

Abstract
Objective
To compare the gene expression patterns of synovial cells from inflamed or normal/reactive areas of synovial membrane obtained from the same patient with osteoarthritis (OA).
Methods
At the time of total knee replacement, synovial tissues were obtained from 12 patients with knee OA. The inflammation status of the synovial membrane was characterized according to macroscopic criteria and classified as normal/reactive or inflamed. Biopsy samples were cultured separately for 7 days. Microarray gene expression profiling was performed on normal/reactive and inflamed areas. Western blot and immunohistochemistry were used to confirm the identified genes that were differentially expressed.
Results
We identified 896 genes that were differentially expressed between normal/reactive and inflamed areas. The key pathways were related to inflammation, cartilage metabolism, Wnt signaling, and angiogenesis. In the inflammation network, the genes TREM1 and S100A9 were strongly up-regulated. The genes MMP3, MMP9, CTSH (cathepsin H), and CTSS (cathepsin S) were significantly up-regulated in the cartilage catabolism pathway, while the most up-regulated anabolism enzyme gene was HAS1. In the Wnt signaling pathway, the genes for Wnt-5a and low-density lipoprotein receptor–related protein 5 were up-regulated, while the gene FZD2 and the gene for Dkk-3 were down-regulated. Finally, STC1, which codes for a protein involved in angiogenesis, was identified as the most up-regulated gene in inflamed compared with normal/reactive areas.
Conclusion
This study is the first to identify different expression patterns between 2 areas of the synovial membrane from the same patient. These differences concern several key pathways involved in OA pathogenesis. This analysis also provides information regarding new genes and proteins as potential targets of treatment.
Osteoarthritis (OA) is the most common joint disease and is a major cause of joint pain and disability in the aging population (1). OA is a degenerative disease associated with an abnormal remodeling of joint tissues principally driven by a host of inflammatory mediators within the affected joint (2). Its etiology is multifactorial (i.e., age, obesity, joint injury, genetic predisposition), and the pathophysiologic process affects the entire joint (articular cartilage, subchondral bone, synovial membrane, menisci and ligaments, muscles and nerves). Destruction of articular cartilage, sclerosis of subchondral bone, and formation of osteophytes, synovial inflammation, and ligament and meniscal damage constitute the main features characterizing OA.
Over the last decade, attention has turned to the importance of synovial inflammation in OA. Synovial inflammation has been proven to contribute to patient symptoms (joint swelling and pain from inflammation), signs (effusion), and disease progression (3). It is involved early in the disease process (4) and is thought to be secondary to osteocartilaginous fragments and inflammatory/catabolic factors entering the synovial cavity (5). Synovial histologic changes in OA include synovial hypertrophy and hyperplasia, with an increase in the number of synovial lining cells often accompanied by infiltration of lymphocytes and macrophages either diffusely or in perivascular aggregates (3,6). The inflammation is mostly confined to areas adjacent to pathologically damaged cartilage and bone (7).
Synovial inflammation is promoted by a large number of soluble mediators of inflammation. Among them, interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα) are the 2 major cytokines involved in the pathogenesis of OA (6). In addition to inducing the synthesis of matrix metalloproteinases (MMPs) and other proteinases, IL-1β and TNFα can not only induce their own production but also stimulate chondrocytes, synovial cells, and T lymphocytes to produce other cytokines (i.e., IL-6, IL-8, IL-15, IL-17, leukemia inhibitory factor) and prostaglandin E2 (2,8). In patients at an early stage of OA, the expression of synovial chemokines was found to be associated with the presence of synovial inflammation (9), and the expression of CCL19 and its receptor CCR7 was associated with most severe symptoms.
Finally, recent studies have implicated alarmins in synovial inflammation. The alarmins S100A8 and S100A9 are crucially involved in synovial activation and cartilage destruction during OA, and their elevated levels may predict joint destruction (10).
Synovial neovascularization is another important feature of the inflamed synovial membrane, resulting from an imbalance between pro- and antiangiogenic factors (11). The neovascularization may be largely driven by synovitis (12). Therefore, angiogenesis and inflammation are 2 key closely integrated and interrelated processes that may affect disease progression and pain (13).
Our research group has developed an original methodology to investigate the cross-talk between angiogenesis and inflammation, comparing different areas of the synovial membrane with various levels of severity of synovitis (14). This method consists of selecting samples from normal/reactive or inflamed areas from the same OA patient using the Ayral synovitis score, in order to culture cells from these areas separately (15). This model led us to observe and compare concomitant pathophysiologic pathways. Our first results showed that synovial cells from an inflamed area produced more proinflammatory cytokines (IL-6 and IL-8), more vascular endothelial growth factor (VEGF), and less thrombospondin 1 (TSP-1) than synovial cells from a normal/reactive area, suggesting an imbalance between pro- and antiangiogenic factors in favor of proangiogenic factors (14). In addition, VEGF production showed a strong positive correlation with IL-6 and IL-8 secretion, while a significant negative correlation was obtained between TSP-1 and IL-8. This last observation indicates the link that exists between inflammation and angiogenesis in the synovial membrane (14).
Transcriptomic analysis is of growing importance in understanding how altered gene expression contributes to complex disease, providing greater insights into biologic pathways and molecular mechanisms that regulate disease development and progression. In the present study, we compared gene expression profiles of synovial cells isolated from inflamed or normal/reactive areas of synovial membrane from OA patients. We identified key pathways involved in OA pathogenesis and, subsequently, new potential targets for OA treatment.
PATIENTS AND METHODS
Patients
Synovial tissue samples from 12 patients with knee OA (10 women and 2 men; mean age 70 years, range 50–83 years) were obtained at the time of total knee joint replacement surgery. All subjects provided informed consent, and ethical approval (ethics committee agreement of Catholic University of Louvain, no. B403201111664) was granted for this study. The inflammation status of the synovial membrane was characterized by the surgeon according to macroscopic criteria established by Ayral (15). These criteria included synovial vascularization, villi formation, and hypertrophic aspect of the tissue and allowed the classification of the synovial membrane into 3 grades: normal, reactive, and inflamed. For the purposes of this study, synovial samples were grouped as normal/reactive or inflamed.
Isolation and culture of synovial cells
Normal/reactive and inflamed synovial biopsy samples were separately digested as previously described (14) with type IA collagenase from Clostridium histolyticum (1 mg/ml; Sigma-Aldrich) in complete medium, consisting of Dulbecco's modified Eagle's medium (Lonza) supplemented with 10 mM HEPES, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mMl-glutamine, and 10% fetal calf serum (Lonza), for 4 hours at 37°C. The cell suspensions were passed through a 70-μm filter to remove any undigested tissue. The filtered cell suspensions were then collected by centrifugation at 800g. Synovial cells from either normal/reactive or inflamed biopsy samples were cultured separately in a T25 cell culture flask in 10 ml of complete medium, at 37°C in a 5% CO2 humidified atmosphere. After 2 days, medium was changed to remove nonadherent cells, and cells were cultured in fresh medium for 5 days. At the end of these 5 days of culture, culture media and cells were collected for further analysis.
RNA extraction
Total RNA was extracted using an RNeasy Mini Kit (Qiagen) and reverse-transcribed with SuperScript III reverse transcriptase according to the instructions of the manufacturer (Invitrogen). The yield of the extracted RNA was determined spectrophotometrically by measuring the optical density at 260 nm. The purity and quality of extracted RNA were evaluated using an Experion RNA StdSens Analysis Kit (Bio-Rad). High-quality RNA with RNA quality indicator scores of >8 was used for microarray experiments.
Microarray
Gene expression profiling was performed using an Illumina multisample format Human HT-12 v4 BeadChip, which contains more than 47,000 probes and which profiles 12 samples simultaneously on a single chip. For each sample, 250 ng of total RNA was labeled using an Illumina TotalPrep-96 RNA Amplification Kit (Ambion) according to the manufacturer's instructions. Briefly, double-stranded complementary DNA was synthesized using T7-oligo(dT) primers, followed by an in vitro transcription reaction to amplify antisense RNA while biotin was incorporated into the synthesized antisense RNA probe. The antisense RNA probe was then purified and quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific). The biotinylated complementary RNA probe was hybridized to the Human HT-12 v4 BeadChip. Labeled antisense RNA (750 ng) was used for hybridization to each array. The hybridization, washing, and scanning were performed according to the manufacturer's instructions. The arrays were scanned using a BeadArray Reader (Illumina). The microarray images were registered and extracted automatically during the scan according to the manufacturer's default settings. Raw microarray intensity data were analyzed with GenomeStudio software and normalized using the quantile normalization method according to the manufacturer's recommendation.
Differential analysis was performed with Biometric Research Branch (BRB) ArrayTools software (available at http://linus.nci.nih.gov/BRB-ArrayTools.html). The quantile-normalized data were loaded as a single file in BRB ArrayTools. They are filtered out based on the variance for the gene across the arrays, a feature included in BRB ArrayTools that allows the removal of unexpressed or equally expressed transcripts. The variance of the log ratios for each gene is compared to the median of all the variances. Those genes not significantly more variable than the median gene are filtered out. Specifically, the quantity (n – 1) Vari/Varmed is computed for each gene i. Vari is the variance of the log intensity for gene i across the entire set of n arrays, and Varmed is the median of these gene-specific variances. This quantity is compared to a percentile of the chi-square distribution with n – 1 degrees of freedom. This is an approximate test of the hypothesis that gene i has the same variance as the median variance. We used the threshold of P < 0.05 for the chi-square test. Of 47,231 probes used on the array, 17,511 passed the filter.
The Class Comparison Tool (BRB ArrayTools) computes the number of genes that are differentially expressed among the classes at the statistical significance level selected in the t-test menu and creates a gene list containing information about the significant genes. The Class Comparison between Groups of Arrays Tool computes a t-test separately for each gene using the normalized log intensities for 1-color oligonucleotide arrays. Since the normal/reactive and inflamed samples came from the same patient, we use the option “paired t-test,” available in BRB ArrayTools, to improve the statistical power of the analysis. The Class Comparison Tool also provides univariate permutation tests for the significance of individual genes. The univariate permutation tests are computed separately for each gene. The output gene list table is ordered by univariate P value with the most significant genes listed first. The false discovery rate (FDR), computed by the Benjamini and Hochberg method, associated with a row of the table is an estimate of the proportion of the genes that represent false-positives and that have univariate P values less than or equal to the P value in that row. We used a paired t-test P value threshold of less than 0.005 in our analysis; we computed an FDR of 0.0975 for the last statistically selected probe.
All data have been deposited in NCBI GEO (16) and are accessible through GEO Series accession no. GSE46750 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46750). The biologic relevance of up- and down-regulated genes was analyzed using IPA (Ingenuity Systems).
Western blotting
Cells were collected at 4°C and lysed for 30 minutes on ice in 100 μl of buffer (25 mM HEPES, 150 mM NaCl, 0.5% Triton X-100, 10% glycerol, and 1 mM dithiothreitol) containing phosphatase inhibitors (1 mM Na3VO4, 25 mM β-glycerophosphate, and 1 mM NaF). Total protein extracts (20 μg; independent protein extracts from 3 different patients) were fractionated by electrophoresis on a polyacrylamide gel (15% for S100A9; 10% for stanniocalcin 1 [STC1], triggering receptor expressed on myeloid cells 1 [TREM-1], α-tubulin, and Wnt-5a) and transferred onto an Immobilon-P membrane (PVDF; Millipore). Membranes were blocked in 1% blocking solution (Western blocking reagent; Roche) overnight at 4°C without shaking. Membranes were then incubated for 2 hours with primary antibodies (against S100A9, Wnt-5a, STC1, TREM-1, and α-tubulin) diluted in 0.5% blocking solution. Mouse monoclonal anti-S100A9 (sc-376772; 1:2,000 dilution), anti–Wnt-5a (sc-365370; 1:100 dilution), and anti–α-tubulin (sc-8035; 1:1,000 dilution) antibodies as well as goat polyclonal anti-STC1 (sc-14346; 1:250 dilution) and anti–TREM-1 (sc-19310; 1:100 dilution) antibodies were purchased from Santa Cruz Biotechnology. Horseradish peroxidase (HRP)–linked anti-goat or anti-mouse IgG antibodies (Dako) were used as secondary antibodies. The reaction was revealed with enhanced chemiluminescence detection reagent according to the manufacturer's instructions (ECL kit; Amersham Pharmacia Biotech). THP-1 cell lysate (sc-2238), HL-60 whole cell lysate (sc-2209), and 293T lysate (sc-124651) were used as positive controls for anti–TREM-1, anti-S100A9, and anti–Wnt-5a antibodies, respectively (data not shown).
Immunohistochemistry
Synovial biopsy samples from normal/reactive or inflamed areas were fixed, dehydrated in xylene, and embedded in paraffin using standard procedures. Five-micrometer sections were cut and deparaffinized according to standard protocol. These sections were stained with anti-STC1 (1:50 dilution), anti-S100A9 (1:100 dilution), and anti–hyaluronan synthase 1 (anti–HAS-1) (sc-23145; 1:50 dilution) antibodies. Antibodies were purchased from Santa Cruz Biotechnology. Secondary antibodies, HRP enzyme conjugate, and 3,3′-diaminobenzidine substrate were used to develop immunostaining in accordance with the instructions of the manufacturer (Dako). Slides were counterstained with hematoxylin and eosin. Negative control experiments were performed with isotype controls for IgG: normal goat IgG for STC1 and HAS-1 (AB-108-C; 1:250 dilution) (R&D Systems) and mouse IgG1 for S100A9 (X0931; 1:50 dilution); (Dako).
RESULTS
Global microarray results
From among 47,000 probes, 17,500 were filtered out. Differential analysis was done from these 17,500 filtered probes. The Class Comparison test between normal/reactive and inflamed areas was based on a paired t-test where normal/reactive and inflamed areas were paired for each patient (n = 12). Probes with a P value less than 0.005 were chosen as up- or down-regulated ones. The 896 genes identified as differentially expressed between normal/reactive and inflamed areas are listed in Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38315/abstract.
Main pathways identified
Based on the standard networks generated by IPA, the analysis was deepened in order to identify specific OA pathways. A significant number of genes that were differentially expressed between normal/reactive and inflamed OA synovial biopsy samples were categorized as belonging to inflammation, cartilage extracellular matrix metabolism, Wnt signaling, and angiogenesis pathways. Inflamed synovium:normal/reactive synovium ratios of >1.5 and ≤0.75 were considered relevant. The 4 key pathways were analyzed in detail.
Inflammation network
As presented in Table1, a large number of mediators of inflammation were demonstrated to be up-regulated in inflamed compared with normal/reactive OA synovial biopsy samples. These mediators belonged to different categories: inflammatory cytokines, chemokines, enzymes, and their related partners. In addition, this allowed the identification of 2 genes of interest in the inflammation process: TREM1 (P = 0.0003929, FDR 0.0783) and the alarmin S100A9 (P = 0.0014609, FDR 0.083).
Table 1.
Genes differentially expressed between normal/reactive and inflamed areas of synovial biopsy samples*
| Pathway, type of compound | Up-regulated | Down-regulated |
|---|---|---|
| Inflammation | ||
| Cytokines | IL8 (4.45), IL6 (3.43), TNFRSF21 (1.99), IFI30 (1.91), TNFAIP6 (1.62), IRF8 (1.61) | – |
| Chemokines | CXCL5 (4.38), CXCL6 (3.89), CXCL16 (2.8), CXCL2 (2.62), CXCL1 (2.52) | – |
| Enzymes | ALOX5AP (3.35), PLD1 (2.64), ALOX5 (2.32), PTGES (2.04), PLCB1 (1.87), SOD2 (1.79), TBXAS1 (1.75), PI3 (1.71), PLA2G4A (1.58) | – |
| Other | TREM1 (3.45), S100A9 (2.68), OSM (1.57), PPARG (1.49) | – |
| Anabolism | HAS1 (2.16), BMP6 (2.48), COLL22A1 (2.05) | COL1A2 (0.75), VIM (0.75), MATN2 (0.74), HABP4 (0.72), HAPLN1 (0.66), HAS3 (0.65), COL16A1 (0.64), CILP (0.63), COL6A3 (0.63), GPC4 (0.58), HAPLN1 (0.58), ACAN (0.42) |
| Catabolism | MMP9 (3.53), MMP3 (2.82), CTSH (2.02), ADAMDEC1 (1.76), CTSS (1.52) | – |
| Angiogenesis | STC1 (5.83), PF4V1 (2.97), EDNRB (2.64), AQP9 (2.58), HBEGF (2.51), BDKRB1 (2.4), RCAN1 (2.31), ECGF1 (1.97), DNER (1.85), RCAN1 (1.84), BDKRB2 (1.7), PECAM1 (1.65) | PDGFC (0.71), RNH1 (0.69) |
Values in parentheses correspond to fold change of expression between normal/reactive and inflamed areas. Genes represented more than once (HAPLN1 and RCAN1) reflect different splice variants or transcript variants.
To validate the differential expression of TREM1 in OA synovial biopsy samples, we performed Western blot analyses. As illustrated in Figure 1, the level of TREM-1 protein was increased in inflamed compared with normal/reactive areas. Similarly, S100A9 was evaluated and an increase of protein production was observed in inflamed compared with normal/reactive areas (Figure 1). These results were confirmed by immunohistochemistry (Figures 2A–D). In inflamed synovial biopsy samples, staining for S100A9 was observed in perivascular and sublining cells. Increased staining was observed in the intima lining layer of inflamed biopsy samples compared with normal/reactive biopsy samples.
Figure 1.
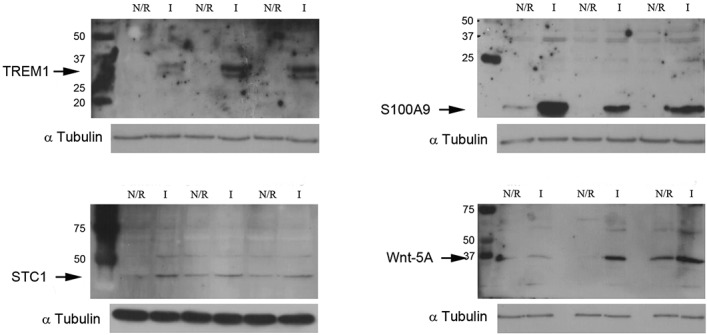
Increase of triggering receptor expressed on myeloid cells 1 (TREM-1), S100A9, stanniocalcin 1 (STC1), and Wnt-5a protein production in inflamed (I) areas compared with normal/reactive (N/R) areas. Total protein extracts from normal/reactive or inflamed areas were analyzed by Western blotting with anti–TREM-1 (30/26 kd), anti-S100A9 (14 kd), anti-STC1 (31 kd), anti–Wnt-5a (42 kd), and anti–α-tubulin (55 kd; control) antibodies. Representative images are shown (independent protein extracts from 3 different patients).
Figure 2.
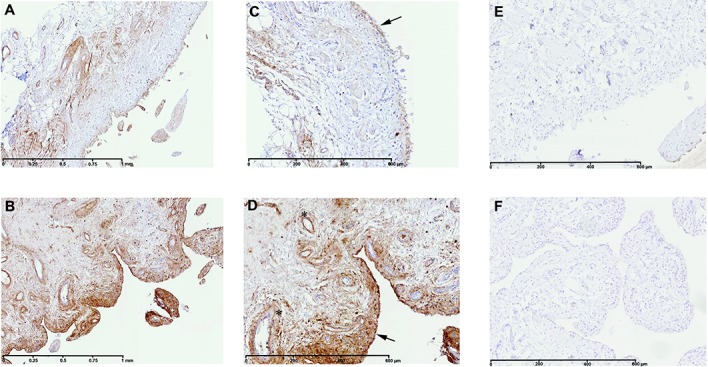
Immunohistochemical detection of S100A9 in normal/reactive (A and C) and inflamed (B and D) synovial biopsy samples. Also shown are negative controls with mouse IgG1 in normal/reactive (E) and inflamed (F) synovial biopsy samples. In inflamed synovial biopsy samples, staining for S100A9 is observed in perivascular and sublining cells. At the time of surgery, synovial biopsy samples from normal/reactive or inflamed areas were macroscopically selected as described in Patients and Methods. Normal/reactive and inflamed synovial biopsy samples were stained with anti-S100A9 antibody. Representative images are shown. Arrows indicate intima lining; asterisks indicate blood vessels. Original magnification × 5 in A and B; × 10 in C–F.
Cartilage metabolism pathway
The comparison between normal/reactive and inflamed synovial biopsy samples highlighted several factors involved in both cartilage anabolism and catabolism (Table1). Interestingly, the differential expression of these genes revealed an imbalance between anabolism and catabolism. Thus, MMP3 (P = 0.0044072, FDR 0.0955), MMP9 (P = 0.0013676, FDR 0.0824), CTSH (cathepsin H) (P = 0.0032275, FDR = 0.0931), and CTSS (cathepsin S) (P = 0.0015613, FDR 0.0837) were significantly up-regulated in inflamed synovial biopsy samples. In contrast, the most highly up-regulated anabolism enzyme gene was HAS1 (P = 0.0006656, FDR 0.0809). This observation was confirmed at the protein level using immunohistochemistry. In inflamed synovial biopsy samples, there was an increase in HAS-1–positive cells with predominant localization in the intima lining (Figures 3A–D).
Figure 3.
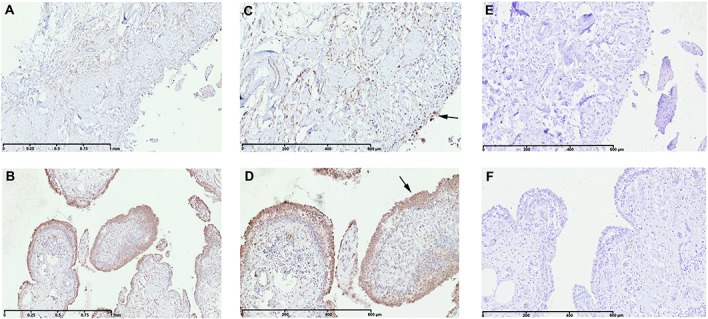
Immunohistochemical detection of hyaluronan synthase 1 (HAS-1) in normal/reactive (A and C) and inflamed (B and D) synovial biopsy samples. Also shown are negative controls with normal goat IgG in normal/reactive (E) and inflamed (F) synovial biopsy samples. In inflamed synovial biopsy samples, there is an increase in HAS-1–positive cells with predominant localization in the intima lining. At the time of surgery, synovial biopsy samples from normal/reactive or inflamed areas were macroscopically selected as described in Patients and Methods. Normal/reactive and inflamed synovial biopsy samples were stained with anti–HAS-1 antibody. Representative images are shown. Arrows indicate intima lining. Original magnification × 5 in A and B; × 10 in C–F.
Wnt signaling pathway
As revealed by the network analysis, the Wnt signaling pathway was differentially activated between normal/reactive and inflamed OA synovial biopsy samples. Indeed, genes for Wnt-5a (P = 0.0030684, FDR 0.0931) and low-density lipoprotein receptor–related protein 5 (P = 0.0023973, FDR 0.0881) were significantly up-regulated (2.11-fold and 1.55-fold, respectively). In contrast, the gene Frizzled family receptor 2 (FZD2) (P = 0.0036821, FDR 0.0931) and the genes for Dkk-3 (P = 0.0049476, FDR 0.0974) and Wnt-5b (P = 0.0014003, FDR 0.083) were significantly down-regulated (0.76-fold, 0.69-fold, and 0.57-fold, respectively).
To confirm the implications of the Wnt signaling pathway in synovial inflammation at the protein level, we studied the production of Wnt-5a, the most highly up-regulated gene of this pathway. As shown in Figure 1, the level of Wnt-5a protein was increased in the inflamed area compared with the normal/reactive area of the OA synovial membrane.
Angiogenesis pathway
The other identified and relevant pathway for OA was angiogenesis. Many genes in this pathway were significantly up- or down-regulated (Table1). Interesting mediators such as aquaporin 9 (AQP9) (P = 0.0003422, FDR 0.0783) and delta/notch-like epidermal growth factor repeat containing (DNER) (P = 0.0019947, FDR 0.0876) were shown to be significantly increased.
Among these genes, one of the most highly up-regulated was STC1 (5.83-fold increased expression; P = 0.0008499, FDR 0.0809). Consistent with the microarray data, a significant increase of STC1 production was observed in inflamed compared with normal/reactive areas by Western blot analysis (Figure 1). These results were also supported by immunohistochemical analysis. Indeed, in the inflamed area (Figures 4B and D), the staining for STC1 in perivascular and sublining cells was more intense than in the normal/reactive area (Figures 4A and C).
Figure 4.
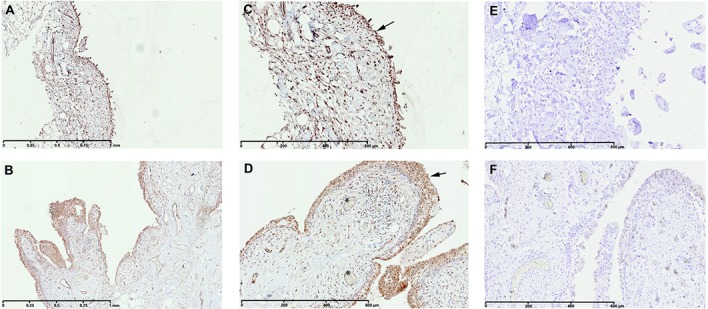
Immunohistochemical detection of stanniocalcin 1 (STC1) in normal/reactive (A and C) and inflamed (B and D) synovial biopsy samples. Also shown are negative controls with normal goat IgG in normal/reactive (E) and inflamed (F) synovial biopsy samples. In inflamed synovial biopsy samples, staining for STC1 is observed in perivascular and sublining cells. At the time of surgery, synovial biopsy samples from normal/reactive or inflamed areas were macroscopically selected as described in Patients and Methods. Normal/reactive and inflamed synovial biopsy samples were stained with anti-STC1 antibody. Representative images are shown. Arrows indicate intima lining; asterisks indicate blood vessels. Original magnification × 5 in A and B; × 10 in C–F.
DISCUSSION
Transcriptome analysis represents a promising approach in the understanding of complex diseases such as OA. In past years, reports of several studies aiming to determine the involvement of gene modulation have been published. These studies focused on human knee and hip cartilage as well as on cultured fibroblasts from knee synovial tissues (17).
The main conceptual questions raised by researchers are how samples from different patients can be pooled together and how the observed level of gene expression should be compared with that in appropriate controls. It is indeed obvious that a wide clinical heterogeneity can be found among patients due to both individual factors and disease characteristics. The comparison of the observed gene expression level with that in an appropriate control group or “healthy subject group” is also subject to debate. Samples considered healthy are commonly obtained from knee joints at necropsy or during arthroscopic examination, from meniscal lesions or from joints appearing normal on magnetic resonance imaging (18). Consequently, depending on the nature of the sample, an important heterogeneity among controls can also be observed.
The principal originality and novelty of the present work was to compare, using microarray analysis, the gene expression pattern of synovial cells from inflamed and normal/reactive areas of synovial membrane from the same OA patient. Methodologically, we used a rigorous procedure including macroscopic and histologic evaluation (15), flow cytometry analysis, and measurement of proinflammatory mediators to confirm the accuracy and reliability of our sampling method. Samples were digested and cultured for 7 days to enable us to use cells at passage 0 for our microarray experiments. In these cultures, flow cytometry analysis demonstrated the lack of contamination with other cell types, especially lymphocytes and endothelial cells that might have interfered with the microarray analysis (normal/reactive area 90.3% CD90+, 1.4% CD14+, and 0.8% CD3+; inflamed area 92.6% CD90+, 1.7% CD14+, and 1.3% CD3+). We also demonstrated that, in our model, the inflammatory profile of synoviocytes from the inflamed area was conserved, since we measured higher levels of IL-6 and IL-8 in culture medium (14).
Our microarray analysis showed that numerous genes involved in the inflammation, cartilage anabolism/catabolism, and angiogenesis pathways are differentially expressed in synovial membrane biopsy samples from inflamed and normal/reactive areas. Regarding the inflammation pathway, we confirmed the up-regulation of a large number of cytokine and chemokine genes, such as IL6, IL8, CXCL5, CXCL6, CXCL16, CXCL2, and CXCL1. In addition, we confirmed the up-regulation of genes for 2 interesting proteins, TREM-1 and S100A9. Since the discovery of TREM-1, much evidence has suggested that it has an important ability to regulate inflammation (19). TREM-1 is a recently identified immunoglobulin-like cell surface receptor with no cytoplasmic signaling domain (20). Its natural ligand is unknown, but its activation leads to the production of multiple proinflammatory cytokines and chemokines. This response can in turn amplify inflammatory responses (21). Our data indicated that TREM-1 expression is increased in inflamed compared with normal/reactive areas of OA synovial membrane, suggesting a potential role of this glycoprotein in OA synovitis. Further, TREM-1 was absent from normal/reactive synovial cell extracts, indicating a focal expression of this factor in inflamed areas of OA synovial membrane. TREM-1 appears to be an interesting therapeutic target and could be a marker of OA synovitis.
S100A9 is a member of the alarmin family (22). This alarmin is a pivotal protein in synovial inflammation and cartilage destruction in OA (10). In cartilage, S100A9 expression is restricted to hypertrophic chondrocytes and plays a major role in the mineralization of the cartilage matrix (23). High levels of S100A9 have been found in synovial fluid from OA patients and patients with rheumatoid arthritis (RA) (10-fold higher levels in synovial fluid from RA patients compared with OA patients) (24). The analysis of arthroscopic synovial biopsy samples from patients with early symptomatic OA in the Cohort Hip and Cohort Knee study also revealed increased levels of S100A9 (messenger RNA [mRNA] and protein), which correlated significantly with synovial lining thickness, cellularity in the subintima, and joint destruction (10).
An imbalance between anabolic and catabolic genes in favor of catabolic genes was also demonstrated. Indeed, the magnitude of fold expression was higher for catabolic genes. This disequilibrium is observed in cartilage during OA. In addition, it has been suggested that synovitis may accelerate the catabolism of articular cartilage and contribute to the progression of chondropathy by the production of a large amount of MMPs (25). Indeed, our results indicated that the genes MMP3 and MMP9 were up-regulated in inflamed compared with normal/reactive areas. Several results reported in the literature are concordant with our results. Yuan et al (25) observed a preferential localization of MMP-3 in pannus-like tissue covering hyaline cartilage, suggesting that this mediator was secreted by OA synovial tissue and acted directly to degrade cartilage. We also showed an up-regulation of bone morphogenetic protein 6 (BMP-6). This protein is classically implicated in cartilage and bone repair in arthritis. However, Lories et al also demonstrated that BMP-6 was expressed in arthritic synovium and strongly up-regulated by proinflammatory cytokines (26).
We previously demonstrated the importance of angiogenesis in synovitis as well as the close link between angiogenesis and inflammation (14). These results are supported by the data generated in the present microarray analysis. We showed that various key mediators of angiogenesis were differently expressed in inflamed and normal/reactive synovial membrane. For example, we observed an up-regulation of the gene for AQP-9. The aquaporins are involved in cell migration and angiogenesis (27). Moreover, Nagahara et al (28) reported that AQP-9 mRNA was detected in synovial tissues from OA and RA patients with hydrarthrosis and that its expression was strongly induced in fibroblast-like synoviocytes by TNFα, suggesting that AQP-9 might be related to synovitis.
Besides these confirmatory results, our study has identified several potential targets of interest for the treatment of OA. We demonstrated for the first time the up-regulation, at both the gene and protein levels, of STC1, a glycoprotein hormone originally discovered in the corpuscles of Stannius of bony fish (29). Recent work has indicated that STC1 may be involved in the control of the angiogenesis process (30). STC1 is described as a vascular-specific angiogenesis-associated protein. It plays roles in “angiogenic sprouting” via the VEGF/VEGF receptor 2 pathway (31). These observations suggest that STC1 could be a key mediator of synovium neovascularization in OA synovitis.
In our microarray analysis, the Wnt signaling pathway was differentially activated in inflamed compared with normal/reactive OA synovial biopsy samples. More particularly, we found an up-regulation of Wnt-5a, a ligand that activates the β-catenin–independent pathway. Wnt-5a was involved in IL-1β–induced MMP expression and chondrocyte differentiation, suggesting that it might be involved in cartilage destruction (32). A better understanding of Wnt signaling and the members of this pathway could be an approach to therapeutic management of OA.
Additionally, we noted a series of genes that were differently regulated in inflamed compared with normal/reactive areas of OA synovial membrane, but have not to date been associated with OA pathogenesis. These include up-regulated genes such as hydroxysteroid (11-beta) dehydrogenase 1 (HSD11B1), the solute carrier organic anion transporter (SLCO) family, or microRNA (MIR1275, 199A2, 221, and 302c) and down-regulated genes such as cysteine-rich protein 2 (CRIP2), lysyl oxidase–like 1 (LOXL1), or AHNAK nucleoprotein. A better understanding of the role played by these genes in OA pathogenesis could lead to the identification of new therapeutic targets.
In conclusion, our microarray analysis has revealed genes that were focally regulated in OA synovial membrane. These genes are associated with important pathophysiologic processes in OA and represent candidate targets for OA treatment. Using a unique culture system, this study is the first to identify different expression patterns between 2 areas of the synovial membrane from the same patient. As expected, these differences concern key pathways involved in OA pathogenesis. This analysis also provided novel information regarding new genes of interest (STC1, S100A9) and provided evidence of the involvement of the Wnt-5a signaling pathway in OA.
Acknowledgments
The authors would like to thank Benoît Hennuy (Groupe Interdisciplinaire de Génoprotéomique Appliquée [GIGA] Genomics Core Facility, University of Liège, Liège, Belgium) for technical assistance with the microarray, Emilie Feyereisen and Isabelle Dasoul (Laboratory of Tumor and Developmental Biology, GIGA Cancer, University of Liège, Liège, Belgium) for technical assistance with immunohistochemistry experiments, and Christelle Sanchez (Bone and Cartilage Research Unit, University of Liège, Liège, Belgium) and Christelle Boileau for kind assiatance with manuscript preparation.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Henrotin had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Lambert, Montell, Vergés, Henrotin.
Acquisition of data. Lambert, Dubuc, Munaut, Noël.
Analysis and interpretation of data. Lambert, Montell, Vergés, Henrotin.
ROLE OF THE STUDY SPONSOR
Bioiberica, Spain had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Bioiberica, Spain.
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Aigner T, Rose J, Martin J, Buckwalter J. Aging theories of primary osteoarthritis: from epidemiology to molecular biology. Rejuvenation Res. 2004;7:134–45. doi: 10.1089/1549168041552964. [DOI] [PubMed] [Google Scholar]
- Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–707. doi: 10.1002/art.34453. [review] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625–35. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64:1263–7. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel-Pelletier J, Pelletier JP. Is osteoarthritis a disease involving only cartilage or other articular tissues? Eklem Hastalik Cerrahisi. 2010;21:2–14. [PubMed] [Google Scholar]
- Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51:249–57. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayral X, Pickering EH, Woodworth TG, Mackillop N, Dougados M. Synovitis: a potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis–results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthritis Cartilage. 2005;13:361–7. doi: 10.1016/j.joca.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Honorati MC, Bovara M, Cattini L, Piacentini A, Facchini A. Contribution of interleukin 17 to human cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthritis Cartilage. 2002;10:799–807. doi: 10.1053/joca.2002.0829. [DOI] [PubMed] [Google Scholar]
- Scanzello CR, McKeon B, Swaim BH, DiCarlo E, Asomugha EU, Kanda V, et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: molecular characterization and relationship to symptoms. Arthritis Rheum. 2011;63:391–400. doi: 10.1002/art.30137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lent PL, Blom AB, Schelbergen RF, Sloetjes A, Lafeber FP, Lems WF, et al. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012;64:1466–76. doi: 10.1002/art.34315. [DOI] [PubMed] [Google Scholar]
- Bonnet CS, Walsh DA. Osteoarthritis, angiogenesis and inflammation. Rheumatology (Oxford) 2005;44:7–16. doi: 10.1093/rheumatology/keh344. [DOI] [PubMed] [Google Scholar]
- Ashraf S, Walsh DA. Angiogenesis in osteoarthritis. Curr Opin Rheumatol. 2008;20:573–80. doi: 10.1097/BOR.0b013e3283103d12. [DOI] [PubMed] [Google Scholar]
- Walsh DA, Bonnet CS, Turner EL, Wilson D, Situ M, McWilliams DF. Angiogenesis in the synovium and at the osteochondral junction in osteoarthritis. Osteoarthritis Cartilage. 2007;15:743–51. doi: 10.1016/j.joca.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Lambert C, Mathy-Hartert M, Dubuc JE, Montell E, Verges J, Munaut C, et al. Characterization of synovial angiogenesis in osteoarthritis patients and its modulation by chondroitin sulfate. Arthritis Res Ther. 2012;14:R58. doi: 10.1186/ar3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayral X. Diagnostic and quantitative arthroscopy: quantitative arthroscopy. Baillieres Clin Rheumatol. 1996;10:477–94. doi: 10.1016/s0950-3579(96)80045-8. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynard LN, Loughlin J. The genetics and functional analysis of primary osteoarthritis susceptibility. Expert Rev Mol Med. 2013;15:e2. doi: 10.1017/erm.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rey MJ, Usategui A, Izquierdo E, Canete JD, Blanco FJ, Criado G, et al. Transcriptome analysis reveals specific changes in osteoarthritis synovial fibroblasts. Ann Rheum Dis. 2012;71:275–80. doi: 10.1136/annrheumdis-2011-200281. [DOI] [PubMed] [Google Scholar]
- Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchon A, Dietrich J, Colonna M. Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–5. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- Tessarz AS, Cerwenka A. The TREM-1/DAP12 pathway. Immunol Lett. 2008;116:111–6. doi: 10.1016/j.imlet.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Perera C, McNeil HP, Geczy CL. S100 calgranulins in inflammatory arthritis. Immunol Cell Biol. 2010;88:41–9. doi: 10.1038/icb.2009.88. [DOI] [PubMed] [Google Scholar]
- Zreiqat H, Howlett CR, Gronthos S, Hume D, Geczy CL. S100A8/S100A9 and their association with cartilage and bone. J Mol Histol. 2007;38:381–91. doi: 10.1007/s10735-007-9117-2. [DOI] [PubMed] [Google Scholar]
- Baillet A, Trocme C, Berthier S, Arlotto M, Grange L, Chenau J, et al. Synovial fluid proteomic fingerprint: S100A8, S100A9 and S100A12 proteins discriminate rheumatoid arthritis from other inflammatory joint diseases. Rheumatology (Oxford) 2010;49:671–82. doi: 10.1093/rheumatology/kep452. [DOI] [PubMed] [Google Scholar]
- Yuan GH, Tanaka M, Masuko-Hongo K, Shibakawa A, Kato T, Nishioka K, et al. Characterization of cells from pannus-like tissue over articular cartilage of advanced osteoarthritis. Osteoarthritis Cartilage. 2004;12:38–45. doi: 10.1016/j.joca.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Lories RJ, Derese I, Ceuppens JL, Luyten FP. Bone morphogenetic proteins 2 and 6, expressed in arthritic synovium, are regulated by proinflammatory cytokines and differentially modulate fibroblast-like synoviocyte apoptosis. Arthritis Rheum. 2003;48:2807–18. doi: 10.1002/art.11389. [DOI] [PubMed] [Google Scholar]
- Nico B, Ribatti D. Aquaporins in tumor growth and angiogenesis. Cancer Lett. 2010;294:135–8. doi: 10.1016/j.canlet.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Nagahara M, Waguri-Nagaya Y, Yamagami T, Aoyama M, Tada T, Inoue K, et al. TNF-α-induced aquaporin 9 in synoviocytes from patients with OA and RA. Rheumatology (Oxford) 2010;49:898–906. doi: 10.1093/rheumatology/keq028. [DOI] [PubMed] [Google Scholar]
- Yeung BH, Law AY, Wong CK. Evolution and roles of stanniocalcin. Mol Cell Endocrinol. 2012;349:272–80. doi: 10.1016/j.mce.2011.11.007. [DOI] [PubMed] [Google Scholar]
- He LF, Wang TT, Gao QY, Zhao GF, Huang YH, Yu LK, et al. Stanniocalcin-1 promotes tumor angiogenesis through up-regulation of VEGF in gastric cancer cells. J Biomed Sci. 2011;18:39. doi: 10.1186/1423-0127-18-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law AY, Wong CK. Stanniocalcin-1 and -2 promote angiogenic sprouting in HUVECs via VEGF/VEGFR2 and angiopoietin signaling pathways. Mol Cell Endocrinol. 2013;374:73–81. doi: 10.1016/j.mce.2013.04.024. [DOI] [PubMed] [Google Scholar]
- Ge XP, Gan YH, Zhang CG, Zhou CY, Ma KT, Meng JH, et al. Requirement of the NF-κB pathway for induction of Wnt-5A by interleukin-1β in condylar chondrocytes of the temporomandibular joint: functional crosstalk between the Wnt-5A and NF-κB signaling pathways. Osteoarthritis Cartilage. 2011;19:111–7. doi: 10.1016/j.joca.2010.10.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.