

Abstract
Aberrant sperm DNA methylation patterns, mainly in imprinted genes, have been associated with male subfertility and oligospermia. Here, we performed a genome-wide methylation analysis in sperm samples representing a wide range of semen parameters. Sperm DNA samples of 38 males attending a fertility centre were analysed with Illumina HumanMethylation27 BeadChips, which quantify methylation of >27 000 CpG sites in cis-regulatory regions of almost 15 000 genes. In an unsupervised analysis of methylation of all analysed sites, the patient samples clustered into a major and a minor group. The major group clustered with samples from normozoospermic healthy volunteers and, thus, may more closely resemble the normal situation. When correlating the clusters with semen and clinical parameters, the sperm counts were significantly different between groups with the minor group exhibiting sperm counts in the low normal range. A linear model identified almost 3000 CpGs with significant methylation differences between groups. Functional analysis revealed a broad gain of methylation in spermatogenesis-related genes and a loss of methylation in inflammation- and immune response-related genes. Quantitative bisulfite pyrosequencing validated differential methylation in three of five significant candidate genes on the array. Collectively, we identified a subgroup of sperm samples for assisted reproduction with sperm counts in the low normal range and broad methylation changes (affecting approximately 10% of analysed CpG sites) in specific pathways, most importantly spermatogenesis-related genes. We propose that epigenetic analysis can supplement traditional semen parameters and has the potential to provide new insights into the aetiology of male subfertility.
Keywords: epigenetic markers, male infertility, methylation arrays, sperm DNA methylation
Introduction
The molecular basis of male infertility remains largely unknown (Gianotten et al., 2004). A number of candidate gene studies revealed that abnormal sperm DNA methylation patterns are associated with reduced sperm count and function (Marques et al., 2008; Kobayashi et al., 2009; Hammoud et al., 2010; Navarro-Costa et al., 2010; Poplinski et al., 2010) as well as outcome of assisted reproductive technologies (ART) (El Hajj et al., 2011). Accumulating evidence suggests that the spermatozoa contributes more to the embryo than the paternal genome (Krawetz, 2005; Carrell & Hammoud, 2010). Epigenetic sperm factors may affect the regulation of essential paternal genes during embryogenesis and further development. Spermatogenesis is a highly coordinated process that involves haploidization and epigenetic reprogramming of the paternal genome. In the primordial germ cells of the foetal germ line, all methylation patterns are essentially erased, restoring totipotency and an equivalent epigenetic state in germ cells of both sexes. The sperm-specific methylation patterns are then established during germ cell differentiation (Hajkova et al., 2002; Carrell, 2012). Remethylation is initiated after prenatal mitotic arrest in prospermatogonia and proceeds in a gene-specific manner until the end of the pachytene spermatocyte stage (Rousseaux et al., 2005; Oakes et al., 2007; Boyano et al., 2008). Whole genome bisulfite sequencing revealed that the promoter regions of most developmentally important genes are hypomethylated in spermatozoa (Hammoud et al., 2009), most likely to ensure their rapid activation in the early embryo. Although most repeats are methylated in spermatozoa to prevent retrotransposition activity, several subfamilies of L1 and ALU transposons appear to be relatively hypomethylated in spermatozoa (Molaro et al., 2011).
Most previous studies on the connection between epigenetic sperm factors and male infertility have used imprinted genes as a model (Marques et al., 2008; Kobayashi et al., 2009; Hammoud et al., 2010; Poplinski et al., 2010; El Hajj et al., 2011). As the germ line methylation imprints are protected against reprogramming after fertilization (Reik et al., 2001; Haaf, 2006), aberrant sperm methylation patterns at imprinted loci may directly interfere with post-zygotic development. Indeed, similar epigenetic abnormalities were reported in abortions after ART and in the paternal sperm samples (Kobayashi et al., 2009). However, epigenetic alterations in non-imprinted genes and regulatory DNA sequences may be equally important for expression of the paternal genome in the early embryo. One study (Houshdaran et al., 2007) demonstrated that poor semen parameters were associated with elevated methylation levels at numerous non-imprinted genes, suggesting improper erasure of somatic methylation patterns in the male germ line. One serious problem in interpreting sperm methylation patterns is that we do not know much about the normal range of epigenetic variation and the impact of paternal (most importantly age) and environmental factors. Microarray-based methylation analyses demonstrated numerous methylation-variable CpG sites in the sperm epigenome. Evidently, each spermatozoa of the same sample exhibits a unique methylation profile (Flanagan et al., 2006). With the notable exception of a few highly penetrant imprinting mutations, the phenotypic consequences of epigenetic differences between sperm samples remain largely unclear. At the end, it takes only one spermatozoa to fertilize the oocyte.
Both genome-wide (Krausz et al., 2012) and candidate gene (Kläver et al., 2013) analyses suggest that DNA methylation patterns in normozoospermic males are relatively stable in different quality sperm subpopulations (swim up and down fractions of the same semen samples) and over time (semen samples collected over a period of >180 days from the same males). So far, there are few genome-wide studies (Aston et al., 2011; Pacheco et al., 2011) comparing the methylation profiles of 43 and 21 sperm samples, respectively, with a wide range of semen parameters. Pacheco et al. (2011) associated low motility spermatozoa with genome-wide DNA hypomethylation, but this did not explain most of the variation in their data set. Aston et al. (2011) identified two samples with abnormal sperm packaging and one sample with a high incidence of abnormal in vitro fertilization (IVF) embryogenesis displaying broad methylation changes. Here, we used the same Illumina Infinium array (assessing >27 000 CpG loci) to analyse sperm samples from 38 men presenting to a reproductive health clinic for infertility treatment. A subgroup of sperm samples with sperm counts in the low normal range was characterized by increased methylation of genes involved in spermatogenesis and decreased methylation of genes involved in inflammation and immune response.
Materials and methods
Sperm samples
The study was approved by the Ethics Committee at the Medical Faculty of Wuerzburg University and informed written consent was obtained from all participating subjects. Sperm samples were collected from couples attending the Fertility Center Wiesbaden and pseudonymized before methylation analysis (Table S1). Because we wanted to study a cohort which is representative of couples undergoing infertility treatment, participating males were not selected on the basis of semen or clinical parameters. Sperm samples (excess materials) were taken from 38 men giving their informed consent within a certain time period. In addition, semen samples of four healthy normozoospermic volunteers were collected as control group. Two consecutive (within 6 weeks) samples of one donor served as biological replicates.
Semen parameters were measured according to the WHO guidelines (World Health Organization, 2010). Male infertility was assumed in males with sperm concentration fewer than 20 × 106/mL (oligozoospermia), fewer than 50% spermatozoa with forward progression (categories a and b) or fewer than 25% spermatozoa with category a movement (asthenozoospermia), and/or fewer than 15% spermatozoa with normal morphology (teratozoospermia). Female infertility was assumed in couples, where the males displayed normal semen parameters and with clear indication for female factors, in particular tubal infertility. Some couples showed both male and female factors (combined infertility) or neither male nor female factors (idiopathic infertility).
Spermatozoa for IVF/intracytoplasmatic sperm injection (ICSI) were selected by the standard swim-up separation technique. The remaining material (swim-down fraction) was frozen until further use. After thawing, sperm cells were purified from lymphocytes, epithelial cells, cell debris, bacteria and seminal fluid using Pure Sperm 40/80 (Nidacon, Molndal, Sweden). Sperm purity was checked through inverted light microscopy. Purified spermatozoa were incubated for 2 h at 56 °C (on a thermomixer) with 100 mm Tris-Cl, 10 mm EDTA, 500 mm NaCl, 1% SDS, 2% β-mercaptoethanol and 100 μL proteinase K (>600 mAU/mL). DNA was isolated with the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) following the recommendations of the manufacturer.
Microarray analysis
Bisulfite conversion of sperm DNA was performed using the EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA). The samples of 38 patients and five controls were hybridized to four Infinium HumanMethylation27 BeadChips (Illumina, San Diego, CA, USA) with 12 arrays each and scanned with an Illumina Bead Array Reader. This assay allows quantitative measurements of DNA methylation at 27 578 different CpG sites in 14 495 genes (mainly in promoter regions). The raw data were analysed using Illumina GenomeStudio software. The final report with the methylation β-values was exported into the statistical framework r version 2.15.1 (http://www.r-project.org) for further analyses.
The gene ontology (GO) annotation from Entrez GeneIDs was assigned to all target genes, resulting in 13 865 different Entrez GeneIDs. The differential methylation information of the analysed CpG sites was summarized into one p-value per gene using the minimal p–value to increase sensitivity. In a GO enrichment analysis implemented in the GOSim package (Froehlich & Beissbarth, 2012), the significant genes were analysed for an enrichment of biological processes compared to all genes present on the chip. All p-values calculated by the GOSim package were adjusted for multiple testing by the false discovery rate. In addition, we split the differentially methylated gene set into GO terms associated with increased vs. those with decreased CpG methylation values.
Bisulfite pyrosequencing
To validate the observed microarray methylation differences, bisulfite pyrosequencing of five differentially methylated genes was performed with 29 sperm DNA samples, using a PyroMark Q96 MD pyrosequencing system and the PyroMark Gold Q96 CDT Reagent kit (Qiagen). Gene-specific polymerase chain reaction (PCR) and sequencing primers (Table S2) were designed using the PyroMark Assay Design 2.0 software (Qiagen). The PCR reaction mixture consisted of 2.5 μL 10× buffer, 20 mm MgCl2, 0.5 μL 10 mm dNTP mix, 1 μL (10 pmol) of each primer, 0.2 μL (1 U) FastStart Taq DNA polymerase (Roche Diagnostics, Mannheim, Germany), 18.8 μL PCR-grade water and 1 μL (∼100 ng) of bisulfite-converted DNA. Amplifications were carried out with an initial denaturation step at 95 °C for 5 min, 35 cycles of 95 °C for 30 sec, primer-specific annealing temperature for 30 sec and 72 °C for 45 sec, and a final extension step at 72 °C for 5 min. The Pyro Q-CpG software (Qiagen) was used for data analysis. The averaged β-values of the pyrosequencing measurements were analysed for methylation differences between the two groups defined by microarray analysis.
Statistical and bioinformatic analysis
All statistical analyses were performed with the statistical framework r version 2.15.1. The Lumi package (Du et al., 2008) was used to read and control the quality of the methylation data. The entire methylation array data set (including 38 patients and five control samples) was normalized and no outliers were observed (Figs S1 & S2). The quality controls also showed a good bisulfite conversion (Fig. S3) and overall performance of the array (Fig. S4). Subsequent analyses were based on the M-values representing the methylated probe intensities divided by the corresponding unmethylated probe intensities (Du et al., 2010). After adjusting the colour bias of the data, a smooth quantile normalization was performed. Hierarchical Ward clustering based on Euclidean distances of the sitewise M-values was performed (Ward, 1963). The robustness of the clustering was validated via bootstrap analysis (with 10 000 bootstraps) as implemented in the pvclust package (Suzuki & Shimodaira, 2011). The cluster dendrogram was visualized by iTOL (Letunic & Bork, 2011). As an alternative clustering method, correspondence analysis (Legendre & Legendre, 1998) was applied as implemented in the Vegan package (Oksanen et al., 2012). Owing to the absence of normally distributed data we measured the degree of association by the Spearman correlation and applied the associated statistical test (Best & Roberts, 1975).
Differentially methylated sites between two groups were tested via linear modelling including an empirical Bayes smoothing of the standard errors models using the Limma package (Smyth, 2004). All derived p-values throughout the whole manuscript were adjusted for multiple testing by the Benjamini–Hochberg procedure (Benjamini & Hochberg, 1995). To estimate the signal–to-noise ratio a β-uniform mixture model (Pounds & Morris, 2003) was fitted to all raw p-values using the BioNet package (Beisser et al., 2009). As pyrosequencing intensities are typically measured as β-values, all pyrosequencing data were analysed using β-values. All pyrosequencing results were tested in a two-group comparison using a Wilcoxon rank sum test.
Results
Unsupervised clustering of methylation array data
At present male factor infertility of couples undergoing ART is mainly defined by abnormal semen parameters (sperm concentration, motility and morphology). Our main goal was to find out whether males attending a fertility centre can also be classified according to epigenetic parameters. To this end, 38 consecutive sperm DNA samples of males attending a fertility centre with a wide range of semen parameters (Table S1) were analysed with Infinium HumanMethylation27K arrays. All samples passed the quality controls which are implemented in the Lumi package (Figs S1 & S2) and the Illumina GenomeStudio Methylation Module v1.0 (Figs S3 & S4). The age of the patients ranged from 27 to 54 years, sperm count from 7 to 120 × 106/mL, sperm motility from 1 to 65% and sperm morphology from 5 to 30% normally shaped spermatozoa. Sperm count, morphology and motility significantly correlated with each other (Spearman correlation test; p < 0.001); however, there was no correlation with age.
First, an unsupervised clustering was performed to identify major effects dividing the data set. Hierarchical clustering (Fig. 1A) and correspondence analysis (Fig. 1B) clearly split the data set into two groups, A with 29 samples and B with 9 samples respectively. These two groups accounted for 13.8% of the entire variation in the data. Clustering of the data supported this grouping with 83% based on 10 000 bootstrap replicates. This grouping remained stable for different cluster algorithms (single, average and complete clustering).
Figure 1.
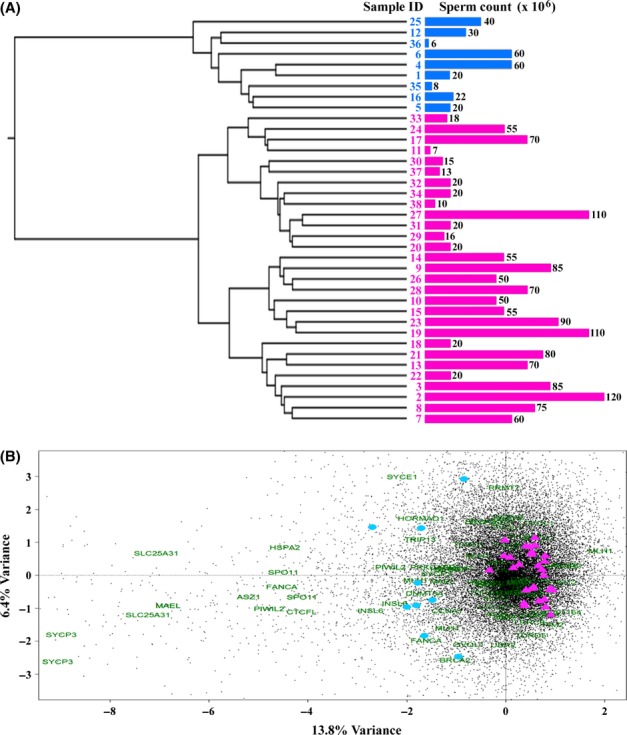
Unsupervised clustering of the 38 patient samples. (A) The dendrogram shows a hierarchical Ward clustering of the 38 analysed sperm DNAs based on Euclidian distances. The top split of the tree clearly divides the two clades into two groups, ‘A’ highlighted in pink and ‘B’ in blue. This grouping is supported by a bootstrap value of 83%. The lengths of the blue and pink bars, respectively, indicate the sperm count of the analysed samples. (B) The correspondence analysis based on the M-values of all CpG sites on the array also shows a clear split of the analysed samples into group A (pink triangles) and B (blue circles), which are consistent with the results of Ward clustering. This grouping accounts for 13.8% of the information in the underlying data set. All sites of the data set are displayed as black dots, while sites of genes in the spermatogenesis-related gene ontology terms (DNA methylation involved in gamete generation, meiotic prophase I and spermatogenesis) are displayed by green gene names.
In addition to the 38 sperm samples from a fertility centre, the four hybridized arrays contained five samples from four healthy normozoospermic volunteers, which can be considered as a control group. Correspondence analysis of the 1000 CpGs sites showing the highest methylation variation in the entire data set with the 38 study samples and five control samples revealed that the control samples clustered with the major group A (Fig. 2). As expected, the two biological replicates (two sperm samples of the same male, separated by 1 month) clustered very closely together.
Figure 2.
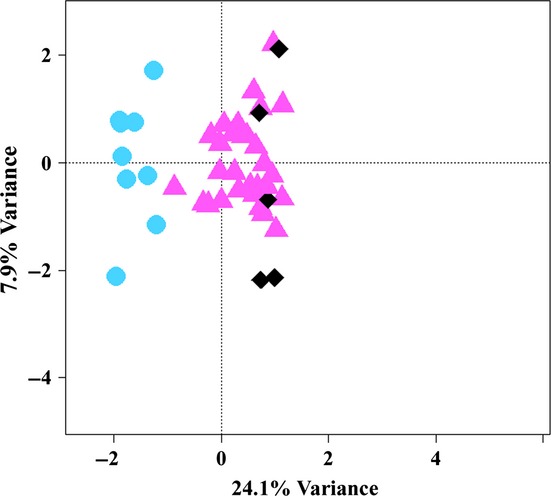
Correspondence analysis of the 1000 CpG sites showing the highest variation in the entire data set with the 38 patients and five control probes. Similar to the CA without controls, it also shows a clear split of the analysed samples between patient group B (blue circles) and patient group A (pink triangles). The controls (black diamonds) cluster with group A. This grouping accounts for 24.1% of the information in the underlying data set. The two neighbouring diamonds in the right bottom quarter are biological replicates.
Correlation analysis
To address possible causes underlying this clustering, semen parameters were correlated with the two identified clusters. Sperm count, morphology and motility, age of the donor and technical factors such as the bisulfite conversion rate were considered cofactors which might influence sperm DNA methylation. Significant negative correlations were found for sperm count (rho = −0.420; p = 0.009) and for the bisulfite conversion efficiency (rho = −0.343; p = 0.035). The box plot diagrams in Fig. 3 show that the sperm counts in group B are in the low normal range (median 22; interquartile range (IQR) 20–40 × 106/mL), whereas the counts in group A are distributed in a wider range (median 55; IQR 20–75 × 106/mL). Sperm motility, morphology and age of the donor were not associated with the observed clustering.
Figure 3.
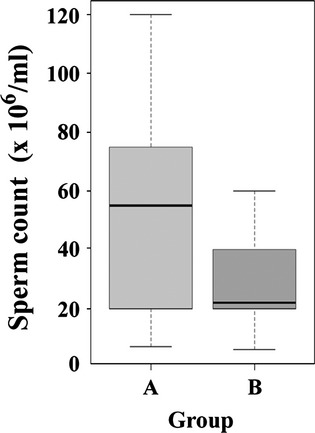
Sperm counts in groups A and B. The box plots show the distribution of sperm counts in 38 patients assigned to groups A (29 samples) vs. B (9 samples). The bottom and the top of the boxes represent the 25th and 75th percentiles respectively. The median is represented by vertical lines. Bars extend from the boxes to at most 1.5 times the height of the box.
Three of nine (33%) ART attempts using the analysed sperm samples in group B and 13 of 29 (45%) in group A resulted in a pregnancy and live birth of a baby (Table S1). No abortions or perinatal complications were reported in group A or B. Although the pregnancy rate in group A was lower, this difference was not statistically significant. According to semen and clinical parameters, male and/or female factor infertility was assumed. There was no significant between-group difference in male vs. female factors.
Differentially methylated genes
Most (72–74%) of the 27 578 tested CpG sites were hypomethylated (β-value <20%) in all analysed sperm samples, 15–18% were hypermethylated (β-value >80% methylation) and 8–13% displayed intermediate (20–80%) methylation levels. Statistical tests based on a moderated t-test (Smyth, 2004) between groups A and B resulted in 2929 significant differentially methylated CpG sites. Sixty-four to 71% of the significant sites represent hypomethylated CpGs, 16–22% hypermethylated CpGs and 7–18% displayed intermediate methylation levels. The distribution of the mean totals is significantly different (chi-squared test; p = 2.15e-05) from that of all CpG sites on the array. Overall, 2096 significant CpG sites (representing 1656 genes) showed higher and 833 CpGs (representing 688 genes) lower methylation levels in group B. Usually, the observed between-group differences were in the order of several percentage points; few CpG sites displayed β-value differences around 20%. There were no CpGs with <20% methylation in one group and >80% in the other. Table 1 presents the top 10 genes with higher and the top 10 with lower methylation levels in group B, compared to group A.
Table 1.
Gene-specific CpG sites with the most significant methylation gain or loss in group B, compared to A
| β-values in group A (%) | β-values in group B (%) | Between-group difference (%) | p-valuea | |
|---|---|---|---|---|
| Higher methylation in group B | ||||
| HNRNPG-T | 5.4 | 26.7 | 21.3 | 1.66e-16 |
| NMUR1 | 6.7 | 18.9 | 12.2 | 2.48e-15 |
| TCL1A | 3.7 | 12.3 | 8.6 | 5.90e-15 |
| DNAI1 | 6.8 | 18.7 | 11.9 | 1.97e-14 |
| ELMO3 | 2.9 | 16.6 | 13.7 | 2.40e-14 |
| HUS1B | 4.1 | 22.5 | 18.4 | 3.64e-14 |
| ALPK3 | 5.1 | 15.0 | 9.9 | 3.64e-14 |
| TPTE | 4.2 | 25.1 | 20.9 | 3.64e-14 |
| FLJ36046 | 5.1 | 18.7 | 13.6 | 3.64e-14 |
| LRFN4 | 5.8 | 19.7 | 13.9 | 7.86e-14 |
| Lower methylation in group B | ||||
| EDG6 | 96.7 | 93.1 | 3.6 | 5.62e-07 |
| TNF | 95.8 | 93.7 | 2.1 | 4.87e-07 |
| TNFRSF4 | 94.2 | 89.5 | 4.7 | 4.85e-07 |
| F2RL3 | 92.3 | 88.7 | 2.5 | 4.38e-07 |
| GAS2L2 | 92.9 | 88.6 | 4.3 | 4.34e-08 |
| S100A4 | 91.0 | 84.3 | 6.7 | 1.14e-08 |
| SLC25A22 | 96.5 | 92.2 | 4.3 | 9.62e-11 |
| TNFRSF18 | 96.2 | 91.8 | 4.4 | 5.95e-11 |
| OSM | 97.3 | 93.5 | 3.8 | 2.22e-12 |
| CSEN | 95.1 | 86.7 | 8.4 | 4.38e-13 |
Adjusted for multiple testing.
To identify the biological processes which were differentially regulated in groups A and B, GO enrichment analysis was performed. In a quantitative GO enrichment each gene was weighted by its lowest p-value with lower p-values having higher effects on the enrichment. Quantitative enrichment revealed a strong association with GO terms for gametogenesis, inflammation and immune response-related processes (Table S3). For a more specific analysis of the 1656 identified genes with increased methylation values and the 688 genes with decreased methylation qualitative GO enrichment was used. GO terms related to spermatogenesis were found to be enriched in the gene set with a gain of methylation: three of the five top terms were specific for (male) germ cell development (Table 2). In contrast, the gene set with lower methylation was enriched for processes involved in inflammation and immune response (6 of the top 10 terms) as well as epigenetic modification (two terms).
Table 2.
Qualitative GO enrichment of differentially methylated genes
| p-valuea | |
|---|---|
| GO terms of genes with higher methylation in group B | |
| DNA methylation involved in gamete generation | 0.0002 |
| Meiotic prophase I | 0.0005 |
| Synaptic transmission | 0.0006 |
| Peptidyl-citrulline biosynthetic process from peptidyl-arginine | 0.0007 |
| Spermatogenesis | 0.0013 |
| GO terms of genes with lower methylation in group B | |
| Response to pain | 0.0005 |
| Negative regulation of sodium-dependent phosphate transport | 0.0017 |
| Negative regulation of histone H4 acetylation | 0.0033 |
| Positive regulation of interleukin-18 production | 0.0033 |
| Negative regulation of type IV hypersensitivity | 0.0033 |
| Transforming growth factor beta receptor complex assembly | 0.0033 |
| Regulation of tumor necrosis factor production | 0.0034 |
| Positive regulation of inflammatory response | 0.0041 |
| Positive regulation of interferon-alpha biosynthetic process | 0.0050 |
| Hypermethylation of CpG island | 0.0050 |
Adjusted for multiple testing.
Validation of differentially methylated genes
To validate the methylation array results, five significant genes with increased methylation levels in group B were analysed by bisulfite pyrosequencing in 29 of the 38 sperm samples on the array; of the remaining patients no more DNA was available. INSL6, MAEL, PIWIL2, SLC25A31 and SPO11 were selected because they each displayed at least two highly significant CpG sites on the array and GO terms related to spermatogenesis. MAEL (Soper et al., 2008) and PIWIL2 (Bak et al., 2011) inhibit retrotransposition activity of repetitive elements during male germ cell development. INSL6 is required for the progression of spermatogenesis at late meiotic prophase (Burnicka-Turek et al., 2009). The nuclear-encoded mitochondrial protein SLC25A31 is important for energy-consuming reactions in the sperm flagellum (Kim et al., 2007). SPO11 initiates meiotic recombination through the formation of DNA double-strand breaks (Keeney, 2001). Genetic polymorphisms in this gene may predispose to idiopathic male infertility (Zhang et al., 2011). The average methylation level of all analysed CpG sites in the pyrosequencing assay (Table S2) was used as a quantitative measure for methylation of a given gene in a given sample. Consistent with the results of array analysis, all five tested genes displayed higher methylation levels in group B (Fig. 4). Three genes, INSL6, MAEL and SLC25A31 showed borderline significance (Table 3).
Figure 4.
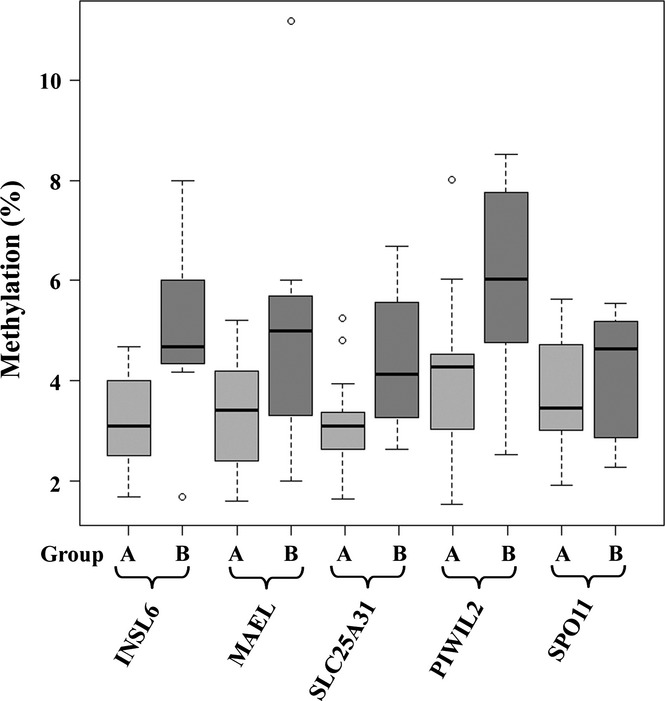
Pyrosequencing results of candidate genes. The box plots show the distribution of INSL6, MAEL, SLC25A3, PIWIL2 and SPO11 methylation values in groups A and B. The bottom and the top of the boxes represent the 25th and 75th percentiles respectively. The median is represented by vertical lines. Bars extend from the boxes to at most 1.5 times the height of the box. Circles indicate outliers.
Table 3.
Pyrosequencing results of candidate genes
| Gene | Microarray analysis | Pyrosequencing analysis | |||
|---|---|---|---|---|---|
| β-values (% difference) | Minimal p-value | β-values (% difference) | p-value | Adjusted p-value | |
| INSL6 | 1.52 | 7.50e-04 | 1.86 | 0.013 | 0.057 |
| MAEL | 3.30 | 1.62e-09 | 1.81 | 0.082 | 0.103 |
| SLC25A31 | 9.64 | 2.28e-11 | 1.35 | 0.033 | 0.057 |
| PIWIL2 | 1.81 | 5.20e-06 | 2.05 | 0.034 | 0.057 |
| SPO11 | 4.30 | 3.60e-08 | 0.34 | 0.575 | 0.575 |
Discussion
In an exploratory methylation array analysis of ART sperm samples, we identified two groups of patients by cluster analysis and correspondence analysis. The smaller group B of patients with sperm counts in the low normal range (median 22; IQR 20–40 × 106/mL) displayed slightly (in the order of several percentage points) higher methylation values in genes related to spermatogenesis and slightly lower methylation values in genes related to inflammation and immune response. Our results revealed significant methylation differences in >10% of analysed CpG sites between the smaller group B and the main group A, although often with small effect size. At the individual level, it may be difficult to estimate the impact of a 1–2% point methylation difference in a given gene on male fertility. However, we propose that similar to genome-wide association studies with genetic markers, even a minor methylation difference between groups can uncover genes and pathways, which may play a major role in sperm quality and developmental potential. In a conceptionally related study (Pacheco et al., 2011) using the same methylation array an effect on a lower dendrogram split was associated with sperm motility. Together these two studies suggest a connection between classical semen parameters and sperm methylation patterns.
Of the 38 patients studied here, 22 were diagnosed with male factor infertility based on semen parameters and 16 were normozoospermic. Because microarray analysis did not detect systematic methylation differences between patient samples with normal and abnormal semen parameters, this does not explain the separation between groups A and B. The observation that sperm samples of presumably fertile volunteers with repeatedly normal semen parameters clustered with the major group A argues in favour of the notion that this group closely resembles the reference sperm methylome (Krausz et al., 2012). Taken together, our results suggest that group B represents a specific subgroup of males with fertility problems, maybe caused by a common aetiology(ies).
In contrast to previous candidate gene studies (Marques et al., 2008; Kobayashi et al., 2009; Hammoud et al., 2010; Poplinski et al., 2010; El Hajj et al., 2011), which found increased rates of imprinting defects in spermatozoa of oligospermic males, and two recent methylation array studies (Aston et al., 2011; Pacheco et al., 2011), the differentially methylated CpGs sites between groups A and B were not enriched in imprinted genes. One possible explanation of the increased methylation of spermatogenesis-related genes in group B may be sperm DNA damage in infertile/subfertile males. Experimental evidence suggests that external factors (i.e. cigarette smoking, pollutants and medical drugs) as well as internal factors (i.e. paternal age and metabolic disorders) can have an effect on sperm DNA integrity (Pacey, 2010). In particular oxidative stress in the male germ line and the resulting DNA damage have been linked to global DNA methylation changes (Tunc & Tremellen, 2009) and male infertility (Gharagozloo & Aitken, 2011). However, so far the clinical relevance of sperm DNA damage testing and therapy (i.e. by antioxidants) on pregnancy rates through natural conception or ART remains unclear (Zini, 2011; Beshay & Bukulmez, 2012). In addition, our study demonstrates a significant reduction in methylation in inflammation and immune response-related genes. This is consistent with expression array studies demonstrating increased transcript levels corresponding to inflammatory activity in testicular biopsies from infertile males (Spiess et al., 2007). It is tempting to speculate that the epigenetic signatures in sperm samples of group B reflect inflammation and/or autoimmune processes interfering with fertility.
With the notable exception of low sperm count, the two groups separated by microarray analysis could not be correlated with clinically relevant parameters, such as male vs. female infertility or ART outcome. Nevertheless, our results suggest that the epigenetic regulation of spermatogenesis-related and other critical pathways is altered in sperm samples of a subgroup of males attending a fertility centre. Three of five differentially methylated genes could be validated by bisulfite pyrosequencing. The borderline significance can be explained by the low sample size and the p-value adjustment. Our results suggest the possibility to develop useful clinical biomarkers for group definition in a diagnostic laboratory. Future studies with higher resolution (>450 000 CpG sites) methylation arrays and larger patient cohorts may allow an improved classification of sperm samples and provide better insights into the association of sperm DNA methylation patterns and ART outcome. Despite numerous efforts, so far genetic defects explain only a minor part of male fertility problems (Gianotten et al., 2004). The role of epimutations is likely to be largely underestimated. Epigenetics provides the most likely molecular mechanism for gene–environment interactions in the male germ line that may adversely affect semen quality and embryonic development. Because in principle, epigenetic changes are reversible, a better understanding of epigenetic dysregulation in the male germ line may open new strategies to restore fertility in male subfertility/infertility cases by modulating DNA methylation, that is, by nutrition or drugs.
Acknowledgments
This study was supported by research grants HA 1374/13-1 (to T.H.) and GR 1547/14-1 (to JG) from the German Research Foundation.
Author contributions
N.E.H., J.G. and T.Hf. designed the study, J.G., T.Hn. and M.S. contributed materials, N.E.H., J.K. and I.N. performed the experiments, B.S., M.D. and T.M. analysed the data and B.S. and T.Hf. wrote the manuscript.
Competing interests
The authors have declared that no competing interests exist.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Distribution of red and green colour intensities (microarray quality control).
Distribution of intensities (microarray quality control).
Efficiency of bisulfite conversion (microarray quality control).
Non-polymorphic control test for assay performance (microarray quality control).
Semen and clinical parameters of the analysed sperm samples.
Primers and conditions for bisulfite pyrosequencing.
Qualitative GO enrichment of differentially methylated genes.
References
- Aston KI, Punj V, Liu L, Carrell DT. Genome-wide sperm deoxyribonucleic acid methylation is altered in some men with abnormal chromatin packaging or poor in vitro fertilization embryogenesis. Fertil Steril. 2011;97:285–292. doi: 10.1016/j.fertnstert.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Bak CW, Yoon TK, Choi Y. Functions of PIWI proteins in spermatogenesis. Clin Exp Reprod Med. 2011;38:61–67. doi: 10.5653/cerm.2011.38.2.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisser D, Klau GW, Dandekar T, Müller T, Dittrich M. BioNet an R-package for the functional analysis of biological networks. Bioinformatics. 2009;26:1129–1130. doi: 10.1093/bioinformatics/btq089. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 1995;57:289–300. [Google Scholar]
- Beshay VE, Bukulmez O. Sperm DNA damage: how relevant is it clinically? Curr Opin Obstet Gynecol. 2012;24:172–179. doi: 10.1097/GCO.0b013e32835211b5. [DOI] [PubMed] [Google Scholar]
- Best DJ, Roberts DE. Algorithm AS 89: the upper tail probabilities of Spearman's rho. J R Stat Soc Ser C (Appl Stat) 1975;24:377–379. [Google Scholar]
- Boyano MD, Andollo N, Zalduendo MM, Aréchaga J. Imprinting of mammalian male gametes is gene specific and does not occur at a single stage of differentiation. Int J Dev Biol. 2008;52:1105–1111. doi: 10.1387/ijdb.072284mb. [DOI] [PubMed] [Google Scholar]
- Burnicka-Turek O, Shirneshan K, Paprotta I, Grzmil P, Meinhardt A, Engel W, et al. Inactivation of insulin-like factor 6 disrupts the progression of spermatogenesis at late meiotic prophase. Endocrinology. 2009;15:4348–4357. doi: 10.1210/en.2009-0201. [DOI] [PubMed] [Google Scholar]
- Carrell DT. Epigenetics of the male gamete. Fertil Steril. 2012;97:267–274. doi: 10.1016/j.fertnstert.2011.12.036. [DOI] [PubMed] [Google Scholar]
- Carrell DT, Hammoud SS. The human sperm epigenome and its potential role in embryonic development. Mol Hum Reprod. 2010;16:37–47. doi: 10.1093/molehr/gap090. [DOI] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM. Lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj N, Zechner U, Schneider E, Tresch A, Gromoll J, Hahn T, et al. Methylation status of imprinted genes and repetitive elements in sperm DNA from infertile males. Sex Dev. 2011;5:60–69. doi: 10.1159/000323806. [DOI] [PubMed] [Google Scholar]
- Flanagan JM, Popendikyte V, Pozdniakovaite N, Sobolev M, Assadzadeh A, Schumacher A, et al. Intra- and interindividual epigenetic variation in human germ cells. Am J Hum Genet. 2006;79:67–84. doi: 10.1086/504729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froehlich H, Beissbarth T. 2012. GOSim: computation of functional similarities between GO terms and gene products; GO enrichment analysis. R package version 1.2.7.2. Available at: http://CRAN.R-project.org/package=GOSim.
- Gharagozloo P, Aitken RJ. The role of sperm oxidative stress in male infertility and the significance of oral antioxidant therapy. Hum Reprod. 2011;26:1628–1640. doi: 10.1093/humrep/der132. [DOI] [PubMed] [Google Scholar]
- Gianotten J, Lombardi MP, Zwinderman AH, Lilford RJ, van der Veen F. Idiopathic impaired spermatogenesis: genetic epidemiology is unlikely to provide a short-cut to better understanding. Hum Reprod Update. 2004;10:533–539. doi: 10.1093/humupd/dmh045. [DOI] [PubMed] [Google Scholar]
- Haaf T. Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol. 2006;310:13–22. doi: 10.1007/3-540-31181-5_2. [DOI] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–478. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Purwar J, Pflueger C, Cairns BR, Carrell DT. Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil Steril. 2010;94:1728–1733. doi: 10.1016/j.fertnstert.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Houshdaran S, Cortessis VK, Siegmund K, Yang A, Laird PW, Sokol RZ. Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. PLoS ONE. 2007;2:e1289. doi: 10.1371/journal.pone.0001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- Kim YH, Haidl G, Schaefer M, Egner U, Mandal A, Herr JC. Compartmentalization of a unique ADP/ATP carrier protein SFEC (Sperm Flagellar Energy Carrier, AAC4) with glycolytic enzymes in the fibrous sheath of the human sperm flagellar principal piece. Dev Biol. 2007;302:463–476. doi: 10.1016/j.ydbio.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kläver R, Tüttelmann F, Bleiziffer A, Haaf T, Kliesch S, Gromoll J. DNA methylation in spermatozoa as a prospective marker in andrology. Andrology. 2013;1:731–740. doi: 10.1111/j.2047-2927.2013.00118.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, et al. DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet. 2009;17:1582–1591. doi: 10.1038/ejhg.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz C, Sandoval J, Sayols S, Chianese C, Giachini C, Heyn H, et al. Novel insights into DNA methylation features in spermatozoa: stability and peculiarities. PLoS ONE. 2012;7:e44479. doi: 10.1371/journal.pone.0044479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawetz SA. Paternal contribution: new insights and future challenges. Nat Rev Gen. 2005;6:633–642. doi: 10.1038/nrg1654. [DOI] [PubMed] [Google Scholar]
- Legendre P, Legendre L. Numerical Ecology. New York: Elsevier, Amsterdam; 1998. [Google Scholar]
- Letunic I, Bork P. Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques CJ, Costa P, Vaz B, Carvalho F, Fernandes S, Barros A, et al. Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol Hum Reprod. 2008;14:67–74. doi: 10.1093/molehr/gam093. [DOI] [PubMed] [Google Scholar]
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell. 2011;146:1029–1041. doi: 10.1016/j.cell.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Costa P, Nogueira P, Carvalho M, Leal F, Cordeiro I, Calhaz-Jorge C, et al. Incorrect DNA methylation of the DAZL promoter CpG island associates with defective human sperm. Hum Reprod. 2010;10:2647–2654. doi: 10.1093/humrep/deq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol. 2007;307:368–379. doi: 10.1016/j.ydbio.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara GL, et al. 2012. Vegan: community ecology package. R package version 2.0-4. Available at: http://CRAN.R-project.org/package=vegan.
- Pacey AA. Environmental and lifestyle factors associated with sperm DNA damage. Hum Fertil (Camb) 2010;13:189–193. doi: 10.3109/14647273.2010.531883. [DOI] [PubMed] [Google Scholar]
- Pacheco SE, Houseman EA, Christensen BC, Marsit CJ, Kelsey KT, Sigman A, et al. Integrative DNA methylation and gene expression analyses identify DNA packaging and epigenetic regulatory genes associated with low motility sperm. PLoS ONE. 2011;6:e20280. doi: 10.1371/journal.pone.0020280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplinski A, Tüttelmann F, Kanber D, Horsthemke B, Gromoll J. Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int J Androl. 2010;33:642–649. doi: 10.1111/j.1365-2605.2009.01000.x. [DOI] [PubMed] [Google Scholar]
- Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19:1236–1242. doi: 10.1093/bioinformatics/btg148. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Rousseaux S, Caron C, Govin J, Lestrat C, Faure AK, Khochbin S. Establishment of male-specific epigenetic information. Gene. 2005;345:139–153. doi: 10.1016/j.gene.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Soper SF, van der Heijden GW, Hardiman TC, Goodheart M, Martin SL, de Boer P, et al. Mouse maelstrom, a component of nuage, is essential for spermatogenesis and transposon repression in meiosis. Dev Cell. 2008;15:2852–2897. doi: 10.1016/j.devcel.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess AN, Feig C, Schulze W, Chalmel F, Cappallo-Obermann H, Primig M, et al. Cross-platform gene expression signature of human spermatogenic failure reveals inflammatory-like response. Hum Reprod. 2007;22:2936–2946. doi: 10.1093/humrep/dem292. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H. 2011. Pvclust: hierarchical clustering with p-values via multiscale bootstrap resampling. R package version 1.2-2. Available at: http://CRAN.R-project.org/package=pvclust.
- Tunc O, Tremellen K. Oxidative DNA damage impairs global sperm DNA methylation in infertile men. J Assist Reprod Genet. 2009;26:537–544. doi: 10.1007/s10815-009-9346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JH. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963;48:236–244. [Google Scholar]
- World Health Organization. WHO Laboratory Manual for the Examination and Processing of Human Semen. Geneva: World Health Organization; 2010. [Google Scholar]
- Zhang J, Zhou DX, Wang HX, Tian Z. An association study of SPO11 gene single nucleotide polymorphisms with idiopathic male infertility in Chinese Han population. J Assist Reprod Genet. 2011;28:731–736. doi: 10.1007/s10815-011-9571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zini A. Are sperm chromatin and DNA defects relevant in the clinic? Syst Biol Reprod Med. 2011;57:78–85. doi: 10.3109/19396368.2010.515704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of red and green colour intensities (microarray quality control).
Distribution of intensities (microarray quality control).
Efficiency of bisulfite conversion (microarray quality control).
Non-polymorphic control test for assay performance (microarray quality control).
Semen and clinical parameters of the analysed sperm samples.
Primers and conditions for bisulfite pyrosequencing.
Qualitative GO enrichment of differentially methylated genes.