

Abstract
The pathogenesis of liver cirrhosis is not completely elucidated. Although in the majority of patients, the risk factors may be identified in B and C viral hepatitis, alcohol intake, drugs or fatty liver disease, there is a small percentage of patients with no apparent risk factors. In addition, the evolution of chronic liver disease is highly heterogeneous from one patient to another. Among patient with identical risk factors, some rapidly progress to cirrhosis and hepatocellular carcinoma (HCC) whereas others have a benign course. Therefore, a genetic predisposition may contribute to the development of cirrhosis and HCC. Evidence supporting the role of genetic factors as a risk for cirrhosis has been accumulating during the past years. In addition to the results from epidemiological studies, polymorphisms studies and data on twins, the concept of telomere shortening as a genetic risk factor for chronic liver disease and HCC has been proposed. Here we review the literature on telomerase mutations, telomere shortening and liver disease including hepatocellular carcinoma.
Keywords: Chromosomes, Telomere, Telomerase, Liver-cirrhosis, Hepatocarcinoma
Core tip: The pathogenesis of liver cirrhosis is not completely elucidated. Genetic predisposition may contribute to the development of cirrhosis and hepatocellular carcinoma (HCC). Evidence supporting the role of genetic factors as a risk for cirrhosis and the concept of telomere shortening as a genetic risk factor for chronic liver disease and HCC has been proposed. Here we review the literature on telomerase mutations, telomere shortening and liver disease including hepatocellular carcinoma.
TELOMERE AND TELOMERASE
Telomeres consist of repetitive DNA sequences (TTAGGG) associated with a specialized protein complex named shelterin and are located at the ends of linear chromosomes. They function as a cap to stabilize and protect chromosomes from erosion and from being mistaken for double-strand DNA breaks[1]. During each cell division, telomeres shorten due to the ‘‘end-replication problem’’ that is the DNA polymerase’s inability to fully replicate the 3’ end of chromosomes. In order to limit telomere attrition, germline and some somatic cells express telomerase, a reverse transcriptase that maintains telomere length by synthesizing new DNA sequences and adding them to the end of the chromosome[2]. Telomerase is an enzymatic protein complex including the telomerase reverse transcriptase (TERT) and the telomerase RNA component (TERC) used as a template to synthesize telomere DNA.
When telomeres are too short, they signal the arrest of cell proliferation resulting in cell senescence or apoptosis. If protective mechanisms, such as the p53 tumor-suppressor gene, are inactive, thus allowing continued proliferation, telomeres become extremely short and dysfunctional; they may cause chromosomes end-to-end fusions and ultimately chromosomal instability. Conversely, cells transfected with the telomerase gene can proliferate indefinitely[3]. Despite telomerase activity, telomere shortening is inevitable, thereby limiting the proliferative lifespan of human cells. As expected, for a given organ, telomere length decreases with the age of the subjects. There are also iatrogenic causes of telomere shortening: for example after bone marrow transplantation when hematopoietic stem cells and progenitor cells are highly proliferative in order to reconstitute hematopoiesis. In addition, telomere attrition may be genetically determined as a result of telomerase complex’s genes mutations leading to an inherited inability to elongate telomeres.
TELOMERE AND DISEASE
Mutations in the TERT and TERC genes are considered the most common cause of inherited human telomere-mediated disease[4]. Even with a mild reduction in telomerase activity, telomere length homeostasis may be altered and results in what has been called a syndrome complex which include different age-dependent disease[5,6]. Telomere-mediated disease has diverse presentations that span the age spectrum. Their type, age of onset, and severity depend on the extent of the telomere length defect (Table 1). Telomerase mutations may have high penetrance and induce, during infancy, severe telomere shortening that manifests as developmental delay, cerebellar hypoplasia, and immunodeficiency, features that are recognized in the rare Hoyeraal-Hreidarsson syndrome[7,8].
Table 1.
Telomere erosion and human disease
| Telomerase mutations as genetic determinants | Telomerase mutations as genetic risk factors | |
| Characteristics | High penetrance | Low penetrance |
| Childhood onset disease | Adult onset disease | |
| Congenital clinical manifestations | Single or multiple organs | |
| Disease | Dyskeratosis congenita | Aplastic anemia |
| Lung fibrosis | ||
| Liver cirrhosis | ||
| Telomere syndromes |
Telomerase mutations may be highly penetrant causing congenital clinical manifestations as in dyskeratosis congenita or may be less penetrant and manifest in adult life inducing single or multiple organ damage[7].
In children and young adults, telomere-mediated disease causes bone marrow failure and at times may be recognized in the mucocutaneous syndrome dyskeratosis congenita, which is defined by a triad of mucocutaneous features: skin hyperpigmentation, dystrophic nails, and oral leukoplakia[9-11]. During adult life, telomerase mutations may represent risk factors rather than genetic determinants and need environmental, epigenetic or other genetic factors to contribute to disease development. These mutations are less penetrant and induce single-organ damage in adults, usually without the classic physical signs of dyskeratosis congenita. This is case of diseases such as aplastic anemia, pulmonary fibrosis and liver cirrhosis. Telomere-mediated disease manifests in adults as isolated or syndromic clustering of idiopathic pulmonary fibrosis (IPF), liver cirrhosis, and bone marrow failure[12]. Mutant TERT and TERC genes account for 8%-15% of familial and 1%-3% of sporadic pulmonary fibrosis cases[13-15]. The co-occurrence of IPF and bone marrow failure within a single family is highly predictive for the presence of a germline telomerase defect[16]. Mutant telomere genes do not cause only isolated cases of IPF, liver cirrhosis or aplastic anemia but they may also cause subclinical diseases in other organs even if one single organ disease manifestations dominate. For example patient with IPF who have a telomerase mutation is at increased risk to develop liver diseases as well as bone marrow failure[13,14]. It has been proposed that their shared short telomere length defect unifies them as a single syndrome continuum[17] (Table 2). This classification is significant because the telomere defect is present in the germline of these patients and thus, even when a single presentation predominates, complications may arise in other organs. The consideration of the telomere syndromes as a single spectrum is important for patient management in different clinical settings.
Table 2.
Telomere syndromes
| Spectrum of features in bone marrow, lung and liver disease |
| Haematologic |
| Macrocytosis |
| Isolated cytopenias |
| Plastic anemias |
| Myelodysplasia |
| Acute myeloid leukemia |
| Pulmonary |
| Asymptomatic restrictive defects on lung function tests |
| Idiopathic pulmonary fibrosis |
| Nonspecific interstitial pneumonia |
| Idiopathic interstitial pneumonias |
| Liver |
| Mildly elevated transaminases |
| Atrophic nodular liver at imaging studies |
| Splenomegaly |
| Cryptogenic liver fibrosis/cirrhosis |
Mutant telomere genes and telomere shortening do not cause only a single organ damage such as idiopathic pulmonary fibrosis (IPF), aplastic anemia or liver cirrhosis but often the affected patient has also subclinical disease concurrently in other organs even if the symptoms of a single organ predominate[17].
TELOMERE AND LIVER DISEASE
The pathogenesis of liver cirrhosis is not completely elucidated. Although in the majority of patients a cause can be identified in viral hepatitis, alcohol intake, drugs or fatty liver disease, there is still a small percentage of patients with no apparent risk factors[18]. In addition, it is not either completely elucidated why patients with identical risk factors have a different clinical manifestations and clinical course; in fact some patients progress to cirrhosis and/or hepatocellular carcinoma (HCC) whereas others have a benign clinical course, suggesting that host factors, different from age and gender, may play a critical role in disease progression. Many attempts to identify possible genetic risk factors for the development of cirrhosis have been done with little fortune. Among others, the concept of telomere shortening as genetic risk factor for cirrhosis has been proposed. Conditions with high cell turnover such as chronic liver injury accelerate telomere shortening, leading to impairment of cell proliferation and senescence once telomeres are critically short. The cellular growth arrest and/or senescence appears to be profibrogenic by as-yet undefined mechanisms. Several studies have investigated the relationship between cirrhosis and telomere shortening, showing that telomeres shortening is a marker of cirrhosis formation correlating with the accumulation of senescent hepatocytes[19]. Kitada et al[20] demonstrated the relationship between telomere shortening and cirrhosis in 1995. They observed that telomere length in tissue from cirrhotic liver was shorter than in liver with chronic hepatitis and both were shorter than telomere length in normal liver tissue. Subsequent studies confirmed that a shortened telomere length was correlated with the degree of fibrosis, suggesting that telomere shortening may contribute to and be a marker of cirrhosis[20-22]. Moreover, studies on telomerase-deficient mice provided experimental evidence that shortened telomeres, in response to chronic liver injury, are associated with impaired liver regeneration and accelerated cirrhosis development. Restoration of telomerase activity into the liver of these mice resulted in reduction of cirrhosis and improved liver function[23]. These findings suggest that reduced telomerase activity may contribute to cirrhosis development.
Mutations in the telomerase complex genes have been implicated in rare human diseases characterized by accelerated telomere shortening and organ failure such as dyskeratosis congenita[7]. Interestingly, patients suffering from these diseases showed an increased frequency of liver pathologies including fibrosis and cirrhosis. Our group was the first to report on the coexistence of cryptogenic cirrhosis (CC) and IPF, not in the setting of dyskeratosis congenita, and telomere disfunction. We described a case report on 48-year-old woman, diagnosed of cryptogenic liver cirrhosis, idiopathic pulmonary fibrosis and diabetes. Both CC and IPF had a rapid progression and after eighteen months the patient died. Sequencing and mutation analysis of TERT and TERC genes demonstrated the presence of a heterozygous TERT mutation (L153M). The TERT L153M variant results in a change of methionine for leucine at amino acid 153, in the protein region that seems to be involved in the template RNA and telomeric DNA binding. Furthermore, leukocyte telomere length was significantly shorter. Our case report not only confirmed the association between IPF and telomere dysfunction, but, more interestingly, gives further evidence of telomere involvement in liver disease progression and suggests a possible link between nonalcoholic fatty liver disease (NAFLD) and CC through telomere shortening[24,25]. Recently, two studies have also investigated the frequency of telomerase mutations in patients with sporadic cirrhosis compared to healthy controls[26,27]. Both studies screened patients for variation in the TERT and TERC genes. In the first study by Calado et al[26] the Authors found missense mutations in the TERT and TERC genes. The frequency of TERT gene mutations in cirrhotic patients was significantly greater than controls. Moreover cirrhosis was also associated with shorter telomeres in peripheral blood leukocytes. Finally, most TERT variants showed reduced telomerase activity in vitro. In the second study Hartmann et al[27] screened patients with sporadic cirrhosis and non-cirrhotic controls for telomerase mutations. Of note, control group was composed of 473 healthy individuals and 127 patients with chronic HCV infection who did not develop cirrhosis during a long follow-up. The data analysis revealed a significantly increased frequency of telomerase mutations in the cirrhosis group compared to the control group. Again, TERT gene mutations in cirrhotic patients were associated with reduced telomerase activity in vitro and shorter telomeres in peripheral blood leukocytes compared to non-cirrhotic patients. Interestingly, Hartmann et al[27] found an increased frequency of end stage liver disease (Child B or C cirrhosis, hepatocellular carcinoma, or evaluation for liver transplantation) in cirrhotic patients harboring telomerase mutations compared to cirrhotic patients without telomerase mutation. This result suggests that telomerase mutations may accelerate liver disease progression to cirrhosis in a context of chronic liver injury. Telomerase mutations were very different between the two studies; the most common TERT variant in the Calado study (A1062 T found in six patients) was not found in the Hartmann study and conversely, the most common TERT variant in the Hartmann study (G1109R also found in six patients) was not found in the study by Calado. Together, the current data would suggest that telomere attrition may play a role in the sequence of events leading to cirrhosis. According to this view chronic liver injury induces hepatocyte regeneration and therefore an elevated cell turnover; hence increased telomere shortening and cell senescence. Eventually, if the injury persists, other cells, such as stellate cells become activated leading to fibrogenesis (Figure 1).
Figure 1.
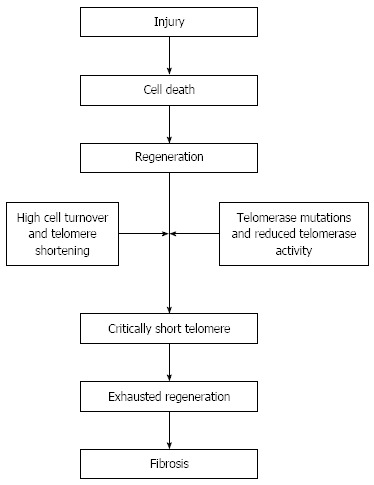
Telomere shortening and liver disease: A possible mechanism. Injuries leading to cell death cause repair and regeneration process which in turn leads to increased cell turnover and therefore to telomere erosion. If a telomerase gene mutation is present, telomerase activity may be reduced and telomere may become critically short leading hepatocyte to senescence. Hepatocyte senescence is a profibrotic state that activates stellate cells responsible for fibrosis[25].
On the same line, however, a recent study[28], conducted on liver sections from 70 patients with NAFLD and 60 controls, found that telomere shortening was observed in NAFLD patients but other indices of cell ageing, such as cell cycle inhibitor p21 and nuclear area were better predictors of disease progression. In fact a higher hepatocyte p21 expression and a greater nuclear area were significantly associated with adverse liver-related outcome but not with the telomere length. Similarly, in paired biopsies p21 expression and nuclear area correlated to fibrosis stage.
Taken together these data show the relation between telomerase mutation and chronic liver disease progression to cirrhosis, probably due to a reduced telomerase activity and therefore an impaired telomere length maintenance. Moreover, since the prevalence of telomerase mutations seems to be to be rather low in the general population, they may not be the major contributing factor to cirrhosis. Probably looking for telomerase genes mutations only, there is the risk to underestimate the real contribution of the telomere system to the development of cirrhosis. In fact other components of the telomere complex have been shown to be important for telomerase activity, these components are proteins such as dyskerin and the telomere binding proteins[29]; mutations in these components can lead to an impairment of telomere function; recently a mutation of the binding protein TIN2 has been involved in the evolution of aplastic anemia[30]. Finally also the mutations in the noncoding sequence of TERC and TERC could be responsible for impairment in the expression of TERC and TERT. Probably the sequence analysis of all components of the “telomere system” will reveal the real contribution of telomere complex genes mutations to the development of liver cirrhosis.
TELOMERE AND HEPATOCELLULAR CARCINOMA
Natural history of cirrhosis is often complicated with the occurrence of hepatocellular carcinoma (HCC). The observation that telomere shortening is an established feature of chronic liver disease has led to suggest that it may play a role in the pathophysiology of HCC. This view stems from a large number of studies indicating that telomere biology is involved in the initiation as well as in the progression of HCC[31]. The telomere hypothesis of cancer holds that telomere shortening results in chromosomal instability (CIS) which drives cancer initiation. In this regard Plentz et al[32] analyzed specimen of a group of HCC and regenerative nodules correlating hepatocyte telomere length with the ploidy grade, taken as a measure of chromosomal instability. These Authors showed that telomeres were shorter in HCC hepatocyte as compared to those in regenerative nodules and normal liver tissue and that, within group of HCC, hepatocyte telomere length of aneuploid was shorter than that of diploid tumors. Farazi et al[33] in order to better elucidate the role of telomere dysfunction in the development of HCC used some experimental models of HCC utilizing telomerase deficient mice, null for the telomerase RNA component, mTERC(-/-). In all HCC models both incidence and HCC lesions were suppressed showing on the histological level a significant increase of early stage neoplastic lesions and a reciprocal reduction of high grade malignancies. These experimental data indicate that telomere dysfunction plays an opposite role in the initiation versus the progression of HCC.
On the other hand progression of neoplastic growth needs an efficient telomere signaling. Initial studies[21] reported a slight increase of telomere length in poorly differentiated as compared to better differentiated HCC, suggesting a reactivation of telomerase and restored chromosomal stability to a level compatible with tumor cell viability. In this regard several reports have shown that telomerase activity was detected in nearly 90% of HCC as compared to only 21% of non-tumor tissue and was paralleled by the increase of TERT mRNA levels implying the possibility that TERT mRNA expression could predict or be a marker of HCC[34-37]. The effective role of the intact telomere/telomerase system has been illustrated in experimental HCC models utilizing telomerase knock-out mice mTERC(-/-) and littermates mTERC(+/-). This study has shown that being the prevalence of short telomere comparable in the liver of the two cohorts, the formation of HCC was strongly suppressed in mTERC(-/-) as compared to mTERC(+/-)[38]. Mechanism of telomerase activation may be diverse in some way depending on the etiology of chronic liver diseases[39]. This is best illustrated by the occurrence of the integration of hepatitis B virus into the human telomerase gene in HCC[40,41]. Moreover the list of recurrent HBV target genes is expanding[42] suggesting the possible mechanisms by which HBV may cause telomere dysfunction leading to initiation as well progression of HCC[43-47]. In conclusion our knowledge of the causes and mechanisms relating telomere dysfunction to the genesis and growth of HCC is increasing and leads to foresee therapeutic approaches, such as combined immune-chemotherapy and gene therapy, for its cure[48,49].
CONCLUSION
Telomere shortening and telomerase regulation play an important role on tissue regeneration during aging, chronic diseases and on cancer promotion and progression. In chronic liver disease the hepatocytes regenerative capacity is limited by telomere shortening, resulting in exhaustion of cell regeneration, fibrosis and cirrhosis formation. Short telomeres increase the risk of HCC but at the same time they limit the progression of cancer. The therapeutic option of blocking senescence, reactivating the telomerase, in order to block the exhaustion of the liver regenerative capacity might be beneficial depending on the effect of such approach on the HCC formation. Further studies are needed in order to better understand telomere biology in human disease and carcinogenesis and to identify potential therapeutic options.
Footnotes
P- Reviewers: Inzaugarat E, Mascitelli L S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 2.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 3.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 4.Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res. 2012;730:52–58. doi: 10.1016/j.mrfmmm.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dokal I. Dyskeratosis congenita. A disease of premature ageing. Lancet. 2001;358 Suppl:S27. doi: 10.1016/s0140-6736(01)07040-4. [DOI] [PubMed] [Google Scholar]
- 7.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 10.Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, Jones A, Hennekam RC, Poustka A, Mason PJ, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999;107:335–339. doi: 10.1046/j.1365-2141.1999.01690.x. [DOI] [PubMed] [Google Scholar]
- 11.Vulliamy T, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–2170. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 12.de la Fuente J, Dokal I. Dyskeratosis congenita: advances in the understanding of the telomerase defect and the role of stem cell transplantation. Pediatr Transplant. 2007;11:584–594. doi: 10.1111/j.1399-3046.2007.00721.x. [DOI] [PubMed] [Google Scholar]
- 13.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 14.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117:5607–5611. doi: 10.1182/blood-2010-11-322149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kodali VP, Gordon SC, Silverman AL, McCray DG. Cryptogenic liver disease in the United States: further evidence for non-A, non-B, and non-C hepatitis. Am J Gastroenterol. 1994;89:1836–1839. [PubMed] [Google Scholar]
- 19.Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, Flemming P, Franco S, Blasco MA, Manns MP, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002;16:935–942. doi: 10.1096/fj.01-0977com. [DOI] [PubMed] [Google Scholar]
- 20.Kitada T, Seki S, Kawakita N, Kuroki T, Monna T. Telomere shortening in chronic liver diseases. Biochem Biophys Res Commun. 1995;211:33–39. doi: 10.1006/bbrc.1995.1774. [DOI] [PubMed] [Google Scholar]
- 21.Urabe Y, Nouso K, Higashi T, Nakatsukasa H, Hino N, Ashida K, Kinugasa N, Yoshida K, Uematsu S, Tsuji T. Telomere length in human liver diseases. Liver. 1996;16:293–297. doi: 10.1111/j.1600-0676.1996.tb00748.x. [DOI] [PubMed] [Google Scholar]
- 22.Aikata H, Takaishi H, Kawakami Y, Takahashi S, Kitamoto M, Nakanishi T, Nakamura Y, Shimamoto F, Kajiyama G, Ide T. Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res. 2000;256:578–582. doi: 10.1006/excr.2000.4862. [DOI] [PubMed] [Google Scholar]
- 23.Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science. 2000;287:1253–1258. doi: 10.1126/science.287.5456.1253. [DOI] [PubMed] [Google Scholar]
- 24.Carulli L, Dei Cas A, Nascimbeni F. Synchronous cryptogenic liver cirrhosis and idiopathic pulmonary fibrosis: a clue to telomere involvement. Hepatology. 2012;56:2001–2003. doi: 10.1002/hep.26089. [DOI] [PubMed] [Google Scholar]
- 25.Chaiteerakij R, Roberts LR. Telomerase mutation: a genetic risk factor for cirrhosis. Hepatology. 2011;53:1430–1432. doi: 10.1002/hep.24304. [DOI] [PubMed] [Google Scholar]
- 26.Calado RT, Brudno J, Mehta P, Kovacs JJ, Wu C, Zago MA, Chanock SJ, Boyer TD, Young NS. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology. 2011;53:1600–1607. doi: 10.1002/hep.24173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartmann D, Srivastava U, Thaler M, Kleinhans KN, N’kontchou G, Scheffold A, Bauer K, Kratzer RF, Kloos N, Katz SF, et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology. 2011;53:1608–1617. doi: 10.1002/hep.24217. [DOI] [PubMed] [Google Scholar]
- 28.Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, Allison M, Coleman N, Alexander G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol. 2013;58:549–556. doi: 10.1016/j.jhep.2012.10.031. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 30.Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satyanarayana A, Manns MP, Rudolph KL. Telomeres and telomerase: a dual role in hepatocarcinogenesis. Hepatology. 2004;40:276–283. doi: 10.1002/hep.20308. [DOI] [PubMed] [Google Scholar]
- 32.Plentz RR, Schlegelberger B, Flemming P, Gebel M, Kreipe H, Manns MP, Rudolph KL, Wilkens L. Telomere shortening correlates with increasing aneuploidy of chromosome 8 in human hepatocellular carcinoma. Hepatology. 2005;42:522–526. doi: 10.1002/hep.20847. [DOI] [PubMed] [Google Scholar]
- 33.Farazi PA, Glickman J, Jiang S, Yu A, Rudolph KL, DePinho RA. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003;63:5021–5027. [PubMed] [Google Scholar]
- 34.Nagao K, Tomimatsu M, Endo H, Hisatomi H, Hikiji K. Telomerase reverse transcriptase mRNA expression and telomerase activity in hepatocellular carcinoma. J Gastroenterol. 1999;34:83–87. doi: 10.1007/s005350050220. [DOI] [PubMed] [Google Scholar]
- 35.Shimojima M, Komine F, Hisatomi H, Shimizu T, Moriyama M, Arakawa Y. Detection of telomerase activity, telomerase RNA component, and telomerase reverse transcriptase in human hepatocellular carcinoma. Hepatol Res. 2004;29:31–38. doi: 10.1016/j.hepres.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Lee CM, Hsu CY, Eng HL, Huang WS, Lu SN, Changchien CS, Chen CL, Cho CL. Telomerase activity and telomerase catalytic subunit in hepatocellular carcinoma. Hepatogastroenterology. 2004;51:796–800. [PubMed] [Google Scholar]
- 37.Kojima H, Yokosuka O, Imazeki F, Saisho H, Omata M. Telomerase activity and telomere length in hepatocellular carcinoma and chronic liver disease. Gastroenterology. 1997;112:493–500. doi: 10.1053/gast.1997.v112.pm9024303. [DOI] [PubMed] [Google Scholar]
- 38.Lechel A, Holstege H, Begus Y, Schienke A, Kamino K, Lehmann U, Kubicka S, Schirmacher P, Jonkers J, Rudolph KL. Telomerase deletion limits progression of p53-mutant hepatocellular carcinoma with short telomeres in chronic liver disease. Gastroenterology. 2007;132:1465–1475. doi: 10.1053/j.gastro.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 39.El Idrissi M, Hervieu V, Merle P, Mortreux F, Wattel E. Cause-specific telomere factors deregulation in hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32:64. doi: 10.1186/1756-9966-32-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paterlini-Bréchot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Bréchot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–3916. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 41.Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, Nagorney DM, Gostout BS, Burgart LJ, Boix L, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22:3813–3820. doi: 10.1038/sj.onc.1206528. [DOI] [PubMed] [Google Scholar]
- 42.Ding D, Lou X, Hua D, Yu W, Li L, Wang J, Gao F, Zhao N, Ren G, Li L, et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet. 2012;8:e1003065. doi: 10.1371/journal.pgen.1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim H, Oh BK, Roncalli M, Park C, Yoon SM, Yoo JE, Park YN. Large liver cell change in hepatitis B virus-related liver cirrhosis. Hepatology. 2009;50:752–762. doi: 10.1002/hep.23072. [DOI] [PubMed] [Google Scholar]
- 44.Oh BK, Kim H, Park YN, Yoo JE, Choi J, Kim KS, Lee JJ, Park C. High telomerase activity and long telomeres in advanced hepatocellular carcinomas with poor prognosis. Lab Invest. 2008;88:144–152. doi: 10.1038/labinvest.3700710. [DOI] [PubMed] [Google Scholar]
- 45.Oh BK, Kim YJ, Park C, Park YN. Up-regulation of telomere-binding proteins, TRF1, TRF2, and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am J Pathol. 2005;166:73–80. doi: 10.1016/S0002-9440(10)62233-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee YH, Oh BK, Yoo JE, Yoon SM, Choi J, Kim KS, Park YN. Chromosomal instability, telomere shortening, and inactivation of p21(WAF1/CIP1) in dysplastic nodules of hepatitis B virus-associated multistep hepatocarcinogenesis. Mod Pathol. 2009;22:1121–1131. doi: 10.1038/modpathol.2009.76. [DOI] [PubMed] [Google Scholar]
- 47.Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26 Suppl 1:144–152. doi: 10.1111/j.1440-1746.2010.06546.x. [DOI] [PubMed] [Google Scholar]
- 48.Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, Ormandy LA, Manns MP, Beaugrand M, Bruix J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer. 2010;10:209. doi: 10.1186/1471-2407-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu Y, Shen Y, Ji B, Wang L, Zhang Z, Zhang Y. Combinational RNAi gene therapy of hepatocellular carcinoma by targeting human EGFR and TERT. Eur J Pharm Sci. 2011;42:387–391. doi: 10.1016/j.ejps.2011.01.004. [DOI] [PubMed] [Google Scholar]