

Abstract
Chronic hepatitis B virus (HBV) infection is one of the most common causes of hepatocellular carcinoma (HCC), a malignant tumor with high mortality worldwide. One remarkable clinical feature of HBV-related HCC is that its incidence is higher in males and postmenopausal females compared to other females. Increasing evidence indicates that HBV-associated HCC may involve gender disparity and that it may be a type of hormone-responsive malignant tumor. Sex hormones, such as androgen and estrogen, have been shown to play very different roles in the progression of an HBV infection and in the development of HBV-related HCC. Through binding to their specific cellular receptors and affecting the corresponding signaling pathways, sex hormones can regulate the transactivation of HBx, cause the chronic release of inflammatory cytokines in the hepatocellular microenvironment, and participate in epigenetic and genetic alternations in hepatocytes. All of these functions may be related to the initiation and progression of HBV-associated HCC. A thorough investigation of the molecular mechanisms underlying the gender-related disparity in HBV-related HCC should provide a new perspective for better understanding its pathogenesis and exploring more effective methods for the prevention and treatment of this disease.
Keywords: Hepatitis B virus, Hepatocellular carcinoma, Gender disparity, Sex hormones
Core tip: Increasing evidence indicates that hepatitis B virus (HBV)-associated hepatocellular carcinoma (HCC) may involve gender disparity and that it may be a type of hormone-responsive malignant tumor. Sex hormones have been shown to play very different roles in the progression of an HBV infection and in the development of HBV-related HCC. The article reviews the reported molecular mechanisms of the gender disparity in HBV-related HCC, with an aim to improve the understanding of the development and progression of HBV-associated HCC and exploring more effective prevention and treatment of this disease.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most prevalent malignant tumors and a leading cause of cancer-related deaths globally[1-3]. Chronic hepatitis B virus (HBV) infection, the strongest factor associated with HCC development in epidemiology, has been found to play an important role in the progression of HCC. Many countries, particularly those in eastern Asia, sub-Saharan Africa and parts of Europe, have high incidence and mortality rates of HBV-associated HCC[2,4,5]. To further investigate the clinical features of HBV-related HCC and develop more effective therapeutic strategies, considerable efforts have recently been exerted in exploring the molecular mechanisms involved in the development and progression of HBV-associated HCC. Many clinical studies have shown that one remarkable clinical feature of HBV-associated HCC is that its incidence generally tends to be higher in males and postmenopausal females than other females, but the exact molecular mechanisms of this disparity have seldom been investigated[6-8]. The article reviews the reported molecular mechanisms associated with the gender disparity in HBV-related HCC to improve the understanding of its role in the development and progression of HBV-associated HCC.
ROLE OF SEX HORMONES IN THE DEVELOPMENT OF HBV-ASSCOCIATED HCC
Most previous studies have demonstrated that there is a sexual disparity in the development of HBV-related HCC. For example, female HBV carriers generally have lower viral loads than male carriers, the risk of HBV-associated HCC is lower in females than in males, and the ratio of the estradiol level to the testosterone level tends to decrease in female patients with HBV-related HCC than in female HBV carriers who do not develop HCC[9-11]. To date, the mechanisms underlying that males are more susceptible to developing HCC after an HBV infection have become an important topic, drawing widespread attention from scientists. An increasing number of studies have suggested that HBV-associated HCC may be a hormone-responsive malignant tumor[12].
It was reported that high levels of serum testosterone in males with an HBV infection are associated with their development of HCC. Even when the condition of their viral hepatitis or alcoholic cirrhosis improved, their risk for HCC was still increasing[9,13]. These results suggested that the androgen receptor (AR) might be involved in HBV-related hepatocarcinogenesis and an active androgen signaling pathway might increase the risk of HBV-associated HCC[14-16]. The AR, a ligand-dependent transcription factor of the nuclear receptor superfamily, was reported to be overexpressed in HCC. After two nuclear processes, including the AR N-C interaction (dimerization step) and the transcriptional activation of the AR N-terminal transactivation domain (NTD) (transactivation step), are completed, the AR is fully activated[14,15]. Interestingly, among male HBV carriers with an increased risk of HCC, both higher androgen levels and more active androgen receptor gene alleles were detected, compared with those of controls[15,16]. Similarly, the ratio of estradiol to testosterone was shown to be significantly lower in female patients with HBV-associated HCC. Wang et al[15] demonstrated that the HBV X viral protein (HBx), a well-known transactivator of HBV transcription, indirectly increases the levels of androgen receptor mRNA by affecting cytosolic signaling pathways[11,17]. HBx can enhance two nuclear processes, including the dimerization and transactivation steps, by regulating two switch kinases: c-Src kinase and GSK-3 kinase. Functionally, c-Src kinase regulates AR NTD activity, and GSK-3 kinase plays an important role in regulating the AR N-C interaction. Further investigation shows that c-Src primarily enhances the transcriptional activity of the AR NTD. However, GSK-3 inactivation contributed to the HBx-enhanced AR N-C interaction[14,18-20]. In male mice with androgen receptor knockout, the occurrence of HCC induced by diethylnitrosamine (DEN) was delayed and fewer tumors formed than in wild type mice[15,21]. In transgenic mice with an HBV infection, the hepatic androgen receptor was found to increase the rate of HBV-induced hepatocarcinogenesis. Through binding directly to the cognate androgen-responsive element (ARE) in enhancer I (Enh I) of the HBV genome, androgen cooperated with the androgen-signaling pathway to increase the transcription and replication of HBV genes[15] (Figure 1). These findings suggest that there may be an interesting positive cross-regulatory loop between the androgen-signaling pathway and the HBx viral protein in the development of HCC in male HBV carriers, as Chen et al[22] demonstrated in clinical reports on male HBV patients. Similarly, Feng et al[23] elucidated another interestingly self-amplifying positive regulatory circuit including AR, cell cycle-related kinase (CCRK) and β-catenin, which were found both in HCC cell lines and immortal human liver cells and further detected in animal models and primary HCC specimens from patients with HBV infection. Through directly binding to androgen-responsive element of the CCRK promoter region, ligand-activated AR could increase CCRK expression by up-regulating its transcription in HCC cell lines. And overexpression of CCRK modulated activation of the β-catenin signaling pathway which positively regulates expression and function of AR in HCC cell lines. Finally, through up-regulating activation of the β-catenin/T cell factor signaling pathway, this vicious loop might play a critical role in tumorigenesis and progression of HBV-associated HCC[23] (Figure 2).
Figure 1.
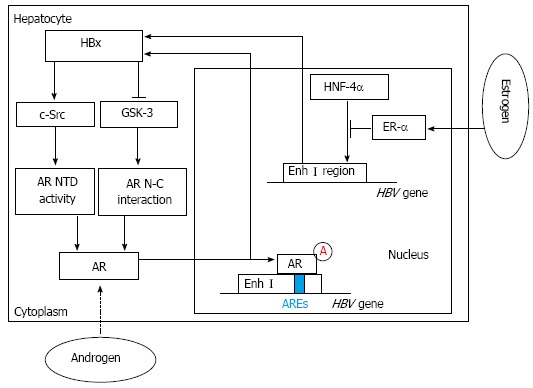
Hepatitis B virus X viral protein expression and sex hormone signaling pathways. By activating c-Src and inactivating glycogen synthase kinase-3 (GSK-3), hepatitis B virus X viral protein (HBx) can enhance androgen receptor (AR) N-terminal transactivation domain (NTD) activity and AR N-C interaction, which contribute to full activation of AR. Then through binding directly to the cognate androgen-responsive element (ARE) in enhancer I (Enh I) of the hepatitis B virus (HBV) genome, androgen can cooperate with the androgen-signaling pathway to increase the transcription and replication of HBV genes and HBx expression. In contrast, estrogen can cooperate with estrogen receptor-α (ER-α) to decrease HBV RNA transcription by suppressing the activity of the HBV Enh I through preventing hepatocyte nuclear factor 4α (HNF-4α) from binding to Enh I.
Figure 2.

The regulation and function of androgen receptor signaling pathway. On one hand, ligand-activated androgen receptor (AR) can increase cell cycle-related kinase (CCRK) expression through directly binding to androgen-responsive element (ARE) of the CCRK promoter region and stimulate its transcription and expression. Ectopic expression of CCRK in immortalized human liver cells activated β-catenin/T cell factor (TCF) signaling to stimulate cell cycle progression and to induce AR expression. On the other hand, ligand-stimulated AR can increase miR-216a transcription by directly binding to the ARE site within the promoter region of pri-miR-216a, which can lead to the elevation of the miR-216a level and the subsequent suppression of its target genes including some tumor suppressors. Finally, the early stage of hepatocarcinogenesis, including increased cell proliferation and enhanced migration and invasive activities of hepatocytes, is partly initiated.
Unlike androgen, estrogen may play an important role in protecting against the progression of HBV infections and the development of HBV-related HCC[7,15]. Almog et al[24] reported that estrogen treatment reduced the level of HBV mRNA. De Maria et al[25] found that variant estrogen receptors (ERs), a primary cause of estrogen unresponsiveness, were expressed more frequently in male and postmenopausal female HCC patients. Moreover, female HBV carriers with a longer exposure to estrogen through taking oral contraceptives or postmenopausal hormone replacement therapy have a lower risk of developing HCC[15,26]. Acting through two nuclear receptors, estrogen receptors α and β (ER-α and ER-β), estrogen plays an important role in many biological processes at the molecular level[7,27]. In contrast to the augmentation of HBV RNA transcription by the AR, the ER was found to decrease HBV RNA transcription by suppressing the activity of the HBV enhancer I that is located in the Enh I region of the HBV genome. Further investigation suggested that the hinge region of ER-α could physically interact with hepatocyte nuclear factor 4α (HNF-4α), which diminished the HNF-4α binding to Enh I region of the HBV genome[26,28]. HNF-4α, a nuclear protein that is abundant in the liver, plays an important role in regulating hepatic transcription and the development of normal liver structures[29,30]. Wang et al[26] demonstrated that the HNF-4α binding site within HBV enhancer I is essential for the down-regulation of HBV transcription by ER-α. It was observed that ER-α can squelch the activity of HNF-4α through its hinge region, preventing HNF-4α from binding to enhancer I (Figure 1). This process finally results in the decreased activity and even the dysfunction of enhancer I in HBV transcription. The hepatic ER-α can work alone even in the absence of estrogen, which is different from the AR. This change may be due to the presence of some of the hepatic ER-α in the nucleus[26]. Moreover, some studies showed that HBx, ER-α and histone deacetylase 1 (HDAC1) could form a complex. Furthermore, Han et al[31] demonstrated that HBx could inhibit the transcriptional activity of ER-α through its interaction with ER-α and its recruitment of HDAC1.
GENDER DISPARITY, INFLAMMATORY CYTOKINES AND HBV-ASSOCIATED HCC
Chronic inflammation is a major contributor to tumorigenesis[12,32]. Hepatitis-related inflammatory cytokines, such as interleukin-6 (IL-6) and interleukin-1β (IL-1β), have been identified as core promoters of susceptibility to an HBV infection, persistence of an HBV infection and the development of HBV-related HCC[33-35]. Many studies have examined the effects of these cytokines on the initiation, promotion, and progression of HCC[35-37]. Interestingly, chemical carcinogens, such as DEN, have been shown to induce more apoptosis, necrosis, and compensatory proliferation of hepatic cells in male mice than in females. Similar gender disparity in liver injury has also been observed after the administration of CCl4[32,38,39]. These results have suggested that the effects of chronic inflammation and inflammatory cytokines on the development and progression of HBV-related HCC may vary in patients partly due to their gender difference.
In some previous animal studies, researchers demonstrated that a persistently high level of serum IL-6 was associated with increased liver injury and HCC development and that IL-6 overexpression could lead to HCC tumorigenesis in mice. Moreover, the ablation of IL-6 reversed the progression of hepatocarcinogenesis[32,40]. Further investigation revealed that, through the activation of IkB kinase β (IKKβ) in Kupffer cells (KCs), exogenous IL-6 augmented DEN-induced damage in both untreated and estradiol-treated male mice. Furthermore, the absence of IL-6 eliminated the gender difference by reducing the extent of injury in males, which suggested that estradiol may attenuate downstream IL-6 signaling[32,39]. Recently, IL-6 secreted from KCs was proposed as a possible explanation for the gender disparity in hepatocarcinogenesis. Naugler et al[32] showed that the estrogen-mediated inhibition of IL-6 production by KCs reduced the risk of HCC in females. Their further investigation revealed that estrogen and estrogen receptor-mediated signaling pathway blocked IL-6 expression in KCs, resulting in decreased HCC incidence in the DEN mouse model. The mechanisms underlying these phenomena were revealed that estrogens played a role through nuclear and membrane proteins, such as the transcription factors including nuclear factor κB (NFκB), STAT3, C/EBPβ and the Toll-like receptor adaptor protein MyD88[32,41]. Taub et al[42] proved that IL-6 could activate the transcription factor STAT3. Naugler et al[32] demonstrated that the activated form of STAT3 was absent in the livers of IL-6-/- mice and that wild-type (WT) female mice exhibited less STAT3 activation than males after DEN administration. Similarly, the sustained activation of JNK was also found to be required for DEN-induced liver injury and hepatocarcinogenesis[12,31]. By decreasing the activity of core factors such as NF-κB, which was associated with the IL-6 promoter activity and required for the production of IL-6 in vivo and in vitro, estrogens and the ER reduced the level of IL-6 and attenuated its induction of liver injury[43] (Figure 3). Finally, suppressing IL-6 signaling might result in the reduction of HBV-related HCC in females.
Figure 3.

Interleukin-6 expression and estrogen receptor signaling pathway. By decreasing the activity of core factors such as NFκB, STAT3 and C/EBPβ, which is associated with the interleukin-6 (IL-6) promoter activity and required for the production of IL-6 in vivo and in vitro, estrogen combines with the estrogen receptor (ER) signaling pathway to decrease the IL-6 level and attenuate its induction of liver injury. NFκB: Nuclear factor κB; STAT3: Signal transducer and activator of transcription 3; C/EBPβ: CCAAT/enhancer-binding protein β.
The IL-1 gene family (including IL-1α, IL-1β and IL-1RN) is a group of inflammatory cytokines that appear to serve as tumor growth factors in hepatocarcinogenesis. Jiang et al[33] demonstrated that IL-1α was an essential factor for the development of HBV-related HCC in males and its persistent expression was a specific predictor of chronic liver inflammation that could potentially be used as a marker in clinical diagnosis. Clinically, dynamic monitoring of IL-1α had been used and was shown to be a potentially valuable tool in detecting the recurrence of HBV-associated HCC and a possible target for pharmaceutical research. Further investigation of estrogen signaling in menopausal females indicated that the inactivation of estrogen signaling might contribute to the up-regulation of IL-1α and IL-6 expression. Moreover, a significant linear correlation and dependence between decreasing ER expression levels and increasing IL-1α expression levels and the lack of ER expression in male tumor tissues were found, which suggested that the protective anti-inflammatory effect mediated by estrogen did not occur in male HCC tumors[33,35]. Further investigation suggested that excessive activation of the inflammatory response and the IL-α-MyD88-IL-6 signaling pathway induces a compensatory proliferative response in normal hepatic cells and leads to hepatocarcinogenesis in tissue adjacent to the tumors of male patients with HBV-related HCC. Using a luciferase reporter assay, it was found that estradiol might suppress IL-1α expression. Moreover, estradiol might inhibit IL-1α transcription by binding to the ER-α-binding site in the IL-1α promoter region[33-35]. Analogously, increased levels of IL-1β in the liver induced by HBV infection might finally lead to hepatocyte damage and the development of HCC[34]. Bamba et al[44] showed that IL-1β could increase the production of prostaglandin E2 and hepatocyte growth factor. By increasing the COX-2 and nitric oxide levels, high levels of IL-1β induced angiogenesis and promoted tumor growth[44,45]. Furthermore, IL-1β could also attenuate the interferon-induced antiviral activity and STAT1 activation in the liver. Hirankarn et al[34] demonstrated that IL-1β-511 C allele polymorphism in the promoter region elevated the binding activity of its transcription initiation factor and the presence of two C alleles (CC) was required to increase the likelihood of HBV-related HCC development[46].
EPIGENETIC AND GENETIC ALTERNATIONS-RELATED GENDER DIFFERENCES IN THE DEVELOPMENT OF HBV-RELATED HCC
HCC development is a multistep tumorigenic process that includes many carcinogenic-related changes at the molecular level that are affected by epigenetic and/or genetic alternations. It has been shown that many of the HCC risk factors are related to epigenetic changes, such as the regulation of mRNA levels by microRNAs (miRNAs), DNA methylation and histone modification. However, the underlying mechanisms leading to HBV-related HCC development and their relationships with the gender disparity of this process have not been fully explored[6,22,47].
MiRNAs are small noncoding RNAs of approximately 20 nucleotides that bind to conserved 3’-untranslated region (3’-UTR) sequences of their target mRNA, thereby inducing either the inhibition of their translation or their degradation. In the tumor cells, the functional roles of some miRNAs in targeting specific oncogenes or tumor suppressor genes are being increasingly identified. In the hepatocarcinogenic process, numerous miRNAs show abnormal expression patterns in the profiles of HCC tissues compared with their paired adjacent nontumorous tissues. Hence, miRNAs have recently been reported to be a group of host genetic factors associated with hepatocarcinogenesis[22,47]. For instance, by analyzing seven miRNAs, Gao et al[48] identified three miRNAs with aberrant expression patterns in precancerous liver tissues, including miR-224, miR-145, and miR-199b. Increasing evidence that sex hormones affected tumorigenesis by regulating specific miRNAs was reported in malignant tumors such as breast cancer and prostate cancer[49,50]. Similarly, Chen et al[22] showed that miRNAs could be the candidates affected by the androgen pathway in the early stage of hepatocarcinogenesis of HBV-related HCC in males and there was a gender disparity in the pattern of expression of the candidate miRNAs, such as miR-216a, in liver tissues in the precancerous stage. Male HBV-related HCC patients with preferentially elevated miR-216a in their precancerous liver tissues were shown to have a higher risk of mortality. This finding suggested that miR-216a levels are associated with patient prognosis. Further studies indicated that the ligand-stimulated androgen pathway can activate and increase miR-216a transcription by directly binding to the ARE site within the promoter region of pri-miR-216a, leading to the elevation of the miR-216a level and the subsequent suppression of its target genes (Figure 2), such as the tumor suppressor in lung cancer-1 gene (TSLC1). Finally, the early stage of hepatocarcinogenesis, including increased cell proliferation and enhanced migration and invasive activities of hepatocytes, was partly initiated[22]. Through high-throughput DNA sequencing and stem-loop polymerase chain reaction conformation assays, Jiang et al[33] demonstrated that miR-22 was highly expressed and correlated with low ER and high IL-1α expression in the tissue adjacent to tumor tissue in male patients. However, in the paired HCC tissues, the results were quite the contrary. The underlying mechanisms were revealed by using the luciferase reporter assay. The results indicated that ER was a candidate target of miR-22. By directly targeting the 3’-UTR region of ER mRNA, miR-22 could suppress ER transcription. Because both estradiol and the ER played an important role in inhibiting IL-1α expression in normal hepatocytes during chronic liver inflammation, increased miR-22 levels might result in the down-regulation of ER expression and the induction and augmentation of IL-1α transcription. Finally, the persistent high level of IL-1α secreted from necrotic hepatocytes might lead to compensatory proliferation and tumorigenesis in normal hepatic cells.
The silencing of tumor suppressor genes induced by hypermethylation had also been found to occur frequently in many human malignant cancers, including HCC. Previous investigations had shown that abnormal hypermethylation in the promoter region might be the main mechanism underlying the dysfunction of p16INK4a in HCC[6]. The p16INK4A gene, which is located on chromosome 9p21, encodes a critical negative regulator of cell cycle progression and is one of the most frequently inactivated tumor suppressor genes in various tumor types[51-53]. By binding to cyclin-dependent kinases, p16INK4A protein inhibited the phosphorylation of retinoblastoma (Rb) protein and led to the G1-phase arrest of tumor cells. In some recent studies, hypermethylation of the p16INK4a promoter was detected not only in tumor tissues but also in cirrhotic liver tissues, implicating this process in the early stage of HBV-mediated hepatocarcinogenesis[51,54]. Wang et al[6] demonstrated a higher frequency of p16INK4a gene hypermethylation in HCC tissues than in the adjacent non-tumor tissues and the disease-free liver controls. Further studies suggested that a persistent HBV infection might play a role in inducing p16INK4A promoter methylation in hepatocarcinogenesis, possibly starting from an early stage[6,51]. Interestingly, it was also found that male patients had a higher frequency of p16INK4a promoter hypermeythylation in their tumor tissues than those of females. However, the size of that study’s cohort was not sufficiently large to allow a consolidated final conclusion. And the relationship between the significant increase of p16INK4a promoter hypermethylation and the susceptibility to HBV-related HCC in male patients required further investigation[6]. Because according to studies done by Zhu et al[55] and Zang et al[56], there was no significant correlation between p16INK4a hypermethylation and gender disparity in HBV-related HCC. Therefore, there has not been a consolidated final conclusion about the gender disparity of p16INK4a hypermethylation in HBV-related HCC. Furthermore, the role p16INK4A hypermethylation in the development and progression of HBV-associated HCC remains largely unknown and also needs more attention.
In recent years, considerable efforts have been made to investigate the genetic alternations involved in the progression of HCC, but the genetic events and the underlying mechanisms that lead to the initiation and progression of HCC remain largely unclear. And many genetic variants have been found and reported to be related to HCC development. DNA copy number alteration (CNA), a type of genetic alternation resulting in the gain or loss of either specific genomic regions or even entire chromosomes, has been supposed to be a major feature of HCC[57,58]. Through comparative genomic hybridization (CGH), Zhu et al[8] demonstrated that there was a significantly different CNA pattern between female and male HBV-HCC patients. In particular, gains of 1q21.3-q22, 11q11 and 19q13.31-q13.32 and loss of 16p11.2 were more frequently observed in female HCC tumors, whereas loss of 11q11 was more frequently observed in male HCC tumors. Further investigations suggested that the 1q gain might predispose humans to chromosomal alterations and was thus one of the early genomic events associated with the development of HCC[8]. Moreover, four growth-related genes, JTB, HAX-1, SHC1 and CKS1B, all of which were located within a 4.79 Mb region of 1q21.3-q22, were shown to be more highly expressed in HCC than in non-cancerous tissues[59,60]. A gender-related difference in the 11q11 copy number changes in HCC tumors was found to coexist with other CNAs, such as the gain of 19q13.31-q13.32. It had been shown that CNAs played a significant role in tumorigenesis through copy number-induced alterations in gene expression levels and other molecular mechanisms[61,62]. Thus, the coexistence of the loss and gain of specific chromosomes might indicate the roles of CNAs in the gender-related differential presentation of HBV-associated HCC. As Fabris et al[63] demonstrated, male 5,10-methylenetetrahydrofolate reductase (MTHFR) 677 TT carriers with alcoholic cirrhosis had an increased risk of developing HBV-related HCC. MTHFR, a key enzyme in the folate cycle, played an important role in regulating the metabolism of the amino-acid homocysteine. Through one of its substrates, MTHFR can increase the incorporation of uracil into DNA and lead to an augmented risk of point mutations and DNA/chromosome breakage. In addition, MTHFR can regulate the level of S-adenosyl-L-methionine by increasing the level of hypomethylation of modified DNA and changing its gene expression[64]. Furthermore, the MTHFR C677T SNP, which is associated with male gender- and alcohol-related liver disease, can increase the risk of the occurrence of HCC by 7.5-fold in males compared with females and alcoholic patients carrying the C/* allele[65]. Further investigation revealed the underlying mechanisms. Through regulation of folate metabolism and the circulating homocysteine levels, the MTHFR C677T SNP lowers the S-adenosyl-L-methionine levels, induces the stand point of DNA hypomethylation and hyperhomocysteinemia and consequently increases steatosis in the liver[63]. Finally, alcoholic males have an increased risk of HCC development. A growing number of clinical and epidemiological studies had shown that chronic HBV infections progressed more rapidly in males than in females from both clinical and virological aspects. Mutations in the surface (S) gene of HBV (S region of HBV), particularly in the major hydrophilic region (MHR) of the S gene (MHR region), were reported to be related to immune evasion by HBV[66-68]. Lee et al[69] found that the acquisition of MHR mutations followed by HBeAg seroconversion in male patients might contribute to the persistent infection that is characteristic of HBeAg-negative hepatitis B. And two types of MHR mutations, the L110I and G145A, were firstly found to contribute to HBV persistence and immune evasion. By inducing the persistent replication of HBV, the acquisition of MHR mutations combined with HBeAg seroconversion in male HBV carriers might lead to development of HBV-related HCC.
More interestingly, further investigations revealed that HBx might play a critical role in epigenetic modifications in HBV-infected liver cells and HBV-related HCC. HBx, a key factor in the molecular and cellular pathogenesis of HBV-related HCC, was proved to be correlated with sex hormone (including androgen and estrogen) signaling pathways[15,22,26,31]. Chen et al[70] showed that HBx could reduce the expression of EGFR protein through up-regulating the expression of miR-7 which targets 3’-UTR of epidermal growth factor receptor (EGFR) mRNA. And the HBx-miR-7-EGFR axis might act as an important controller in regulation of growth rate of HCC cells. Likewise, the newly identified HBX/miR–15b/FUT2/Globo H axis suggests one possible molecular mechanism of HCC cell proliferation and represents a new potential therapeutic target for HCC[71]. Through reviewing the current research about HBx-induced epigenetic changes, Wu et al[72] suggested that the epigenetic events such as aberrant miRNA expression, DNA methylation and histone modifications related to HBx play important roles in the development of HBV-related HCC. And all these findings further indicate that epigenetic and genetic alternations in HBV-related HCC may associate with gender difference and play a vital role in the development and progression of this disease. But the exact mechanisms through which HBx disrupts these epigenetic and genetic changes and the relationship between HBx and gender disparity remain largely unknown and need deeper explorations.
CONCLUSION
Gender disparity may play an important role in the initiation and progression of HBV-related HCC depending on a series of different pathogenic determinants, including the levels of sex hormones and inflammatory cytokines and epigenetic and genetic alternations. This review indicates that estrogen may have a protective effect against the progression of chronic liver disease and the development of HBV-associated HCC through decreasing HBV RNA transcription and inflammatory cytokine levels. In contrast, male patients may have a higher risk of HBV-associated HCC development through excessive activation of the androgen signaling pathway. Moreover, epigenetic and genetic alternations can also affect the impact of gender differences on the risk of HBV-related HCC development, but the exact mechanisms remain largely unknown and require further investigations.
Although gender disparity has been proven to be a factor that strongly influences the risk of development of HBV-related HCC, the effects of the clinical application of endocrine therapies or hormone-blocking therapies are not ideal. Several studies showed that therapeutic methods targeting sex hormone pathways using drugs such as tamoxifen and flutamide failed to achieve the desired effect in treating HCC. Endocrine therapies using tamoxifen, a type of drug that exerts a relatively more estrogenic than anti-estrogenic effect in liver tissues, exhibited only modest beneficial effects in a relatively small population of patients with advanced HCC and sometimes resulted in a worse survival rate and increased negative impacts. Anti-androgenic drugs, such as flutamide, failed to provide a remarkable survival advantage when applied in hormone-blocking treatments for HCC. The expression of variant ERs in patients with advanced HCC, which can cause estrogen unresponsiveness, may partly explain these bewildering results. But the exact mechanism has not been fully investigated, and the use of hormonal drugs in patients with advanced HCC is not currently recommended[12,30,73-75].
Therefore, studies taking a more in-depth look at the mechanisms that underlie the gender-associated differences in HBV-related HCC are urgently needed, which may provide more useful information for understanding, detecting and treating this disease. Especially, we should focus on investigating genetic abnormalities and epigenetic alterations associated with gender disparity found in HBV-related HCC. Since certain epigenetic alterations precede disease pathology, they have the potential to serve as excellent biomarkers for diagnosis, prognosis, and monitoring. The epigenetic abnormalities associated with gender disparity in HCC would make them excellent targets for epigenetic therapy, which are currently approved for the treatment of a few hematological malignancies. Future work, including obtaining a greater understanding of the mechanisms of gender disparity in HBV-related HCC, is necessary to determine the extent of their utility in treating HCC.
Footnotes
Supported by National Natural Science Foundation of China, Nos. 81372552 and 81172349/H1617
P- Reviewers: Liang L, Wong GLH S- Editor: Wen LL L- Editor: A E- Editor: Wu HL
References
- 1.Li Z, Yuan W, Ning S, Li J, Zhai W, Zhang S. Role of leptin receptor (LEPR) gene polymorphisms and haplotypes in susceptibility to hepatocellular carcinoma in subjects with chronic hepatitis B virus infection. Mol Diagn Ther. 2012;16:383–388. doi: 10.1007/s40291-012-0008-1. [DOI] [PubMed] [Google Scholar]
- 2.Jin YJ, Chung YH, Kim JA, Park WH, Lee D, Seo DD, Ryu SH, Jang MK, Yu E, Lee YJ. Factors predisposing metastatic tumor antigen 1 overexpression in hepatitis B virus associated hepatocellular carcinoma. Dig Dis Sci. 2012;57:2917–2923. doi: 10.1007/s10620-012-2296-z. [DOI] [PubMed] [Google Scholar]
- 3.Patton ME, Su JR, Nelson R, Weinstock H; Centers for Disease Control and Prevention (CDC) Hepatocellular carcinoma - United States, 2001-2006. MMWR Morb Mortal Wkly Rep. 2010;59:517–520. [PubMed] [Google Scholar]
- 4.Chu CM, Lin CC, Chen YC, Jeng WJ, Lin SM, Liaw YF. Basal core promoter mutation is associated with progression to cirrhosis rather than hepatocellular carcinoma in chronic hepatitis B virus infection. Br J Cancer. 2012;107:2010–2015. doi: 10.1038/bjc.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373:582–592. doi: 10.1016/S0140-6736(09)60207-5. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Cheng J, Xu C, Liu S, Jiang S, Xu Q, Chen X, Zhuang H, Lu F. Quantitative methylation analysis reveals gender and age differences in p16INK4a hypermethylation in hepatitis B virus-related hepatocellular carcinoma. Liver Int. 2012;32:420–428. doi: 10.1111/j.1478-3231.2011.02696.x. [DOI] [PubMed] [Google Scholar]
- 7.Baig S. Gender disparity in infections of Hepatitis B virus. J Coll Physicians Surg Pak. 2009;19:598–600. [PubMed] [Google Scholar]
- 8.Zhu ZZ, Wang D, Cong WM, Jiang H, Yu Y, Wen BJ, Dong H, Zhang X, Liu SF, Wang AZ, et al. Sex-related differences in DNA copy number alterations in hepatitis B virus-associated hepatocellular carcinoma. Asian Pac J Cancer Prev. 2012;13:225–229. doi: 10.7314/apjcp.2012.13.1.225. [DOI] [PubMed] [Google Scholar]
- 9.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273.e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang YT, Jen CL, Yang HI, Lee MH, Su J, Lu SN, Iloeje UH, Chen CJ. Lifetime risk and sex difference of hepatocellular carcinoma among patients with chronic hepatitis B and C. J Clin Oncol. 2011;29:3643–3650. doi: 10.1200/JCO.2011.36.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farinati F, De Maria N, Marafin C, Fagiuoli S, Della Libera G, Naccarato R. Hepatocellular carcinoma in alcoholic cirrhosis: is sex hormone imbalance a pathogenetic factor? Eur J Gastroenterol Hepatol. 1995;7:145–150. [PubMed] [Google Scholar]
- 12.Ruggieri A, Barbati C, Malorni W. Cellular and molecular mechanisms involved in hepatocellular carcinoma gender disparity. Int J Cancer. 2010;127:499–504. doi: 10.1002/ijc.25298. [DOI] [PubMed] [Google Scholar]
- 13.Yu MW, Chen CJ. Elevated serum testosterone levels and risk of hepatocellular carcinoma. Cancer Res. 1993;53:790–794. [PubMed] [Google Scholar]
- 14.Yang WJ, Chang CJ, Yeh SH, Lin WH, Wang SH, Tsai TF, Chen DS, Chen PJ. Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3beta kinase pathways. Hepatology. 2009;49:1515–1524. doi: 10.1002/hep.22833. [DOI] [PubMed] [Google Scholar]
- 15.Wang SH, Yeh SH, Lin WH, Wang HY, Chen DS, Chen PJ. Identification of androgen response elements in the enhancer I of hepatitis B virus: a mechanism for sex disparity in chronic hepatitis B. Hepatology. 2009;50:1392–1402. doi: 10.1002/hep.23163. [DOI] [PubMed] [Google Scholar]
- 16.Yu MW, Yang YC, Yang SY, Cheng SW, Liaw YF, Lin SM, Chen CJ. Hormonal markers and hepatitis B virus-related hepatocellular carcinoma risk: a nested case-control study among men. J Natl Cancer Inst. 2001;93:1644–1651. doi: 10.1093/jnci/93.21.1644. [DOI] [PubMed] [Google Scholar]
- 17.Chiu CM, Yeh SH, Chen PJ, Kuo TJ, Chang CJ, Chen PJ, Yang WJ, Chen DS. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc Natl Acad Sci USA. 2007;104:2571–2578. doi: 10.1073/pnas.0609498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouchard MJ, Wang L, Schneider RJ. Activation of focal adhesion kinase by hepatitis B virus HBx protein: multiple functions in viral replication. J Virol. 2006;80:4406–4414. doi: 10.1128/JVI.80.9.4406-4414.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology. 2004;39:1683–1693. doi: 10.1002/hep.20245. [DOI] [PubMed] [Google Scholar]
- 20.Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–170. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Ma WL, Hsu CL, Wu MH, Wu CT, Wu CC, Lai JJ, Jou YS, Chen CW, Yeh S, Chang C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 2008;135:947–955, 955.e1-5. doi: 10.1053/j.gastro.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen PJ, Yeh SH, Liu WH, Lin CC, Huang HC, Chen CL, Chen DS, Chen PJ. Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology. 2012;56:632–643. doi: 10.1002/hep.25695. [DOI] [PubMed] [Google Scholar]
- 23.Feng H, Cheng AS, Tsang DP, Li MS, Go MY, Cheung YS, Zhao GJ, Ng SS, Lin MC, Yu J, et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives β-catenin/T cell factor-dependent hepatocarcinogenesis. J Clin Invest. 2011;121:3159–3175. doi: 10.1172/JCI45967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almog Y, Klein A, Adler R, Laub O, Tur-Kaspa R. Estrogen suppresses hepatitis B virus expression in male athymic mice transplanted with HBV transfected Hep G-2 cells. Antiviral Res. 1992;19:285–293. doi: 10.1016/0166-3542(92)90010-3. [DOI] [PubMed] [Google Scholar]
- 25.De Maria N, Manno M, Villa E. Sex hormones and liver cancer. Mol Cell Endocrinol. 2002;193:59–63. doi: 10.1016/s0303-7207(02)00096-5. [DOI] [PubMed] [Google Scholar]
- 26.Wang SH, Yeh SH, Lin WH, Yeh KH, Yuan Q, Xia NS, Chen DS, Chen PJ. Estrogen receptor α represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4α. Gastroenterology. 2012;142:989–998.e4. doi: 10.1053/j.gastro.2011.12.045. [DOI] [PubMed] [Google Scholar]
- 27.Hervouet E, Cartron PF, Jouvenot M, Delage-Mourroux R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics. 2013;8:237–245. doi: 10.4161/epi.23790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong S. Hepatitis B virus, a sex hormone-responsive virus. Gastroenterology. 2012;142:696–699. doi: 10.1053/j.gastro.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen ML, Lee KD, Huang HC, Tsai YL, Wu YC, Kuo TM, Hu CP, Chang C. HNF-4α determines hepatic differentiation of human mesenchymal stem cells from bone marrow. World J Gastroenterol. 2010;16:5092–5103. doi: 10.3748/wjg.v16.i40.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han J, Ding L, Yuan B, Yang X, Wang X, Li J, Lu Q, Huang C, Ye Q. Hepatitis B virus X protein and the estrogen receptor variant lacking exon 5 inhibit estrogen receptor signaling in hepatoma cells. Nucleic Acids Res. 2006;34:3095–3106. doi: 10.1093/nar/gkl389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 33.Jiang R, Deng L, Zhao L, Li X, Zhang F, Xia Y, Gao Y, Wang X, Sun B. miR-22 promotes HBV-related hepatocellular carcinoma development in males. Clin Cancer Res. 2011;17:5593–5603. doi: 10.1158/1078-0432.CCR-10-1734. [DOI] [PubMed] [Google Scholar]
- 34.Hirankarn N, Kimkong I, Kummee P, Tangkijvanich P, Poovorawan Y. Interleukin-1beta gene polymorphism associated with hepatocellular carcinoma in hepatitis B virus infection. World J Gastroenterol. 2006;12:776–779. doi: 10.3748/wjg.v12.i5.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, Ng EK, Chan FK, Sung JJ, Chan HL. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. Int J Cancer. 2009;124:2766–2770. doi: 10.1002/ijc.24281. [DOI] [PubMed] [Google Scholar]
- 36.Ben-Ari Z, Mor E, Papo O, Kfir B, Sulkes J, Tambur AR, Tur-Kaspa R, Klein T. Cytokine gene polymorphisms in patients infected with hepatitis B virus. Am J Gastroenterol. 2003;98:144–150. doi: 10.1111/j.1572-0241.2003.07179.x. [DOI] [PubMed] [Google Scholar]
- 37.Nieters A, Yuan JM, Sun CL, Zhang ZQ, Stoehlmacher J, Govindarajan S, Yu MC. Effect of cytokine genotypes on the hepatitis B virus-hepatocellular carcinoma association. Cancer. 2005;103:740–748. doi: 10.1002/cncr.20842. [DOI] [PubMed] [Google Scholar]
- 38.Nakatani T, Roy G, Fujimoto N, Asahara T, Ito A. Sex hormone dependency of diethylnitrosamine-induced liver tumors in mice and chemoprevention by leuprorelin. Jpn J Cancer Res. 2001;92:249–256. doi: 10.1111/j.1349-7006.2001.tb01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa H, Maeda S. Inflammation- and stress-related signaling pathways in hepatocarcinogenesis. World J Gastroenterol. 2012;18:4071–4081. doi: 10.3748/wjg.v18.i31.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 43.Xing D, Oparil S, Yu H, Gong K, Feng W, Black J, Chen YF, Nozell S. Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β. PLoS One. 2012;7:e36890. doi: 10.1371/journal.pone.0036890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bamba H, Ota S, Kato A, Matsuzaki F. Nonsteroidal anti-inflammatory drugs may delay the repair of gastric mucosa by suppressing prostaglandin-mediated increase of hepatocyte growth factor production. Biochem Biophys Res Commun. 1998;245:567–571. doi: 10.1006/bbrc.1998.8436. [DOI] [PubMed] [Google Scholar]
- 45.Rahman MA, Dhar DK, Yamaguchi E, Maruyama S, Sato T, Hayashi H, Ono T, Yamanoi A, Kohno H, Nagasue N. Coexpression of inducible nitric oxide synthase and COX-2 in hepatocellular carcinoma and surrounding liver: possible involvement of COX-2 in the angiogenesis of hepatitis C virus-positive cases. Clin Cancer Res. 2001;7:1325–1332. [PubMed] [Google Scholar]
- 46.Tian Z, Shen X, Feng H, Gao B. IL-1 beta attenuates IFN-alpha beta-induced antiviral activity and STAT1 activation in the liver: involvement of proteasome-dependent pathway. J Immunol. 2000;165:3959–3965. doi: 10.4049/jimmunol.165.7.3959. [DOI] [PubMed] [Google Scholar]
- 47.Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 48.Gao P, Wong CC, Tung EK, Lee JM, Wong CM, Ng IO. Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis. J Hepatol. 2011;54:1177–1184. doi: 10.1016/j.jhep.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 49.Xuan P, Han K, Guo M, Guo Y, Li J, Ding J, Liu Y, Dai Q, Li J, Teng Z, et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS One. 2013;8:e70204. doi: 10.1371/journal.pone.0070204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Y, Bao-Han W, Lv X, Su Y, Zhao X, Yin Y, Zhang X, Zhou Z, MacNaughton WK, Wang H. MicroRNA-34a mediates the autocrine signaling of PAR2-activating proteinase and its role in colonic cancer cell proliferation. PLoS One. 2013;8:e72383. doi: 10.1371/journal.pone.0072383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Hui AM, Sun L, Hasegawa K, Torzilli G, Minagawa M, Takayama T, Makuuchi M. p16INK4A hypermethylation is associated with hepatitis virus infection, age, and gender in hepatocellular carcinoma. Clin Cancer Res. 2004;10:7484–7489. doi: 10.1158/1078-0432.CCR-04-1715. [DOI] [PubMed] [Google Scholar]
- 52.Hui AM, Sakamoto M, Kanai Y, Ino Y, Gotoh M, Yokota J, Hirohashi S. Inactivation of p16INK4 in hepatocellular carcinoma. Hepatology. 1996;24:575–579. doi: 10.1002/hep.510240319. [DOI] [PubMed] [Google Scholar]
- 53.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 54.Kaneto H, Sasaki S, Yamamoto H, Itoh F, Toyota M, Suzuki H, Ozeki I, Iwata N, Ohmura T, Satoh T, et al. Detection of hypermethylation of the p16(INK4A) gene promoter in chronic hepatitis and cirrhosis associated with hepatitis B or C virus. Gut. 2001;48:372–377. doi: 10.1136/gut.48.3.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu YZ, Zhu R, Fan J, Pan Q, Li H, Chen Q, Zhu HG. Hepatitis B virus X protein induces hypermethylation of p16(INK4A) promoter via DNA methyltransferases in the early stage of HBV-associated hepatocarcinogenesis. J Viral Hepat. 2010;17:98–107. doi: 10.1111/j.1365-2893.2009.01156.x. [DOI] [PubMed] [Google Scholar]
- 56.Zang JJ, Xie F, Xu JF, Qin YY, Shen RX, Yang JM, He J. P16 gene hypermethylation and hepatocellular carcinoma: a systematic review and meta-analysis. World J Gastroenterol. 2011;17:3043–3048. doi: 10.3748/wjg.v17.i25.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walther Z, Jain D. Molecular pathology of hepatic neoplasms: classification and clinical significance. Patholog Res Int. 2011;2011:403929. doi: 10.4061/2011/403929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mínguez B, Tovar V, Chiang D, Villanueva A, Llovet JM. Pathogenesis of hepatocellular carcinoma and molecular therapies. Curr Opin Gastroenterol. 2009;25:186–194. doi: 10.1097/MOG.0b013e32832962a1. [DOI] [PubMed] [Google Scholar]
- 59.Midorikawa Y, Tsutsumi S, Nishimura K, Kamimura N, Kano M, Sakamoto H, Makuuchi M, Aburatani H. Distinct chromosomal bias of gene expression signatures in the progression of hepatocellular carcinoma. Cancer Res. 2004;64:7263–7270. doi: 10.1158/0008-5472.CAN-04-1275. [DOI] [PubMed] [Google Scholar]
- 60.Wong N, Chan A, Lee SW, Lam E, To KF, Lai PB, Li XN, Liew CT, Johnson PJ. Positional mapping for amplified DNA sequences on 1q21-q22 in hepatocellular carcinoma indicates candidate genes over-expression. J Hepatol. 2003;38:298–306. doi: 10.1016/s0168-8278(02)00412-9. [DOI] [PubMed] [Google Scholar]
- 61.Kuiper RP, Ligtenberg MJ, Hoogerbrugge N, Geurts van Kessel A. Germline copy number variation and cancer risk. Curr Opin Genet Dev. 2010;20:282–289. doi: 10.1016/j.gde.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fabris C, Toniutto P, Falleti E, Fontanini E, Cussigh A, Bitetto D, Fornasiere E, Fumolo E, Avellini C, Minisini R, et al. MTHFR C677T polymorphism and risk of HCC in patients with liver cirrhosis: role of male gender and alcohol consumption. Alcohol Clin Exp Res. 2009;33:102–107. doi: 10.1111/j.1530-0277.2008.00816.x. [DOI] [PubMed] [Google Scholar]
- 64.Miner SE, Evrovski J, Cole DE. Clinical chemistry and molecular biology of homocysteine metabolism: an update. Clin Biochem. 1997;30:189–201. doi: 10.1016/s0009-9120(96)00172-5. [DOI] [PubMed] [Google Scholar]
- 65.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 66.Lok AS, McMahon BJ. Chronic hepatitis B: update of recommendations. Hepatology. 2004;39:857–861. doi: 10.1002/hep.20110. [DOI] [PubMed] [Google Scholar]
- 67.Zuckerman AJ, Zuckerman JN. Molecular epidemiology of hepatitis B virus mutants. J Med Virol. 1999;58:193–195. doi: 10.1002/(sici)1096-9071(199907)58:3<193::aid-jmv1>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 68.Zuckerman AJ. Effect of hepatitis B virus mutants on efficacy of vaccination. Lancet. 2000;355:1382–1384. doi: 10.1016/S0140-6736(00)02132-2. [DOI] [PubMed] [Google Scholar]
- 69.Lee SA, Cho YK, Lee KH, Hwang ES, Kook YH, Kim BJ. Gender disparity in distribution of the major hydrophilic region variants of hepatitis B virus genotype C according to hepatitis B e antigen serostatus. J Med Virol. 2011;83:405–411. doi: 10.1002/jmv.21988. [DOI] [PubMed] [Google Scholar]
- 70.Chen YJ, Chien PH, Chen WS, Chien YF, Hsu YY, Wang LY, Chen JY, Lin CW, Huang TC, Yu YL, et al. Hepatitis B Virus-Encoded X Protein Downregulates EGFR Expression via Inducing MicroRNA-7 in Hepatocellular Carcinoma Cells. Evid Based Complement Alternat Med. 2013;2013:682380. doi: 10.1155/2013/682380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tian Y, Yang W, Song J, Wu Y, Ni B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol Cell Biol. 2013;33:2810–2816. doi: 10.1128/MCB.00205-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu CS, Yen CJ, Chou RH, Chen JN, Huang WC, Wu CY, Yu YL. Downregulation of microRNA-15b by hepatitis B virus X enhances hepatocellular carcinoma proliferation via fucosyltransferase 2-induced Globo H expression. Int J Cancer. 2014;134:1638–1647. doi: 10.1002/ijc.28501. [DOI] [PubMed] [Google Scholar]
- 73.Llovet JM. Updated treatment approach to hepatocellular carcinoma. J Gastroenterol. 2005;40:225–235. doi: 10.1007/s00535-005-1566-3. [DOI] [PubMed] [Google Scholar]
- 74.Pompili M, Francica G, Ponziani FR, Iezzi R, Avolio AW. Bridging and downstaging treatments for hepatocellular carcinoma in patients on the waiting list for liver transplantation. World J Gastroenterol. 2013;19:7515–7530. doi: 10.3748/wjg.v19.i43.7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37:429–442. doi: 10.1053/jhep.2003.50047. [DOI] [PubMed] [Google Scholar]