

Abstract
Synthetic biology holds promise as both a framework for rationally engineering biological systems and a way to revolutionize how we fundamentally understand them. Essential to realizing this promise is the development of strategies and tools to reliably and predictably control and characterize sophisticated patterns of gene expression. Here we review the role that RNA can play towards this goal and make a case for why this versatile, designable, and increasingly characterizable molecule is one of the most powerful substrates for engineering gene expression at our disposal. We discuss current natural and synthetic RNA regulators of gene expression acting at key points of control – transcription, mRNA degradation, and translation. We also consider RNA structural probing and computational RNA structure predication tools as a way to study RNA structure and ultimately function. Finally, we discuss how next-generation sequencing methods are being applied to the study of RNA and to the characterization of RNA's many properties throughout the cell.
Keywords: Gene regulation, Next-generation sequencing, Non-coding RNA, RNA structure, Synthetic biology
1 Introduction
The field of synthetic biology is emerging to solve some of the fundamental challenges that arise when we try to harness the powerful, yet extremely complex, capabilities of natural biomolecular systems. The applications that synthetic biologists are pursuing are diverse and range from optimizing the biosynthetic capabilities of cells to produce fuels and drugs, to harnessing the ability of cells to act as environmental biosensors, to manipulating cellular communities and tissues for medical purposes [1, 2]. While the challenges raised by these applications demand a sophistication of engineering on many levels, at the heart of all of these applications is the precise control of gene expression – turning the right genes on at the right times and at the right levels. Because of this central role of gene expression, one of the grand challenges of synthetic biology has become the development of an engineering framework that allows the design, construction, and characterization of genetic networks that reliably implement specified genetic control.
While great strides have been made towards this goal by focusing on protein systems (reviewed in [2, 3]), there is a case to be made for the power of RNA-based gene regulators as fundamental components of the synthetic biology toolbox. To start with, RNA's role as a versatile regulator of cellular genomes is being uncovered at an accelerating pace [4], and there are now well studied RNA mechanisms that regulate almost all aspects of gene expression [5–9]. We also have a rapidly emerging biophysical understanding of the relationship between RNA structure and function that has served as an excellent starting point for computational RNA design [10–14]. But perhaps even more compelling is our ability to characterize RNAs in unprecedented breadth and scale with the advent of next-generation sequencing (NGS). In fact, there has been an explosion of sequencing-based tools that can characterize almost all aspects of RNAs – from cellular abundances, to locations of protein and ribosome binding, to structures and interactions [15–24] – all with variations on an experiment that is rapidly becoming cheaper and faster [25].
We therefore hypothesize that RNA represents one of the most powerful substrates at our disposal with which to develop a discipline of engineering gene expression inside cells, because it is versatile, designable, and easily characterizable. It is our goal in this review to present the case for RNA by focusing on these three features in turn. We start with a survey of what is known about natural RNA regulatory mechanisms and an update on their latest engineering. We then give a brief introduction and overview of computational and experimental RNA structure determination techniques and how they fit into the RNA engineering design paradigm. Finally, we provide an introduction to several important NGS characterization techniques for RNA, with pointers to resources for starting these experiments, and thoughts on how they can be applied to RNA engineering problems. To make our discussion concrete, we focus on prokaryotic RNA-based regulation, although much progress has been made in engineering RNA regulation in eukaryotes [26–32].
2 Natural and engineered RNA mechanisms for control of gene expression
Sixty years of molecular biology has taught us that gene expression can be thought of as a series of biochemical conversions from DNA to RNA and in many cases to protein (Fig. 1). While this is a simplified model, it is a useful framework for establishing the control points of basic gene expression. In prokaryotes, the most obvious points of control are RNA synthesis (transcription), RNA degradation, protein synthesis (translation), and protein degradation. Choosing different points of control can have different ramifications for the dynamics and steady-state levels of gene expression [33]. There are also a variety of different mechanisms that can regulate each point of control. These factors, combined with potential issues of compatibility between different mechanisms, create a rich optimization challenge for the biomolecular engineer wishing to precisely control gene expression. While proteins play fundamental roles in gene expression, RNAs have also been found to regulate multiple points of control, namely transcription, RNA degradation, and translation. For each control point, we discuss the main natural classes and mechanisms of RNA-based gene regulation and those that have been recently engineered by synthetic biologists (Fig. 2A and B).
Figure 1.
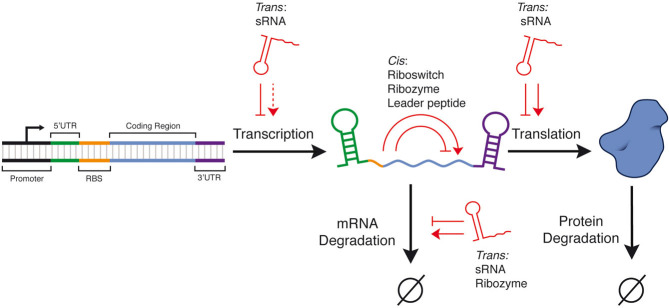
The central synthesis and degradation steps of prokaryotic gene expression. The points of control where cis- and trans-acting RNA regulators exert their regulation are highlighted. Pointed arrows indicate activation while blunt arrows indicate repression. Dashed arrow indicates a gap in regulatory mechanisms. RBS, ribosome binding site; UTR, untranslated region; sRNA, small RNA.
Figure 2.

The major classes of natural and synthetic RNA regulators, organized into (A) transcriptional and translational regulators and (B) regulators of mRNA degradation.
2.1 Translational control of gene expression by RNA
2.1.1 General roles of RNA structure in translation
Translation initiation has long been known to be a rate-limiting step of gene expression [34, 35]. Translation is initiated through an interaction between the 16S ribosomal RNA (rRNA) and a ribosome binding site (RBS) that lies in the 5′ untranslated region (5′ UTR) of a prokaryotic messenger RNA (mRNA). Evidence suggests that the ribosome makes contacts within a ∼52 nucleotide mRNA region [36], within which is an eight nucleotide Shine-Dalgarno sequence (SD) that is partially complementary to the 3′ end of the 16S rRNA [37, 38]. It is well established that variations of the SD sequence away from a complementary match to the 16S 3′ end have an effect on translation rate. Moreover, it has also long been known that RNA secondary structures, defined by the pattern of canonical base pairs (A pairs to U, G pairs to C or U), in the region surrounding the RBS and the start codon can alter RBS accessibility and thus tune translation initiation rate [39]. Recent evidence by Kudla et al. [40] suggests that this may be a general principle of translation regulation. In this work, the authors showed that for a library of 154 synonymously coded green fluorescent protein (GFP) variants, the strongest positive correlate of high GFP expression was weaker predicted RNA structure near the RBS and start codon [40].
More recently, synthetic biologists have sought strategies to minimize the effect of 5′ UTR structures. Mutalik et al. [41] recently demonstrated one such strategy that uses two SD sequences in the mRNA – one that controls translation of a leader peptide, and a second that controls translation of the downstream gene. Co-translation of the leader peptide disrupts secondary structures within the downstream RBS, thus minimizing secondary structure effects that arise from different combinations of genetic elements. Alternative strategies have sought to physically remove excess 5′ UTR sequences of mRNAs by cleavage via ribozymes [42], or with ribonucleases derived from the bacterial clustered regulatory interspaced short palindromic repeats (CRISPR) defense system [43]. Moreover, as discussed in Section 3, computational tools are now being developed to design 5′ UTR sequences with RNA folding in mind so that translation initiation rates can be predictably tuned [10].
While the presence of general RNA structure in the 5′ UTR can adjust RBS accessibility, many natural and synthetic RNA regulators of translation specifically target the RBS and start codon with either trans-acting RNA-RNA interactions or cis-acting RNA structures that can change conformations and thus switch translation on or off, as described below.
2.1.2 Translational control: Small RNAs
Small RNAs (sRNAs) are a diverse and abundant class of short (50–100 nucleotides) transcripts that directly target specific mRNAs through intermolecular base-pairing interactions [6, 44] (Fig. 2A). They can either be cis-encoded antisense RNAs, and thus fully complementary to their targets, or trans-encoded at a distant location with only partial target complementarity [5]. In the latter case, these sRNAs often require the RNA chaperone protein Hfq hypothesized to stimulate base-pairing by recruiting both sRNA and target mRNA to its surface, induce structural rearrangements in either the sRNA or mRNA, stabilize bound sRNA from ribonuclease degradation and stimulate degradation of sRNA and mRNA hybrids via recruitment of ribonucleases [45].
By far the most abundant natural mechanism of gene regulation by sRNA is at the post-transcriptional level. The simplest mechanism involves sRNA binding and occlusion of the RBS or start codon of a target mRNA [46] (Fig. 2A), however binding outside of this region can also alter translation [47–48]. Figure 2A highlights several sRNA translational regulators and the natural genes they target such as IS10 RNA-OUT/RNA-IN [49], and sok/hok [50].
In addition to acting as repressors, in a few cases sRNAs have been shown to be translational activators [51]. Generally known as the anti-antisense mechanism, translational activation occurs when sRNA-mRNA base-pairing interactions induce the formation of alternative mRNA structures that expose the RBS. Notable examples include the sRNA RNAIII activation of hla in Staphylococcus aureus [52] and DsrA activation of rpoS in E. coli [53] (Fig. 2A).
One of the earliest strategies of engineering synthetic sRNA-like regulation was the development of riboregulators [54] (Fig. 2A). Riboregulators are in essence a cleaner implementation of the natural sRNA regulation principle. Target mRNAs were designed to be cis-repressed RNA (crRNA) by the inclusion of a 5′ intrinsic stem-loop structure that encompassed the RBS. Activation of translation was achieved by the introduction of an sRNA analog called a trans-activating RNA (taRNA) designed to target and hybridize this stem-loop to expose the RBS. Moreover, these riboregulators have been further engineered to provide orthogonal crRNA and taRNA pairs that have been effectively combined in higher-order devices such as a four input genetic switchboard [55] and a synthetic counter [56]. This approach was recently extended by Rodrigo et al. [12], who developed an algorithm to computationally search for additional crRNA/taRNA pairs using a structure-guided design.
Additional recent strategies have been to create variants of natural sRNAs. For example, Mutalik et al. [57] engineered the copy number control element from the transposon IS10, consisting of the sRNA (RNA-OUT) and the target mRNA (RNA-IN; Fig. 2A). In a mechanistically driven approach, RNA-IN and RNA-OUT variants were generated by mutating the sequences known to govern the specificity of the interaction. By gathering gene expression data for a 52 × 52 matrix of different RNA-IN/RNA-OUT pairs, the authors were able to construct a statistical model to find sequence elements and predicted structural features that were critical to function. In particular, they found that the overall hybridization energy of the RNA-IN/RNA-OUT pair, and a 5 bp region that seeds the hybridization, were key features that correlated with function [57].
Synthetic Hfq-binding sRNAs have also been designed by using existing sRNAs as scaffolds to provide Hfq binding sites and transcriptional terminators while modifying the mRNA target binding sequence. Sharma et al. [58] screened libraries of randomized target binding sequence to generate sRNAs able to repress a variety of endogenous E. coli genes. Alternatively, Na et al. [59] rationally designed target sequences complementary to either the translation initiation or coding regions of target mRNAs. These synthetic sRNAs were used in a metabolic engineering approach to combinatorially repress the expression of chromosomal genes and identify E. coli strains with the highest production of a desired metabolite. One of the major advantages of using sRNA for the repression of endogenous genes is that no chromosomal modification is required to generate knock-down strains [59].
Going further, natural sRNAs have also recently been engineered to change structure and function in response to external ligands (Fig. 2A). Qi et al. [60] fused the IS10 RNA-OUT hairpin to a well characterized RNA aptamer – an RNA structure that can bind to a specific molecule. In this design, the loop of the aptamer hairpin was made complementary to the RNA-OUT loop to form a pseudoknot interaction in the absence of ligand, rendering the antisense non-functional. Ligand binding to the aptamer prevented pseudoknot formation, thus allowing the RNA-OUT to fold properly and repress its target. This trans-acting, ligand-dependent conditional folding strategy was actually inspired by natural cis-acting RNA regulators called riboswitches, which we discuss next.
2.1.3 Translational control: Riboswitches
Riboswitches are a class of RNA regulators that control gene expression in response to ligands, and appear to be used ubiquitously across bacteria (Fig. 2A) [61]. Simple riboswitches are composed of two domains: an aptamer and a gene expression platform [62]. These domains are structurally linked, with ligand-dependent rearrangements in the aptamer domain causing RNA conformational changes within the expression platform that lead to different gene expression outcomes [62]. Riboswitches that regulate translation have expression platforms that fold to either occlude or expose the RBS, and can be either activators such as the S-adenosylhomocysteine (SAH) riboswitch ahcY [63] (Fig. 2A), or repressors as in the case of the adenosylcobalam [64] and thiamine [65] riboswitches (Fig. 2A). Some riboswitches such as the glycine riboswitch have tandem copies of the same aptamer before the expression platform, allowing cooperative response to ligands [66]. There are also examples of riboswitches with two different aptamers that allow integration of multiple inputs as in the metE riboswitch [67].
As suggested by their ubiquity and utility in nature, synthetic riboswitches hold great potential for engineering biological systems that can sense and respond to environmental or intracellular inputs. Interestingly, natural riboswitches were only discovered after they were first engineered in the laboratory by combining an aptamer that binds to ATP and a self-cleaving ribozyme [68]. Since then, progress has been made in developing systematic selection and engineering strategies to fuse different aptamers to appropriate expression platforms to make synthetic riboswitches (Fig. 2A). For example, Desai and Gallivan [69] screened a library consisting of the theophylline aptamer upstream of a variable linker sequence and an RBS to find a theophylline riboswitch with 36-fold activation. In another example, Suess et al. [70] rationally designed a theophylline riboswitch that had alternative stem-loop folds in close proximity to the RBS. Further progress has been aided by the development of high-throughput functional screening assays to rapidly screen sequence variants including colorimetric [71] and flow-cytometry [72] based screens, and even selection strategies based on cell motility [73]. These approaches should aid in the development of riboswitches from aptamers discovered from the systematic evolution of ligands by exponential enrichment (SELEX) method [74–76], an in vitro technique that has allowed the discovery of aptamers that bind to a wide array of ligands [76]. In fact, riboswitches have already been constructed from a number of different aptamers including those responsive to antibiotics [77, 78], proteins [79] and other small molecules [68, 78, 80].
2.1.4 Translational control: Ribozymes
Ribozymes are naturally occurring RNA sequences that have the ability to catalyze the cleavage or ligation of RNA strands (for review see [81]). Most synthetic ribozymes have utilized the hammer-head ribozyme, which has also been fused with aptamers to create ligand-controlled ribozymes called aptazymes [68]. For example, Wieland and Hartig [82] developed a theophylline-responsive aptazyme that cleaved structures occluding the RBS, thereby activating gene expression (Fig. 2A). More recently, ribozymes have been developed that self-cleave in response to trans-acting sRNAs that are functional in vivo, providing the potential for coupling ribozymes to existing and synthetic sRNAs for the development of RNA networks [83] (Fig. 2A).
2.2 Control of gene expression by mRNA degradation
2.2.1 General roles of RNA structure in mRNA degradation
In bacteria, mRNA degradation is primarily achieved by the RNA degradosome, a protein complex composed of RNase E, polynucleotide phosphorylase (PNPase), and RhlB RNA helicase [84]. Degradation is a multiple step process, with initial RNase E cleavage being the most rate limiting. Interestingly, since RNase E cleaves single-stranded RNA, secondary structures can inhibit its activity. Indeed, this has been effectively used to engineer specific secondary structures within the 5′ UTR of mRNAs to increase stability, and therefore overall gene expression, over a 10-fold range [85].
2.2.2 mRNA degradation control: sRNA
Natural sRNAs that regulate translation also affect mRNA degradation. In fact, sRNA-mRNA interaction is typically followed by rapid degradation by RNase E or RNase III [5]. Moreover, Hfq has been shown to interact with components of the degradosome, highlighting the possibility of Hfq assisted degradation [86]. Some sRNAs such as RhyB targeting sodB mRNA [87] and SgrS targeting ptsG mRNA primarily block translation initiation [88], though for many mRNAs, concomitant degradation makes repression irreversible [45]. In other cases such as MicC sRNA, degradation is the sole route of repression [89] (Fig. 2B). Although the vast majority of known sRNAs destabilize mRNA, two exceptions are the GadY and VR-RNA sRNAs that exert a stabilization effect on the gadX and colA mRNAs respectively [5, 90, 91] (Fig. 2B). The degradation effect exerted by natural sRNA has also been emulated by synthetic versions of sRNA [92]. In this work, Man et al. [92] developed a series of synthetic sRNAs designed to target 5′ UTRs to trigger RNase E-mediated degradation and showed that the inclusion of natural Hfq-binding sites stabilized the synthetic sRNA in vivo and facilitated gene silencing.
2.2.3 mRNA degradation control: Ribozymes
The cleavage of mRNA catalyzed by ribozymes has also been shown to effect mRNA degradation. For example members of the glmS class combine ligand response to glucosamine-6-phosphate (GlcN6P) with self-cleavage and thus can be considered as a type of riboswitch [93] (Fig. 2B). The cleavage catalyzed by the glmS ribozyme results in rapid degradation of cleaved products [93]. Relying on a similar mechanism, Carothers et al. [11] engineered aptazyme and ribozyme dependent cleavage of mRNA to control degradation for the regulation of biosynthetic pathways (Fig. 2B).
2.2.4 mRNA degradation control: Riboswitches
The inhibition of translation caused by riboswitches has been shown to indirectly induce mRNA degradation. As translating ribosomes can protect mRNA species from decay, when translation is inhibited by ligand binding to a riboswitch, an indirect consequence is mRNA degradation as hypothesized for the thiM and btuB riboswitches [94]. However, recently the E. coli lysC riboswitch was shown to directly induce mRNA degradation in addition to inhibiting translation by ligand-dependent rearrangements resulting in exposure of an RNase E site [95].
2.3 Transcriptional control of gene expression by RNA
Bacterial transcriptional regulation at the RNA level occurs primarily through transcription termination. Unlike for translation and mRNA degradation, specific RNA sequences and structures are required to terminate transcription. In rho-mediated termination, the protein factor rho binds to specific binding sites of mRNAs and traverses the mRNA in a 5'–3′ direction, eventually causing actively transcribing polymerase to abort [96]. More common is intrinsic termination, where hairpin structures placed immediately upstream of extended polyU sequences fold while the polymerase is paused at the polyU, causing polymerase to abort [97, 98]. While these mechanisms are ubiquitous in nature, RNA-mediated transcription regulation is just being recognized as a particularly powerful point of control in synthetic biology [99]. Indeed, engineering RNA-mediated transcriptional regulators holds the promise of constructing RNA-only genetic networks that require no protein intermediates [99].
While this has only been shown as a proof-of-concept, potential advantages of such RNA-only networks over protein-based networks are faster signal propagation due to quick RNA degradation rates, and a smaller genetic footprint as regulatory RNAs can be encoded in fewer nucleotides than proteins. Additionally, in certain instances RNA-only networks possibly offer lower metabolic burden since amino acids are not consumed for translation; however, this has yet to be fully explored. For all the potential advantages, work in this area is only just beginning, as discussed below.
2.3.1 Transcriptional control: sRNA
Transcription attenuation is a repressive sRNA-based mechanism where an antisense sRNA targets an attenuator sequence that lies in the 5′ UTR of the target mRNA (Fig. 2A). The attenuator sequence can adopt two mutually exclusive confirmations depending on the base-pairing to sRNA: in the presence of sRNA, a terminator structure is formed in the mRNA thereby causing premature termination of transcription before the coding sequence is reached, while in the absence of sRNA, anti-terminator structures are formed that inhibit terminator formation and enable transcriptional read-through [5]. To date, this mechanism has been found for cis-encoded sRNAs in plasmids and phages of Gram-positive bacteria [5, 6] and has been shown to function in Gram-negative bacteria [5].
Of these natural mechanisms, the S. aureus plasmid pT181 attenuator [100] has been shown to be a versatile regulatory building block for RNA-based genetic networks [99]. In particular, independently acting, or orthogonal, antisense/attenuator pairs were engineered through mutations and shown to independently regulate GFP and RFP targets in the same cell. Following the example of natural riboswitches, these orthogonal attenuators were also placed in tandem on the same transcript and shown to evaluate genetic NOR logic [99]. Importantly, these attenuators were also used to construct the first example of an RNA-only network, a two stage transcriptional cascade, whereby signals were propagated directly as RNA molecules with no intermediate protein signals. In addition, the same aptamer fusion strategy highlighted above was used to produce aptamer-pT181 antisense variants that respond to theophylline and the MS2 coat protein, opening the door for creating sophisticated RNA circuitry that dynamically responds to environmental and intracellular cues [60] (Fig. 2A). Recently, large libraries of orthogonal pT181 antisense/attenuator variants were created through a chimeric fusion strategy to translational sRNAs, thus removing a critical barrier to making larger RNA-based genetic circuits from this system a reality [101].
2.3.2 Transcriptional control: Riboswitches
Transcriptional regulation by riboswitches involves expression platforms that fold into terminator hairpins or alternative anti-terminator structures much like the transcriptional attenuators discussed above (Fig. 2A). In addition, a class of riboswitches that regulates gene expression via rho-dependent transcriptional termination has recently been discovered [102]. They are generally repressive, such as thiamin pyrophosphate (TPP) and flavin mononucleotide (FMN) riboswitches from Bacillus subtilis [103], and utilize the ligand dependent formation of terminator structures [62] (Fig. 2A). There are also a few known examples of activators such as the adenine and glycine riboswitches [66, 104] (Fig. 2A). Although riboswitches are widely present in nature, the development of synthetic variants of riboswitches has been limited, and work has mainly focused on translational control due, in part to the limited structural characterization of transcriptional expression platforms. Recently, however, Wachsmuth et al. [13] used an in silico guided approach to screen variants of riboswitch designs that varied in a spacer sequence between the aptamer and a synthetic transcription expression platform. Several variants in the spacer sequence were shown to functionally activate transcription in E. coli in the presence of the theophylline ligand. Another approach harnessed the modular nature of aptamers and expression platforms. Ceres et al. [105] showed that naturally occurring and synthetic aptamers could be effectively fused with three different naturally occurring expression modules of riboswitches from B. subtilis to create chimeric transcriptional riboswitches that respond to tetracycline and theophylline.
2.3.3 Transcriptional control: Leader peptides
Leader peptides are naturally occurring cis-regulatory elements that couple translation of a small coding sequence to the transcription of downstream genes (Fig. 2A). Two notable examples of natural leader peptide regulators integrate tryptophan-dependent translation of a leader peptide to the control of downstream genes in the trp and tna operons. In the trp operon, a 14 amino acid leader peptide coding sequence contains two tandem tryptophan codons, and is directly upstream of a sequence that can form either a transcriptional terminator or an anti-terminator based on the concentration of charged tryptophan tRNAs [106] (Fig. 2A). In contrast, the tna operon relies upon ribosome stalling in a leader peptide to prevent transcriptional termination by physical occlusion of a Rho termination factor-binding site (rut site) [106] (Fig. 2A). Recently, these mechanisms have been engineered to sense the presence of unnatural amino acids, and to expand the repertoire of sRNA-mediated transcriptional regulators available to synthetic biologists [107, 108]. In the first example, Lui et al. [107] designed variants of the leader peptide sequence to contain “blank codons” that do not naturally encode amino acids, thereby inducing ribosomal stalling in both the trp and tna leader peptides (Fig. 2A). By introducing tRNAs to decode these blank codons, and aminoacyl-tRNA-synthetases to charge these tRNAs with unnatural amino acids, they showed that they could couple the presence of unnatural amino acids to either transcriptional activation (tna leader peptide) or repression (trp leader peptide). In the second example, Liu et al. [108] demonstrated that sRNAs could be used to regulate the translation of the leader peptides themselves, thus converting this translational regulation into transcriptional regulation of the downstream sequence (Fig. 2A).
2.3.4 Transcriptional control: CRISPRs systems
While natural CRISPR systems are involved in complex bacterial defense systems, recently they have been engineered to provide a powerful mechanism of RNA-meditated transcription control. In an exciting new advance, Qi et al. [109] describe what they call CRISPR interference (CRISPRi), a system engineered from the type II CRISPR of Streptococcus pyogenes (Fig. 2A). In this system, the Cas9 protein forms a complex with a small guide RNA (sgRNA) that contains a region complementary to a target genomic DNA sequence. In the native system, the Cas9:sgRNA complex binds to the target DNA and the nuclease activity of Cas9 then cleaves the DNA. However, in this case, the authors used a catalytically dead Cas9 mutant (dCas9), which still binds to the target and was shown to block RNA polymerase (RNAP) binding during either transcription initiation or elongation. This work effectively opens another route for creating RNA-only genetic circuitry by configuring networks of sgRNAs that control their own transcription in sophisticated patterns, and could be synergistic with the sRNA-based networks described above.
2.4 Regulation of protein function by RNA
In addition to the regulation of gene expression, RNA is able to regulate post-translationally by directly interacting with proteins. Modulation of protein function is typically achieved by sequestration: for example, the E. coli σ70 RNAP holoenzyme can be sequestered by the 6S RNA, which is thought to mimic the conformation of σ70 DNA promoters elements [7]. Similarly the CsrA protein involved in mRNA translation and degradation can be sequestered by the CsrB RNA [7].
RNA-protein interactions have successfully been engineered using the ability of RNA aptamers to recognize and bind a variety of proteins. For example, aptamers able to bind the TetR transcriptional regulator have been successfully generated and placed in the 5′ UTR of mRNAs to confer post-transcriptional regulation of gene expression by TetR [110]. Alternatively, these aptamer-protein interactions have been harnessed to build one and two-dimensional RNA scaffolds in vivo that include binding sites for tagged proteins, thus enabling intracellular localization of multiple proteins [111]. These RNA scaffolds were used to organize and enhance the production of a hydrogen-producing enzymatic pathway [111].
3 Tools for engineering the RNA sequence-structure-function relationship
It is clear that the rich versatility of RNA gene regulatory function is intimately tied to RNA structure. In fact, it is precisely the ability of single stranded RNA molecules to fold back on themselves with strong base-pairing interactions that allows fundamental processes of gene expression to be blocked or allowed as discussed above. In addition, unpaired nucleotides can fold into complex three-dimensional topologies that create specific binding sites for other cellular RNAs, small molecules, and proteins that, when bound, can alter the underlying fold and regulatory function of the RNA [4]. Thus our capacity to engineer with RNA boils down to our ability to engineer its fundamental sequence-structure–function relationship.
Fortunately, we have a rapidly emerging understanding of this relationship based on a strong biophysical foundation that has already led to the beginnings of a system of computational RNA design. Much like for proteins, a given RNA structure is divided into secondary and tertiary structures, the latter defined as the three-dimensional arrangement of its atoms [112]. Unlike for proteins, however, secondary structures often form faster and with more stable free energies than tertiary structures [113]. This critical observation has allowed the decoupling of the RNA secondary and tertiary structure folding problems, and great progress has been made in predicting the ensemble of secondary structures of an RNA sequence using experimentally determined thermodynamic base-pairing parameters [112, 114].
Since RNA-based gene regulation often involves the conditional formation of specific base-pairing patterns, RNA secondary structure prediction algorithms can be immediately converted into computational design tools for engineering RNA-based gene regulators. In addition, recent advances in biochemically characterizing RNA structures and interactions have created experimental approaches that can improve structural modeling and enable observation of ligand-mediated structural switching, but these advances are only just beginning to be used for RNA engineering. In this section we give an overview of these computational and experimental methods in the context of engineering RNA-based gene regulation.
3.1 Computational methods for predicting RNA structure
The most well established methods for computationally predicting RNA structures focus on calculating the secondary structure partition function for an RNA sequence. The partition function, defined from statistical thermodynamics as the Boltzmann-weighted sum over all possible RNA folds, mathematically characterizes the ensemble of structures in which a given RNA sequence can exist in solution. Once calculated, the partition function is the key to equilibrium structural properties of the RNA molecule and can be used to determine the minimum free energy (MFE) RNA structure, suboptimal folds that are close to the MFE, the probability that a given region is base paired over the ensemble of folds [112], and can even be used to statistically sample folds that occur in the ensemble [115]. There are several algorithms that perform these calculations that differ mainly in implementation details and thermodynamic parameter sets. The most commonly used algorithms that also have convenient web servers include RNAStructure [116], ViennaRNA [117], and Unafold [114].
The ability to model RNA secondary structures has been applied to allow more rational engineering of RNA regulators. Most recently, Rodrigo et al. [12] used a search algorithm based on ViennaRNA to assess potential designs of taRNA/crRNA riboregulator pairs as discussed previously. By searching through sequences and modeling their folding and interactions, they were able to generate six orthogonal taRNA/crRNA pairs that showed up to 11-fold activation of translation. Similarly, in order to reduce the number of constructs that had to be experimentally tested, Qi et al. [42] used the M-fold algorithm [118] to assess the folding of designed aptamer and non-coding RNA (ncRNA) fusions to find versions that were predicted to interfere with the structure of the ncRNA in the absence of ligand [60]. Additionally, rational design algorithms have used folding algorithms to develop synthetic riboswitches that activate transcription [13]. In this work, Wachsmuth et al. [13] computationally generated designs that varied within a spacer and a expression platform sequence, while maintaining aptamer sequence, and assessed the MFE structure based on several criteria designed to measure functionality. Finally, folding algorithms have been used to develop design tools focused on the role of general RNA structure in gene regulation. In particular, Salis et al. [10] developed the RBS calculator, an algorithm that uses the Nupack suite of RNA folding algorithms [119] to calculate free energies of competing RNA structures that could prevent ribosome binding to the RBS. Using this information, the RBS calculator models the effective translation initiation rate of a given mRNA sequence competing for ribosome binding within a pool of cellular mRNAs [10]. The RBS calculator was shown to predict measured strength of GFP expression from a library of 5′ UTRs within a factor of 2.3 over a 100 000-fold range and could be used in a forward design mode to design an RBS within an mRNA context to produce a specified amount of protein expression [10].
While excellent progress has been made in using computational RNA structure prediction tools to engineer RNA gene regulatory mechanisms, we emphasize that their inherent approximations should be kept in mind when applying them. First and foremost, most of these algorithms only take into account secondary structures formed from canonical base-pairing interactions, although programs such as MC-Fold and MC-Sim have begun to address this problem [120]. Even then, most algorithms do not predict the formation of pseudoknots, which occur when nucleotides in a loop pair with a region outside the local helices that close the loop [121]. Although a new wave of algorithms such as Turboknot [122] and HotKnots [123] allow pseudoknot formation, these algorithms still do not incorporate the rich set of non-canonical base-pairing interactions known to form functional motifs in a wide array of natural RNAs [124]. The prediction and design of these motifs remain at the forefront of computational RNA structure methods, so such motifs are not currently designable using these tools.
While these approximations should be kept in mind when using secondary structure folding algorithms to engineer RNA folds, it is also important to realize that they model folding conditions where the RNAs have reached thermodynamic equilibrium. Other frontiers in RNA structure-prediction include taking into account co-transcriptional folding kinetics. Co-transcriptional folding has been shown to dynamically induce structural rearrangements in the mRNA being transcribed and is particularly important for structural switches such as riboswitches, ribozymes, and leader peptide attenuators [125]. Computational methods such as Kinefold have begun to provide tools to study RNA folding pathways during transcription, albeit with some of the same inherent limitations regarding approximate thermodynamic parameters and non-canonical base pairs discussed above [126]. Recently, Kinefold was used in an RNA engineering context to design appropriate sequence contexts for proper folding of ribozymes and aptazymes engineered to control the mRNA degradation rate, and thus enzyme levels, in a biosynthetic pathway [11]. We anticipate increased application of co-transcriptional folding algorithms to designing RNA folds, particularly those that dynamically change state to control gene expression.
3.2 Experimental methods for characterization of mRNA structure
Despite the many advances in computational structure methods, the approximations discussed above mean that predicted structures are only a model of what is actually present inside the cell. Although numerous RNA species such as riboswitches [127, 128] and ribozymes have been resolved structurally by X-ray crystallography and NMR, due to the low-throughput nature and the specialization required for these methods, the number of structures is limited, and to date there are no crystal structures of sRNAs. Experimental chemical and enzymatic probing methods offer an alternate way to infer RNA structures and interactions with other molecules, and the potential to couple these methods with next-generation sequencing (NGS) opens the door to ultra high-throughput RNA structural determination (see Section 4).
Experimental RNA structure probing relies on the use of enzymes or chemicals that either cleave or chemically modify RNA in a structure-dependent fashion (Fig. 3). A wide variety of these chemical and enzymatic probes exist, each with different selectivities for a particular RNA sequence or structure [129, 130]. These techniques are commonly used on RNAs that have been folded in vitro, the structure of which is determined by quantifying the number of modification or cleavage events at each nucleotide, which is inherently linked to the underlying structure of the RNA. Modified or cleaved positions can be identified directly by gel electrophoresis or through reverse transcription, which is blocked by modification or cleavage [131]. Reverse transcription creates a pool of truncated DNA species whose 3′ ends marks the location of a chemical modification or cleavage [132]. These reverse transcription products can be read off using a variety of techniques, and there has been considerable effort in making chemical probing experimental protocols relatively straightforward and high-throughput by using capillary electrophoresis [133] and even NGS [17, 18, 20].
Figure 3.

Chemical probing of RNA structure. Specific chemicals covalently modify folded RNAs at unpaired and flexible nucleotide positions. These modified RNAs are converted into DNA via reverse transcription, which is blocked by the modifications, thus creating truncated single-stranded DNA (ssDNA) species. These ssDNA species are then sequenced, and locations of chemical modifications identified to derive a measure of nucleotide “reactivity” to the chemical. Reactivities are then used to infer structural properties of the input RNAs.
Once reverse transcription products have been quantified, there are now robust methods of converting the distributions of modification position into an estimate of the “reactivity” of each nucleotide to the chemical probe [19, 134]. Reactivities can then be used to infer RNA structures and RNA-ligand interactions. Recent progress in this area has utilized experimental reactivities as constraints in RNA-folding algorithms by first converting them into pseudo-free energies that are incorporated along with the thermodynamic free energy parameters [135]. The implementation of this approach in the RNAStructure package has been shown to increase the accuracy of secondary structure predictions [135], and work is being done to establish best practices when utilizing and interpreting this information [136, 137]. Additionally, reactivity constraints have now been included in versions of the RNAStructure's pseudoknot prediction tool [121] as well as the SeqFold structure predication algorithm [138].
An alternative to chemical and enzymatic probing is in-line probing, a technique that harnesses the ability of RNA to non-enzymatically cleave itself at differential rates according to the RNA structure [139]. RNA molecules such as riboswitches are transcribed and folded in vitro and incubated in the presence or absence of ligand for around 48 h. Cleaved products can then be analyzed directly by gel electrophoresis and the structure elicited [139].
Although the application of experimental structure probing to RNA engineering is just beginning, the current research does highlight the enormous potential for structural and functional insight. First of all, these experiments provide validation and improvements of computational predictions, thus increasing the accuracy of structural information forming the basis for rational design. For example, Liu et al. [108], in their design of tna leader peptide adaptors, used experimental structure determination to provide structural validation that secondary structures of the wild-type system could be readily disrupted and still produce a functional regulator. Secondly, experimental structure probing is sensitive to more than just secondary structure, and can also be used to analyze RNA-ligand interactions. In fact, one common chemical probing technique called selective 2′ -hydroxyl acylation analyzed by primer extension (SHAPE) produces lower reactivities at positions that participate in RNA-ligand interactions, and has been used to identify small-molecule-aptamer interactions in riboswitches [140] and RNA-protein interactions [141]. In a recent use of this feature in an engineering context, Qi et al. [60] showed how SHAPE probing could give structural mechanistic validation of the engineered IS10 RNA-OUT-theophylline aptamer fusion.
Perhaps the two most promising aspects of structural probing for RNA engineering are the adaption of these techniques to high-throughput sequencing platforms and the improvements in the chemistry and range of application of the probes. In particular, recent innovations in NGS have enabled the multiplexing of chemical probing experiments, thus significantly increasing the throughput of structural probing experiments. These high-throughput approaches include parallel analysis of RNA structure (PARS) [17] fragmentation sequencing (FragSeq) [18], and selective 2′ -hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) [20] and are discussed in detail in Section 4. In addition, newer generations of SHAPE probes such as 2-methylnicotinic acid imidazolide (NAI) and 2-methyl-3-furoic acid imidazolide (FAI) allow reactions to be carried out at physiological conditions and in vivo [142]. This opens the door to characterize engineered RNA regulators within the myriad of potential interactions in the cellular milieu.
Thus chemical probing has the power to not only improve the accuracy of structural modeling, but also to aid in our understanding of more complex RNA interactions not accessible through computation alone. Experimental structural probing can help elucidate key mechanisms of ligand binding, intermolecular interaction, and conformational switching that lie at the heart of engineering complex modes of RNA regulation. We anticipate these technologies to be instrumental in future RNA engineering efforts.
4 Characterizing RNA gene regulators with next-generation sequencing
NGS refers to a suite of technologies that have created an explosion in the throughput of DNA sequencing (for a review of the technology see [25]). At their core is a sequencing-by-synthesis process that can be spatially arrayed and imaged for millions of molecules at a time. In fact, the high-throughput of NGS has led to the emergence of a whole family of creative applications, called “sequence census” methods, that use the capability to count DNA molecules to ask questions about biological systems [143].
The principle of sequence census methods is simple: (i) use biochemical processes to convert a question of interest into a pool of DNA molecules, (ii) “count” this pool with NGS, (iii) statistically correct for any biases in the conversion and sequencing, and (iv) analyze the corrected counts to answer the question. For example, if the question is “what RNAs are being expressed in a cell population?” then the biochemical conversion would consist of extracting RNA from the population and using reverse transcription to convert it into DNA, while statistical analysis might include normalizing counts to internal references. In fact, such a technique, called RNA-Seq, is well established [15, 16] and similar techniques are applicable to measuring RNA structures and interactions with a wide array of important cellular molecules (Fig. 4).
Figure 4.

Next-generation sequencing can characterize RNA abundances, structures and interactions across the cell. Names of established NGS-techniques that characterize these features are highlighted next to the interactions examined.
4.1 Characterizing RNA abundance
RNA-Seq uses NGS to measure RNA abundance [15, 16] and involves isolation of an RNA population, fragmentation of RNA, and conversion into DNA by random priming. After platform-specific library preparation steps, the library of fragments is then sequenced using the chosen NGS platform. The experimental steps are well established in a number of vendor-specific and published protocols [144], and there are robust computational pipelines available for analyzing read counts to quantify RNA abundance and differential gene expression among different conditions [145].
RNA-Seq is an obvious and powerful component of the synthetic biology characterization toolbox, especially for engineered RNA gene regulators. Since all cellular RNAs are examined simultaneously, RNA-Seq has the potential to characterize both introduced synthetic genetic circuitry and host gene expression patterns, far beyond what the typically used fluorescent protein reporters allow. This capability will allow synthetic biologists to monitor and even engineer interactions with host networks, which could become critical as the field moves to engineer larger and more sophisticated genetic networks. In addition, RNA-Seq has been used to characterize RNA abundance pools generated by SELEX to enable more efficient generation of RNA aptamers [146].
4.2 Characterizing RNA structures
Beyond measuring the abundance of a particular RNA sequence, NGS is providing a platform to generate structural information. As mentioned above, there currently exist three distinct approaches to the high-throughput characterization of RNA structure; PARS [17, 147] FragSeq [18], and SHAPE-Seq [19, 20, 148]. PARS and FragSeq are similar in that they use structure dependent nuclease cleavage to create a distribution of RNA fragments that are sequenced with a variant of the RNA-Seq protocol. By comparing fragment distributions generated from nucleases with different structural sensitivity, structural signatures can be discerned for thousands of RNA species in vitro. In contrast, SHAPE-Seq utilizes a more sensitive chemical probe, influenced by both secondary and tertiary RNA structure, to first chemically modify RNAs in vitro, followed by conversion to cDNA and sequencing as described above [19, 20, 148]. Furthermore, the SHAPE-Seq protocol and data analysis method [148] should be generally applicable to other chemical probes that interrogate other aspects of RNA structure such as solvent accessibility [149] and in vivo structures. Despite these differences, it should be mentioned that each of these techniques are in their infancy, and we fully expect dramatic improvements in accuracy, applicability and throughput.
4.3 Characterizing RNA transcription and translation
Ribosomal profiling is an NGS-based experiment that determines the number of ribosomes translating a particular mRNA species in vivo [21]. The technique is based on capturing actively translating ribosomes with affinity pull-downs, using exonucleases to chew up RNA sequences not bound by protein, then releasing ribosome-bound sequences for subsequent library preparation and sequencing (for a protocol see [150]). Analyzing where ribosome-bound sequences fall on each mRNA in the transcriptome allows the quantification of the relative translation rate on an mRNA species at a given time [151], and also allows researchers to characterize sites of non-canonical translation [152]. Similarly, the position and orientation of RNAP and therefore the effective transcription rate can be determined by global run-on sequencing (GRO-Seq) [22], an analogous technique to ribosomal profiling that targets actively transcribing RNAPs. In an engineering context, profiling ribosome and RNAP positions could be a powerful tool for synthetic biology. In particular, these techniques can be used to understand and troubleshoot network dynamics by quantifying the translation and transcription rate of individual nodes in a network. Moreover, by comparison to endogenous RNA species they can help assess the burden of an implemented network on the population of RNAP and ribosomes, giving complementary information to RNA-Seq experiments. Finally, these methods can be used to functionally characterize RNA regulators of gene expression by measuring the effect of RNA regulators on transcription and translation rates of target mRNAs.
4.4 Characterizing RNA interactions
Much like for ribosomal profiling, RNA's interaction with other cellular proteins can be determined using NGS. RNA immunoprecipitation (RIP-Seq) uses immunoprecipitation of an RNA-protein complex and subsequent reverse transcription and sequencing to identify which RNA sequences interact with a protein of interest [23]. In addition, intermolecular and intramolecular RNA-RNA interactions can be resolved using cross-linking, ligation, and sequencing of hybrids (CLASH) [24]. The CLASH method relies upon purification of RNA-RNA hybrids mediated by cross-linking to a protein-binding partner. These RNA-protein complexes are then immunoprecipitated and the RNA-RNA hybrids ligated, reverse transcribed and sequenced. Because this technique relies upon a protein interaction to the RNA-RNA hybrid it has so far only been used to study ribonucleoprotein complexes. Expansion of this technique to enable generic purification of RNA-RNA hybrids could dramatically increase its potential use for synthetic biology.
In summary, NGS provides an array of high-throughput tools to study RNA within the context of the cell. Although its application to RNA engineering is yet to be fully realized, we believe it is an immensely diverse tool that has the power to fundamentally change the approach to characterizing RNA-based gene regulatory systems.
5 Conclusion
We reviewed and presented the case for RNA's powerful role in the engineering of biological systems. We believe that the combination of being a versatile, designable, and now broadly characterizable regulator of gene expression places RNA in a unique and central position as a master substrate for engineering gene expression. Although much work remains to be done to link these three powerful features and realize RNA's potential, as reviewed above, important strides have been made. While conceivably RNA synthetic biology is still mainly in the phase of engineering basic regulatory parts, its emerging transition toward engineering networks and higher-order regulatory systems holds great promise for synthetic biology as a whole.
Acknowledgments
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1144153 (M.K.T., K.E.W., and S.M.) and by the Defense Advanced Research Projects Agency Young Faculty Award (DARPA YFA) [N66001-12-1-4254 to J.B.L.]. J.B.L. is an Alfred P. Sloan Research Fellow.
The authors declare no financial or commercial conflict of interest.
Glossary
- CRISPR
clustered regulatory interspaced short palindromic repeats
- crRNA
cis-repressed RNA
- FragSeq
fragmentation sequencing
- GFP
green fluorescent protein
- MFE
minimum free energy
- mRNA
messenger RNA
- NGS
next-generation sequencing
- PARS
parallel analysis of RNA structure
- RBS
ribosome binding site
- RNAP
RNA polymerase
- SD
Shine-Dalgarno
- sgRNA
small guide RNA
- SHAPE
selective 2′-hydroxyl acylation analyzed by primer extension
- SHAPE-seq
selective 2′-hydroxyl acylation analyzed by primer extension sequencing
- sRNA
small RNA
- taRNA
trans-activating
- 5′ UTR
5′ untranslated region
Biography
 Founded in July 2011 at Cornell University, the Lucks Laboratory comprises post-docs, graduate students and undergraduate students from a wide range of backgrounds including chemical engineering, biochemistry, biological and environmental engineering, chemistry and physics. Our research interests are in unraveling the design principles that underlie the relationship between the sequence, structure and function of RNA molecules. Once thought to be a passive carrier of genetic information, RNAs are now understood to perform essential functions in the cell through sophisticated three-dimensional structures. Our central goal is to understand and design these structures so that we may utilize RNA function to engineer biomolecular systems as solutions to challenging problems in biology, medicine and biotechnology. To do this, we also create technologies that allow for high-throughput characterization of RNA structures, which we use as a diagnostic tool in RNA engineering. This technology in turn opens new doors through which we can ask fundamental biological questions such as how specific RNA structures mediate cellular processes. With these fundamental investigations, we learn new RNA design principles that then feed back into our engineering methodology. Pictured are (back row) James Chappell, Julius B. Lucks, David Loughrey; (front row) Sarai Meyer, Melissa K. Takahashi, and Kyle E. Watters.
Founded in July 2011 at Cornell University, the Lucks Laboratory comprises post-docs, graduate students and undergraduate students from a wide range of backgrounds including chemical engineering, biochemistry, biological and environmental engineering, chemistry and physics. Our research interests are in unraveling the design principles that underlie the relationship between the sequence, structure and function of RNA molecules. Once thought to be a passive carrier of genetic information, RNAs are now understood to perform essential functions in the cell through sophisticated three-dimensional structures. Our central goal is to understand and design these structures so that we may utilize RNA function to engineer biomolecular systems as solutions to challenging problems in biology, medicine and biotechnology. To do this, we also create technologies that allow for high-throughput characterization of RNA structures, which we use as a diagnostic tool in RNA engineering. This technology in turn opens new doors through which we can ask fundamental biological questions such as how specific RNA structures mediate cellular processes. With these fundamental investigations, we learn new RNA design principles that then feed back into our engineering methodology. Pictured are (back row) James Chappell, Julius B. Lucks, David Loughrey; (front row) Sarai Meyer, Melissa K. Takahashi, and Kyle E. Watters.
References
- 1.Khalil AS, Collins JJ. Synthetic biology: Applications come of age. Nat. Rev. Genet. 2010;11:367–379. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purnick PE, Weiss R. The second wave of synthetic biology: From modules to systems. Nat. Rev. Mol. Cell. Biol. 2009;10:410–422. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
- 3.Cheng AA, Lu TK. Synthetic biology: An emerging engineering discipline. Annu. Rev. Biomed. Eng. 2012;14:155–178. doi: 10.1146/annurev-bioeng-071811-150118. [DOI] [PubMed] [Google Scholar]
- 4.Sharp PA. The centrality of RNA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr. Opin. Microbiol. 2007;10:102–109. doi: 10.1016/j.mib.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Storz G, Opdyke JA, Wassarman KM. Regulating bacterial transcription with small RNAs. Cold Spring Harb. Symp. Quant. Biol. 2006;71:269–273. doi: 10.1101/sqb.2006.71.033. [DOI] [PubMed] [Google Scholar]
- 7.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: Expanding frontiers. Mol. Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Georg J, Hess WR. cis-Antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lioliou E, Romilly C, Romby P, Fechter P. RNA-mediated regulation in bacteria: From natural to artificial systems. N. Biotechnol. 2010;27:222–235. doi: 10.1016/j.nbt.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carothers JM, Goler JA, Juminaga D, Keasling JD. Model-driven engineering of RNA devices to quantitatively program gene expression. Science. 2011;334:1716–1719. doi: 10.1126/science.1212209. [DOI] [PubMed] [Google Scholar]
- 12.Rodrigo G, Landrain TE, Jaramillo A. De novo automated design of small RNA circuits for engineering synthetic riboregulation in living cells. Proc. Natl. Acad. Sci. USA. 2012;109:15271–15276. doi: 10.1073/pnas.1203831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachsmuth M, Findeiss S, Weissheimer N, Stadler PF, Morl M. De novo design of a synthetic riboswitch that regulates transcription termination. Nucleic Acids Res. 2013;41:2541–2551. doi: 10.1093/nar/gks1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xayaphoummine A, Viasnoff V, Harlepp S, Isambert H. Encoding folding paths of RNA switches. Nucleic Acids Res. 2007;35:614–622. doi: 10.1093/nar/gkl1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 17.Kertesz M, Wan Y, Mazor E, Rinn JL, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Underwood JG, Uzilov AV, Katzman S, Onodera CS, et al. FragSeq: Transcriptome-wide RNA structure probing using high-throughput sequencing. Nat. Methods. 2010;7:995–1001. doi: 10.1038/nmeth.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aviran S, Trapnell C, Lucks JB, Mortimer SA, et al. Modeling and automation of sequencing-based characterization of RNA structure. Proc. Natl. Acad. Sci. USA. 2011;108:11069–11074. doi: 10.1073/pnas.1106541108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucks JB, Mortimer SA, Trapnell C, Luo S, et al. Multiplexed RNA structure characterization with selective 2′ -hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc. Natl. Acad. Sci. USA. 2011;108:11063–11068. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao J, Ohsumi TK, Kung JT, Ogawa Y, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc. Natl. Acad. Sci. USA. 2011;108:10010–10015. doi: 10.1073/pnas.1017386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzker ML. Sequencing technologies – the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 26.Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science. 2011;333:1307–1311. doi: 10.1126/science.1205527. [DOI] [PubMed] [Google Scholar]
- 27.Bayer TS, Smolke CD. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat. Biotechnol. 2005;23:337–343. doi: 10.1038/nbt1069. [DOI] [PubMed] [Google Scholar]
- 28.Weigand JE, Suess B. Tetracycline aptamer-controlled regulation of pre-mRNA splicing in yeast. Nucleic Acids Res. 2007;35:4179–4185. doi: 10.1093/nar/gkm425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culler SJ, Hoff KG, Smolke CD. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science. 2010;330:1251–1255. doi: 10.1126/science.1192128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YY, Jensen MC, Smolke CD. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc. Natl. Acad. Sci. USA. 2010;107:8531–8536. doi: 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tigges M, Denervaud N, Greber D, Stelling J, Fussenegger M. A synthetic low-frequency mammalian oscillator. Nucleic Acids Res. 2010;38:2702–2711. doi: 10.1093/nar/gkq121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leisner M, Bleris L, Lohmueller J, Xie Z, Benenson Y. Rationally designed logic integration of regulatory signals in mammalian cells. Nat Nanotechnol. 2010;5:666–670. doi: 10.1038/nnano.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alon U. Network motifs: Theory and experimental approaches. Nat. Rev. Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 34.Jacques N, Dreyfus M. Translation initiation in Escherichia coli: Old and new questions. Mol. Microbiol. 1990;4:1063–1067. doi: 10.1111/j.1365-2958.1990.tb00679.x. [DOI] [PubMed] [Google Scholar]
- 35.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 36.Huttenhofer A, Noller HF. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 1994;13:3892–3901. doi: 10.1002/j.1460-2075.1994.tb06700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shine J, Dalgarno L. Identical 3'-terminal octanucleotide sequence in 18S ribosomal ribonucleic acid from different eukaryotes. A proposed role for this sequence in the recognition of terminator codons. Biochem. J. 1974;141:609–615. doi: 10.1042/bj1410609a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steitz JA, Jakes K. How ribosomes select initiator regions in mRNA: Base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1975;72:4734–4738. doi: 10.1073/pnas.72.12.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Smit MH, van Duin J. Secondary structure of the ribosome binding site determines translational efficiency: A quantitative analysis. Proc. Natl. Acad. Sci. USA. 1990;87:7668–7672. doi: 10.1073/pnas.87.19.7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mutalik VK, Guimaraes JC, Cambray G, Lam C, et al. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- 42.Lou C, Stanton B, Chen YJ, Munsky B, Voigt CA. Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol. 2012;30:1137–1142. doi: 10.1038/nbt.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. RNA processing enables predictable programming of gene expression. Nat. Biotechnol. 2012;30:1002–1006. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- 44.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desnoyers G, Bouchard MP, Masse E. New insights into small RNA-dependent translational regulation in prokaryotes. Trends Genet. 2013;29:92–98. doi: 10.1016/j.tig.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 47.Bouvier M, Sharma CM, Mika F, Nierhaus KH, Vogel J. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol. Cell. 2008;32:827–837. doi: 10.1016/j.molcel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 48.Darfeuille F, Unoson C, Vogel J, Wagner EG. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell. 2007;26:381–392. doi: 10.1016/j.molcel.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 49.Kittle JD, Simons RW, Lee J, Kleckner N. Insertion sequence IS10 anti-sense pairing initiates by an interaction between the 5′ end of the target RNA and a loop in the anti-sense RNA. J. Mol. Biol. 1989;210:561–572. doi: 10.1016/0022-2836(89)90132-0. [DOI] [PubMed] [Google Scholar]
- 50.Gerdes K, Thisted T, Martinussen J. Mechanism of post-segregational killing by the hok/sok system of plasmid R1: Sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid-free cells. Mol. Microbiol. 1990;4:1807–1818. doi: 10.1111/j.1365-2958.1990.tb02029.x. [DOI] [PubMed] [Google Scholar]
- 51.Frohlich KS, Vogel J. Activation of gene expression by small RNA. Curr. Opin. Microbiol. 2009;12:674–682. doi: 10.1016/j.mib.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 52.Morfeldt E, Taylor D, von Gabain A, Arvidson S. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 1995;14:4569–4577. doi: 10.1002/j.1460-2075.1995.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc. Natl. Acad. Sci. USA. 1998;95:12462–12467. doi: 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Isaacs FJ, Dwyer DJ, Ding C, Pervouchine DD, et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 2004;22:841–847. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 55.Callura JM, Cantor CR, Collins JJ. Genetic switchboard for synthetic biology applications. Proc. Natl. Acad. Sci. USA. 2012;109:5850–5855. doi: 10.1073/pnas.1203808109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Friedland AE, Lu TK, Wang X, Shi D, et al. Synthetic gene networks that count. Science. 2009;324:1199–1202. doi: 10.1126/science.1172005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mutalik VK, Qi L, Guimaraes JC, Lucks JB, Arkin AP. Rationally designed families of orthogonal RNA regulators of translation. Nat. Chem. Biol. 2012;8:447–454. doi: 10.1038/nchembio.919. [DOI] [PubMed] [Google Scholar]
- 58.Sharma V, Yamamura A, Yokobayashi Y. Engineering artificial small RNAs for conditional gene silencing in Escherichia coli. ACS Synth. Biol. 2012;1:6–13. doi: 10.1021/sb200001q. [DOI] [PubMed] [Google Scholar]
- 59.Na D, Yoo SM, Chung H, Park H, et al. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 2013;31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- 60.Qi L, Lucks JB, Liu CC, Mutalik VK, Arkin AP. Engineering naturally occurring trans-acting non-coding RNAs to sense molecular signals. Nucleic Acids Res. 2012;40:5775–5786. doi: 10.1093/nar/gks168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 62.Breaker RR. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012;4:1–15. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang JX, Lee ER, Morales DR, Lim J, Breaker RR. Riboswitches that sense S-adenosylhomocysteine and activate genes involved in coenzyme recycling. Mol. Cell. 2008;29:691–702. doi: 10.1016/j.molcel.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nou X, Kadner RJ. Adenosylcobalamin inhibits ribosome binding to btuB RNA. Proc. Natl. Acad. Sci. USA. 2000;97:7190–7195. doi: 10.1073/pnas.130013897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winkler W, Nahvi A, Breaker RR. Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature. 2002;419:952–956. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 66.Mandal M, Lee M, Barrick JE, Weinberg Z, et al. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 67.Sudarsan N, Hammond MC, Block KF, Welz R, et al. Tandem riboswitch architectures exhibit complex gene control functions. Science. 2006;314:300–304. doi: 10.1126/science.1130716. [DOI] [PubMed] [Google Scholar]
- 68.Tang J, Breaker RR. Rational design of allosteric ribozymes. Chem. Biol. 1997;4:453–459. doi: 10.1016/s1074-5521(97)90197-6. [DOI] [PubMed] [Google Scholar]
- 69.Desai SK, Gallivan JP. Genetic screens and selections for small molecules based on a synthetic riboswitch that activates protein translation. J. Am. Chem. Soc. 2004;126:13247–13254. doi: 10.1021/ja048634j. [DOI] [PubMed] [Google Scholar]
- 70.Suess B, Fink B, Berens C, Stentz R, Hillen W. A theophylline responsive riboswitch based on helix slipping controls gene expression in vivo. Nucleic Acids Res. 2004;32:1610–1614. doi: 10.1093/nar/gkh321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lynch SA, Desai SK, Sajja HK, Gallivan JP. A high-throughput screen for synthetic riboswitches reveals mechanistic insights into their function. Chem. Biol. 2007;14:173–184. doi: 10.1016/j.chembiol.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lynch SA, Gallivan JP. A flow cytometry-based screen for synthetic riboswitches. Nucleic Acids Res. 2009;37:184–192. doi: 10.1093/nar/gkn924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Topp S, Gallivan JP. Guiding bacteria with small molecules and RNA. J. Am. Chem. Soc. 2007;129:6807–6811. doi: 10.1021/ja0692480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 75.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 76.Stoltenburg R, Reinemann C, Strehlitz B. SELEX – a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007;24:381–403. doi: 10.1016/j.bioeng.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 77.Hanson S, Bauer G, Fink B, Suess B. Molecular analysis of a synthetic tetracycline-binding riboswitch. RNA. 2005;11:503–511. doi: 10.1261/rna.7251305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ogawa A. Rational design of artificial riboswitches based on ligand-dependent modulation of internal ribosome entry in wheat germ extract and their applications as label-free biosensors. RNA. 2011;17:478–488. doi: 10.1261/rna.2433111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stripecke R, Oliveira CC, McCarthy JE, Hentze MW. Proteins binding to 5′ untranslated region sites: A general mechanism for translational regulation of mRNAs in human and yeast cells. Mol. Cell. Biol. 1994;14:5898–5909. doi: 10.1128/mcb.14.9.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sinha J, Reyes SJ, Gallivan JP. Reprogramming bacteria to seek and destroy an herbicide. Nat. Chem. Biol. 2010;6:464–470. doi: 10.1038/nchembio.369. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Serganov A, Patel DJ. Ribozymes, riboswitches and beyond: Regulation of gene expression without proteins. Nat. Rev. Genet. 2007;8:776–790. doi: 10.1038/nrg2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wieland M, Hartig JS. Improved aptazyme design and in vivo screening enable riboswitching in bacteria. Angew. Chem. Int. Ed. Engl. 2008;47:2604–2607. doi: 10.1002/anie.200703700. [DOI] [PubMed] [Google Scholar]
- 83.Klauser B, Hartig JS. An engineered small RNA-mediated genetic switch based on a ribozyme expression platform. Nucleic Acids Res. 2013;41:5542–5552. doi: 10.1093/nar/gkt253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carpousis AJ. The RNA degradosome of Escherichia coli: An mRNA-degrading machine assembled on RNase E. Annu. Rev. Microbiol. 2007;61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 85.Carrier TA, Keasling JD. Library of synthetic 5′ secondary structures to manipulate mRNA stability in Escherichia coli. Biotechnol. Prog. 1999;15:58–64. doi: 10.1021/bp9801143. [DOI] [PubMed] [Google Scholar]
- 86.Morita T, Maki K, Aiba H. RNase E-based ribonucleoprotein complexes: Mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev. 2005;19:2176–2186. doi: 10.1101/gad.1330405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Afonyushkin T, Vecerek B, Moll I, Blasi U, Kaberdin VR. Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res. 2005;33:1678–1689. doi: 10.1093/nar/gki313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kawamoto H, Morita T, Shimizu A, Inada T, Aiba H. Implication of membrane localization of target mRNA in the action of a small RNA: Mechanism of post-transcriptional regulation of glucose transporter in Escherichia coli. Genes Dev. 2005;19:328–338. doi: 10.1101/gad.1270605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pfeiffer V, Papenfort K, Lucchini S, Hinton JC, Vogel J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 2009;16:840–846. doi: 10.1038/nsmb.1631. [DOI] [PubMed] [Google Scholar]
- 90.Opdyke JA, Kang JG, Storz G. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J. Bacteriol. 2004;186:6698–6705. doi: 10.1128/JB.186.20.6698-6705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Obana N, Shirahama Y, Abe K, Nakamura K. Stabilization of Clostridium perfringens collagenase mRNA by VR-RNA-dependent cleavage in 5′ leader sequence. Mol. Microbiol. 2010;77:1416–1428. doi: 10.1111/j.1365-2958.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- 92.Man S, Cheng R, Miao C, Gong Q, et al. Artificial trans-encoded small non-coding RNAs specifically silence the selected gene expression in bacteria. Nucleic Acids Res. 2011;39:1–15. doi: 10.1093/nar/gkr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Collins JA, Irnov I, Baker S, Winkler WC. Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 2007;21:3356–3368. doi: 10.1101/gad.1605307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deana A, Belasco JG. Lost in translation: The influence of ribosomes on bacterial mRNA decay. Genes Dev. 2005;19:2526–2533. doi: 10.1101/gad.1348805. [DOI] [PubMed] [Google Scholar]
- 95.Caron MP, Bastet L, Lussier A, Simoneau-Roy M, et al. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc. Natl. Acad. Sci. USA. 2012;109:3444–3453. doi: 10.1073/pnas.1214024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Richardson JP. Rho-dependent termination and ATPases in transcript termination. Biochim. Biophys. Acta. 2002;1577:251–260. doi: 10.1016/s0167-4781(02)00456-6. [DOI] [PubMed] [Google Scholar]
- 97.Farnham PJ, Platt T. Rho-independent termination: Dyad symmetry in DNA causes RNA polymerase to pause during transcription in vitro. Nucleic Acids Res. 1981;9:563–577. doi: 10.1093/nar/9.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Larson MH, Greenleaf WJ, Landick R, Block SM. Applied force reveals mechanistic and energetic details of transcription termination. Cell. 2008;132:971–982. doi: 10.1016/j.cell.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lucks JB, Qi L, Mutalik VK, Wang D, Arkin AP. Versatile RNA-sensing transcriptional regulators for engineering genetic networks. Proc. Natl. Acad. Sci. USA. 2011;108:8617–8622. doi: 10.1073/pnas.1015741108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kumar CC, Novick RP. Plasmid pT181 replication is regulated by two countertranscripts. Proc. Natl. Acad. Sci. USA. 1985;82:638–642. doi: 10.1073/pnas.82.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Takahashi MK, Lucks JB. A modular strategy for engineering orthogonal chimeric RNA transcription regulators. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt452. DOI: 10.1093/nar/gkt452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hollands K, Proshkin S, Sklyarova S, Epshtein V, et al. Riboswitch control of Rho-dependent transcription termination. Proc. Natl. Acad. Sci. USA. 2012;109:5376–5381. doi: 10.1073/pnas.1112211109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mironov AS, Gusarov I, Rafikov R, Lopez LE, et al. Sensing small molecules by nascent RNA: A mechanism to control transcription in bacteria. Cell. 2002;111:747–756. doi: 10.1016/s0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 104.Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 2004;11:29–35. doi: 10.1038/nsmb710. [DOI] [PubMed] [Google Scholar]
- 105.Ceres P, Garst AD, Marcano-Velazquez JG, Batey RT. Modularity of select riboswitch expression platforms enables facile engineering of novel genetic regulatory devices. ACS Synth. Biol. 2013;2:463–472. doi: 10.1021/sb4000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gong F, Yanofsky C. Instruction of translating ribosome by nascent peptide. Science. 2002;297:1864–1867. doi: 10.1126/science.1073997. [DOI] [PubMed] [Google Scholar]
- 107.Liu CC, Qi L, Yanofsky C, Arkin AP. Regulation of transcription by unnatural amino acids. Nat. Biotechnol. 2011;29:164–168. doi: 10.1038/nbt.1741. [DOI] [PubMed] [Google Scholar]
- 108.Liu CC, Qi L, Lucks JB, Segall-Shapiro TH, et al. An adaptor from translational to transcriptional control enables predictable assembly of complex regulation. Nat. Methods. 2012;9:1088–1094. doi: 10.1038/nmeth.2184. [DOI] [PubMed] [Google Scholar]
- 109.Qi LS, Larson MH, Gilbert LA, Doudna JA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Belmont BJ, Niles JC. Engineering a direct and inducible protein–RNA interaction to regulate RNA biology. ACS Chem. Biol. 2010;5:851–861. doi: 10.1021/cb100070j. [DOI] [PubMed] [Google Scholar]
- 111.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Organization of intracellular reactions with rationally designed RNA assemblies. Science. 2011;333:470–474. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 112.Seetin MG, Mathews DH. RNA structure prediction: An overview of methods. Methods Mol. Biol. 2012;905:99–122. doi: 10.1007/978-1-61779-949-5_8. [DOI] [PubMed] [Google Scholar]
- 113.Onoa B, Tinoco I., Jr RNA folding and unfolding. Curr. Opin. Struct. Biol. 2004;14:374–379. doi: 10.1016/j.sbi.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 114.Mathews DH, Turner DH. Prediction of RNA secondary structure by free energy minimization. Curr. Opin. Struct. Biol. 2006;16:270–278. doi: 10.1016/j.sbi.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 115.Chan CY, Lawrence CE, Ding Y. Structure clustering features on the Sfold Web server. Bioinformatics. 2005;21:3926–3928. doi: 10.1093/bioinformatics/bti632. [DOI] [PubMed] [Google Scholar]
- 116.Reuter JS, Mathews DH. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129–138. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011;32:170–173. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- 120.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 121.Hajdin CE, Bellaousov S, Huggins W, Leonard CW, et al. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl. Acad. Sci. USA. 2013;110:5498–5503. doi: 10.1073/pnas.1219988110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Seetin MG, Mathews DH. TurboKnot: Rapid prediction of conserved RNA secondary structures including pseudoknots. Bioinformatics. 2012;28:792–798. doi: 10.1093/bioinformatics/bts044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ren J, Rastegari B, Condon A, Hoos HH. HotKnots: Heuristic prediction of RNA secondary structures including pseudoknots. RNA. 2005;11:1494–1504. doi: 10.1261/rna.7284905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leontis NB, Westhof E. Analysis of RNA motifs. Curr. Opin. Struct. Biol. 2003;13:300–308. doi: 10.1016/s0959-440x(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 125.Pan T, Sosnick T. RNA folding during transcription. Annu. Rev. Biophys. Biomol. Struct. 2006;35:161–175. doi: 10.1146/annurev.biophys.35.040405.102053. [DOI] [PubMed] [Google Scholar]
- 126.Xayaphoummine A, Bucher T, Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005;33:605–610. doi: 10.1093/nar/gki447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lu C, Ding F, Chowdhury A, Pradhan V, et al. SAM recognition and conformational switching mechanism in the Bacillus subtilis yitJ S box/SAM-I riboswitch. J. Mol. Biol. 2010;404:803–818. doi: 10.1016/j.jmb.2010.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Q, Kang M, Peterson RD, Feigon J. Comparison of solution and crystal structures of preQ1 riboswitch reveals calcium-induced changes in conformation and dynamics. J. Am. Chem. Soc. 2011;133:5190–5193. doi: 10.1021/ja111769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weeks KM. Advances in RNA structure analysis by chemical probing. Curr. Opin. Struct. Biol. 2010;20:295–304. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Knapp G. Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol. 1989;180:192–212. doi: 10.1016/0076-6879(89)80102-8. [DOI] [PubMed] [Google Scholar]
- 131.Krol A, Carbon P. A guide for probing native small nuclear RNA and ribonucleoprotein structures. Methods Enzymol. 1989;180:212–227. doi: 10.1016/0076-6879(89)80103-x. [DOI] [PubMed] [Google Scholar]
- 132.Brunel C, Romby P. Probing RNA structure and RNA–ligand complexes with chemical probes. Methods Enzymol. 2000;318:3–21. doi: 10.1016/s0076-6879(00)18040-1. [DOI] [PubMed] [Google Scholar]
- 133.Mortimer SA, Weeks KM. Time-resolved RNA SHAPE chemistry: Quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat. Protoc. 2009;4:1413–1421. doi: 10.1038/nprot.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Karabiber F, McGinnis JL, Favorov OV, Weeks KM. QuShape: Rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA. 2013;19:63–73. doi: 10.1261/rna.036327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Low JT, Weeks KM. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52:150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kladwang W, VanLang CC, Cordero P, Das R. Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry. 2011;50:8049–8056. doi: 10.1021/bi200524n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ramachandran S, Ding F, Weeks KM, Dokholyan NV. Statistical analysis of SHAPE-directed RNA secondary structure modeling. Biochemistry. 2013;52:596–599. doi: 10.1021/bi300756s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ouyang Z, Snyder MP, Chang HY. SeqFold: Genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res. 2013;23:377–387. doi: 10.1101/gr.138545.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Regulski EE, Breaker RR. In-line probing analysis of riboswitches. Methods Mol. Biol. 2008;419:53–67. doi: 10.1007/978-1-59745-033-1_4. [DOI] [PubMed] [Google Scholar]
- 140.Steen KA, Malhotra A, Weeks KM. Selective 2′ -hydroxyl acylation analyzed by protection from exoribonuclease. J. Am. Chem. Soc. 2010;132:9940–9943. doi: 10.1021/ja103781u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gherghe C, Lombo T, Leonard CW, Datta SA, et al. Definition of a high-affinity Gag recognition structure mediating packaging of a retroviral RNA genome. Proc. Natl. Acad. Sci. USA. 2010;107:19248–19253. doi: 10.1073/pnas.1006897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Spitale RC, Crisalli P, Flynn RA, Torre EA, et al. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wold B, Myers RM. Sequence census methods for functional genomics. Nat. Methods. 2008;5:19–21. doi: 10.1038/nmeth1157. [DOI] [PubMed] [Google Scholar]
- 144.Wilhelm BT, Marguerat S, Goodhead I, Bahler J. Defining transcribed regions using RNA-seq. Nat. Protoc. 2010;5:255–266. doi: 10.1038/nprot.2009.229. [DOI] [PubMed] [Google Scholar]
- 145.Trapnell C, Roberts A, Goff L, Pertea G, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schutze T, Wilhelm B, Greiner N, Braun H, et al. Probing the SELEX process with next-generation sequencing. PLoS ONE. 2011;6:e29604. doi: 10.1371/journal.pone.0029604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wan Y, Qu K, Ouyang Z, Chang HY. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat. Protoc. 2013;8:849–869. doi: 10.1038/nprot.2013.045. [DOI] [PubMed] [Google Scholar]
- 148.Mortimer SA, Trapnell C, Aviran S, Pachter L, Lucks JB. SHAPE-Seq: High-throughput RNA structure analysis. Curr. Prot. Chem. Biol. 2012;4:275–297. doi: 10.1002/9780470559277.ch120019. [DOI] [PubMed] [Google Scholar]
- 149.Weeks KM, Mauger DM. Exploring RNA structural codes with SHAPE chemistry. Acc. Chem. Res. 2011;44:1280–1291. doi: 10.1021/ar200051h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Steitz JA. Polypeptide chain initiation: Nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature. 1969;224:957–964. doi: 10.1038/224957a0. [DOI] [PubMed] [Google Scholar]
- 152.Li GW, Oh E, Weissman JS. The anti-Shine–Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]