serum concentrations of copeptin (and also vasopressin) correlated positively with both serum osmolality as well as total kidney size in a total of 102 ADPKD patients.8 Moreover, there was a negative correlation with GFR and effective renal blood flow (both measured with the 125I-iothalamate/131I-hippuran method), and a consistent correlation of copeptin levels with serum osmolality was also found.8 Most intriguingly, a recent study showed that copeptin is a candidate prognostic marker for subsequent decline of renal functions as well as need for RRT in ADPKD patients.10
Based on the data available to date, there are two major reasons for the deregulation of the neurohypophysis–kidney axis. First, changes in kidney architecture or even preceding changes in cell polarity may cause a urinary concentration deficit with subsequently elevated plasma osmolalities. Already in the 1960s, a urinary concentration deficit in the disease was observed.14 Moreover, it is well known that, in the pediatric cystic kidney disease nephronophthisis, a urine concentration deficit is usually among the first clinical symptoms.15 In ADPKD, however, it is important to distinguish an early and rather mild urinary concentration deficit from the severe urinary concentration deficit associated with many forms of ESRD in the late course of the disease. In fact, as shown by Zittema et al.11 and Gabow et al.16 in large patient cohorts, maximal urine concentration capacities (maximal urine osmolality after overnight thirsting) are reduced but remain well above serum osmolality. In addition, Zittema et al.11 showed that patients carrying a polycystic kidney disease 1 (PKD1) mutation have early urine concentration deficits and increased vasopressin/copeptin levels.11 Second, the deregulation of the neurohypophysis–kidney axis could be an altered central release of vasopressin, because a syndrome of inappropriate antidiuretic hormone secretion-like phenotype was observed in PKD1 haploinsufficient rodents, which was shown by Ahrabi et al.17 Importantly, these mice were able to reabsorb abnormally high amounts of water, thereby supporting the idea of a nontubular basis of the urinary concentration deficit.17 In addition, a recent clinical study suggested that both components (i.e., an increased vasopressin secretion as well as an early urinary concentration deficit) are already present in children carrying the PKD mutation when compared with age-matched controls.18 Most importantly, the urine concentration deficit did not depend on kidney size or cyst number. However, these data have to be carefully evaluated given the fact that cysts may not be detectable using ultrasound with lower resolution compared with magnetic resonance imaging. It is conceivable that very small cysts beyond resolution limits may disrupt the architecture of the inner medulla and thus, interfere with the generation of the countercurrent multiplier system responsible for urinary concentration. Taken together, a strong body of evidence has been provided that shows that there is a pathologically hyperactive vasopressin/vasopressin receptor system in patients with ADPKD.
Canonical V2R-Dependent Signaling Pathway through cAMP and Protein Kinase A
In the human kidney tubule, the V2R is expressed in at least three different segments. V2Rs are predominantly found in the collecting duct.19 In addition, these receptors are also expressed and functional in the medullary thick ascending limb (mTAL) of the loop of Henle as well as the distal convoluted tubule with species- and sex-specific distribution patterns.20–22 The canonical V2R-dependent signaling pathway and its major role in water reabsorption in the renal collecting duct were revealed after the breakthrough cloning of aquaporin (AQP).23 Here, Neilsen et al.23 predominantly used native tissues or relied on ex vivo tubular preparations or in vivo stimulation in Brattleboro rats that lack endogenous arginine vasopressin (AVP) production by the neurohypophysis. Subsequent in vitro studies analyzing V2R function also used immortalized tubular cell culture models in which V2Rs are expressed24,25 or primary cultured rat inner medullary collecting duct (IMCD) cells.26–28
The canonical V2R signaling pathway involves activation of a stimulatory G protein followed by activation of adenylate cyclase 6 and the generation of the secondary messenger cAMP. cAMP drives the activation of protein kinase A (PKA), which results in the phosphorylation of several intracellular targets, including the water channel AQP2 at S256 and the transcription factor cAMP response element-binding protein. These events seem to be crucial for transcription and phosphorylation-dependent trafficking of the water channel AQP2, a prerequisite for water reabsorption and urine concentration in the collecting duct system.29,30 Interestingly, a recent study has been found that additional basophilic kinases, such as Calmodulin-dependent kinase 2, may be involved in activation of cAMP response element-binding protein and the water channel AQP2; in fact, these basophilic kinases may even have a more prominent role in regulating these physiologic processes.31
The generation of cAMP by V2R activation is generally believed to also be a key driving force of renal epithelial cell proliferation.9 Elevated cAMP levels are found in a variety of proliferating tissues, even without detectable G protein–coupled receptor activation. In metanephric embryonic kidney cultures, cAMP is able to induce massive cyst development.32 V2R-generated cAMP levels seem to peak early after short-term incubation with vasopressin, and both cell- and tissue-dependent differences of the impact of increased cAMP levels in response to long-term stimulation were reported.33,34 In fact, a diminished cAMP response compared with acute stimulation is at least partially attributed to the internalization of the V2R in response to prolonged AVP stimulation. Recent studies showed that cAMP levels remain elevated in response to the synthetic V2R-specific AVP analog dDAVP (Desmopressin) in cultured collecting duct cells34 and that this process activated the guanine exchange protein directly activated by cAMP. Exchange protein directly activated by cAMP can activate the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase (ERK) in a cAMP-dependent manner in collecting duct cells.35 However, the physiologic response to vasopressin seems to be a decrease of MAPK activity.
Moreover, increased levels of cAMP result in the activation of a number of epithelial transporters that may contribute to secretion of fluids into the cysts in ADPKD.36 Interestingly, cyst secretion preferably occurs in conjunction with nucleotides, such as ATP.37 Elegant studies showed that these nucleotides are released in response to physiologic concentrations of AVP in the cortical and inner medullary collecting ducts at very high local concentrations of up to 0.3 µM.38 In cultured cells, vasopressin-induced cAMP formation increases chloride secretion as well as sodium excretion through various transporters and channels.39 The pathophysiology of regulation of secretion of fluids and electrolytes into the cyst lumen has been reviewed in detail elsewhere.36 However, cAMP-dependent regulation of transport activity of other segments of the nephron, such as the mTAL of the Henle loop, are not well studied in ADPKD, although mTAL is volumetrically the predominant structure in the outer medulla.40–42 V2R activation may lead to a significant increase in activity or abundance of the active sodium transporters, the apical membrane type 3 Na+/H+ exchanger and Na+:K+:2Cl− cotransporter cotransporter in vivo.40,42 These transporters are crucially involved in maintaining corticomedullary osmolality gradient as well as urine concentration. Consequently, alterations in osmolality again are expected to modulate various stress kinase pathways in the cells that, therefore, may directly regulate the proliferative tone of kidney cells. This connection has not yet been studied in greater detail.
V2R-Mediated Calcium/Calmodulin-Dependent Kinase Signaling
Recent exciting evidence suggests that additional pathways may contribute to the pathogenic role of V2R in ADPKD. A quantitative phosphoproteomic analysis using stable isotope labeling by amino acids in mpk cortical collecting duct (mpkCCD) cells showed that vasopressin significantly induces phosphorylation of multiple phosphorylation sites in various proteins.43 Interestingly, bioinformatic analyses of the amino acid sequence surrounding these residues revealed that about 70% of all phosphopeptides increased by vasopressin treatment contained arginine or lysine residues at the -3 position43—a finding that is consistent with phosphorylation by basophilic kinases, including PKA. Basophilic kinases are kinases of the AGC family, such as PKA, protein kinase G, and protein kinase C, as well as calmodulin-dependent kinases. In fact, the study confirmed the important role of PKA in the cellular responses to vasopressin stimulation. In addition to PKA, calmodulin-dependent kinases are activated through V2R, which may cause important additional signaling activities. Calcium/calmodulin-dependent signaling is triggered by vasopressin through the release of intracellular calcium by ryanodine receptors.44 Vasopressin-dependent calcium changes follow a distinct oscillative pattern.45 Calmodulin activation leads to myosin light chain kinase activation subsequent to phosphorylation of myosin light chain at T8/T9.46 This process is involved in cytoskeletal reorganization connecting the V2R response with cytoskeletal regulation.46 In addition, analysis of the vasopressin V2R-dependent phosphoproteomic changes dataset revealed that calmodulin-dependent kinase 2 is partially involved in vasopressin signaling.43 Based on transcript levels, this kinase is one of the most abundant kinases in collecting duct.47 A downstream target of this kinase is stathmin. Phosphorylation at S16 of stathmin is increased by V2R activation.36 Interestingly, this protein is involved in microtubule stabilization,48,49 and phosphorylation at S16 can only be observed during formation of the mitotic spindle.48 However, the role of V2R activation in microtubule control is still elusive, although some initial studies have been performed in cultured cells.50,51 Taken together, recent data indicate that V2R-dependent pathways may involve not only canonical cAMP-dependent PKA activity but also, the activation of additional basophilic kinases and downstream mediators.
V2R-Dependent MAPK Signaling
It has been speculated for a long time that vasopressin may modulate MAPK and cyclin-dependent kinase signaling. However, until recently, the role of V2R signaling in MAPK activation remained elusive, which changed with a recent systems biology study using high-throughput quantitative phosphoproteomic analyses after V2R activation in mpkCCD cells.36 V2R stimulation causes a decrease of the activity of various MAPKs, at least under physiologic conditions. Quantitative global phosphoproteomic analyses using both native tissue, as well as mpkCCD cells, revealed that treatment with a V2R agonist caused a significant decrease in phosphorylation of proline-directed phosphorylation motifs, thereby suggesting inhibited activity of MAPKs.43,52–54 This effect is a quantitatively important effect. In primary cultured IMCD cells, p38 activity is decreased in response to cAMP and dDAVP.55 Moreover, V2R stimulation in native collecting duct tissue inhibited activation and phosphorylation of ERK and c-Jun N-terminal kinase.54 In addition, increased levels of cAMP, as well as vasopressin V2R activation, were associated with diminished phosphorylation at proline-directed phosphorylation sites in mTAL.34
The decreased activity of MAPKs after physiologic vasopressin stimulation is also associated with decreased phosphorylation and activity of known MAPK-dependent transcription factors, including activating transcription factor 2 and the tumor suppressor protein p53.36 In addition, nuclear abundance of JunD is significantly decreased in response to dDAVP.56 Also, phosphorylation of c-Jun in the nucleus is decreased in response to dDAVP.57 In conclusion, all of these studies suggest an important but ill-defined role for V2R-dependent modulation of MAPK signaling in the renal epithelium.
Interestingly, a different effect is observed in cells derived from ADPKD cysts, which may suggest a critical phenotypical switch in PKD (Figure 2). In contrast to wild-type collecting duct cells, stimulation of cyst-derived cells with vasopressin at a supraphysiological dose resulted in markedly increased ERK phosphorylation.58 These data are consistent with studies in undifferentiated human embryonic kidney cells expressing transfected V2R.59 ERK activation is known to be a key player in proliferating epithelia.56 Most importantly, basal cAMP levels are not elevated in rodent models of ADPKD.60 However, the molecular correlate for this switch is not yet entirely identified and may require additional proteomics studies. Currently, there is evidence that decreased intracellular calcium is partly responsible for this switch and that the elevation of calcium within cystic cells leads to a normalized proliferation response in response to cAMP.58,61 In addition, a Ca-dependent modulation of cAMP signaling may involve different isoforms of adenylate cyclases 1, 2, 3, and 6 and phosphodiesterases.61–63
Figure 2.
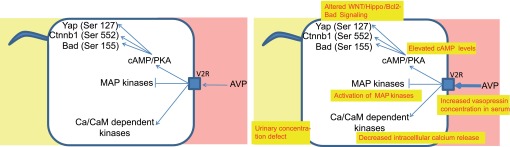
Comparison of physiologic vasopressin signaling and the pathologic vasopressin response in ADPKD. The yellow boxes indicate findings in ADPKD cystic epithelia. dep, dependent.
V2R-Dependent Signaling Pathways Are Involved in Controlling Apoptosis and Proliferation
Several of the recently described canonical and noncanonical V2R signaling pathways may have essential roles in not only the regulation of cell proliferation but also, the assurance of cell survival and the prevention of apoptosis in vivo. Mice with a genetic deletion of the gene encoding for the water channel AQP1, which is predominantly expressed in the proximal nephron and essential for generating the countercurrent gradient by significantly increasing water permeability in the thin descending limb of Henle,64 show an altered proliferation rate of tubular cells in response to vasopressin.65 Furthermore, a recent phosphoproteomic study in vasopressin-responsive native tubular cells revealed that already short-term vasopressin leads to a slight decrease of cleaved caspases.53 This study also confirmed decreased activity of MAPKs in response to vasopressin. Interestingly, studies in cell culture as well as native IMCD cells revealed a PKA-dependent phosphorylation of the proapoptotic Bcl2 family member Bcl2-agonist of cell death at three PKA sites (S112, S136, and S155).53 Phosphorylation at these residues within the BH3 domain is believed to reduce the cellular driving force for apoptosis through control of the release of Bcl2.66 In addition, Stat5a phosphorylation is increased by V2R activation at the activating site Ser779.53 Stat5a is known to increase Bcl2 expression. One recent study found a net decrease in apoptosis rates by even physiologic concentrations of vasopressin in cell culture systems.67 Interestingly, in polycystic Han:SPRD rats, expression of Bcl-XL is decreased, whereas Bcl-2 seems to be suppressed, suggesting a deregulation of apoptosis.68 Finally, phosphorylation of Akt at an activating site is increased by activation of the Vasopressin-V2R receptor, which was shown in isolated IMCD tubules.54 This mechanism may contribute to a direct antiapoptotic effect of vasopressin response.
V2R-Dependent Modulation of Members of the Canonical Wnt Signaling Pathway
One of the unanticipated findings in a number of quantitative phosphoproteomic studies analyzing V2R-dependent signaling was an increased phosphorylation of β-catenin at S552. β-Catenin is a well studied protein and an essential mediator of the highly conserved canonical Wnt signaling pathway.42 Deregulated Wnt signaling has been implicated in the pathogenesis of cystic kidney diseases in numerous studies.69 Induced phosphorylation of β-catenin at S552 was consistently found in a variety of vasopressin-responsive tissues, including whole–inner medullary lysates of Brattleboro rats,70 preparations of rat IMCD tubules53 as well as rat mTAL tubules,41 and mpkCCD cells.43 This phosphorylation site is a typical basophilic phosphorylation site, and it is phosphorylated by PKA, leading to the intriguing hypothesis that canonical V2R signaling may be interconnected with Wnt signaling.71 S552 phosphorylation mediates interaction between β-catenin and TCF,72 which has potential impact on gene expression controlling growth and proliferation of cells.73 In fact, pharmacological inhibition of PKA was shown to inhibit phosphorylation at this site.70 Moreover, β-catenin accumulated in the nucleus of cells stimulated with vasopressin, which was revealed by a recent proteomic study.74 In this study, 3987 proteins were analyzed and quantified using stable isotope labeling by amino acids in the nucleus in the absence and presence of vasopressin in mpkCCD cells. β-Catenin (specifically, its phosphorylated form) was among the proteins with the highest nuclear trafficking rate in response to vasopressin. In addition, GSK3β, another member of the Wnt signaling system, is also connected to vasopressin signaling. Knockdown of the kinase resulted in diminished cAMP levels and reduced urinary concentration ability.75 Thus, an increased activity of this kinase, which may be observed in cystic epithelia, may, therefore, even be involved in the modulation of cAMP response.
Benefits of V2R Antagonism—What Are the Crucial Components?
V2R blockade, as well as water loading, lead to polyuria and thus, an altered tubular flow rate. Thus, it is conceivable that flow-dependent effects transmitted through the stimulation of various cilia-dependent pathways, such as suppression of mTor signaling,76 might be involved in the beneficial effect of V2R antagonists. The results obtained in PKD models could be further complicated by the fact that V2R may be missorted to the apical surface of the tubular cells.77 However, the V2R antagonist-mediated flow increase would most likely be in the very distal section of the collecting duct (in the IMCD). Most cysts, however, do not originate from this segment. There would also be no beneficial effect on larger cyst populations, which are no longer connected to the tubular system; however, this result seems to be likely. Future studies need, therefore, to address the issue of water consumption compared with V2R signaling alone to assess any potential benefit in treating ADPKD patients.
Conclusion
Recent exciting evidence has shown that V2R activation contributes to the progression of kidney failure in ADPKD patients. More importantly, pharmacological antagonism of V2R signaling is now emerging as the most promising way to treat ADPKD.1,2 The inhibition of V2R in vivo slows down cyst growth and preserves kidney function. Progress has been made to describe the pathophysiological feedback loops in the vasopressin system in ADPKD patients. Now, it is time to go back to the bench. Much of the beneficial effect may be not only attributed to classic canonical V2R signaling but also, the result of previously ill-described modulation of signaling pathways. Important questions should now be addressed. What are the signaling networks that are affected on V2R inhibition in ADPKD patients? What are the molecular components of the phenotypical switch in response to cAMP? We believe that modification of noncanonical pathways, such as the Wnt signaling system, is highly conceivable and that the concept of using V2R inhibition can be even more powerful if we identified these noncanonical V2R-dependent pathways.
Disclosures
T.B. has received honoraria for scientific presentations from Novartis, Amgen, Otsuka, and Hexal and serves as a scientific expert on an Otsuka-funded European Polycystic Kidney Disease Advisory Board.
Acknowledgments
Polycystic kidney disease research, as well as studies into vasopressin-2 receptor signaling, have benefited from the contributions of many groups all over the world. We apologize to those colleagues whose work could not be cited in this article because of space limitations.
M.M.R. was supported by the Fritz-Scheler Stipendium by the KfH Stiftung for Präventivmedizin. This work was supported by Deutsche Forschungsgemeinschaft Grants SCHE1562-2 (to B.S.) and SFB832 (to B.S. and T.B.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, Horie S, Nutahara K, Ouyang J, Krasa HB, Czerwiec FS, TEMPOFormula and 156-05-002 Study Investigators : Tolvaptan in autosomal dominant polycystic kidney disease: Three years’ experience. Clin J Am Soc Nephrol 6: 2499–2507, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres VE: Role of vasopressin antagonists. Clin J Am Soc Nephrol 3: 1212–1218, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Gattone VH, 2nd, Wang X, Harris PC, Torres VE: Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9: 1323–1326, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Wang CJ, Creed C, Winklhofer FT, Grantham JJ: Water prescription in autosomal dominant polycystic kidney disease: A pilot study. Clin J Am Soc Nephrol 6: 192–197, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, TEMPO 3:4 Trial Investigators : Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367: 2407–2418, 2012. 23121377 [Google Scholar]
- 6.Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Hörl WH, Obermüller N, Arns W, Pavenstädt H, Gaedeke J, Büchert M, May C, Gschaidmeier H, Kramer S, Eckardt K-U: Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 363: 830–840, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wüthrich RP: Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 363: 820–829, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Meijer E, Bakker SJL, van der Jagt EJ, Navis G, de Jong PE, Struck J, Gansevoort RT: Copeptin, a surrogate marker of vasopressin, is associated with disease severity in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 361–368, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meijer E, Boertien WE, Zietse R, Gansevoort RT: Potential deleterious effects of vasopressin in chronic kidney disease and particularly autosomal dominant polycystic kidney disease. Kidney Blood Press Res 34: 235–244, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Boertien WE, Meijer E, Zittema D, van Dijk MA, Rabelink TJ, Breuning MH, Struck J, Bakker SJL, Peters DJM, de Jong PE, Gansevoort RT: Copeptin, a surrogate marker for vasopressin, is associated with kidney function decline in subjects with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 27: 4131–4137, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Zittema D, Boertien WE, van Beek AP, Dullaart RPF, Franssen CFM, de Jong PE, Meijer E, Gansevoort RT: Vasopressin, copeptin, and renal concentrating capacity in patients with autosomal dominant polycystic kidney disease without renal impairment. Clin J Am Soc Nephrol 7: 906–913, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Morgenthaler NG, Struck J, Alonso C, Bergmann A: Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem 52: 112–119, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Morgenthaler NG, Müller B, Struck J, Bergmann A, Redl H, Christ-Crain M: Copeptin, a stable peptide of the arginine vasopressin precursor, is elevated in hemorrhagic and septic shock. Shock 28: 219–226, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Maldonado M, Yium JJ, Eknoyan G, Suki WN: Adult polycystic kidney disease: Studies of the defect in urine concentration. Kidney Int 2: 107–113, 1972 [DOI] [PubMed] [Google Scholar]
- 15.Krishnan R, Eley L, Sayer JA: Urinary concentration defects and mechanisms underlying nephronophthisis. Kidney Blood Press Res 31: 152–162, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Gabow PA, Kaehny WD, Johnson AM, Duley IT, Manco-Johnson M, Lezotte DC, Schrier RW: The clinical utility of renal concentrating capacity in polycystic kidney disease. Kidney Int 35: 675–680, 1989 [DOI] [PubMed] [Google Scholar]
- 17.Ahrabi AK, Terryn S, Valenti G, Caron N, Serradeil-Le Gal C, Raufaste D, Nielsen S, Horie S, Verbavatz J-M, Devuyst O: PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J Am Soc Nephrol 18: 1740–1753, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Ho TA, Godefroid N, Gruzon D, Haymann J-P, Maréchal C, Wang X, Serra A, Pirson Y, Devuyst O: Autosomal dominant polycystic kidney disease is associated with central and nephrogenic defects in osmoregulation. Kidney Int 82: 1121–1129, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Park F, Mattson DL, Skelton MM, Cowley AW, Jr.: Localization of the vasopressin V1a and V2 receptors within the renal cortical and medullary circulation. Am J Physiol 273: R243–R251, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Carmosino M, Brooks HL, Cai Q, Davis LS, Opalenik S, Hao C, Breyer MD: Axial heterogeneity of vasopressin-receptor subtypes along the human and mouse collecting duct. Am J Physiol Renal Physiol 292: F351–F360, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl W, Bachmann S: Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol 293: F1166–F1177, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Fenton RA, Brønd L, Nielsen S, Praetorius J: Cellular and subcellular distribution of the type-2 vasopressin receptor in the kidney. Am J Physiol Renal Physiol 293: F748–F760, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Nielsen S, Chou CL, Marples D, Christensen EI, Kishore BK, Knepper MA: Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci U S A 92: 1013–1017, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaeggeler H-P, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, Horisberger J-D, Rossier BC: Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol 16: 878–891, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Duong Van Huyen J, Bens M, Vandewalle A: Differential effects of aldosterone and vasopressin on chloride fluxes in transimmortalized mouse cortical collecting duct cells. J Membr Biol 164: 79–90, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Henn V, Edemir B, Stefan E, Wiesner B, Lorenz D, Theilig F, Schmitt R, Vossebein L, Tamma G, Beyermann M, Krause E, Herberg FW, Valenti G, Bachmann S, Rosenthal W, Klussmann E: Identification of a novel A-kinase anchoring protein 18 isoform and evidence for its role in the vasopressin-induced aquaporin-2 shuttle in renal principal cells. J Biol Chem 279: 26654–26665, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Stefan E, Wiesner B, Baillie GS, Mollajew R, Henn V, Lorenz D, Furkert J, Santamaria K, Nedvetsky P, Hundsrucker C, Beyermann M, Krause E, Pohl P, Gall I, MacIntyre AN, Bachmann S, Houslay MD, Rosenthal W, Klussmann E: Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol 18: 199–212, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Edemir B, Pavenstädt H, Schlatter E, Weide T: Mechanisms of cell polarity and aquaporin sorting in the nephron. Pflugers Arch 461: 607–621, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Lu HJ, Matsuzaki T, Bouley R, Hasler U, Qin Q-H, Brown D: The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am J Physiol Renal Physiol 295: F290–F294, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice WL, Zhang Y, Chen Y, Matsuzaki T, Brown D, Lu HAJ: Differential, phosphorylation dependent trafficking of AQP2 in LLC-PK1 cells. PLoS One 7: e32843, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Douglass J, Gunaratne R, Bradford D, Saeed F, Hoffert JD, Steinbach PJ, Knepper MA, Pisitkun T: Identifying protein kinase target preferences using mass spectrometry. Am J Physiol Cell Physiol 303: C715–C727, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, Calvet JP: Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J Am Soc Nephrol 17: 3424–3437, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Maeda Y, Terada Y, Nonoguchi H, Knepper MA: Hormone and autacoid regulation of cAMP production in rat IMCD subsegments. Am J Physiol 263: F319–F327, 1992 [DOI] [PubMed] [Google Scholar]
- 34.Kortenoeven MLA, Trimpert C, van den Brand M, Li Y, Wetzels JFM, Deen PMT: In mpkCCD cells, long-term regulation of aquaporin-2 by vasopressin occurs independent of protein kinase A and CREB but may involve Epac. Am J Physiol Renal Physiol 302: F1395–F1401, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Klein JD, Blount MA, Martin CF, Kent KJ, Pech V, Wall SM, Sands JM: Epac regulates UT-A1 to increase urea transport in inner medullary collecting ducts. J Am Soc Nephrol 20: 2018–2024, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terryn S, Ho A, Beauwens R, Devuyst O: Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta 1812: 1314–1321, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Buchholz B, Teschemacher B, Schley G, Schillers H, Eckardt K-U: Formation of cysts by principal-like MDCK cells depends on the synergy of cAMP- and ATP-mediated fluid secretion. J Mol Med (Berl) 89: 251–261, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Odgaard E, Praetorius HA, Leipziger J: AVP-stimulated nucleotide secretion in perfused mouse medullary thick ascending limb and cortical collecting duct. Am J Physiol Renal Physiol 297: F341–F349, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, Wallace DP: Tolvaptan inhibits ERK-dependent cell proliferation, Cl⁻ secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol 301: F1005–F1013, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giménez I, Forbush B: Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278: 26946–26951, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Gunaratne R, Braucht DWW, Rinschen MM, Chou C-L, Hoffert JD, Pisitkun T, Knepper MA: Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci U S A 107: 15653–15658, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ecelbarger CA, Kim GH, Wade JB, Knepper MA: Regulation of the abundance of renal sodium transporters and channels by vasopressin. Exp Neurol 171: 227–234, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Rinschen MM, Yu M-J, Wang G, Boja ES, Hoffert JD, Pisitkun T, Knepper MA: Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc Natl Acad Sci U S A 107: 3882–3887, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou CL, Yip KP, Michea L, Kador K, Ferraris JD, Wade JB, Knepper MA: Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J Biol Chem 275: 36839–36846, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Yip K-P, Sham JSK: Mechanisms of vasopressin-induced intracellular Ca2+ oscillations in rat inner medullary collecting duct. Am J Physiol Renal Physiol 300: F540–F548, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou C-L, Christensen BM, Frische S, Vorum H, Desai RA, Hoffert JD, de Lanerolle P, Nielsen S, Knepper MA: Non-muscle myosin II and myosin light chain kinase are downstream targets for vasopressin signaling in the renal collecting duct. J Biol Chem 279: 49026–49035, 2004 [DOI] [PubMed] [Google Scholar]
- 47.Uawithya P, Pisitkun T, Ruttenberg BE, Knepper MA: Transcriptional profiling of native inner medullary collecting duct cells from rat kidney. Physiol Genomics 32: 229–253, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.le Gouvello S, Manceau V, Sobel A: Serine 16 of stathmin as a cytosolic target for Ca2+/calmodulin-dependent kinase II after CD2 triggering of human T lymphocytes. J Immunol 161: 1113–1122, 1998 [PubMed] [Google Scholar]
- 49.Daub H, Gevaert K, Vandekerckhove J, Sobel A, Hall A: Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J Biol Chem 276: 1677–1680, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Sabolić I, Katsura T, Verbavatz JM, Brown D: The AQP2 water channel: Effect of vasopressin treatment, microtubule disruption, and distribution in neonatal rats. J Membr Biol 143: 165–175, 1995 [DOI] [PubMed] [Google Scholar]
- 51.Hartwig JH, Ausiello DA, Brown D: Vasopressin-induced changes in the three-dimensional structure of toad bladder apical surface. Am J Physiol 253: C707–C720, 1987 [DOI] [PubMed] [Google Scholar]
- 52.Miller ML, Jensen LJ, Diella F, Jørgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li S, Cesareni G, Pawson T, Turk BE, Yaffe MB, Brunak S, Linding R: Linear motif atlas for phosphorylation-dependent signaling. Sci Signal 1: ra2, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffert JD, Pisitkun T, Saeed F, Song JH, Chou CL, Knepper MA: Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol Cell Proteomics 11: M111.014613, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pisitkun T, Jacob V, Schleicher SM, Chou C-L, Yu M-J, Knepper MA: Akt and ERK1/2 pathways are components of the vasopressin signaling network in rat native IMCD. Am J Physiol Renal Physiol 295: F1030–F1043, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nedvetsky PI, Tabor V, Tamma G, Beulshausen S, Skroblin P, Kirschner A, Mutig K, Boltzen M, Petrucci O, Vossenkämper A, Wiesner B, Bachmann S, Rosenthal W, Klussmann E: Reciprocal regulation of aquaporin-2 abundance and degradation by protein kinase A and p38-MAP kinase. J Am Soc Nephrol 21: 1645–1656, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, Germino GG, Pandolfi PP, Boletta A: Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol 29: 2359–2371, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolger SJ, Hurtado PAG, Hoffert JD, Saeed F, Pisitkun T, Knepper MA: Quantitative phosphoproteomics in nuclei of vasopressin-sensitive renal collecting duct cells. Am J Physiol Cell Physiol 303: C1006–C1020, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP, Grantham JJ: cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 57: 1460–1471, 2000 [DOI] [PubMed] [Google Scholar]
- 59.Oligny-Longpré G, Corbani M, Zhou J, Hogue M, Guillon G, Bouvier M: Engagement of β-arrestin by transactivated insulin-like growth factor receptor is needed for V2 vasopressin receptor-stimulated ERK1/2 activation. Proc Natl Acad Sci U S A 109: E1028–E1037, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE: Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19: 102–108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamaguchi T, Hempson SJ, Reif GA, Hedge A-M, Wallace DP: Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17: 178–187, 2006 [DOI] [PubMed] [Google Scholar]
- 62.Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE: Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease [published online ahead of print October 24, 2013]. J Am Soc Nephrol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pinto CS, Reif GA, Nivens E, White C, Wallace DP: Calmodulin-sensitive adenylyl cyclases mediate AVP-dependent cAMP production and Cl- secretion by human autosomal dominant polycystic kidney cells. Am J Physiol Renal Physiol 303: F1412–F1424, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chou CL, Knepper MA, Hoek AN, Brown D, Yang B, Ma T, Verkman AS: Reduced water permeability and altered ultrastructure in thin descending limb of Henle in aquaporin-1 null mice. J Clin Invest 103: 491–496, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai Q, McReynolds MR, Keck M, Greer KA, Hoying JB, Brooks HL: Vasopressin receptor subtype 2 activation increases cell proliferation in the renal medulla of AQP1 null mice. Am J Physiol Renal Physiol 293: F1858–F1864, 2007 [DOI] [PubMed] [Google Scholar]
- 66.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ: Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619–628, 1996 [DOI] [PubMed] [Google Scholar]
- 67.Miller RL, Sandoval PC, Pisitkun T, Knepper MA, Hoffert JD: Vasopressin inhibits apoptosis in renal collecting duct cells. Am J Physiol Renal Physiol 304: F177–F188, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ecder T, Melnikov VY, Stanley M, Korular D, Lucia MS, Schrier RW, Edelstein CL: Caspases, Bcl-2 proteins and apoptosis in autosomal-dominant polycystic kidney disease. Kidney Int 61: 1220–1230, 2002 [DOI] [PubMed] [Google Scholar]
- 69.Benzing T, Simons M, Walz G: Wnt signaling in polycystic kidney disease. J Am Soc Nephrol 18: 1389–1398, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Bansal AD, Hoffert JD, Pisitkun T, Hwang S, Chou C-L, Boja ES, Wang G, Knepper MA: Phosphoproteomic profiling reveals vasopressin-regulated phosphorylation sites in collecting duct. J Am Soc Nephrol 21: 303–315, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO: Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem 281: 9971–9976, 2006 [DOI] [PubMed] [Google Scholar]
- 72.Taurin S, Sandbo N, Yau DM, Sethakorn N, Dulin NO: Phosphorylation of β-catenin by PKA promotes ATP-induced proliferation of vascular smooth muscle cells. Am J Physiol Cell Physiol 294: C1169–C1174, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z: Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282: 11221–11229, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schenk LK, Bolger SJ, Luginbuhl K, Gonzales PA, Rinschen MM, Yu M-J, Hoffert JD, Pisitkun T, Knepper MA: Quantitative proteomics identifies vasopressin-responsive nuclear proteins in collecting duct cells. J Am Soc Nephrol 23: 1008–1018, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rao R, Patel S, Hao C, Woodgett J, Harris R: GSK3beta mediates renal response to vasopressin by modulating adenylate cyclase activity. J Am Soc Nephrol 21: 428–437, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weimbs T: Polycystic kidney disease and renal injury repair: Common pathways, fluid flow, and the function of polycystin-1. Am J Physiol Renal Physiol 293: F1423–F1432, 2007 [DOI] [PubMed] [Google Scholar]
- 77.Saigusa T, Reichert R, Guare J, Siroky BJ, Gooz M, Steele S, Fenton RA, Bell PD, Kolb RJ: Collecting duct cells that lack normal cilia have mislocalized vasopressin-2 receptors. Am J Physiol Renal Physiol 302: F801–F808, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]