

Abstract
Chemical mapping experiments offer powerful information about RNA structure but currently involve ad hoc assumptions in data processing. We show that simple dilutions, referencing standards (GAGUA hairpins), and HiTRACE/MAPseeker analysis allow rigorous overmodification correction, background subtraction, and normalization for electrophoretic data and a ligation bias correction needed for accurate deep sequencing data. Comparisons across six noncoding RNAs stringently test the proposed standardization of dimethyl sulfate (DMS), 2′-OH acylation (SHAPE), and carbodiimide measurements. Identification of new signatures for extrahelical bulges and DMS “hot spot” pockets (including tRNA A58, methylated in vivo) illustrates the utility and necessity of standardization for quantitative RNA mapping.
Structure mapping, also known as footprinting, provides a rapid means for probing nucleic acid conformation at single-nucleotide resolution. New modification chemistries, higher-throughput readouts, multidimensional expansions, error analysis, and resources for sharing data are advancing the approach.1 Despite powerful insights from separate data sets, ad hoc choices in data processing have precluded robust comparison of chemical reactivities across RNAs and readouts.2−7 For example, “hot spots” that might signal specific noncanonical features6,7 in one RNA cannot be confidently established in other RNAs without universal reactivity scales, analogous to problems in nuclear magnetic resonance chemical shift analysis prior to the adoption of referencing samples.8
In principle, establishing reactivities should be unambiguous. Modification fractions ri of nucleotides i can be directly computed from the numbers of “raw” observed products Fi by
| 1 |
(derivation in the Supporting Information). While F0, the number of “full-length” products without chemical modification, is visible for RNA domains of up to 500 nucleotides, accurate quantitation is typically precluded by detector saturation of this strong band in electrophoresis data or by ligation biases in deep sequencing data. Our lab’s previous likelihood framework for F0 depended on a priori reactivity distributions that were approximate.2 Aviran et al. explored setting F0 to zero when it could not be measured,5 a poor assumption under typical “single-hit” conditions. Karabiber et al. proposed equalizing reactivities observed in the 5′ half versus the 3′ half of the data,3,4 a generally inaccurate approximation. Several recent studies have not applied eq 1.9 Further complicating cross-experiment comparisons are differences in whether eq 1 is applied to no-modifier control samples, in sequence alignment tools, in error estimation, and in normalization procedures,2,3,5 as well as a lack of validation protocols.
To address these issues, we implemented two straightforward standardization strategies: (1) dilution comparisons to mitigate saturation and (2) use of universal internal controls (Figure 1A,B). To illustrate, Figure 1C gives capillary electrophoresis (CE) data of primer extension products for the P4–P6 domain of the Tetrahymena ribozyme probed with dimethyl sulfate (DMS) to methylate exposed N1/N3 atoms of A/C nucleotides.10 The saturated peak shape for the fully extended product is apparent; 10-fold dilution of the same sample gave a weaker signal-to-noise ratio overall but an unsaturated, Gaussian shape for the F0 peak (Figure 1D; further dilutions verified the lack of saturation). Automated scaling of these dilution data allowed unbiased measurement of F0 (Figure 1E,F). Application of eq 1, background subtraction, and normalization (see below) gave the reactivity profile in Figure 1F. The final results agreed within error with averaged data collected by different experimenters (Figure 1F and Methods and Figure 1 of the Supporting Information). Further, as expected (but not assumed), DMS reactivities at G and U nucleotides were within error of zero. Tests comparing data from 8-fold variations of DMS and reagents 1-cyclohexyl(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate (CMCT, modifying G/U)10 and 1-methyl-7-N-isatoic anhydride (1M7, modifying 2′-OH; SHAPE3,4) further confirmed this standardization (Figure 2 of the Supporting Information).
Figure 1.
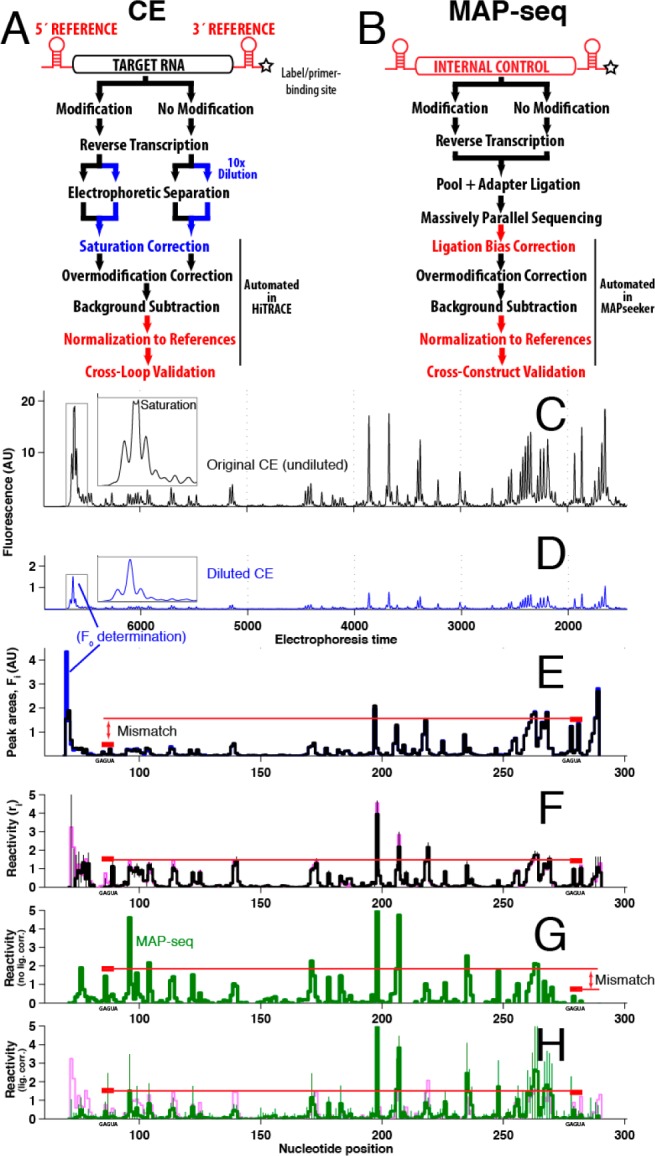
Proposed steps to standardize chemical mapping experiments (red and blue text) read out by (A) capillary electrophoresis and (B) deep sequencing (MAP-seq). CE profiles for the P4–P6–2HP RNA probed with DMS at (C) standard dilution and (D) 10-fold dilution. (E) Automated scaling matches diluted sample data to undiluted data. (F) Final reactivity profile (black), validated by data taken at 4-fold lower DMS concentrations (green, nearly indistinguishable) and equality at GAGUA referencing hairpins (red). MAP-seq data for P4–P6 RNA without (F) and with (G) ligation bias correction determined from internal referencing. (H) Overlay of CE and MAP-seq data; errors are standard deviations of replicates (Figure 1 of the Supporting Information).
Independent validation of this procedure came from incorporating “reference” hairpins in 5′ and 3′ flanking cassettes.3,4 GAGUA hairpin loops (Figure 2a) give strong signals for DMS (at the A’s), CMCT (at the bulge U), and 1M7 (all five residues). “Raw” Fi counts were 5-fold lower at the 5′ GAGUA than at the 3′ GAGUA (red bars in Figure 1E), as reverse transcriptases encountered stops in between those segments (“overmodification”, also called attenuation or signal decay). The equality of the GAGUA final reactivities ri confirmed accurate overmodification correction and background subtraction of these data (red bars in Figure 1F) and supported use of the GAGUA data as normalization standards.
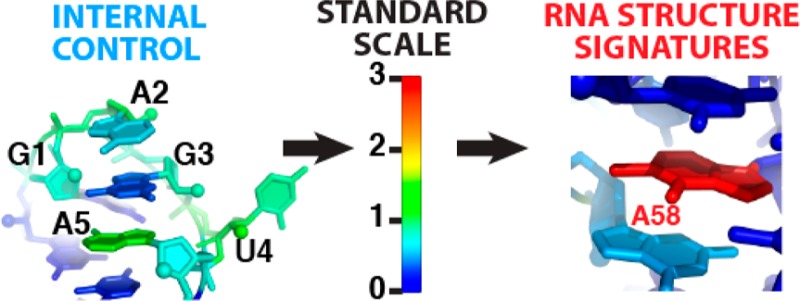
Figure 2.

Three-dimensional environments associated with high chemical reactivity to Watson–Crick edge modifiers [DMS for A/C and CMCT for G/U (base color)] and/or 2′-OH acylation [1M7 (backbone color)]. (A) GAGUA hairpin sets the normalization scale for DMS (A2 and A5), CMCT (U4), and 1M7 (all nucleotides). (B) L6b from the P4–P6 domain. (C) Interdomain linker from the glycine riboswitch. (D) Bulge in the ligand binding pocket of the adenine riboswitch. (E–G) Pockets promoting high adenosine N1 reactivity and low 2′-OH reactivity in tRNA (N1-methyl shown) (E) and the P4–P6 domain (F and G). Hot spot nucleotides are labeled in panels B–G. Protein Data Bank entries are listed in Table 1 of the Supporting Information.
An alternative readout, MAP-seq (multiplexed accessibility probing), follows nucleic acid modification and primer extension with ligation of an Illumina adapter and deep sequencing, without bias-introducing polymerase chain reaction amplification (Methods of the Supporting Information).11 We previously observed (through CE) that ligation yields were systematically low for full-length cDNA products. This effect led to underestimation of F0 and to an apparent discordance between the 5′ and 3′ GAGUA references (red bars, Figure 1G). Nevertheless, the requirement of equality at these sequences allowed automated estimation of a ligation bias correction factor [0.18 in this case (Methods of the Supporting Information)]. Despite involving rather different protocols, the CE and MAP-seq results then agreed within errors estimated from replicates (Figure 1H, and see below).
To comprehensively test the standardization protocol, we took measurements with DMS, CMCT, and 1M7, using both CE and MAP-seq protocols on several structured RNAs, including ligand-bound riboswitches and rRNA domains (Figures 3–8 of the Supporting Information).2,10 In the MAP-seq experiment, data for the P4–P6–2HP domain established the ligation bias correction factor and normalization for the coloaded RNAs. The agreement within error between reactivities at GAGUA reference hairpins across all constructs and general agreement between CE and MAP-seq data sets confirmed the accuracy of the proposed standardization (Figure 1 of the Supporting Information). No length bias was detected for MAP-seq, but a residual sequence bias was seen in reactive purine-rich segments; these mostly occurred in flanking sequences outside the structured RNA domains (Figures 3–8 of the Supporting Information). In both CE and MAP-seq data, normalization to GAGUA references exposed limitations of prior heuristics that normalize based on high percentile values within each RNA (or in 5′ and 3′ halves);2−4,9,10 these values in fact vary by >2-fold across the different RNAs.
The standardization procedures allowed the identification of 33 hot spot nucleotides, defined here as those giving DMS, CMCT, or 1M7 reactivity of >1.5, well above control values (1.0) established by GAGUA references (Table 2 of the Supporting Information). First, in agreement with conventional use of these data to infer secondary structure,10 all 16 cases of high DMS/CMCT/1M7 reactivities observed within stretches of more than two residues corresponded to apical loops (Figure 2B) or unpaired “linkers” (Figure 2C). Second, three isolated adenosines with high 1M7 but low DMS reactivity were stacked on one face, a structural feature previously requiring differential SHAPE measurements for identification.6 Third, all seven isolated highly CMCT/1M7-reactive uridines and two highly 1M7-reactive adenosines were extrahelical bulges7 (Figure 2D), a powerful signature for guiding or validating tertiary structure modeling.12 Most intriguing were five adenosines with DMS reactivities of >1.5 but negligible 1M7 reactivity (Figure 2E–G). Each of these adenosines showed Hoogsteen edge burial and nucleobase stacking on both faces; such burial information should be useful in tertiary structure modeling. The most DMS-reactive nucleotide, A58 in Saccharomyces cerevisiae tRNA(phe) (Figure 2E), is also methylated at the N1 position in vivo.13 The pocket around DMS hot spot nucleotides may thus be under selection for electronegativity to enhance enzymatic reaction or hydrogen bonding to partners. As further examples, A198 and A207 (Figure 2F,G) in the isolated P4–P6 domain are buried, but N1 atoms are available for contacts in the full Tetrahymena ribozyme or recognition by protein partners. These signatures could not be identified unambiguously in prior work because of uncertain data scaling.
The inclusion of dilution samples and referencing hairpins allows standardization, validation, and deeper analysis of structure mapping experiments at negligible additional cost. For CE studies, obtaining the necessary data simply involves diluting the prepared samples into running buffer and repeating electrophoresis and HiTRACE/HiTRACE-Web analysis14 (Figure 1A). Inclusion of GAGUA hairpins was used here to test the overmodification correction and normalize CE data but was only strictly necessary in MAP-seq experiments. In fact, just a single construct with flanking reference hairpins needs to be doped into the MAP-seq RNA pool; standardization is then automated via MAPseeker analysis11 (Figure 1B). The general adoption of simple standardization steps, and their extension to very long transcripts and to other solution conditions and modifiers, should help RNA structure mapping data become more accurate and more transferrable between molecules and experiments.
Acknowledgments
We thank P. Cordero, C. Cheng, B. Stoner, and Das lab members for helpful discussions and S. Mortimer and F. V. Cochran for assistance with 1M7 synthesis.
Supporting Information Available
Derivation of eq 1, experimental methods, CE/MAP-seq comparisons, and a table of sequences, Protein Data Bank entries, and RNA Mapping Database entries for deposited data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
W.K. and T.H.M. contributed equally to this work.
Work was funded by the Burroughs-Wellcome Foundation (CASI 1007236.01 to R.D.), a Stanford Graduate Fellowship (S.T.), National Research Foundation of Korea (No. 2011-0009963 to S.Y.), the National Institutes of Health (5T32GM007276 to T.H.M. and R01GM102519 to R.D.), and the Keck Foundation.
The authors declare no competing financial interest.
This Rapid Report was published ASAP on May 7, 2014. Reference 5 has been updated and the corrected version was reposted on May 8, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Peattie D. A.; Gilbert W. (1980) Proc. Natl. Acad. Sci. U.S.A. 77, 4679–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]; Regulski E. E.; Breaker R. R. (2008) Methods Mol. Biol. 419, 53–67. [DOI] [PubMed] [Google Scholar]; Kladwang W.; Das R. (2010) Biochemistry 49, 7414–7416. [DOI] [PubMed] [Google Scholar]; Rocca-Serra P.; Bellaousov S.; Birmingham A.; Chen C.; Cordero P.; Das R.; Davis-Neulander L.; Duncan C. D.; Halvorsen M.; Knight R.; Leontis N. B.; Mathews D. H.; Ritz J.; Stombaugh J.; Weeks K. M.; Zirbel C. L.; Laederach A. (2011) RNA 17, 1204–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cordero P.; Lucks J. B.; Das R. (2012) Bioinformatics 28, 3006–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W.; Vanlang C. C.; Cordero P.; Das R. (2011) Biochemistry 50, 8049–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabiber F.; McGinnis J. L.; Favorov O. V.; Weeks K. M. (2013) RNA 19, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E. J.; Wilkinson K. A.; Coughlan J. L.; Weeks K. M. (2005) J. Am. Chem. Soc. 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- Aviran S., Lucks J. B., and Pachter L. (2011) Proceedings of the 49th Allerton Conference on Communication, Control, and Computing, 1743−1750.
- Steen K. A.; Rice G. M.; Weeks K. M. (2012) J. Am. Chem. Soc. 134, 13160–13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis J. L.; Dunkle J. A.; Cate J. H.; Weeks K. M. (2012) J. Am. Chem. Soc. 134, 6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschbacher T.; Schubert M.; Allain F. H. (2012) J. Biomol. NMR 52, 179–190. [DOI] [PubMed] [Google Scholar]
- Kertesz M.; Wan Y.; Mazor E.; Rinn J. L.; Nutter R. C.; Chang H. Y.; Segal E. (2010) Nature 467, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ding Y.; Tang Y.; Kwok C. K.; Zhang Y.; Bevilacqua P. C.; Assmann S. M. (2014) Nature 505, 696–700. [DOI] [PubMed] [Google Scholar]; Kwok C. K.; Ding Y.; Tang Y.; Assmann S. M.; Bevilacqua P. C. (2013) Nat. Commun. 4, 2971. [DOI] [PubMed] [Google Scholar]; Rouskin S.; Zubradt M.; Washietl S.; Kellis M.; Weissman J. S. (2014) Nature 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero P.; Kladwang W.; VanLang C. C.; Das R. (2012) Biochemistry 51, 7037–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetin M. G.; Kladwang W.; Bida J. P.; Das R. (2014) Methods Mol. Biol. 1086, 95–117. [DOI] [PubMed] [Google Scholar]
- Sripakdeevong P.; Kladwang W.; Das R. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 20573–20578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta R.; Vainauskas S.; Yarian C.; Sochacka E.; Malkiewicz A.; Guenther R. H.; Koshlap K. M.; Agris P. F. (2000) Nucleic Acids Res. 28, 1374–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S.; Kim J.; Hum J.; Kim H.; Park S.; Kladwang W.; Das R. (2011) Bioinformatics 27, 1798–1805. [DOI] [PubMed] [Google Scholar]; Kim H.; Cordero P.; Das R.; Yoon S. (2013) Nucleic Acids Res. 41, W492–W498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.