

Abstract
Herein we report a direct and efficient method for the synthesis of four new carboxylate-isostere analogs of daptomycin. The side chain carboxylic acid moieties of the aspartic acids (Asp-3, Asp-7 and Asp-9) and β-methyl glutamic acid (MeGlu-12) were all converted into the corresponding carboxylate isosteres using direct synthetic procedures. The present study also describes an esterification protocol to overcome the possible backbone cyclization of the activated side chain carboxylic acid group of either Asp or Glu, onto the backbone amide.
Introduction
Bacterial resistance to clinically available antibiotics continues to be a growing health problem.1 Additionally, multi-drug resistant bacterial infections are becoming increasingly difficult to treat with even the most recently approved antibiotics.1 Therefore, it is of high priority to identify new antibacterial agents to treat such infections. A practical approach to antibacterial drug discovery could involve identification of derivatives of currently known antibacterial agents; perhaps, if a desired modification allows one to access structural analogs in a readily fashion. We initiated a program to identify chemical methods to modify daptomycin (Cubicin®, 1).
Daptomycin is a clinically used lipodepsipeptide antibiotic for the treatment of Gram-positive bacterial infections, including several drug-resistant infections.2,3,4 Despite daptomycin’s remarkable antibacterial property, it is not approved for the treatment of pulmonary infections due to its high affinity to pulmonary surfactants.5 The overall anionic nature of daptomycin appears to play a major role in such surfactant interactions.6 Hence, there has been an increased interest in identifying novel daptomycin derivatives with reduced surfactant interactions and improved potency.7,8,9
Typically, pore-forming antimicrobial peptides have several cationic residues that enable these peptides to adhere to the anionic bacterial membrane.10 Daptomycin and similar calcium binding lipopeptides are functionally quite opposite, because they are overall anionic peptides. We initially hypothesized that conversion of these anionic residues into other functional groups; perhaps into amines would be a useful transformation. However, the complexity of daptomycin makes the selective chemical modification of a particular amino acid residue a challenging task. Since the four carboxylic acids in daptomycin seem to play an important role in the biological activity,11 we decided to develop a direct and efficient method to convert the acid groups into carboxylate isosteres.12 Such modification may allow us to study the relationship between the anionic nature of these residues and the bioactivity and lung surfactant interactions.
We envisioned that modifying the carboxylic acid groups into the corresponding methyl esters (2), primary amides (3), N,O-dimethyl-amides (4) and hydroxamic acids (5) would provide a selection of previously unknown carboxylate isostere analogs of daptomycin (Fig. 2).
Fig. 2.
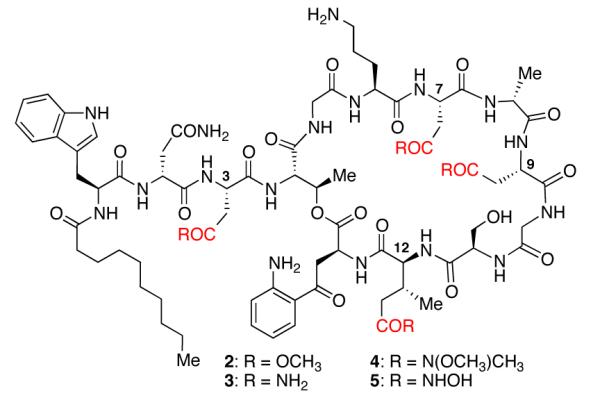
Structures of proposed carboxylate isostere analogs
Results and discussion
Our synthetic approach began with a Boc-protection of ornithine residue (Orn-6) to yield Boc-protected daptomycin (8). Our initial attempts to synthesize 2 and other ester analogs of daptomycin yielded an unexpected result, wherein various approaches to activate the carboxylic acid gave a mixture of side products. We identified these derivatives as a statistical mixture of dehydrated daptomycin analogs. We rationalized that a dehydration reaction is possible if the activated side chain carboxylates undergo a backbone cyclization, which could yield either an imide (6) or an oxazolone derivative (7).
To gain more understanding of this unanticipated dehydration reaction, we have purposely prepared the tetra-dehydrated analog of daptomycin by reacting 8 with HBTU and i-Pr2NEt at 30 °C. The product has been subjected to acid and base hydrolysis as well as to reactions with alcohol and amine nucleophiles. However, the dehydrated product appeared to be surprisingly stable. Although we have no direct evidence, we postulate the formation of imide moieties under the reported reaction conditions.13 After screening several conditions, we developed a protocol that considerably minimized the dehydration side reaction. We activated 8 using pivalic anhydride at 0 °C, then treated with catalytic DMAP and excess methanol to convert the mixed anhydride into the methyl ester (Scheme 2). In the absence of DMAP, very low conversion to the desired product was observed, with formation of a mixture of dehydrated derivatives. The reaction afforded the desired tetra-methyl ester derivative in 48% yield. Since esterification of side chain carboxylic acids found in peptide natural products seems to yield undesired products, the reported strategy allows access to ester derivatives of such peptide natural products more efficiently.
Scheme 2.

Reagents and conditions: Condition A: (i) pivalic anhydride, i-Pr2NEt, DMF, DMAP, MeOH, 0 °C – 23 °C, 48%; (ii) TFA/CH2Cl2, 82%. Condition B: (i) HBTU, i-Pr2NEt, DMF, NH4Cl, 0 °C – 23 °C, 78%; (ii) TFA/CH2Cl2, 85%. Condition C: (i) HBTU, i-Pr2NEt, DMF, HN(OMe)Me•HCl, 0 °C – 23 °C, 75%; (ii) TFA/CH2Cl2, 88%.
Additionally, we have also developed a direct and general method to convert the carboxylic acid groups into amides and hydroxamic acids. For the synthesis of 3, Boc-daptomycin was activated using HBTU at 0 °C, and subsequently reacted with NH4Cl (Scheme 2). The reaction cleanly provides the desired tetra-modified derivative in full conversion. We observed that even with amidation, the dehydration becomes a competitive side reaction at room temperature. We were able to suppress the dehydration considerably by performing the amidation at 0 ° C for the first 60 minutes and then allowing it to go to completion at room temperature. In the next step, the Boc-group of ornithine was deprotected using trifluoroacetic acid (TFA) to yield 3. The Weinreb-amide analog (4) was readily synthesized using a similar procedure in high yield as well (Scheme 2).
The synthesis of the tetra-hydroxamic acid analog 5 required more optimization of reaction conditions compared to the amidation reactions. When we attempted the reaction of 8 with unprotected hydroxylamine, a complex mixture was obtained. After screening several O-protected hydroxylamines, we finally arrived at O-(tetrahydro-pyran-2-yl)-hydroxylamine (THP-ONH2) as a suitable reagent. Since THP-group is acid labile, we anticipated a one-pot deprotection of both THP- and Boc-groups to afford the desired daptomycin derivatives. Under optimized conditions, 8 reacts with THP-ONH2, in the presence of HBTU and i-Pr2NEt to afford the desired product 9 (Scheme 3). During the deprotection step, we noticed that the use of either methanol or ethanol as a nucleophilic additive causes decomposition of 9. After optimization, we have found that a two step procedure worked exceptionally well, where a 2:1 TFA/2-propanol (IPA) mixture allows us to remove the THP-groups cleanly. In a subsequent step, the Boc-group is removed using a mixture of 1:1 TFA/CH2Cl2. This protocol allowed us to prepare either the Bocprotected tetra-hydroxamic acid analog (10) or the fully deprotected analog (5) in high yield (Scheme 3).
Scheme 3.

Reagents and conditions: (i) HBTU, i-Pr2NEt, DMF, THPONH2, 0 °C – 23 °C; (ii) TFA/IPA (1:2), 0 °C – 23 °C, 29% (over two steps); (iii) TFA/CH2Cl2 (1:1), 23 °C, 74%.
The amide analog 3 is structurally interesting, as the Asp-3, Asp-7, Asp-9 and MeGlu-12 residues were all converted into the corresponding Asn or MeGln residues, yielding a polyamide natural product analog of daptomycin. The hydroxamic acid analog 5 may also present interesting possibilities, for example, perhaps as a ligand for metal ions, in particular ferric ion (Fe3+).14 Thus, the hydroxamic acid analog 5 may resemble siderophore analog of daptomycin.
As the developed protocol allowed us to perform a global modificaton of the acidic groups in Asp-2, Asp-7, Asp-9 and MeGlu-12 residues, we were able to confirm the identity of the products through RP-HPLC and high-resolution UPLC-MS analyses. To further confirm the site of modification, we decided to perform an UPLC-MS/MS fragmentation analysis of these analogs. For this, we envisioned to hydrolyze the macrolactone ring of daptomycin analogs to obtain the linear derivatives, which can be fragmented for sequence analysis. In order to validate the hydrolysis condition and subsequent LC-MS/MS protocol, we hydrolyzed the macrolactone ring of daptomycin (1) using aqueous NaOH (pH~9) to obtain the linear daptomycin, and confirmed the structure using LC-MS/MS analysis. The UPLC-MS/MS fragmentation analysis of the daptomycin derivatives is shown in Fig. 3, where the fragmentation pattern is indicated.
Fig. 3.
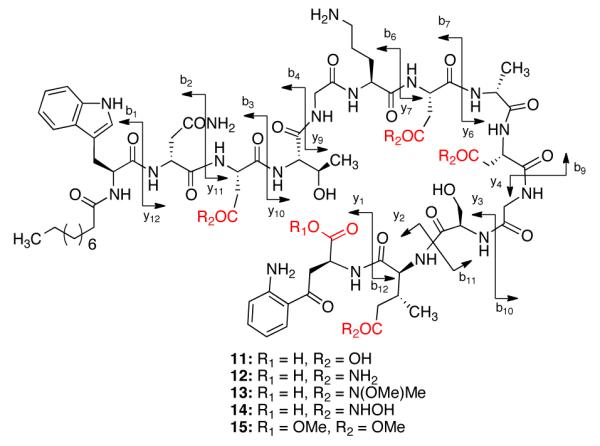
Structures of linear daptomycin derivatives and their MS/MS fragmentation pattern
Under analogous conditions, the tetra-amide 3 was also hydrolyzed to synthesize the linear product. Interestingly, we noticed that the reaction conditions afforded two different products by RP-HPLC and LC-MS analysis in a 1:1 mixture, during this process. One of them was identified as the linear tetraamide and the second product was found to be a tri-amide derivative, where one of the side chains has been hydrolyzed to the carboxylic acid. The UPLC-MS/MS fragmentation analysis of the tetra-amide analog is shown in Table 1, where the fragmentation pattern and corresponding molecular ions are indicated. The analysis of the tri-amide analog indicates that the modification at residue 9 (Asp-9) is highly labile to basic hydrolysis and yields the Asp-9 residue during this process (see supporting information). Using a similar approach, we sequenced the other daptomycin analogs as well (Table 1). The UPLC-MS/MS approach enabled us to unambiguously determine the structure of all of the tetra-modified daptomycin analogs (Fig. 3).
Table 1.
LC-MS/MS fragmentation ions observed during the sequence analysis of 11–15.
| ion | 11(m/z) | 12(m/z) | 13(m/z) | 14(m/z) | 15(m/z) |
|---|---|---|---|---|---|
| b1 | 341.2 | 341.2 | 341.2 | 341.2 | 341.2 |
| b2 | 455.3 | 455.3 | 455.3 | 455.3 | 455.3 |
| b3 | 570.3 | 569.3 | 613.3 | 585.3 | 584.3 |
| b4 | 671.3 | 670.3 | 714.3 | 686.3 | 685.3 |
| b5 | - | - | 771.4 | 743.4 | 742.3 |
| b6 | - | - | - | 857.4 | - |
| b7 | 957.5 | 955.5 | 1043.5 | 987.5 | 985.5 |
| b8 | 1028.5 | 1026.6 | 1114.6 | 1058.5 | 1056.5 |
| b9 | 1143.5 | 1140.6 | 1272.7 | 1188.5 | 1185.5 |
| b10 | 1200.5 | 1197.6 | 1329.7 | - | 1242.6 |
| b11 | 1287.6 | 1284.7 | 1416.8 | - | 1329.6 |
| b12 | 1430.6 | 1426.7 | 1602.8 | 1490.7 | 1486.7 |
| y1 | 209.1 | 209.1 | 209.1 | 209.1 | 223.1 |
| y2 | 352.1 | 351.2 | 395.2 | 367.1 | 380.2 |
| y3 | 439.2 | 438.2 | 482.2 | 454.2 | - |
| y4 | 496.2 | 495.2 | 539.3 | 511.2 | 524.2 |
| y5 | 611.2 | 609.3 | 697.3 | 641.2 | 653.3 |
| y6 | 682.2 | 680.3 | 768.4 | - | 724.3 |
| y7 | 797.3 | 794.3 | 1040.5 | - | 853.3 |
| y8 | 911.4 | 908.4 | - | 956.4 | 967.4 |
| y9 | 968.4 | 965.5 | 1097.5 | - | 1024.4 |
| y10 | 1069.4 | 1066.5 | 1198.6 | 1114.5 | 1125.5 |
| y11 | 1184.4 | 1180.6 | - | 1244.6 | 1254.5 |
| y12 | 1298.5 | 1294.6 | 1470.6 | 1358.5 | 1368.6 |
In addition, we characterized all four analogs using a combination of one- and two-dimensional NMR experiments. Daptomycin is used as the standard to assign the proton chemical shifts for each amino acids (see supporting information). The synthetic analogs were characterized using a comparative analysis of the 1H-13C gHSQC correlation experimental data (see supporting information ). The overall chemical shift patterns of all analogs are preserved relative to daptomycin. The aromatic C-H correlations remained unaffected, while in the aliphatic region, subtle shifts of the β-proton of Asp residues are observed. This is consistent with the modification of these residues. In addition, C-H resonances for the extra –OMe and –NMe groups in 2 and 4 are observed in the aliphatic region (see supporting information).
Conclusions
In conclusion, we report a direct and efficient method to access several carboxylate isostere analogs of the lipopeptide antibiotic, daptomycin. During this study, we have developed an improved synthetic protocol that suppresses the often-observed backbone-cyclization side reaction that occurs when modifying Asp or Glu residues found in complex peptide natural products. This method allowed us to prepare an ester analog of daptomycin efficiently. Additionally, we report the synthesis of three other unique carboxylate isosteres, which include a primary amide, a Weinrebamide and a hydroxamic acid analogs. In addition to being novel tetra-modified carboxylate isostere analogs, these substrates are potential precursors for further chemical derivatization. Finally, the reported synthetic strategy tolerates a wide range of functional groups found in daptomycin, providing a potentially useful and practical approach that could be applicable to the derivatization of other complex peptide natural products known in literature. Finally, the reported UPLC-MS/MS technique may be highly applicable for the characterization of cyclic macrolactone containing peptide natural products and their derivatives.
Experimental
General
All commercially available reagents were purchased and used without further purification, unless otherwise stated. Air and moisture sensitive reactions were done under nitrogen atmosphere employing flame-dried glassware. Commercially available solvents (> 99.0% purity) were used for column chromatography without any further purification. Liquid chromatography-mass spectrometry (LC-MS) was performed on a Waters Acquity instrument equipped with dual atmospheric pressure chemical ionization (API)/electrospray ionization (ESI), a SQ mass spectrometer, and a photodiode array detector. High-resolution liquid chromatography-mass spectrometry (HR-LC/MS) was performed on a Waters XEVO instrument equipped with ESI, a QToF mass spectrometer, and a photodiode array detector. Analytical high-performance liquid chromatography (HPLC) was performed on an Agilent 1100 Series instrument equipped with a diode array detector. Preparative HPLC was performed on a Gilson instrument equipped with a diode array detector and liquid handler. Medium-performance liquid chromatography (MPLC) was performed on a Biotage SP4 instrument equipped with a diode array detector and liquid handler.
HR-LC/MS and HR-LC/MS/MS Method
Method 1: Column = Waters Acquity UPLC® BEH C18 1.7 μm, 2.1 × 100 mm; Temperature = 25 °C Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 0.8 mL/min. Start at 20% B, ramped to 100% B over 3 min, held at 100% B for 1 min.
HPLC Methods
Method 2: Column = Phenomenex Luna 5 μm C18 (2), 250 × 10 mm; Temperature = 25 °C Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 3.0 mL/min; gradient: held at 5% B for 3 min, ramped to 30% B over 2 min, ramped to 95% B over 30 min, held at 95% B for 2 min, ramped to 5% B over 3 min and held for 5 min. Method 3: Column = Phenomenex Luna 5 μm C18 (2), 250 × 10 mm; Temperature = 25 °C Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 3.0 mL/min; gradient: held at 5% B for 3 min, ramped to 20% B over 2 min, ramped to 60% B over 30 min, ramped to 95% B over 2 min, held at 95% B for 2 min, ramped to 5% B over 3 min and held for 5 min. Method 4: Column = SunFire™ Prep C18 OBD™ 5 μm, 19 × 250 mm; Temperature = 25 °C Solvent A = H2O (0.1% formic acid); Solvent B = MeCN (0.1% formic acid); Flow Rate = 19.0 mL/min; gradient: held at 5% B for 3 min, ramped to 20% B over 2 min, ramped to 60% B over 30 min, ramped to 95% B over 2 min, held at 95% B for 2 min, ramped to 5% B over 3 min and held for 5 min.
MPLC Methods
Method 5: Column: SNAP-C18 120 g column with a 12 g samplet; solvent A = H2O (0.1% formic acid), solvent B = MeCN (0.1% formic acid), flow = 45 mL/min, λ = 210 nm; gradient: held at 5% B for 2 CV, ramped to 95% B over 21 CV, held at 95% B for 3 CV.
Synthesis of Compound 2
To a solution of 8 (60 mg, 0.0349 mmol, 1 equiv) and i-Pr2NEt (25 μL, 0.143 mmol, 4 equiv) in DMF (1 mL) at 0 °C, pivalic anhydride (42.5 μL, 0.209 mmol, 6 equiv) was added and stirred for 20 min. At this time, DMAP (4.3 mg, 0.0349 mmol, 1 equiv) and MeOH (0.3 mL) were added to the reaction mixture and continued to stir at 0 °C for another 30 min. Then, the reaction mixture was allowed to warm to 23 °C and stirred for another 4 h. Analysis of reaction mixture by RP-HPLC (Method 2) indicated complete consumption of 8. The crude reaction mixture was diluted with water/MeCN (1:1) and purified by preparative RPHPLC (Method 4). The Boc-protected tetra-methyl ester derivative of daptomycin was obtained as a white solid (30 mg, 48%). HPLC retention time (Method 2): 25.7 min; m/z (UPLC-MS) calcd for C81H118N17O28 1776.83, found 1776.68 [MH]+. The sample was dissolved in a mixture of CH2Cl2 (1 mL) and TFA (1 mL) and stirred for 2 h at 23 °C. The crude reaction mixture was loaded onto a C18 samplet, and purified by MPLC (Method 5). The fractions were concentrated to remove volatile organic solvents and lyophilized to obtain the product (2) as a white solid (23 mg, 82%). HPLC retention time (Method 3): 25.5 min; m/z (LC-MS) calcd for C76H110N17O26 1676.78, found 1676.59 [MH]+.
Synthesis of Compound 3
To a stirring solution of 8 (50 mg, 0.0291 mmol, 1 equiv) and i-Pr2NEt (20 μL, 0.115 mmol, 4 equiv) in DMF (1 mL) at 0 °C, HBTU (55 mg, 0.145 mmol, 5 equiv) was added and stirred for 5 min. At this time, NH4Cl (9.5 mg, 0.178 mmol, 6 equiv) and i-Pr2NEt (30.5 μL, 0.174 mmol, 6 equiv) were added to the reaction mixture and continued to stir at 0 °C for another 30 min. Then, the reaction mixture was allowed to warm to 23 °C and stirred for 4 h. Analysis of reaction mixture by RP-HPLC (Method 2) indicated complete consumption of 8. The crude reaction mixture was diluted with a mixture of water/MeCN (1:1) containing 0.1% formic acid and purified by preparative RP-HPLC (Method 4). The Boc-protected tetra-amide derivative of daptomycin was obtained as a white solid (39 mg, 78%). HPLC retention time (Method 2): 19.8 min; m/z (LC-MS) calcd for C77H114N21O24 1716.83, 1716.69 [MH]+. The sample was dissolved in a mixture of CH2Cl2 (1 mL) and TFA (1 mL) and stirred for 2 h at 23 °C. The crude reaction mixture was loaded onto a C18 samplet, and purified by MPLC (Method 5). The fractions were concentrated to remove volatile organic solvents and lyophilized to obtain the product (3) as a white solid (31 mg, 85%). HPLC retention time (Method 3): 21.4 min; m/z (LC-MS) calcd for C72H106N21O22 1616.78, found 1616.65 [MH]+.
Synthesis of compound 4
To a stirring solution of 8 (50 mg, 0.0291 mmol, 1 equiv) and i-Pr2NEt (20 μL, 0.115 mmol, 4 equiv) in DMF (1 mL) at 0 °C, HBTU (55 mg, 0.145 mmol, 5 equiv) was added and stirred for 5 min. At this time, HN(OMe)Me•HCl (17 mg, 0.174 mmol, 6 equiv) and i-Pr2NEt (30.5 μL, 0.174 mmol, 6 equiv) were added to the reaction mixture and continued to stir at 0 °C for another 30 min. Then, the reaction mixture was allowed to warm to 23 °C and stirred for another 3 h. Analysis of reaction mixture by RPHPLC (Method 2) indicated complete consumption of 8. The crude reaction mixture was diluted with a mixture of water/MeCN (1:1) containing 0.1% formic acid and purified by preparative RP-HPLC (Method 4). The Boc-protected tetra-Weinreb amide derivative of daptomycin was obtained as a pale yellow solid (41 mg, 75%). HPLC retention time (Method 2): 23.2 min; m/z (LC-MS) calcd for C85H130N21O28 1892.94, found 1892.82 [MH]+. The sample was dissolved in a mixture of CH2Cl2 (1 mL) and TFA (1 mL) and stirred for 2 h at 23 °C. The crude reaction mixture was loaded onto a C18 samplet, and purified by MPLC (Method 5). The fractions were concentrated to remove volatile organic solvents and lyophilized to obtain the product (4) as a white solid (34 mg, 88%). HPLC retention time (Method 3): 24.4 min; m/z (LC-MS) calcd for C80H122N21O26 1792.89, found 1792.84 [MH]+.
Synthesis of compound 5
To a stirring solution of 8 (50 mg, 0.0291 mmol, 1 equiv) and i-Pr2NEt (20 μL, 0.115 mmol, 4 equiv) in DMF (1 mL) at 0 °C, HBTU (55 mg, 0.145 mmol, 5 equiv) was added and stirred for 5 min. At this time, H2NOTHP (20.4 mg, 0.174 mmol, 6 equiv) was added to the reaction mixture and continued to stir at 0 °C for another 30 min. Then, the reaction mixture was allowed to warm to 23 °C and stirred for another 6 h. Analysis of reaction mixture by RP-HPLC (Method 2) indicated complete consumption of 8. The crude reaction mixture was diluted with a mixture of water/MeCN (1:1) containing 0.1% formic acid and purified by MPLC (Method 5). The daptomycin derivative 9 was obtained as a semi-pure, pale yellow residue. HPLC retention time (Method 2): 26.3 min; m/z (LC-MS) calcd for C97H147N21O32 1059.02, found 1059.00 [(M+2H)/2]+. Compound 9 was dissolved in a mixture of IPA (2 mL) and TFA (1 mL) and stirred at 23 °C for 3 h. LC-MS analysis indicated complete conversion of 9 to 10. The crude reaction mixture was loaded onto a C18 samplet, and purified using MPLC (Method 5). The fractions were concentrated in vacuo to obtain the product (10) as a semi-pure, pale yellow residue, which was further purified using preparative RP-HPLC (Method 4) to give the pure product (15 mg, 29% over 2 steps). HPLC retention time (Method 2): 19.6 min; m/z (LC-MS) calcd for C77H114N21O28 1780.81, found 1780.82 [MH]+. Compound 10 was dissolved in a mixture of CH2Cl2 (1 mL) and TFA (1 mL) and stirred for 2 h at 23 °C. The crude reaction mixture was loaded onto a C18 samplet, and purified by MPLC (Method 5). The fractions were concentrated to remove volatile organic solvents and lyophilized to obtain the product (5) as a pale yellow solid (10.5 mg, 74%). HPLC retention time (Method 3): 21.3 min; m/z (LC-MS) calcd for C72H106N21O26 1680.76, found 1680.72 [MH]+.
Synthesis of compound 8
To a stirring mixture of daptomycin (5.0 g, 3.08 mmol, 1 equiv) in water (40 mL) was added cold 1 M NaOH(aq) (6.0 mL, 6.16 mmol) dropwise to adjust the pH to 10. Then, Boc2O (1.68g, 7.71 mmol) was added to the solution in portions, while maintaining the reaction mixture at pH~9 by addition of 1 M NaOH(aq) to the reaction mixture. The reaction mixture was monitored in 30 min. intervals and the pH was adjusted to 9.0. After 5 h, the reaction was complete as indicated by RP-HPLC analysis. At this time, the reaction mixture was acidified with cold 1 M HCl(aq) to pH~3 and concentrated to yield a yellow solid. The crude material was dissolved in methanol (0.75 g/5 mL), loaded onto a C18 MPLC samplet and purified using MPLC (Method 5). The fractions containing 8 were combined, concentrated, and dried under high vacuum to yield the product as a yellow solid (4.2 g, 79%). HPLC retention time (Method 2): 21.6 min.; m/z (LC-MS) calcd for C77H110N17O28 1720.77, found 1720.67 [MH]+.
Supplementary Material
Fig. 1.
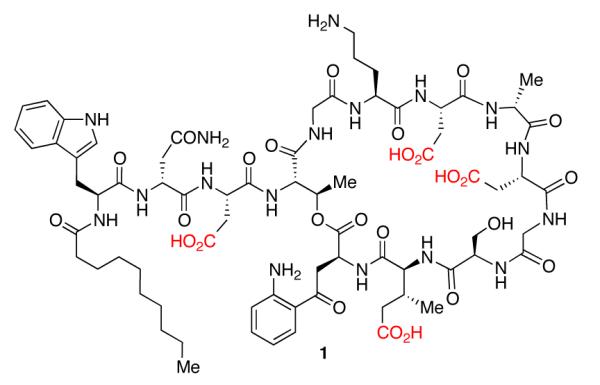
Structure of daptomycin
Scheme 1.
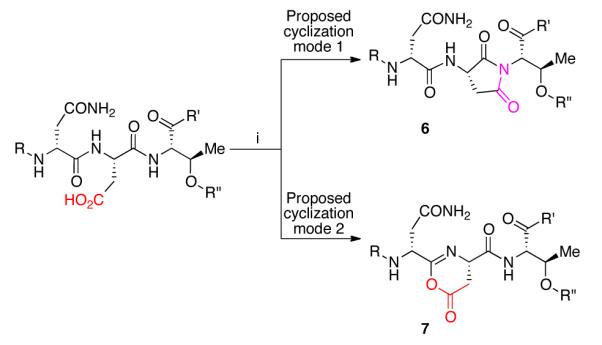
Reagents and conditions: (i) HBTU, i-Pr2NEt, DMF.
Acknowledgement
We thank Cubist Pharmaceuticals, Inc. for providing samples of daptomycin and for financial support. We are also grateful to the National Institutes of Health and Yale University for partial support of this effort.
Footnotes
Electronic Supplementary Information (ESI) available: [Spectroscopic and HPLC data available]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Fischbach MA, Walsh CT. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Debono M, Barnhart M, Carrell CB, Hoffmann JA, Occolowitz JL, Abbott BJ, Fukuda DS, Hamill RL, Biemann K, Herlihy WC. J. Antibiot. 1987;40:761–777. doi: 10.7164/antibiotics.40.761. [DOI] [PubMed] [Google Scholar]
- 3.Steenbergen JN, Alder J, Thorne GM, Tally FP. J. Antimicrob. Chemother. 2005;55:283–288. doi: 10.1093/jac/dkh546. [DOI] [PubMed] [Google Scholar]
- 4.Vilhena C, Bettencourt A. Mini-Rev. Med. Chem. 2012;12:202–209. doi: 10.2174/1389557511209030202. [DOI] [PubMed] [Google Scholar]
- 5.Silverman JA, Perlmutter NG, Shapiro HM. Antimicrob. Agents Chemother. 2003;47:2538–2544. doi: 10.1128/AAC.47.8.2538-2544.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman JA, Mortin LI, VanPraagh ADG, Li TC, Alder J. J. Infect. Dis. 2005;191:2149–2152. doi: 10.1086/430352. [DOI] [PubMed] [Google Scholar]
- 7.Hill J, Siedlecki J, Parr I, Morytko M, Yu X, Zhang YZ, Silverman J, Controneo N, Laganas V, Li TC, Lai JJ, Keith D, Shimer G, Finn J. Bioorg. Med. Chem. Lett. 2003;13:4187–4191. doi: 10.1016/j.bmcl.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 8.He Y, Li J, Yin N, Herradura PS, Martel L, Zhang YZ, Pearson AL, Kulkarni V, Mascio C, Howland K, Silverman JA, Keith DD, Metcalf CA. Bioorg. Med. Chem. Lett. 2012;22:6248–6251. doi: 10.1016/j.bmcl.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Robbel L, Marahiel MA. J. Biol. Chem. 2010;285:27501–27508. doi: 10.1074/jbc.R110.128181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Straus SK, Hancock REW. Biochimica et Biophysica Acta. 2006;1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Grunewald J, Sieber SA, Mahlert C, Linne U, Marahiel MA. J. Am. Chem. Soc. 2004;126:17025–17031. doi: 10.1021/ja045455t. [DOI] [PubMed] [Google Scholar]
- 12.Lemke TL, Williams DA, Roche VF, Zito SW. Foye’s Principles of Medicinal Chemistry. 7th edn. Lippincott, Williams and Wilkins; 2013. pp. 52–53. For a medicinal chemistry perspective on the use of the term “isostere,” see: [Google Scholar]
- 13.Zhang YZ, Boyer R, Sun XC, Paschal J, Chen SH. Bioorg. Med. Chem. Lett. 2000;10:775–778. doi: 10.1016/s0960-894x(00)00096-2. [DOI] [PubMed] [Google Scholar]
- 14.Sandy M, Butler A. Chem. Rev. 2009;109:4580–4595. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.