

Abstract
Objective
Myositis is associated with muscle-targeted inflammation and is observed in some Treg cell–deficient mouse models. Because an autoimmune pathogenesis has been strongly implicated, the aim of this study was to investigate the hypothesis that abnormal exposure to muscle antigens, as observed in muscle injury, can induce autoimmune-mediated myositis in susceptible hosts.
Methods
FoxP3 mutant (scurfy) mice were mated to synaptotagmin VII (Syt VII) mutant mice, which resulted in a new mouse strain that combines impaired membrane resealing with Treg cell deficiency. Lymphocyte preparations from double-mutant mice were adoptively transferred intraperitoneally, with or without purified Treg cells, into recombination-activating gene 1 (RAG-1)–null recipients. Lymph node cells from mice with the FoxP3 mutation were transferred into RAG-1–null mice either 1) intraperitoneally in conjunction with muscle homogenate or purified myosin protein or 2) intramuscularly with or without cotransfer of purified Treg cells.
Results
FoxP3-deficient mouse lymph node cells transferred in conjunction with myosin protein or muscle homogenate induced robust skeletal muscle inflammation. The infiltrates consisted predominantly of CD4+ and CD8+ T cells, a limited number of macrophages, and no B cells. Significant inflammation was also seen in similar experiments using lymph node cells from FoxP3/Syt VII double-mutant mice but was absent in experiments using adoptive transfer of FoxP3 mutant mouse cells alone. The cotransfer of Treg cells completely suppressed myositis.
Conclusion
These data, derived from a new, reproducible model, demonstrate the critical roles of Treg cell deficiency and aberrant muscle antigen exposure in the priming of autoreactive cells to induce myositis. This mouse system has multifaceted potential for examining the interplay in vivo between tissue injury and autoimmunity.
Idiopathic inflammatory myopathies are a group of systemic autoimmune diseases characterized by chronic muscle inflammation resulting in weakness (1) and a variety of clinical manifestations (2). Because inflammation of muscle tissue is the underlying mechanism of disease pathology in each condition, current treatments include broad-spectrum immunosuppressive agents and, more recently, targeted immune therapy directed against inflammatory cells and inflammatory mediators (3,4). Although these treatments can be relatively effective, they can result in significant complications due to systemic immunologic suppression (5–7). However, the development of more specific therapies for immune-mediated myositis requires more data regarding pathogenesis (8). Animal models can provide insight into the pathogenesis of these diseases and facilitate the identification of new pathways that can be targeted therapeutically.
Synaptotagmin VII (Syt VII) is a member of the synaptotagmin family of membrane-trafficking proteins (9). Mice that have mutations affecting both copies of the Syt VII allele develop an inflammatory myositis, presumably as a consequence of exposure to endogenous muscle tissue antigen through impaired membrane resealing (10). Thus, it has been proposed that Syt VII deficiency can cause inflammation by activation and expansion of lymphocytes through exposure to endogenous antigens not normally encountered during the initial establishment of immune tolerance (11).
The FoxP3 gene product scurfin is an X-linked transcription factor involved in the maturation and activation of Treg cells (12). Treg cells actively suppress autoreactive cells and control the immune response (13). In humans, immune dysregulation, polyendocrinopathy, and enteropathy, X-linked syndrome is caused by a FoxP3 mutation that leads to a severe multiorgan autoinflammatory condition frequently resulting in death within the first 2 years of life (14). Scurfy (FoxP3 mutant) mice devoid of functional Treg cells also succumb to multiorgan inflammation primarily affecting the skin, lungs, and liver (12). We previously demonstrated that intraperitoneal adoptive transfer of scurfy mouse lymph node cells into recombination-activating gene 1 (RAG-1)–null mice not only recapitulated these manifestations but also induced inflammation in the colon, salivary glands, and lacrimal glands (15). Hence, although scurfy mice display autoimmune responses against only a few target organs, transfer of scurfy mouse lymphocytes to RAG-1–null mice induces severe inflammation in some of the organs that were spared in the de novo inflammatory response (16). This reveals the presence of a repertoire of autoreactive immune cells against more organs than initially observed in the scurfy mouse.
In both scurfy mice and RAG-1–null mice that underwent adoptive transfer, muscle tissue was spared from an inflammatory response. To examine the role of antigens to muscle tissue in this phenomenon, we administered intramuscular injections of scurfy mouse lymph node cells into RAG-1–null recipients, thus inflicting a muscle injury and inducing severe myopathy (16). We hypothesized that despite a complete deficiency of Treg cells, scurfy mice and the RAG-1–null recipients of scurfy mouse cells still fail to develop muscle inflammation due to a lack of critical exposure to muscle antigens.
In the present study, we tested this hypothesis by comparing FoxP3/Syt VII double-mutant mice with Syt VII mutant mice to determine the influence of Treg cells on suppression of muscle inflammation. Our results indicate that Syt VII deficiency mediates the generation of primed immune cells in donor mice that are capable of targeting muscle, which, when combined with Treg cell deficiency, results in the induction of severe myositis. Conversely, Treg cell supplementation can effectively suppress this autoimmune inflammatory response. Furthermore, myosin is identified as a muscle antigen capable of priming the immune response in this model. Taken together, these results outline a new mouse model of immune-mediated myositis and suggest the critical roles of abnormal autoantigen exposure and loss of Treg cell suppression in the development of muscle tissue inflammation.
MATERIALS AND METHODS
Mice and breeding
All mice were housed at The Ohio State University Wexner Medical Center (OSUWMC) in a Biosafety Level 3 barrier facility. Housing conditions included a 12-hour light/dark cycle, with chow and water available ad libitum. The facilities were maintained at 22–23°C with 30–50% relative humidity. All animal maintenance protocols were approved by the Institutional Animal Care and Use Committee through University Laboratory Animal Resources at OSUWMC.
Male wild-type (WT) C57BL/6 (B6) mice, female B6.Cg-FoxP3sf/x/J (FoxP3+/−) mice, and B6.129S7-Rag1tm1Mom/J (RAG-1–null) mice were obtained from The Jackson Laboratory. Female B6.129S1-Syt7tm1Nan/J (Syt VII+/−) mice were a generous gift from Dr. Norma Andrews (University of Maryland). Female FoxP3+/− mice were mated with male WT B6 mice to generate male FoxP3−/Y (scurfy) mice. Female Syt VII+/− mice were mated with male WT B6 mice to generate male Syt VII+/− mice. The Syt VII+/− mice were then crossbred to produce Syt VII−/− mutant mice. The FoxP3 and Syt VII mutations were combined by mating male FoxP3+/Y/Syt VII+/− mice with female FoxP3+/−/Syt VII+/− mice.
Genotyping
The tails of all mice in litters with FoxP3 or Syt VII mutations were clipped at 22 (±2) days of age. DNA was extracted according to a published protocol (The Jackson Laboratory) using a DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions. Polymerase chain reaction (PCR) amplification was performed for both genes, using The Jackson Laboratory protocol. The primer sequences used for amplification were as follows: for WT Syt VII, forward 5′-CATCCTCCACTGGCCATGAATG-3′ and reverse 5′-GCTTCACCTTGGTCTCCAG-3′; for mutant Syt VII, forward 5′-CTTGGGTGGAGAGGCTATTC-3′ and reverse 5′-AGGTGAGATGACAGGAGATC-3′; for WT FoxP3, forward 5′-CTCAGGCCTCAATGGACAAG-3′; for mutant FoxP3, forward 5′-TCAGGCCTCAATGGACAAAA-3′. A common reverse primer (5′-CATCGGATAAGGGTGGCATA-3′) was used for FoxP3. The PCR products were electrophoresed on 2% agarose gels, and the bands were visualized with ethidium bromide.
Adoptive transfer of lymph node cells
FoxP3−/Y (scurfy) mice with the Syt VII+/+, Syt VII+/−, or Syt VII−/− genotype were killed at 25 (±2) days of age by cervical dislocation, under anesthesia. Single cell suspensions of 0.7–1.5 × 107 cells in 200 μl of sterile phosphate buffered saline (PBS) (pH 7.3–7.4) from pooled lymph nodes (tonsillar, submandibular, axillary, and inguinal locations) were injected intraperitoneally into male RAG-1–null mice.
Muscle protein homogenates were prepared by resecting thigh skeletal muscle (quadriceps femoris) tissue from WT B6 mice. The muscle tissue was mechanically dissociated in sterile PBS (pH 7.3–7.4) at a 30% (weight/volume) ratio and passed through a cell strainer. Following a freeze–thaw cycle, adoptive transfer was carried out by intraperitoneal injection of a FoxP3−/Y/Syt VII+/+lymph node preparation of 0.7–1.5 × 107 cells plus with either 200 μl of the muscle homogenate or 1.11 mg of purified myosin protein (Sigma-Aldrich) adjusted with PBS to a volume of 200 μl. Intramuscular injections (0.7–1.5 × 107 cells in 200 μl) of FoxP3−/Y/Syt VII+/+ lymph node preparations were administered simultaneously into the quadriceps femoris muscle of male RAG-1–null littermates. All male RAG-1–null mice were monitored every other day for ∼4 weeks. Monitoring included assessment of body weights and physical signs of disease progression.
Treg cell purification and adoptive transfer
CD4+ CD25+ Treg cells were isolated from the lymph nodes of WT B6 mice by immunomagnetic sorting, using a commercial CD4+CD25+ Treg cell isolation kit (Miltenyi Biotec) according to the manufacturer's protocol. Treg cell purification was confirmed by flow cytometry (BD FACSCalibur platform; BD Biosciences) to be >80% CD4+/CD25+, using antibodies targeting the lymphocyte markers CD3 (eBioscience), CD4 (eBioscience), and CD25 (eBioscience) according to the manufacturer's instructions. Flow cytometry data were acquired using CellQuest Pro software version 5.1 (BD Biosciences) and analyzed further with FlowJo version 7.6.5 software (Tree Star). Enriched Treg cells (0.5 × 106) and/or FoxP3−/Y lymph node cells (5.0 × 106) in sterile PBS (pH 7.3–7.4) were transferred intravenously (200 μl) into the tail veins of male RAG-1–null mice. Additionally, Treg cells (0.5 × 106) and/or FoxP3−/Y/Syt VII−/− lymph node cells (5.0 × 106) in sterile PBS (pH 7.3–7.4) were transferred intraperitoneally (200 μl) into male RAG-1–null mice. The final Treg cell:lymph node cell supplementation ratio in both experimental conditions was 1:10.
Tissue collection and processing for histologic examination
FoxP3−/Y mice with the Syt VII+/+, Syt VII+/−, or Syt VII−/− genotype were killed on day 16 or day 26. FoxP3+/Y mice with the Syt VII+/+, Syt VII+/−, or Syt VII−/− genotype were killed at 4, 8, 12, or 18 weeks. Recipient RAG-1–null mice were killed on day 28. All mice were killed by cervical dislocation, under anesthesia. The quadriceps femoris muscle from each mouse was resected and flash-frozen in liquid nitrogen–cooled isopentane followed by cryosectioning, or was fixed immediately by immersion in neutral buffered 10% formalin and then processed into paraffin. Serial paraffin sections were stained with hematoxylin and eosin (H&E) (Leica Microsystems) or labeled by indirect immunohistochemistry to detect CD3 (Dako), B220 (CD45R; BD Biosciences), and F4/80 (AbD Serotec). Serial frozen sections were used for H&E staining and indirect immunohistochemistry to localize CD4 and CD8 (both from BD Biosciences). All staining was performed on tissue sections cut at 4 μm, according to the manufacturer's protocol.
Microscope slide imaging and analysis
Glass slides were scanned at 200× or 400× resolution on an Aperio ScanScope XT eSlide capture device and analyzed using Aperio ImageScope digital analysis version 9.1 software. To quantify the extent of inflammation and lymphocyte localization, an Aperio positive pixel count algorithm was run to determine cellular nuclei or positively labeled membranes using calibrated hue, saturation, and intensity values following previously described methods of computer-assisted image analysis (17). From each mouse, 10 measurements of identical total surface area of muscle tissue were quantitated to define a mean value of strong positive pixel intensity. All digitization was verified by manual slide analysis and histopathologic scoring, as described below.
Histopathologic scoring
H&E-stained paraffin sections of skeletal muscle were submitted for independent histopathologic analysis by a board-certified veterinary pathologist (BB). Microscopic lesions in the muscle indicating degeneration and inflammation were subjected to blinded histopathologic analysis using the 10× objective of a bright-field light microscope and a tiered, semiquantitative scoring scheme with predefined criteria (0 = within normal limits, 1 = minimal change, 2 = mild change, 3 = moderate change, and 4 = marked change).
Statistical analysis
All data were expressed as the mean ± SD. Statistical significance was determined by Student's paired 2-tailed t-tests using Microsoft Excel version 2010 software. P values less than or equal to 0.05 were considered significant.
RESULTS
Effect of Syt VII deficiency in mice without Treg cells
Syt VII deficiency results in membrane leakage and is reported to induce spontaneous myositis (11), but the extent of inflammation is limited compared with that observed in our scurfy mouse lymphocyte adoptive transfer model (16). To investigate the role of Treg cells in the suppression of spontaneous myositis, we introduced Treg cell deficiency onto the Syt VII mutant background by generating FoxP3/Syt VII double-mutant mice, in order to examine the combined effects of these 2 mutations on the development of myositis.
In muscle tissue from mice with the Syt VII deficiency alone (Syt VII−/−), the leukocyte infiltrate was mild but diffuse and was observed in both necrotic and non-necrotic areas. This inflammation was evident at 4 weeks but decreased slightly at later time points (Figure 1A). No distinguishable histologic difference was observed between Syt VII+/+ and Syt VII+/− genotypes at any time point (data not shown). Histopathologic scores for muscle inflammation in Syt VII−/− mice were statistically significantly higher than those in Syt VII+/− mice only at the 4-week time point, with mean ± SD scores (4-point scale) of 1.7 ± 0.58 and 0.33 ± 0.58, respectively (Figure 1B).
Figure 1.
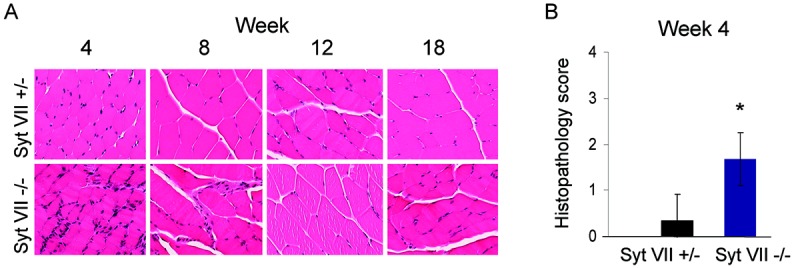
Muscle tissue inflammation in mice with the synaptotagmin VII (Syt VII) mutation is marked at 4 weeks but subsides over time. A, Muscle tissue inflammation in heterozygous (Syt VII+/−) and mutant (Syt VII−/−) mice. Mice were killed at the indicated times, and paraffin-embedded skeletal muscle tissue sections were stained with hematoxylin and eosin. All images were cropped for enhanced resolution and are representative of trends observed in at least 3 mice at each time point in independent experiments. Original magnification × 200. B, Extent of myositis in Syt VII−/− mice at the 4-week time point, as determined by blinded histopathologic scoring. Values are the mean ± SD (n = 3 mice of each genotype). ∗ = P ≤ 0.05 versus Syt VII+/−.
Skeletal muscle tissue was harvested from FoxP3/Syt VII double-mutant (FoxP3−/Y/Syt VII−/−) mice at 16 days or 26 days, in order to observe inflammation. Although the FoxP3 mutation alone (FoxP3−/Y) was not sufficient to cause an inflammatory response directed toward muscle, a deficiency of Syt VII in combination with the FoxP3 mutation (FoxP3−/Y/Syt VII−/−) resulted in significant infiltration of muscle at both 16 days and 26 days after birth (Figure 2A). The inflammation followed a histologic pattern representative of polymyositis in humans, with the endomysial leukocyte infiltrates targeting areas of non-necrotic, morphologically normal muscle fibers. However, both perifascicular inflammation and perivascular inflammation were also observed. Histopathologic scoring of the H&E-stained sections at the 4-week time point showed a mean ± SD score of 2.6 ± 0.55 for double-mutant mice (FoxP3−/Y/Syt VII−/−), which was significantly higher than the scores for single-mutant mice (0.33 ± 0.58 for Syt VII+/− [Figure 1B] and 0.6 ± 0.55 for FoxP3−/Y [Figure 2B]).
Figure 2.
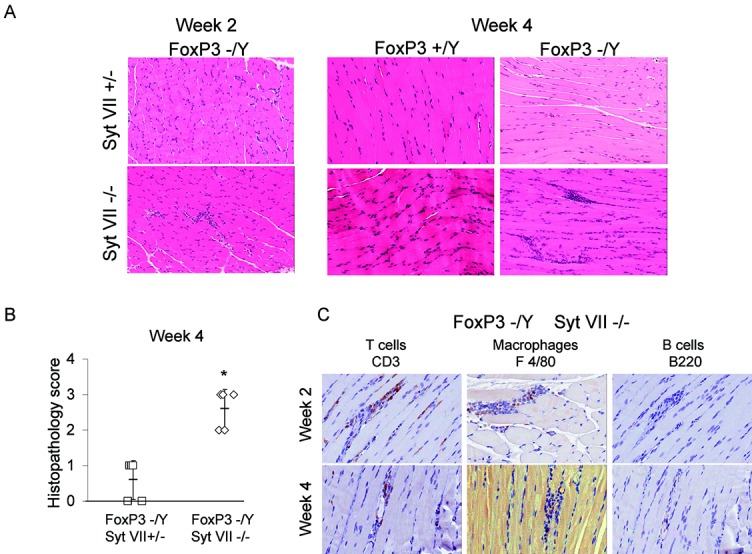
FoxP3 and synaptotagmin VII (Syt VII) double-mutant mice develop significant myositis. Male Syt VII+/− mice were bred with female FoxP3+/− mice to produce male wild-type, Syt VII mutant (Syt VII−/−), FoxP3 mutant (FoxP3−/Y), and double-mutant (Foxp3−/Y/Syt VII−/−) mice. A, Hematoxylin and eosin–stained muscle sections obtained from mice killed at 2 weeks or 4 weeks (±2 days). Original magnification × 200. B, Extent of myositis in Foxp3−/Y/Syt VII+/− and Foxp3−/Y/Syt VII−/− mice at the 4-week time point. Each symbol represents an individual mouse. Bars show the mean ± SD. C, Immunohistochemical staining for CD3 (T cell marker), F4/80 (macrophage cell marker), and B220 (B cell marker) in muscle sections obtained from FoxP3/Syt VII double-mutant mice at the 2-week and 4-week time points. Original magnification × 400. Results are representative of observations made in at least 3 mice at each time point and were independently verified in subsequent experiments. ∗ = P ≤ 0.005 versus Syt VII+/−.
Immunohistochemical staining was performed on the sections obtained from FoxP3−/Y/Syt VII−/− mice at 4 weeks. Application of a pan–B cell marker (B220 [CD45R]) revealed no detectable positive infiltrate in the muscle tissue, while the pan T cell marker CD3 clearly showed the presence of many CD3+ T cells in the same areas (Figure 2C). Staining for markers to identify plasmacytoid and myeloid dendritic cells was negative (data not shown). Taken together, these results demonstrate not only that aberrant antigen exposure in an environment lacking Treg cells results in more robust inflammation than that observed under conditions of Syt VII deficiency alone, but also that this response consists largely of T cells.
Induction of severe inflammation in the skeletal muscle of RAG-1–null recipients by adoptive transfer of lymph node cells from FoxP3−/Y/Syt VII−/− mice
We previously showed that adoptive transfer of scurfy (FoxP3−/Y) mouse lymph node cells into RAG-1–null recipients via either intraperitoneal or intravenous injection was unable to induce inflammation of skeletal muscle, while intramuscular injection of scurfy mouse cells did trigger myositis (16). Adoptive transfer of lymph node preparations from male scurfy mice with and those without Syt VII deficiency were delivered via intraperitoneal injection into RAG-1–null mice to determine whether the priming of lymph node cells in the double-mutant mice, in which myositis develops spontaneously, imparts the ability of these cells to induce inflammation despite the absence of muscle injury or Syt VII mutation in the recipient. Adoptive transfer of FoxP3−/Y/Syt VII−/− mouse lymph node cells by intraperitoneal injection resulted in robust muscle tissue inflammation (mean ± SD histopathologic score 3.0 ± 0.63) compared with that following adoptive transfer of FoxP3−/Y/Syt VII+/− mouse cells (mean ± SD 0.9 ± 0.57) (Figures 3A and B). The histology and extent of inflammation did not differ significantly between transfers of FoxP3−/Y/Syt VII+/+ and FoxP3−/Y/Syt VII+/− mouse lymph node preparations (data not shown).
Figure 3.

Intraperitoneal (IP) adoptive transfer of FoxP3/synaptotagmin VII (Syt VII) double-mutant mouse lymph node cells induces robust myositis in recombination-activating gene 1 (RAG-1)–null recipients. FoxP3-deficient (FoxP3−/Y) lymph node preparations from mice with either a heterozygous or mutant Syt VII genotype were made at the time the mice were killed, and transferred by intraperitoneal (IP) injection into RAG-1–null mice. A, Tissue inflammation in muscle sections obtained 4 weeks after adoptive transfer of FoxP3−/Y/Syt VII+/− mouse cells or FoxP3−/Y/Syt VII−/− mouse cells. Hematoxylin and eosin (H&E) stained; original magnification × 80 (left) and × 200 (right). B, Histopathologic scores of the H&E-stained sections, as determined by blinded analysis. Each symbol represents an individual mouse. Bars show the mean ± SD. C, Immunohistochemical staining for CD4, CD8, B220, and F4/80 in muscle sections obtained from FoxP3−/Y/Syt VII−/− mice 4 weeks after adoptive transfer. All images are representative of trends observed in 6 mice (n = 2 individual experiments). Original magnification × 200. D, Results of digital image analysis showing the pixel intensity values for CD4 T cells, CD8 T cells, F4/80 macrophages, and B220 B cells. Values are the mean ± SD. ∗ = P ≤ 0.001 versus Syt VII+/− (B) and versus all other cell subtype markers (D).
Immunohistochemical analysis further revealed that the response included both CD4+ and CD8+ T cells and F4/80+ macrophages, while B cells were again absent in the inflammatory infiltrate (Figure 3C). Additionally, staining for interleukin 17 (IL-17)–secreting cells revealed little or no positive detection (data not shown). Digital image analysis demonstrated that CD4+ T cells were detected at significantly higher levels relative to the other cell types in the muscle tissue infiltrates (Figure 3D). These data demonstrated that leukocytes educated in the FoxP3/Syt VII double-mutant mouse are sufficient to cause severe myositis in an adoptive transfer model.
Role of lymph node cell priming with abnormal muscle antigens in the induction of myositis in the RAG-1–null mouse adoptive transfer model
In order to further explore the postulate that endogenous antigen exposure leads to myositis development, adoptive transfer of Treg cell–deficient (FoxP3−/Y) mouse lymph node cells into RAG-1–null mice was performed under several conditions that aberrantly exposed these cells to muscle antigens. Because muscle tissue homogenate (18–22) and myosin protein (23) have been previously shown to induce muscle-specific inflammatory responses when coadministered with immunoadjuvant, FoxP3 mutant mouse lymph node cells were injected intraperitoneally into RAG-1–null mice in conjunction with muscle tissue homogenate or purified myosin protein. This resulted in pronounced myositis, and the resulting inflammation was similar to that observed when intramuscular injection was used as a positive control (Figure 4A). Quantitation (using digital image analysis) of the inflammatory response induced by intramuscular injection showed significant infiltration compared with that induced by intraperitoneal injection (Figure 4B). In addition, the mean ± SD histopathologic score resulting from intramuscular injection was 3.0 ± 0.0, compared with scores of 3.4 ± 0.55 and 2.6 ± 0.55 resulting from intraperitoneal coinjection with muscle tissue homogenate and intraperitoneal coinjection with purified myosin protein, respectively (Figure 4C). All of these scores were significantly higher than the score for intraperitoneal adoptive transfer of FoxP3-deficient mouse lymph node cells alone (0.9 ± 0.57) (Figure 4C).
Figure 4.
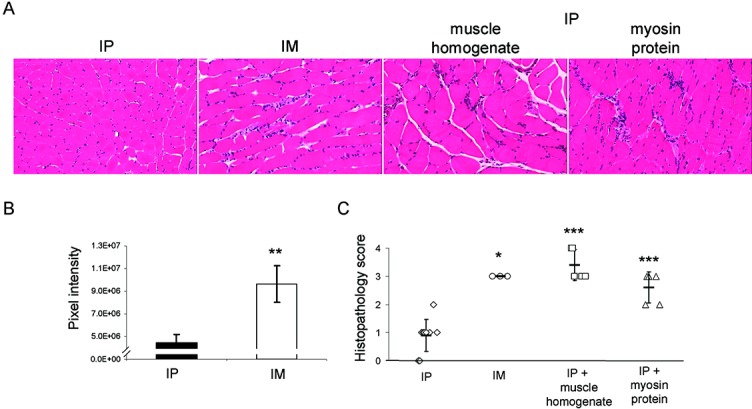
Myositis is induced in RAG-1–null mouse recipients of FoxP3-deficient mouse lymph node cells exposed to muscle antigen. Lymph nodes from FoxP3-deficient mice were prepared and transferred by intraperitoneal injection, intramuscular (IM) injection, intraperitoneal coinjection with muscle tissue homogenate, or intraperitoneal coinjection with purified myosin protein. A, H&E-stained muscle tissue sections obtained from mice in the 4 treatment groups 4 weeks after adoptive transfer. Original magnification × 200. B, Results of digital image analysis showing the pixel intensity values for the inflammatory response induced by intraperitoneal and intramuscular injections. Values are the mean ± SD. C, Histopathologic scores of the H&E-stained muscle tissue sections, as determined by blinded analysis. Each symbol represents an individual mouse. Bars show the mean ± SD. Results are representative of trends observed in at least 3 mice (n = 2 individual experiments). ∗ = P < 0.05; ∗∗ = P < 0.01; ∗∗∗ = P < 0.005 versus intraperitoneal injection. See Figure 3 for other definitions.
To characterize the cell subtypes present in these muscle tissue infiltrates, immunohistochemistry was performed on sections obtained from RAG-1–null mice following these adoptive transfers. As with transfer of FoxP3−/Y/Syt VII−/− mouse lymph node cells, concurrent introduction of FoxP3−/Y mouse lymph node cells plus muscle protein resulted in inflammatory responses consisting largely of T cells with a few macrophages, while B cells were absent. Specifically, the infiltrates were composed primarily of CD4+ T cells, with CD8+ T cells and F4/80+ macrophages present to a lesser extent (Figure 5A). Digital image analysis showed that CD4+ T cells were present at significantly higher levels than CD8+ T cells, macrophages, or B cells (Figure 5B). A comparison across all experimental conditions showed that intraperitoneal transfer of FoxP3-mutant mouse lymph node cells in combination with muscle tissue homogenate attracted significantly more CD4+ T cells (Figure 5B), a circumstance reflected by the highest histopathologic score. Taken together, these results demonstrated that aberrant exposure to endogenous muscle tissue antigens profoundly influenced the specificity of a predominantly T cell–mediated inflammatory response in this adoptive transfer model.
Figure 5.
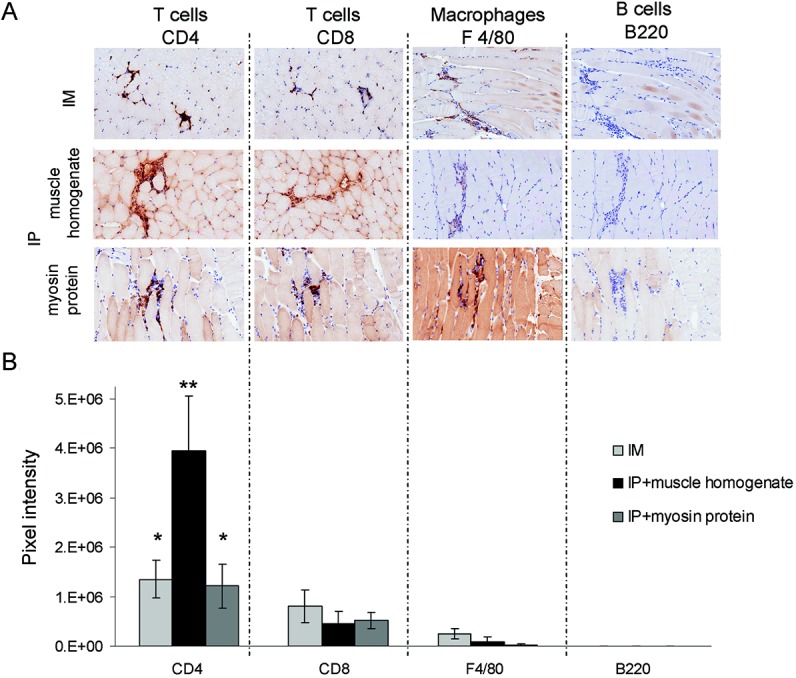
Inflammatory responses in myositis induced by exposure to muscle protein are predominated by CD4+ cells. Preparations of lymph node cells from FoxP3-deficient mice were adoptively transferred into recombination-activating gene 1–null recipients by intramuscular (IM) or intraperitoneal (IP) injection, with or without coadministration of muscle homogenate or purified myosin protein. A, Expression of CD4, CD8, F4/80, and B220 within inflammatory infiltrates, as determined by immunohistochemistry. Original magnification × 200. B, Results of digital image analysis showing the pixel intensity values in areas of infiltration for each leukocyte marker. Results are representative of similar trends observed in at least 5 mice (n = 4 independent experiments). Values are the mean ± SD. ∗ = P < 0.05; ∗∗ = P < 0.0001 versus all other markers in the same experimental group.
Effect of Treg cells in myositis induced by adoptive transfer of inflammatory cells targeting muscle tissue
The suppressive potential of Treg cells toward immune-mediated myositis was investigated in the RAG-1–null mouse adoptive transfer model to determine whether normal mice harbor Treg cells capable of suppressing inflammation induced by this aberrant exposure to muscle antigens. RAG-1–null recipients were given intramuscular injections of FoxP3−/Y mouse lymph node cells or intraperitoneal injections of FoxP3−/Y/Syt VII−/− mouse lymph node cells supplemented with either additional lymph node cells or purified Treg cells from WT B6 mice. Treg cell supplementation suppressed muscle inflammation in RAG-1–null mice that received intramuscular injections of FoxP3-deficient mouse lymph node cells (Figure 6A) and in RAG-1–null mice that received intraperitoneal injections of lymph node cells from FoxP3/Syt VII double-mutant mice (Figure 6B). The extent of inflammation was reduced significantly by the addition of Treg cells, as assessed both quantitatively by digital image analysis and semiquantitatively by coded histopathologic scoring (from a mean ± SD of 3.0 ± 0.0 without Treg cell supplementation to 0.9 ± 0.57 with Treg cell supplementation) (Figure 6C). These results demonstrate the extensive Treg cell repertoire of WT mice and the ability of these cells to modulate the immune response in this model of myositis.
Figure 6.
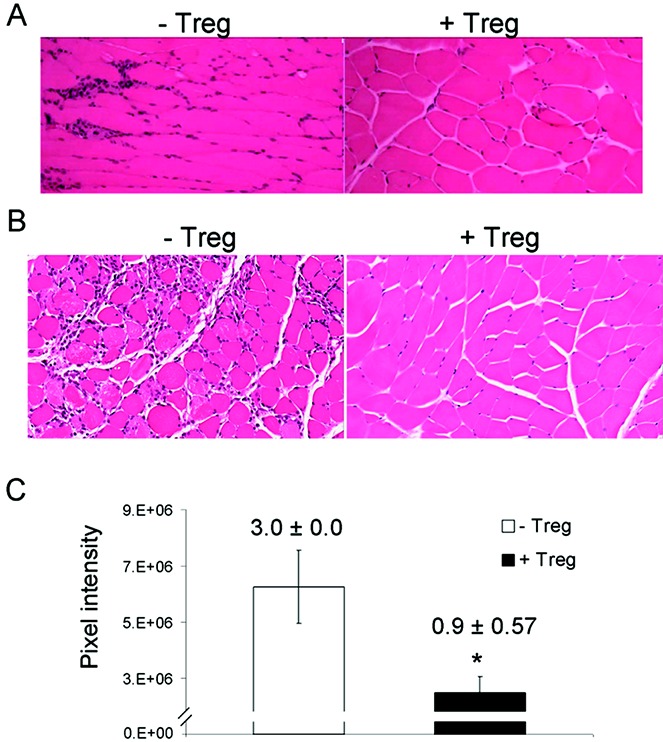
Treg cells suppress immune-mediated muscle tissue inflammation. CD4+CD25+ Treg cells from wild-type C57BL/6 mice were isolated by immunomagnetic sorting, and the purity was confirmed by flow cytometry to be >80%. A and B, Representative hematoxylin and eosin–stained paraffin-embedded muscle tissue sections from recombination-activating gene 1–null mice that received intramuscular injections of lymph node cells from Foxp3-deficient mice (A) or intraperitoneal injections of lymph node cells from Foxp3/Syt VII double-mutant mice (B), either with or without CD4+CD25+ Treg cells. Original magnification × 200. C, Extent of inflammation induced by adoptive transfer, with and without Treg cell supplementation, as assessed quantitatively by digital image analysis and semiquantitatively by histopathologic scoring. Bars show the mean ± SD; values above the bars are the mean ± SD histopathologic scores (n = 3 independent experiments). ∗ = P < 0.001 versus no Treg cell supplementation.
DISCUSSION
Because an autoimmune pathogenesis of myositis has been strongly implicated, this remains the focus of both standardized and emerging therapeutic strategies (24). Recent work has also shown that patients with dermatomyositis (DM) have decreased numbers of Treg cells in both peripheral blood and skin lesions when compared with healthy subjects (25). Furthermore, FoxP3+ Treg cells have been identified at the sites of muscle inflammation in humans, and the association of Treg cells with myoreactive Teff cells has also been demonstrated (26). To our knowledge, the present study is the first to investigate the contribution of abnormal muscle antigen availability to the pathogenesis of myositis in a Treg cell–deficient environment.
FoxP3-deficient mice display intense autoimmune inflammatory processes involving multiple organs but exhibit very little evidence of myositis (16). In contrast, Syt VII mutant mice develop a mild, self-limiting inflammatory response involving the muscles (11). Here, it is possible that muscle antigen–specific Treg cells develop, which at later stages effectively resolve the inflammation. Our findings show that the deficiency of both genes resulted in the development of significant muscle tissue inflammation, which can be attributable to the lack of Treg cell suppression in the presence of abnormal muscle antigen availability. The robust myositis induced by intraperitoneal adoptive transfer of lymph node cells from FoxP3/Syt VII double-mutant mice into RAG-1–null mice demonstrates that the Syt VII deficit is necessary for the generation and expansion of effector immune cells that can target muscle but is not required in the affected organ.
Antigens from muscle tissue have been shown to elicit an inflammatory response when given in conjunction with adjuvant stimulation (18–23). Because immunoadjuvant injections in animal models can elicit well-defined mechanisms of inflammation, even when given alone, and have also been shown to lead to aspecific immune cell activation (27), we examined the role of intracellular muscle proteins as a means of promoting myositis in our Treg cell–deficient model. Given the strong association of Treg cells with myositis (26), the conclusions supported by our data may therefore ultimately hold more physiologic relevance and therapeutic potential than conclusions based on data derived from adjuvant-based models. Transfer of Treg cell–deficient lymph node cells into RAG-1–null mice by intraperitoneal injection with either muscle tissue homogenate or purified myosin protein caused significant muscle inflammation. Although the greatest inflammatory responses were observed with coadministration of muscle tissue homogenate, myosin protein by itself stimulated notable muscle tissue inflammation. Considering that anti-myosin autoantibodies have been previously detected in patients with DM (28), this indicates that myosin is at least one antigen capable of generating myositis. Therefore, the neutralization of extracellular myosin represents a potential therapeutic target for minimizing autoinflammation.
Coadministration of lymph node cells from FoxP3−/Y mice with muscle tissue homogenate resulted in an enhanced inflammatory response compared with coadministration with myosin protein, which implies that additional intracellular autoantigens are presumably functioning pathogenically as well. Consistent with these data, Hsp60 in inflamed muscle tissue has been shown to be the target of regulatory autoreactive T cells in patients with juvenile DM (29), and histidyl–transfer RNA synthetase has been shown to be a target of human γ/δ T cell receptor in the lesions of patients with polymyositis (30,31). Another muscle-specific protein, chromatin remodeler Mi-2, is involved in muscle regeneration and has also been identified as a potential autoantigen (32). Furthermore, autoantibodies to acetylcholine receptor and muscle-specific kinase have been observed in patients with myasthenia gravis (33). Taken together, these findings suggest that antigen-specific immunotherapy may require an individualized patient approach despite similar histologic and clinical presentations.
In the present study, the muscle inflammatory infiltrate was devoid of B cells but included macrophages and a significant number of T cells in all cases. This pattern is consistent with prior mouse models of myositis, in which inflammatory infiltrates consisted predominantly of T cells (chiefly CD4+ cells with some CD8+ cells) and occasional macrophages, but no B cells (34). In addition, dendritic cells and IL-17–secreting cells were either present in insignificant numbers or were absent from the muscle tissue infiltrates in all experimental conditions. This is consistent with our previous studies in the scurfy mouse model, which demonstrated that the majority of the tissue-infiltrating cells are CD4+ Th1 or Th2 cells (35,36).
In samples obtained from patients with juvenile DM, inflamed muscle tissue was shown to contain autoreactive T cells that significantly proliferate when exposed to self antigen (29). Furthermore, our model has shown that adoptive transfer of CD4+ cells alone is sufficient to recapitulate muscle tissue pathology (16). In addition, we recently showed that CD4+ T cells from Treg cell–deficient mice highly express several receptors involved in the trafficking of T cells to the sites of inflammation (35,36), indicating that injury or stress to muscle tissue may result in up-regulation of ligands to these trafficking receptors and therefore play an important role in the recruitment of muscle antigen–primed T cells to muscle. Given that the pathogenesis of myositis most likely derives from T cells stimulated by self antigens to induce autoimmunity (37), and that a subset of CD4+ and CD8+ T cells can be detected in muscle tissue despite immunosuppressive therapy (38), we conclude that effector T cells are essential in both the development of and the sustained inflammation involved in disease pathology.
With Treg cell supplementation at a 1:10 ratio, minimal muscle tissue infiltrate was observed. Similarly, the suppressive role of polyclonal Treg cells expanded in vitro in mediating experimental autoimmune myositis was recently demonstrated in a reproducible mouse model (34). Because there are no definitive data to date demonstrating the involvement of Treg cells in human myositis, our results underscore the need for such investigations. Recent studies in type 1 diabetes mellitus demonstrated that patients with normal levels of circulating Treg cells have a Treg cell defect locally in the pancreas and lymph nodes (39); this suggests that Treg cells in the muscles should be analyzed more closely to better understand myositis pathology. Additionally, the therapeutic application of Treg cells was shown to resolve already-established inflammatory disease in mouse models of acute renal (40) and lung injury (41). Furthermore, it has been suggested that Treg cells contribute to healing in addition to immunosuppression (42). Considering the translational research being pursued in the use of Treg cell immunotherapy to treat Crohn's disease (43) and graft-versus-host disease (44), our results suggest that Treg cells may also be of critical importance when exploring future therapeutic avenues for treating autoimmune-mediated myositis.
Mechanical stress and drug-induced injury to muscle cells may lead to reversible cell degeneration or myofiber death, both of which may allow the abnormal release of endogenous muscle antigens that potentially can invoke an autoimmune myopathy. Importantly, under normal physiologic conditions, these damaged muscle cells should be rapidly cleared by the tissue-resident and circulating macrophages in order to avoid an autoimmune response. Natural Treg cells specific for endogenous antigens should also be present or induced from naive T cells to suppress this potential inflammatory response. Dysregulation of this system could then result in an autoimmune response. Accordingly, our in vivo mouse system will be valuable in studying the myopathy associated with drug-induced injury, particularly because in some cases the spectrum of myopathy following a muscle injury involves an immune mechanism that is responsive to steroid treatment (45). Furthermore, this model could be used to demonstrate that specific nonimmune injury to a particular organ may be the means by which systemic autoimmune diseases can target a specific organ. Taken together, these data demonstrate interplay between aberrant antigen exposure, Treg cell deficiency, and autoimmunity.
Acknowledgments
We would like to acknowledge Alan Flechtner and Kristin Kovach (OSUWMC) for assistance with the preparation and staining of muscle tissue. We also extend our thanks to Dr. Norma Andrews (University of Maryland) for providing the Syt VII heterozygous mice used in this study as well as Nitish Aggarwal and Michael Bruss (OSUWMC) for assistance with immunohistochemistry quantification.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Jarjour had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Young, Sharma, Jarjour.
Acquisition of data. Young, Friedman, Kaffenberger, Bolon, Jarjour.
Analysis and interpretation of data. Young, Sharma, Kaffenberger, Bolon, Jarjour.
REFERENCES
- Figarella-Branger D, Civatte M, Bartoli C, Pellissier JF. Cytokines, chemokines, and cell adhesion molecules in inflammatory myopathies. Muscle Nerve. 2003;28:659–82. doi: 10.1002/mus.10462. [DOI] [PubMed] [Google Scholar]
- Christopher-Stine L, Plotz PH. Myositis: an update on pathogenesis. Curr Opin Rheumatol. 2004;16:700–6. doi: 10.1097/01.bor.0000141925.21941.d8. [DOI] [PubMed] [Google Scholar]
- Sontheimer RD. The management of dermatomyositis: current treatment options. Expert Opin Pharmacother. 2004;5:1083–99. doi: 10.1517/14656566.5.5.1083. [DOI] [PubMed] [Google Scholar]
- Greenberg SA. Inflammatory myopathies: evaluation and management. Semin Neurol. 2008;28:241–9. doi: 10.1055/s-2008-1062267. [DOI] [PubMed] [Google Scholar]
- Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. RIM Study Group. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–24. doi: 10.1002/art.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastmalchi M, Grundtman C, Alexanderson H, Mavragani CP, Einarsdottir H, Helmers SB, et al. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann Rheum Dis. 2008;67:1670–7. doi: 10.1136/ard.2007.077974. [DOI] [PubMed] [Google Scholar]
- Rowin J, Amato AA, Deisher N, Cursio J, Meriggioli MN. Mycophenolate mofetil in dermatomyositis: is it safe? Neurology. 2006;66:1245–7. doi: 10.1212/01.wnl.0000208416.32471.c0. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–27. doi: 10.1038/ncprheum0140. [published erratum appears in Nat Clin Pract Rheumatol 2006;2:398]. [DOI] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–8. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Andrews NW. Membrane repair and immunological danger. EMBO Rep. 2005;6:826–30. doi: 10.1038/sj.embor.7400505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, et al. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol. 2003;162:543–9. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Sung SS, Fu SM, Ju ST. Regulation of multi-organ inflammation in the regulatory T cell-deficient scurfy mice. J Biomed Sci. 2009;16:20. doi: 10.1186/1423-0127-16-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Cava A. The busy life of regulatory T cells in systemic lupus erythematosus. Discov Med. 2009;8:13–7. [PubMed] [Google Scholar]
- Le Bras S, Geha RS. IPEX and the role of Foxp3 in the development and function of human Tregs. J Clin Invest. 2006;116:1473–5. doi: 10.1172/JCI28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Zheng L, Guo X, Fu SM, Ju ST, Jarjour WN. Novel animal models for Sjögren's syndrome: expression and transfer of salivary gland dysfunction from regulatory T cell-deficient mice. J Autoimmun. 2006;27:289–96. doi: 10.1016/j.jaut.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Jarjour WN, Zheng L, Gaskin F, Fu SM, Ju ST. Large functional repertoire of regulatory T-cell suppressible autoimmune T cells in scurfy mice. J Autoimmun. 2007;29:10–9. doi: 10.1016/j.jaut.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapczynski M, Gagne GD, Morgan SJ, Larson KJ, Leroy BE, Blomme EA, et al. Computer-assisted imaging algorithms facilitate histomorphometric quantification of kidney damage in rodent renal failure models. J Pathol Inform. 2012;3:20. doi: 10.4103/2153-3539.95456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakulas BA. In vitro destruction of skeletal muscle by sensitized cells. Nature. 1966;210:1115–8. doi: 10.1038/2101115a0. [DOI] [PubMed] [Google Scholar]
- Kalden JR, Williamson WG, Irvine WJ. Experimental myasthenia gravis, myositis and myocarditis in guinea-pigs immunized with subcellular fractions of calf thymus or calf skeletal muscle in Freund's complete adjuvant. Clin Exp Immunol. 1973;13:79–88. [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, MacLennan IC. Experimental myositis in rats. I. Histological and creatine phosphokinase changes, and passive transfer to normal syngeneic rats. Clin Exp Immunol. 1974;17:139–50. [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, MacLennan IC. Experimental myositis in rats. II. The sensitivity of spleen cells to syngeneic muscle antigen. Clin Exp Immunol. 1975;19:513–20. [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NL, Ringel SP, Kotzin BL. Experimental autoimmune myositis in SJL/J mice. Clin Exp Immunol. 1987;68:117–29. [PMC free article] [PubMed] [Google Scholar]
- Nemoto H, Bhopale MK, Constantinescu CS, Schotland D, Rostami A. Skeletal muscle myosin is the autoantigen for experimental autoimmune myositis. Exp Mol Pathol. 2003;74:238–43. doi: 10.1016/s0014-4800(03)00003-0. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Inflammatory muscle diseases: a critical review on pathogenesis and therapies. Curr Opin Pharmacol. 2010;10:346–52. doi: 10.1016/j.coph.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Antiga E, Kretz CC, Klembt R, Massi D, Ruland V, Stumpf C, et al. Characterization of regulatory T cells in patients with dermatomyositis. J Autoimmun. 2010;35:342–50. doi: 10.1016/j.jaut.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Waschbisch A, Schwab N, Ruck T, Stenner MP, Wiendl H. FOXP3+ T regulatory cells in idiopathic inflammatory myopathies. J Neuroimmunol. 2010;225:137–42. doi: 10.1016/j.jneuroim.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Billiau A, Matthys P. Modes of action of Freund's adjuvants in experimental models of autoimmune diseases. J Leukoc Biol. 2001;70:849–60. [PubMed] [Google Scholar]
- Wada K, Ueno S, Hazama T, Ogasahara S, Kang J, Takahashi M, et al. Radioimmunoassay for antibodies to human skeletal muscle myosin in serum from patients with polymyositis. Clin Exp Immunol. 1983;52:297–304. [PMC free article] [PubMed] [Google Scholar]
- Elst EF, Klein M, de Jager W, Kamphuis S, Wedderburn LR, van der Zee R, et al. Hsp60 in inflamed muscle tissue is the target of regulatory autoreactive T cells in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;58:547–55. doi: 10.1002/art.23202. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Engel AG, Ii K, Harper MC. Polymyositis mediated by T lymphocytes that express the γ/δ receptor. N Engl J Med. 1991;324:877–81. doi: 10.1056/NEJM199103283241303. [DOI] [PubMed] [Google Scholar]
- Bruder J, Siewert K, Obermeier B, Malotka J, Scheinert P, Kellermann J, et al. Target specificity of an autoreactive pathogenic human γδ-T cell receptor in myositis. J Biol Chem. 2012;287:20986–95. doi: 10.1074/jbc.M112.356709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A. Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum. 2009;60:3784–93. doi: 10.1002/art.24977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriggioli MN, Sanders DB. Muscle autoantibodies in myasthenia gravis: beyond diagnosis? Expert Rev Clin Immunol. 2012;8:427–38. doi: 10.1586/eci.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allenbach Y, Solly S, Gregoire S, Dubourg O, Salomon B, Butler-Browne G, et al. Role of regulatory T cells in a new mouse model of experimental autoimmune myositis. Am J Pathol. 2009;174:989–98. doi: 10.2353/ajpath.2009.080422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Sharma PR, Kim YC, Leitinger N, Lee JK, Fu SM, et al. IL-2–controlled expression of multiple T cell trafficking genes and Th2 cytokines in the regulatory T cell-deficient scurfy mice: implication to multiorgan inflammation and control of skin and lung inflammation. J Immunol. 2011;186:1268–78. doi: 10.4049/jimmunol.1002677. [published erratum appears in J Immunol 2011;186:5012-3]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Sung SS, Gaskin F, Fu SM, Ju ST. A novel function of IL-2: chemokine/chemoattractant/retention receptor genes induction in Th subsets for skin and lung inflammation. J Autoimmun. 2012;38:322–31. doi: 10.1016/j.jaut.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201:591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasth AE, Dastmalchi M, Rahbar A, Salomonsson S, Pandya JM, Lindroos E, et al. T cell infiltrates in the muscles of patients with dermatomyositis and polymyositis are dominated by CD28null T cells. J Immunol. 2009;183:4792–9. doi: 10.4049/jimmunol.0803688. [DOI] [PubMed] [Google Scholar]
- Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L, et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes. 2011;60:2903–13. doi: 10.2337/db11-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandolfo MT, Jang HR, Bagnasco SM, Ko GJ, Agreda P, Satpute SR, et al. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76:717–29. doi: 10.1038/ki.2009.259. [DOI] [PubMed] [Google Scholar]
- D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest. 2009;119:2898–913. doi: 10.1172/JCI36498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, et al. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J. 2013;27:2207–19. doi: 10.1096/fj.12-225201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn's disease. Gastroenterology. 2012;143:1207–17.e2. doi: 10.1053/j.gastro.2012.07.116. [DOI] [PubMed] [Google Scholar]
- Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–8. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17:194–200. doi: 10.1016/j.nmd.2006.10.007. [DOI] [PubMed] [Google Scholar]