

Abstract
Cell membrane chromatography (CMC) derived from pathological tissues is ideal for screening specific components acting on specific diseases from complex medicines owing to the maximum simulation of in vivo drug-receptor interactions. However, there are no pathological tissue-derived CMC models that have ever been developed, as well as no visualized affinity comparison of potential active components between normal and pathological CMC columns. In this study, a novel comparative normal/failing rat myocardium CMC analysis system based on online column selection and comprehensive two-dimensional (2D) chromatography/monolithic column/time-of-flight mass spectrometry was developed for parallel comparison of the chromatographic behaviors on both normal and pathological CMC columns, as well as rapid screening of the specific therapeutic agents that counteract doxorubicin (DOX)-induced heart failure from Acontium carmichaeli (Fuzi). In total, 16 potential active alkaloid components with similar structures in Fuzi were retained on both normal and failing myocardium CMC models. Most of them had obvious decreases of affinities on failing myocardium CMC compared with normal CMC model except for four components, talatizamine (TALA), 14-acetyl-TALA, hetisine, and 14-benzoylneoline. One compound TALA with the highest affinity was isolated for further in vitro pharmacodynamic validation and target identification to validate the screen results. Voltage-dependent K+ channel was confirmed as a binding target of TALA and 14-acetyl-TALA with high affinities. The online high throughput comparative CMC analysis method is suitable for screening specific active components from herbal medicines by increasing the specificity of screened results and can also be applied to other biological chromatography models.
As one of the oldest materials of traditional medicine practiced in the world, herbal medicines (HMs) have a history of several thousand years. They are commonly composed of one or several medicinal herbs following specific medical theoretical guides. Their worldwide use has increased in recent decades.1,2 It has been generally acknowledged that the clinical efficacies of HMs are derived from active components, while others may be useless or even have adverse effects.3 Thus, the confirmation of active components in HMs is of great significance to ensure their quality and curative effects and also to elucidate their complicated pharmacodynamic mechanisms.4 However, it is still difficult and time-consuming to recognize and confirm active components with specific effects from hundreds of chemicals even in a single herb.
Biological affinity chromatography combined with advanced detectors has the advantages of high throughput and good selectivity, which is widely used for screening bioactive molecules from complex matrixes5−9 and also for studying binding interactions between molecules and specific receptors.10−12 Cell membrane chromatography (CMC) is a well developed biological chromatographic technique in which cell membranes containing certain receptors are used as the stationary phase.13−15 This method had been extensively reviewed.16,17 It is a practical method for screening active components from complex HMs18−21 and also for investigating drug-receptor interactions.22−24 In order to efficiently characterize and identify the active ingredients that have an affinity to cell membrane stationary phase (CMSP), several two-dimensional liquid chromatography (2D-LC) methods have been established for rapid investigation of complex HMs.18,19,25−28 However, some problems remain: (1) The CMC preparation procedures are time-consuming, especially the membrane extraction steps, and some key parameters, e.g., content of maximum binding protein, lack precise control. (2) Pathological tissue-derived CMC models to screen specific active components against the specific diseases from HMs have rarely been developed. (3) Using one receptor highly expressing cell lines as cell membrane source is a practical way to screen receptor’s agonists and antagonists,13,15,26 but this method neglects the multitarget property of HMs and thus is insufficient in the comprehensive characterization of active components in HMs. While other groups have immobilized multiple membrane proteins of various tissues and cells11,26,29 and showed that these CMC models could be used as a representation of the targeted family of receptors, a comparison between normal and pathological states was not carried out to selectively screen components of HMs which act on the specific diseases.
Among all these cell membrane sources for CMC, the pathological tissues from human or animal after exposure to pathogenic factors are the most ideal materials for screening components from HMs because of the maximum simulation of in vivo drug-receptor interactions. This method has rarely been applied, mainly because of the difficulty of preparing pathological tissue derived CMC models and technical limitations of large scale comparative CMC analysis between normal and pathological CMC models. The comparison between the two states using the CMC method does have its advantages, such as characterization of specific components acting on pathological membranes through comparative procedures and study of the variation of target proteins between normal and pathological states using positive drugs. Fortunately, owing to the development of column selection,30 peak aligning,31 and data processing techniques,32 the comprehensive 2D-LC has gained increasing popularity as a technique because of its powerful separation and analysis abilities with the integration of orthogonal columns and advanced detectors.33,34 In our previous research, a comprehensive two-dimensional CMC system was established for rapid screening of antitumor components from 28 HMs.21
Thus, in this study, a novel strategy of comparative CMC analysis based on column selection and comprehensive 2D-LC techniques was proposed for reliable screening components acting on specific diseases from complex HM samples. Cell membrane sources were derived from the left ventricle of rat hearts. Normal hearts were compared with hearts that were failing by pretreatment with doxorubicin (DOX), which has cumulative and dose-dependent cardiotoxicity and has been used as an ideal modeling drug for inducing acute or chronic cardiomyopathy in experimental animals.35 The direct cardiomyocyte membrane damage, reflected in impairing membrane integrity and stability responsible for cell apoptosis and death, is one of the major causes of DOX-induced heart failure.36,37 The function of multiple membrane receptors is subsequently impaired.38,39Acontium carmichaeli (Fuzi) is a famous herbal medicine and is widely used as a therapeutic agent in East and Southeast Asia. Our group has previously used Fuzi as a major ingredient in a herbal formula for effective treatment of DOX-induced cardiomyopathy40 and myocardial infarction.41,42 However, the definite active components in Fuzi related to its cardioprotective effects are still ambiguous.
Therefore, the proposed comparative CMC analysis method based on the establishment of normal myocardium CMC (NM/CMC) and DOX-induced failing myocardium CMC (FM/CMC) models was applied to screening specific components of Fuzi that can counteract DOX-induced heart failure. The methodology of this new system was elaborately investigated and optimized to reproduce different batches. A series of validation experiments including cell viability assay, competitive displacement assay, and molecular docking modeling were conducted to confirm the cardioprotective effects and binding targets of the screened active components. The automated and high throughput comparative CMC analysis method is suitable for screening active components from HMs on comparing their chromatographic affinity behaviors between normal and pathological CMC columns.
Experimental Section
Ethics Statement
All animal experiments were approved by the Administrative Committee of Experimental Animal Care and Use of Second Military Medical University (SCXK(Hu)2007-0005) and conformed to the National Institute of Health guidelines on the ethical use of animals.
Chemicals and Materials
Doxorubicin hydrochloride, tetracycline, dexamethasone, oxymetazoline, tamsulosin, benzoylaconitine, and 4-aminopyridine (4-AP) were purchased from the National Institute for the Pharmaceutical and Biological Products of China (Beijing, China), and the purities were all over 98%. 25 mg of talatizamine (TALA) was isolated and purified from the roots of A. carmichaeli by the authors. The structure of TALA was unambiguously identified by 1H NMR and MS spectra, and its purity was over 98% determined by HPLC-UV. A. carmichaeli (collection in Sichuan, China) were purchased from Shanghai Leiyunshang Medicine Corp. (Shanghai, China). Rat cardiac H9c2 cell line was obtained from American Type Culture Collection (Rockville, MD, USA). Dulbecco minimal essential medium (DMEM) was purchased from Invitrogen Corporation (Grand Island, NE, USA) and supplemented with 10% fetal calf serum (FBS) obtained from Gibco Co. (Rockville, MD, USA). Dimethyl sulfoxide (DMSO), penicillin streptomycin, and trypsin were purchased from Gibco Co. The silica gel (5 μm, 200 Å) was obtained from Meigao Materials, Inc. (Qingdao, China). HPLC-grade acetonitrile was purchased from Merck Co. (Darmstadt, Germany). MS-grade ammonia acetate was obtained from Sigma Co. (St. Louis, MO, USA). Ultrapure water was prepared with a Milli-Q water purification system (Millipore, Bedford, MA, USA). Other reagents were of analytical grade.
Preparation of Sample and Standard Solutions
The crude drug of Fuzi (200 g) was immersed in 2.4 L of water for 1 h and then decocted to boil for 2 h. The decoction was filtered through four layers of gauze. Next, the dreg was boiled once again for 1 h with 2 L of water, and the decoction was filtrated out with the above method. Afterward, the successive decoctions were merged and condensed by a rotatory evaporator under reduced pressure. Finally, the Fuzi extraction solution was made to a concentration of 1.0 g/mL (expressed as the weight of crude drug). For CMC analysis, the extract was added with 4-fold volume of ethanol to precipitate the polysaccharide and protein. After standing overnight, the supernatant was filtered and condensed to 0.3 g crude drug/mL (the concentrations of main components were more than 50 μM) for injection (5 μL). According to our previous published papers,43 24 components of Fuzi extract were successfully identified by HPLC-TOF/MS. A mixed standards solution of oxymetazoline, nifedipine, and dexamethasone (0.5 mM each) was prepared in 70% methanol. Doxorubicin hydrochloride was immediately dissolved in ddH2O to make a 5 mM solution before use. TALA was dissolved in 0.15% HCl (v/v) to make a 10 mM stock solution. 4-AP was dissolved in 5 mM ammonia acetate or culture medium before use.
Animal Experiments
All animal experiments were performed at the Centre of Laboratory Animals of the Second Military Medical University (Shanghai, China) in accordance with the relevant national legislation and local guidelines. Sprague-Dawley rats, 180–220 g weight, were purchased from the Shanghai Laboratory Animal Co. Mice were housed in a temperature range from 20 to 25 °C and humidity range from 50 to 60%. The rats were fed with commercial aseptic food and tap water continuously available throughout the experimental period. The animals were randomly divided into three groups (control, DOX, and DOX plus Fuzi). For the DOX group (n = 9) and DOX plus Fuzi group (n = 6), DOX was administered intraperitoneally in six equal injections (each containing 2.5 mg/kg) over a period of 2 weeks, with a total cumulative dosage of 15 mg/kg body weight.44 The rats in the DOX plus Fuzi group were treated with Fuzi extract 2 g/kg/BW i.p. for 14 consecutive days after the first administration of DOX. The administration dose in the current study is in accordance with clinical use and had been confirmed to have no cardiotoxicity by our group.45 For the control group (n = 9), saline (0.4 mL) was injected in the same manner. At day 15, after hemodynamics assessments, the hearts of 3 rats in control and DOX groups, respectively, were excised for preparation of CMC columns. The rest of the 6 rats from each of the three groups were sacrificed, and their hearts were rapidly excised and immersed in 10% formaldehyde for hematein-eosin (HE) staining.
NM/CMC and FM/CMC Model
The rat myocardium cell membrane was prepared using our previous method with a few modifications.20,21 In brief, the heart of rats from the normal or DOX-treated group was harvested immediately and the heart atrium was completely removed except the left ventricle part. The cardiac muscular tissue was immerged into precooled saline to wash out the blood. After cutting into pieces in 10 mL of precooled normal saline, 200 mg of tissue was put into a glass homogenizer for homogenizing. The homogenate was then centrifuged at 3000g for 10 min, and 10 mL of phosphate buffered solution (PBS, pH 7.4, 10 mM) was added to the pellet to produce a cell suspension, which was ruptured on ice by an ultrasonic cell disruptor (Scientz biotechnology co., Ningbo, China) at 400 W, 2 s, 5 times with 20 s intervals. The resulting suspension was vortex-mixed and clarified by centrifugation at 1000g for 10 min. The pellet was discarded, and the supernatant was centrifuged at 12 000g for 20 min at 4 °C. The pellet was collected and washed with PBS and centrifuged at 12 000g for another 20 min. Then, 5 mL of saline was added to the pellet and the myocardium cell membrane suspension was obtained. Cell membrane stationary phase (CMSP) was prepared by the adsorption of cell membrane suspension on silica (0.04 g, dried at 120 °C for 2 h before use) under vacuum and agitation conditions at 4 °C. After standing overnight, the myocardium CMSP was washed with 10 mL of PBS 3 times and packed into a column (10 mm × 2 mm i.d.) using a wet packing procedure.
Comprehensive Two-Dimensional Comparative CMC System
The comprehensive 2D comparative CMC system was composed of commercial available modules. As shown in Figure 1, the comprehensive 2D system was performed on an Agilent 1200 series HPLC system consisting of unitary (Pump 1) and binary (Pump 2 and Pump 3) solvent delivery systems, a thermostatically controlled column apartment, an online degasser, and an autosampler controlled by Agilent MassHunter Workstation (Agilent Technologies, Palo Alto, CA, USA). In order to realize the synchronization of the two dimensions, the proper configuration of parameters was determined by the aid of a home-written program in Visual Basic 6.0 (Microsoft, Redmond, WA, USA). CMC columns (n = 6) including both three normal and pathological myocardium models were simultaneously equipped with a position selection valve (Agilent 1100 series, G1159A) for rapid comparative analysis. An electronically controlled 10-port dual-position valve (MXP9960-000, Rheodyne, Rohnert park, CA, USA) equipped with two 500 μL sampling loops was used as the interface between the two dimensions. Each fraction from the first dimension (column 1) was stored in the parallel sampling loops and further introduced in the second dimensional column alternatively every 2.5 min. In total, 12 fractions were collected, and thus, 30 min of the first dimension was conducted for comprehensive two-dimensional analysis. The eluent of the second dimensional column was introduced to a 6220 TOF mass spectrometer (Agilent Technologies) equipped with an electrospray ionization interface, and the signals were collected by the Agilent MassHunter Workstation (Agilent Technologies). When the 12th fraction of column 1 was being analyzed, the position selection valve was switched to column 2 for the equilibrium procedure (Figure 1C).
Figure 1.
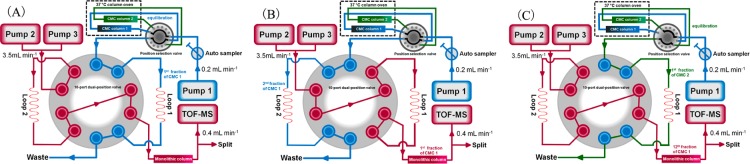
Block scheme of 2D comparative CMC/monolithic column/TOFMS system. The position selection valve is able to load 6 different columns (there are only two columns displayed in the figure for brevity), and all these modules are controlled by online software. (A) Ten-port dual position valve was at position 1. CMC column 1 was equilibrated, and the 1st fraction was collected in loop 1. (B) Ten-port dual position valve was switched to position 2. The 1st fraction was being analyzed by a monolithic column coupled with TOFMS while the 2nd fraction was collected in loop 2. (C) When the 12th fraction of CMC column 1 was being analyzed, the position selection valve was switched to the CMC column 2 for equilibration and collection procedures.
Normal and failing myocardium CMC columns (10 mm × 2 mm I.D.) were applied as the first dimensional columns. The mobile phase was 5 mM ammonia acetate, and the flow rate was 0.2 mL/min. For the second dimension separation, a Chromolith Performance RP-18e monolithic silica column was used (100 mm × 4.6 mm I.D., Merck, Darmstadt, Germany). Column temperature was 30 °C. The mobile phase was composed of solvent A [0.1% formic acid (v/v)] and solvent B (acetonitrile) by a linear gradient elution program as follows: 0–5 min, from 5% B to 25% B; 5–9 min, from 25% B to 75% B; 9–9.5 min, from 75% B to 5% B; 9.5–10 min, 5% B. The flow rate was 3.5 mL/min. The eluent then was split, and 0.4 mL/min was introduced into the TOFMS.
The TOFMS analysis was performed using our previous conditions.21 The original data were exported and imported into MATLAB 7.10.0 (The MathWorks, Sherborn, MA, USA) to conduct baseline correction and peak aligning and then transferred into the 3D plots by a home-written program in MATLAB 7.10.0.
Molecular Docking Study
Molecular docking studies were performed to explore the underlying mechanisms of myocardial protective effect of TALA and other components. Docking calculations were carried out using a Surflex-Dock GeomX module of Sybyl-X1.3.46 The crystal structure of K+ channel protein was retrieved from the Protein Data Bank (PDB) with entry 1J95. The water molecules and the complex inhibitor were removed, and hydrogen atoms were added. The receptor and the ligand files were prepared by using our published protocols.47,48 The flexible ligands were docked to the rigid receptor protein using default settings for all parameters.
Competitive Displacement Assay
The competitive displacement assay was conducted according to our previously reported method with slight modifications.21 Briefly, one FM/CMC column was equilibrated with 5 mM ammonium acetate until steady baseline was obtained, and the retention time of TALA (10 mM, injection volume 1 μL) was recorded by TOFMS. Then, the CMC system was rebalanced with the mobile phase containing 4-AP until the platform of each breakthrough curve could be observed. Afterward, another injection of TALA was performed. Totally, five concentrations of 4-AP in the mobile phase (1, 2, 5, 10, and 25 μM) were tested for revealing the binding target of TALA.
Hemodynamics Assessments, H9c2 Cell Viability Assay, Measurement of Excellular LDH Content, Flow Cytometric Detection of Cell Apoptosis and Death, and Western Blot Analysis
The detailed procedures are presented in the Supporting Information.
Results and Discussion
Cardioprotective Effect of Fuzi on DOX-Induced Heart Failure
Although the pharmacological effects of Fuzi on the cardiovascular system were extensively reported,49 we conducted further in vivo experiments to evaluate the quality of DOX-induced heart failure models, in addition to the cardioprotective effect of Fuzi extract. Table S-1, Supporting Information, shows the arterial pressure parameters obtained from baroreflex sedated animals. Left ventricle end-diastolic pressure (LVEDP) was significantly increased while left ventricle systolic pressure (LVSP) and the maximum rate of pressure increase (+ dP/dt max) were dramatically decreased in DOX-treated rats compared with controls (p < 0.05). However, in Fuzi-treated rats, LVEDP was obviously decreased and LVSP and + dP/dt max were increased compared with the DOX group (p < 0.05). The pathological sections of left ventricle stained with HE indicated that, after DOX modeling, the arrangement of cardiac muscle bundles was disordered. The myocardial cells revealed impaired characteristics including hemorrhage and necrosis. When treated with Fuzi extract, these lesions could be obviously reversed (Figure S-1A–C, Supporting Information). These results clearly demonstrated that the heart failure in rats was successfully induced by cumulative injections of DOX. The pharmacodynamic effect of Fuzi against DOX-induced heart failure was also confirmed. Therefore, the pathological rat hearts were confirmed as ideal CMC tissue sources for screening specific components from Fuzi extract that counteract DOX-induced heart failure.
Optimization of Procedures for CMC Preparation
The methodology of CMC was elaborately investigated and optimized for construction of the pathological tissue-derived CMC model. According to the previous research13 and our experiences, the precise control of cell disruption and the content of maximum binding protein on the column are the major influences on the quality of CMC columns. In this study, an ultrasonic cell disruptor was applied for rapid cell membrane collection. When the probe was directly put in 10 mL of cell suspension or tissue homogenate and was run at 400 W, 2 s, 5 times with 20 s intervals, approximately 80% of the cells could be observed to be broken, which was an ideal condition for coating membrane on silica gel. Before immobilization, one fraction of collected membrane protein was then absolutely lysed and precisely quantified by bicinchoninic acid (BCA) protein assay reagent. The membrane suspension was then adjusted to 0.60 ± 0.05 mg/mL, 5 mL/column, in order to make an excess membrane suspension for reaching the maximum coating content. After the immobilization procedure, the membrane proteins were stripped from the silica by immersed in enhanced RIPA lysis buffer and shaking for 45 min at 4 °C, and the content of maximum binding protein on CMSP was precisely quantified as 0.31 ± 0.05 mg/0.04 g silica/column by the BCA protein assay, which can make a moderate column pressure of 12–15 bar at the flow rate of 0.2 mL/min. According to these parameters, the quality of each CMC column in this research was well controlled and good reproducibility was obtained.
Suitability of the 2D Comparative CMC/Monolithic Column/TOFMS System
As shown in Figure 1A–C, six CMC columns including three NM and FM/CMC columns, respectively, were simultaneously loaded in a 37 °C column oven through a position selection valve with an alternative arrangement. After equilibrium for 15 min, the first retention fraction recognized in the CMC column 1 was enriched into a 500 μL sample loop, and after the 10-port valve switched, the enriched components were pumped into a monolithic analytical column and TOFMS for qualitative analysis. At the same time, the second retention fraction was collected into another sample loop. Due to the poor separating capacity of the CMC column, it is necessary to collect at least two fractions across a first dimension peak for second dimension analysis in order to obtain reliable and comprehensive results. Thus, the modulation period was set at 2.5 min for more accurate analysis and a total of 30 min; 12 fractions of the first dimension were collected for comprehensive 2D analysis. Considering the large trapped volume of each fraction (500 μL), a high flow rate monolithic column (3500 μL/min) was applied as the second dimension for rapid delivery and efficient separation of fractions, and a precise split of 400 μL/min was introduced into TOFMS for real-time accurate mass analysis. Other columns, such as reverse-phase liquid chromatography, ultrapressure liquid chromatography, and porous graphitic carbon column, were also investigated as the second dimension, which could not achieve ideal results containing good peak shapes with short analytical periods. When the last fraction of CMC column 1 was being analyzed, the position selection valve was switched to CMC column 2 for equilibration before the sample injection. All these procedures were controlled by Agilent online acquisition and Rheodyne TitanMX control software.
A mixed standard solution containing negative and positive drugs was used to confirm the selectivity and reproducibility of the myocardium 2D comparative NM/FM CMC analysis system. As shown in Figure 2, dexamethasone (binding to intracellular glucocorticoid receptor) was selected as the negative control drug, which had little retention behavior. Oxymetazoline (α1-adrenergic receptor agonist) and nifedipine (calcium channel blocker) were positive control drugs acting on myocardial cell membrane receptors. From Figure 2A, we can see that nifedipine displayed affinity on the NM/CMC columns while the affinity significantly decreased on the FM/CMC columns (Figure 2B), which is in accordance with the fact that calcium channel density is significantly decreased in DOX-induced cardiomyopathy50 and other animal heart models.51 That is the reason why calcium channel blockers had never been used to treat chronic heart failure. In addition, the retention behavior of oxymetazoline was not decreased in FM/CMC columns compared with NM/CMC columns indicating the high activity of α1-adrenergic receptor in heart failure tissue, reflecting on the adaptive roles in the heart and protecting against the development of heart failure.52 On the other hand, in order to validate the column to column reproducibility, the capacity factors of the three standard drugs in the respective three NM and FM/CMC columns were recorded and the relative standard derivations (RSDs) within groups were all calculated to be less than 10%. These results indicated that the proposed 2D comparative NM/FM CMC analysis system had good selectivity and reproducibility for recognizing target components counteracting heart failure.
Figure 2.

Typical 3D plot of mixed standards obtained by the comprehensive 2D (A) NM/CMC column and (B) FM/CMC column coupled with monolithic column/TOFMS system. 1D: CMC columns (10 mm × 2 mm i.d.); Injection volume, 1 μL of mixed standards (0.5 mM each); Flow rate, 0.2 mL/min; Mobile phase, 5 mM ammonia acetate; Temperature, 37 °C. 2D: monolithic column (100 mm × 4.6 mm i.d.); Mobile phase, 0.1% formic acid (v/v)) and acetonitrile; Gradient elution: 0–4 min, from 5% ACN to 75% ACN; 4–4.5 min, from 75% ACN to 5% ACN; 4.5–5 min, 5% ACN; Flow rate, 3.5 mL/min; Temperature, 30 °C.
Practical Application
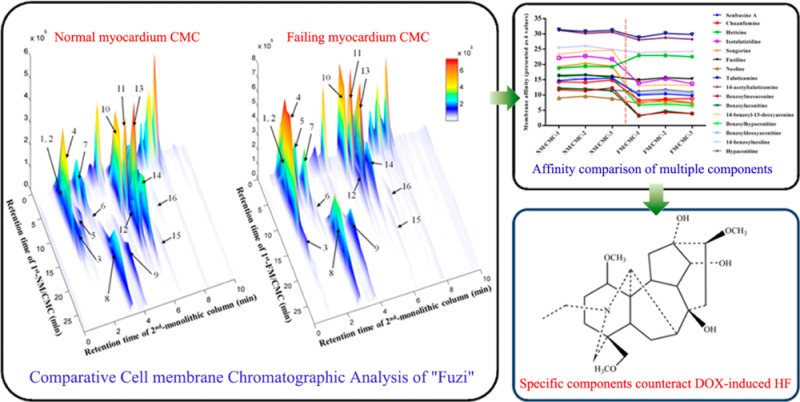
The typical spectra of Fuzi extracts obtained by independent analysis of NM/CMC-TOF/MS and monolithic column-TOF/MS are shown in Figure S-2A,B, Supporting Information, respectively. When applying the online comparative 2D CMC system, the direct recognition of the first dimension could be omitted and the entire retention behaviors of all constituents in Fuzi extract could be visually characterized by the 3D spectrum obtained from TOFMS data (Figure 3). It is the first time both normal and pathological CMC models were simultaneously loaded for studying different chromatographic behaviors of potential active components. Figure 3A,B shows the typical 3D spectra of Fuzi acting on NM/CMC and FM/CMC models, respectively. A total of 16 components in Fuzi were observed as having retention properties on both NM/CMC and FM/CMC models, which were unambiguously identified by matching the accurate mass data obtained by TOFMS with our previous reports (listed in Table 1).43 The chemical structures of 16 potential active components were shown in Figure S-3, Supporting Information. In order to characterize the different retention behaviors of the 16 potential components between two myocardium CMC models, the capacity factors (k) of each component on 6 different columns were calculated by eq 1. The variation trends of membrane affinity on different CMC columns are plotted in Figure 4.
| 1 |
where tR is the retention time of retention component and t0 is the dead time determined by the nonretained compound.
Figure 3.

3D plots of Fuzi extracts obtained by the comprehensive 2D (A) NM/CMC and (B) FM/CMC combined with monolithic column/TOFMS system. 1D: NM/CMC column (10 mm × 2 mm i.d.); Injection volume, 5 μL of Fuzi extract (0.3 g crude drug/mL); Flow rate, 0.2 mL/min; Mobile phase, 5 mM ammonia acetate; Temperature, 37 °C. 2D: monolithic column (100 mm × 4.6 mm i.d.); Mobile phase, 0.1% formic acid (v/v) and acetonitrile; Gradient elution: 0–5 min, from 5% ACN to 25% ACN; 5–9 min, from 25% ACN to 75% ACN; 9–4.5 min, from 75% ACN to 5% ACN; 9.5–10 min, 5% ACN; Flow rate, 3.5 mL/min; Temperature, 30 °C. For peak identification, see Table 1.
Table 1. 16 Potentially Active Components in Fuzi Characterized by the Comparative NM/FM CMC Analysis System.
| [M + H]+m/z |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| peak no. | tR (1st NM/CMC, min) | RSD % of k values, n = 3 (NM/CMC) | tR (1st FM/CMC, min) | RSD % of k values, n = 3 (FM/CMC) | tR (2nd RPLC, min) | detected | expected | error (ppm) | formula | identification |
| 1 | 2.5–15a | 2.70 | 0.5–10a | 2.98 | 0.91 | 424.2685 | 424.2694 | –2.04 | C23H37NO6 | senbusine A |
| 2 | 2.5–15 | 2.83 | 0.5–10 | 3.51 | 1.03 | 394.2596 | 394.2588 | 2.03 | C22H35NO5 | chuanfumine |
| 3 | 7.5–15 | 1.57 | 7.5–20 | 0.99 | 1.35 | 330.2052 | 330.2064 | –3.54 | C20H27NO3 | hetisine |
| 4 | 5–17.5 | 2.22 | 2.5–15 | 5.98 | 2.05 | 408.2761 | 408.2744 | 4.04 | C23H37NO5 | isotalatizidine |
| 5 | 5–17.5 | 3.21 | 2.5–7.5 | 3.96 | 2.2 | 358.2389 | 358.2377 | 3.43 | C22H31NO3 | songorine |
| 6 | 5–15 | 1.21 | 5–15 | 2.57 | 2.45 | 454.2791 | 454.2799 | –1.82 | C24H39NO7 | fuziline |
| 7 | 2.5–10 | 4.47 | 2.5–7.5 | 8.19 | 2.68 | 438.2833 | 438.2850 | –3.91 | C24H39NO6 | neoline |
| 8 | 12.5–30b | 0.96 | 10–30b | 6.39 | 3.22 | 422.2911 | 422.2901 | 2.31 | C24H39NO5 | talatizamine |
| 9 | 12.5–30b | 1.61 | 10–30b | 3.31 | 4.08 | 464.3024 | 464.3007 | 3.47 | C26H41NO6 | 14-acetyltalatizamine |
| 10 | 2.5–15 | 4.18 | 0.5–7.5 | 12.86 | 5.11 | 590.2936 | 590.2960 | –4.02 | C31H43NO10 | benzoylmesaconine |
| 11 | 2.5–12.5 | 3.46 | 0.5–7.5 | 20.10 | 5.50 | 604.3111 | 604.3116 | –0.87 | C32H45NO10 | benzoylaconitine |
| 12 | 7.5–20 | 2.93 | 2.5–12.5 | 1.49 | 5.54 | 558.3059 | 558.3061 | –0.44 | C31H43NO8 | 14-benzoyl-13-deoxyaconine |
| 13 | 5–20 | 3.19 | 0.5–7.5 | 4.46 | 5.78 | 574.3036 | 574.3011 | 4.43 | C31H43NO9 | benzoylhypaconitine |
| 14 | 5–20 | 4.41 | 2.5–12.5 | 4.26 | 6.20 | 588.3174 | 588.3167 | 1.18 | C32H45NO9 | benzoyldeoxyaconitine |
| 15 | 10–25 | 2.31 | 7.5–25 | 6.81 | 6.51 | 542.3086 | 542.3112 | –4.85 | C31H43NO7 | 14-benzoylneoline |
| 16 | 2.5–12.5 | 3.28 | 2.5–12.5 | 3.62 | 6.94 | 616.3126 | 616.3116 | 1.59 | C33H43NO10 | hypaconitine |
Retention time of CMC demonstrated as time points of the start and the end of the elution curve.
Peak that was not completely flushed out by 1st-CMC column within 30 min.
Figure 4.
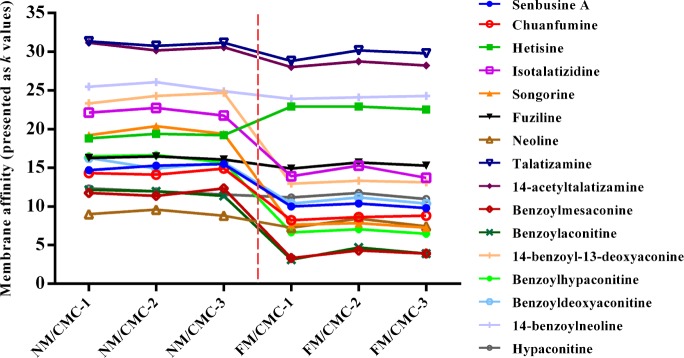
CMC affinity comparison (presented as k values) of 16 potentially active components on NM/CMC (n = 3) and FM/CMC models (n = 3).
From Figure 4, we can see that most of the components have less affinity on the FM/CMC model compared with the NM/CMC model, including the three major alkaloids, benzoylmesaconine (peak No. 10), benzoylaconitine (11), and benzoylhypaconitine (13) in Fuzi. That was mainly due to the complicated cardiotoxicity mechanism of DOX which results in the totally different membrane environments of normal and pathological myocardium in vivo.53 By comparing the difference of affinity on the NM/CMC and FM/CMC model, those components with good retention on NM/CMC, but poorer on FM/CMC, should be considered as nonspecific components for counteracting DOX-induced heart failure. Fewer efforts could be devoted for their further pharmacodynamic validation. On the other hand, those components with good affinity on both NM/CMC and FM/CMC or even better retention on FM/CMC should receive more attention because the high affinity on pathological cell membrane might indicate good selectivity and efficacy. Thus, only four components, hetisine (3), TALA (8), 14-acetyl-TALA (9), 14-benzoylneoline (15), which had high affinities on both NM/CMC and FM/CMC (decrease of k values <3), were focused by the comparative 2D CMC system and considered to counteract DOX-induced heart failure.
Effects on H9c2 Cell Viability and Integrity of Membrane
TALA was isolated from Fuzi for further pharmacodynamic validation because it had the highest affinity on the NM/CMC and FM/CMC model (k = 31.5–32.3) and was at a higher content in Fuzi than three other potential active components (isolation still in progress). DOX-induced H9c2 cell apoptosis and death is an ideal in vitro model for screening drugs that counteract heart failure54 and studying the associations of different molecular indicators.55 Rat cardiac H9c2 cells were treated with TALA (1, 2, 5, 10, 20 μM) in the absence of DOX for 24 h, and then, the rates of cell viability were evaluated by CCK-8. As shown in Figure 5A, TALA at each of these concentrations alone had a slight promoting proliferation but there were no significant differences from the control group (p > 0.05). To analyze the effects of TALA and benzoylaconitine on DOX-induced cytotoxicity in H9c2 cells, cell viability was examined after incubation with TALA or benzoylacontine in the presence of DOX (2 μM). TALA (2, 5, 10, 20 μM) pretreatments provided good protective effects on DOX-mediated cell death in a dose-dependent manner at low doses (p < 0.05, compared to DOX group), while benzoylacontine, a component with good retention on NM/CMC but poor on FM/CMC, showed little protective effects at the same concentrations of TALA (p > 0.05, compared to DOX group). These results confirmed the cardioprotective effect and noncytotoxicity of TALA in vitro and the selectivity of the comparative 2D CMC system. The integrity of plasma membranes was determined by monitoring the activity of cytoplasmic enzyme LDH in the extracellular incubation medium, which represents a common procedure to determine membrane leakage and cellular damage. As shown in Figure 5B, compared with normal cells, the released amount of LDH was increased when exposed to DOX. Compared with the DOX group, TALA (5, 10 μM) significantly decreased the amount of released LDH (p < 0.05). These results suggest that TALA could prevent cardiomyocytes from DOX-induced toxicity partly by the property of stabilizing cell membranes.
Figure 5.
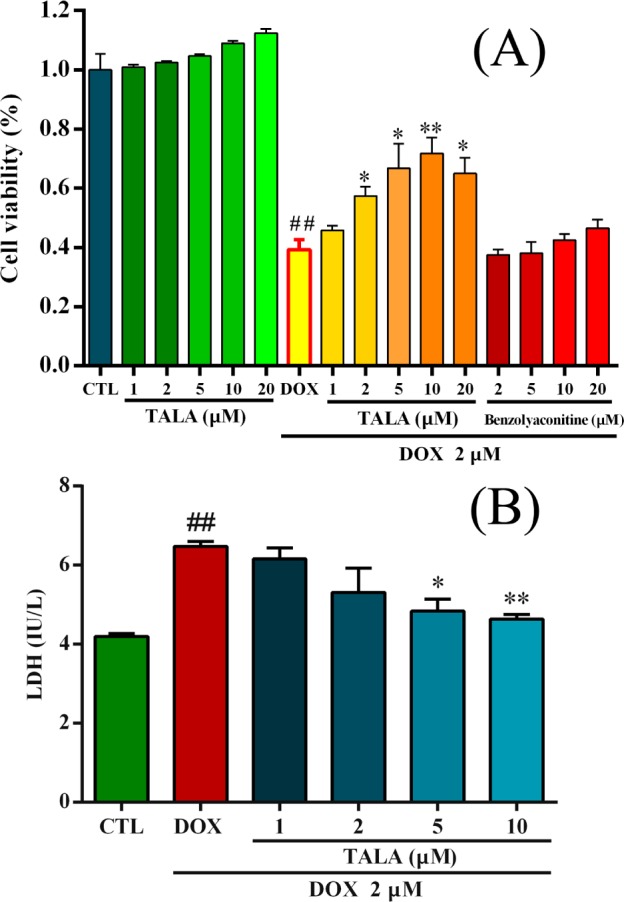
Effects of TALA on DOX-induced injury in rat cardiac H9c2 cells. (A) Cells were treated with TALA and benzolyaconitine (1, 2, 5, 10, 20 μM) for 2 h, followed by incubation with or without DOX for 24 h, and then the rates of cell viability were evaluated by CCK-8. (B) H9c2 cells were treated with TALA (1, 2, 5, and 10 μM) for 2 h, followed by incubation with 2 μM DOX for another 24 h. After collecting cell culture supernatants, extracellular LDH content was measured using a rat LDH ELISA kit. All data were expressed as means ± SEM (n = 3). ##p < 0.01 compared to the negative control group; *p < 0.05 and **p < 0.01 compared to DOX group.
Binding Target of TALA and 14-Acetyl-TALA
Previous researchers claimed that TALA is a selective voltage-gated K+ (Kv) channel blocker with high binding affinity,56,57 which may play a significant role in regulating the cardiac action potential. We first ran 4-AP (a selective Kv channel blocker) on both NM and FM CMC columns. Figure 6A shows that 4-AP has no significant change of activity in the FM/CMC model compared with the NM/CMC model. This was in accordance to our Western blot results of Kv 4.2, which is the major expressed subtype in the left ventricle. Thus, the high affinities of TALA and 14-acetyl-TALA on both NM and FM/CMC models can be explained. Next, a competitive displacement assay was applied to confirm the affinity of TALA on Kv channels in the FM/CMC model. As shown in Figure 6B, retentions of TALA decreased with the increase of concentration of 4-AP in the mobile phase, which indicated that TALA at least has one same binding site with 4-AP. These two drugs were also run on activated silica column alone, and no retention was observed.
Figure 6.
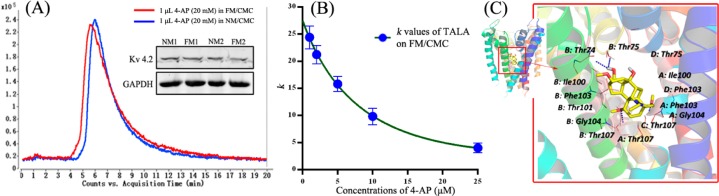
(A) Affinities of 4-AP (Kv channel blocker) on NM and FM/CMC columns and expression levels of Kv 4.2 and GAPDH proteins in nornal and failing myocardium. The chromatographic curves are obtained by extraction ion window of m/z 95.04–95.08. (B) Competitive displacement assay of TALA on FM/CMC columns (presented as k values) with different concentraions of 4-AP (1, 2, 5, 10, and 25 μM) added in mobile phase. Each point with a bar represents the mean ± SEM (n = 3). (C) Molecular docking study of TALA in the active site of K+ channel. Three-dimensional structure of the complex between TALA and the binding site of K+ channel pore (PDB: 1J95). The TALA is separately colored according to atom types (carbon, yellow; oxygen, red; nitrogen, blue). The residues of binding sites of K+ channel that interact with TALA are shown through hydrophobic interactions and hydrogen bonds. Red dotted lines stand for hydrogen bonds, and key residues are labeled in black.
To explore further the underlying mechanisms, docking studies were conducted to investigate the interaction of TALA and 14-acetyl-TALA, two similar compounds in the significant activity counteracting DOX-induced heart failure, with the crystal structure of Kv channel from Streptomyces lividans using Sybyl-X1.3. The docking results were manually checked to ensure that the binding mode of the compound had reasonable interaction and geometry fitting. The predicted interactions between TALA and the binding site of Kv channel are shown in Figure 6C (14-acetyl-TALA in Figure S-4, Supporting Information). Residues Thr75 (chains: B and D), Thr74 (chain: B and D), Ile100 (chain: B, C, and D), Phe103 (chains: A and B), Gly104 (chains: B), and Thr107 (chain: A and D) form hydrophobic cleft around TALA and 14-acetyl-TALA. The specific hydroxyl (−OH) and methoxy (−OMe) groups of the two compounds show the hydrogen-bonding interactions with the backbone of Ile100 (chain: A) and Thr107 (chain: D), which were located in the selectivity filter of K+ channel. In particular, Thr107 was also one of the binding sites of the selective K+ channel blocker 4-AP.58 Recent reports have suggested that the binding pocket and the pore region of K+ channel consist of key residues (Thr, Ile, Gly, Phe, and Ala), which form the selectivity filter of the K+ channel.59 Our docking results suggest that 14-acetyl-TALA and TALA block the Kv channel by entering the selectivity filter to suppress the K+ efflux, thus protecting myocardial cells from apoptosis. Given the similar structure and chromatographic behaviors of the two compounds, 14-acetyl-TALA (score 9.73) was screened as a new selective Kv channel blocker with a higher affinity than TALA (score 8.90).
Synergistic Effect with K+ Channel Blocker
Blockade of sarcolemmal Kv channels can certainly inhibit the apoptotic volume decrease and attenuates apoptosis in many cell types, as in a detailed review by Bortner and Cidlowski.60 As the K+ currents are up-regulated in failing ventricles,61 components with high affinity on failing heart membrane might be good leading compounds for treatment of heart failure. The 4-AP is a classical Kv channel blocker, often used for reducing K+ current.62 The prevention of cell apoptosis and death effect by 4-AP was also extensively studied in many cell types, including myeloblastic leukemia cells,63 pulmonary artery smooth muscle cells,64 cerebellar granule neurons,65 and the rat cardiac H9c2 cell line.66 Except one same binding site, Thr107 (1J95 PDB structure) as TALA, Ala 111 is a specific active binding site of 4-AP,58 which is located in the intracellular mouth of K+ pores.67 In order to further demonstrate the active binding sites and mechanism of TALA on preventing DOX-induced cell apoptosis and death, the synergistic effect of TALA added with the specific Kv channel blocker 4-AP was investigated by flow cytometric analysis of apoptosis and death in FITC-Annexin V-PI double stained H9c2 cells. As shown in Figure S-5, Supporting Information, when treated with 1 μM DOX for 24 h, 36.73% cells of late stages of apoptosis and death was observed. The ratios of DOX-induced cell apoptosis and death can be reduced to 19.92% and 24.70% by treatment with 10 μM TALA or 1 mM 4-AP, respectively. When cotreated with the two drugs, only 14.52% apoptotic and dead cells were observed, which indicated the significant synergistic effect of TALA and 4-AP on prevention of DOX-induced H9c2 cell death. These results confirm that the counteracting DOX-induced heart failure effect of TALA partially relies on blocking Kv channels by binding to different sites of 4-AP, which significantly regulate the loss of intracellular potassium and cell shrinkage in response to regulatory volume decrease, thereby preventing cell apoptosis.60 Because the alkaloids in Fuzi have the same parent nucleus and similar structures, the blocking of Kv channels by Fuzi should be one of the major contributors for its counteraction of DOX-induced heart failure.
Concluding Remarks
We have demonstrated the affinity comparison of major components from Fuzi on normal and failing rat myocardium cell membrane using an integrated column selection and online comprehensive 2D chromatography analytical system for screening of specific components that counteract DOX-induced heart failure. With a comprehensive methodological investigation and optimization, the better quality and reproducibility of each CMC column derived from normal or failing heart tissue were achieved for large scale comparative analysis. Four components were screened and considered as specific active components that counteract DOX-induced heart failure. TALA, having the highest affinity and content, was isolated for further in vitro pharmacodynamic validation to confirm the screen results. According to assays on cell viability, the cardioprotective effect of TALA was confirmed. In addition, Kv channel was successfully identified as the high affinity drug target of TALA and 14-acetyl-TALA by the competitive displacement assay and molecular docking calculation. Consequently, the active components and cardiotonic mechanism of Fuzi were further understood. The established 2D CMC analysis system for comparison of normal and pathological models is suitable for efficiently screening active components from HMs that act on specific diseases by increasing the specificity of the screened results and saving a lot of labor on subsequent pharmacodynamic validation.
In the present myocardium CMC model, hundreds of cell membrane and plasma membrane receptors exist. By characterizing the complicated interactions between multiple components and multiple receptors, the proposed CMC analysis system can be applied as a preliminary screening tool for discovering active leading components from HMs or other complex medicine systems. It could also be applied for the determination of the dissociation constant of analytes using the previous reported multiple receptor CMSP method.29
Acknowledgments
This work was supported by the National Natural Science Fund of China (No. 81273472) and Project of Modernization of Traditional Chinese Medicine of Shanghai (No. 12401900802). The authors would like to acknowledge the financial support for the laboratory at University of Pittsburgh from the NIH R01DA025612 and HL109654 (X.-Q. Xie).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ X. Chen and Y. Cao contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hao H.; Cui N.; Wang G.; Xiang B.; Liang Y.; Xu X.; Zhang H.; Yang J.; Zheng C.; Wu L.; Gong P.; Wang W. Anal. Chem. 2008, 80, 8187–8194. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Bauer R.; Hendry B. M.; Fan T. P.; Zhao Z.; Duez P.; Simmonds M. S.; Witt C. M.; Lu A.; Robinson N.; Guo D. A.; Hylands P. J. BMC Complementary Altern. Med. 2013, 13, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. W.; Vederas J. C. Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- Wang B.; Deng J.; Gao Y.; Zhu L.; He R.; Xu Y. Fitoterapia 2011, 82, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Moaddel R.; Rosenberg A.; Spelman K.; Frazier J.; Frazier C.; Nocerino S.; Brizzi A.; Mugnaini C.; Wainer I. W. Anal. Biochem. 2011, 412, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitabatake T.; Moaddel R.; Cole R.; Gandhari M.; Frazier C.; Hartenstein J.; Rosenberg A.; Bernier M.; Wainer I. W. Anal. Chem. 2008, 80, 8673–8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco Vanzolini K.; Jiang Z.; Zhang X.; Vieira L. C.; Correa A. G.; Cardoso C. L.; Cass Q. B.; Moaddel R. Talanta 2013, 116, 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haginaka J.; Kitabatake T.; Hirose I.; Matsunaga H.; Moaddel R. Anal. Biochem. 2013, 434, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M.; Wilson D. R.; Fugmann S. D.; Moaddel R. Anal. Chem. 2011, 83, 7400–7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habicht K. L.; Frazier C.; Singh N.; Shimmo R.; Wainer I. W.; Moaddel R. J. Pharm. Biomed. Anal. 2013, 72, 159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moaddel R.; Musyimi H. K.; Sanghvi M.; Bashore C.; Frazier C. R.; Khadeer M.; Bhatia P.; Wainer I. W. J. Pharm. Biomed. Anal. 2010, 52, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moaddel R.; Oliveira R. V.; Kimura T.; Hyppolite P.; Juhaszova M.; Xiao Y.; Kellar K. J.; Bernier M.; Wainer I. W. Anal. Chem. 2008, 80, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L.; Wang S.; Geng X. Chromatographia 2001, 54, 71–76. [Google Scholar]
- Zeng C. M.; Zhang Y.; Lu L.; Brekkan E.; Lundqvist A.; Lundahl P. Biochim. Biophys. Acta 1997, 1325, 91–98. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Xiao Y.; Kellar K. J.; Wainer I. W. Anal. Biochem. 1998, 264, 22–25. [DOI] [PubMed] [Google Scholar]
- He L. C.; Wang S. C.; Yang G. D.; Zhang Y. M.; Wang C. H.; Yuan B. X.; Hou X. F. Drug Discovery Ther. 2007, 1, 104–107. [PubMed] [Google Scholar]
- Moaddel R.; Wainer I. W. Nat. Protoc. 2009, 4, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Sun M.; Zhang Y.; Du H.; He L. J. Chromatogr., A 2010, 1217, 5246–5252. [DOI] [PubMed] [Google Scholar]
- Hou X.; Wang S.; Hou J.; He L. J. Sep. Sci. 2011, 34, 508–513. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Chen X.; Lv D.; Dong X.; Zhang G.; Chai Y. J. Pharm. Anal. 2011, 21, 81–91. [Google Scholar]
- Chen X.; Cao Y.; Lv D.; Zhu Z.; Zhang J.; Chai Y. J. Chromatogr., A 2012, 1242, 67–74. [DOI] [PubMed] [Google Scholar]
- Du H.; Ren J.; Wang S.; He L. Anal. Bioanal. Chem. 2011, 400, 3625–3633. [DOI] [PubMed] [Google Scholar]
- Du H.; He J.; Wang S.; He L. Anal. Bioanal. Chem. 2010, 397, 1947–1953. [DOI] [PubMed] [Google Scholar]
- Zeng A.; Yuan B.; Wang C.; Yang G.; He L. J. Chromatogr., B 2009, 877, 1833–1837. [DOI] [PubMed] [Google Scholar]
- Wang L.; Ren J.; Sun M.; Wang S. J. Pharm. Biomed. Anal. 2010, 51, 1032–1036. [DOI] [PubMed] [Google Scholar]
- Sun M.; Ren J.; Du H.; Zhang Y.; Zhang J.; Wang S.; He L. J. Chromatogr., B 2010, 878, 2712–2718. [DOI] [PubMed] [Google Scholar]
- Wang C.; He L.; Wang N.; Liu F. J. Chromatogr., B 2009, 877, 3019–3024. [DOI] [PubMed] [Google Scholar]
- Hou X.; Zhou M.; Jiang Q.; Wang S.; He L. J. Chromatogr., A 2009, 1216, 7081–7087. [DOI] [PubMed] [Google Scholar]
- Moaddel R.; Cloix J. F.; Ertem G.; Wainer I. W. Pharm. Res. 2002, 19, 104–107. [DOI] [PubMed] [Google Scholar]
- Jover E.; Matamoros V.; Bayona J. M. J. Chromatogr., A 2009, 1216, 4013–4019. [DOI] [PubMed] [Google Scholar]
- Adcock J. L.; Adams M.; Mitrevski B. S.; Marriott P. J. Anal. Chem. 2009, 81, 6797–6804. [DOI] [PubMed] [Google Scholar]
- Pierce K. M.; Kehimkar B.; Marney L. C.; Hoggard J. C.; Synovec R. E. J. Chromatogr., A 2012, 1255, 3–11. [DOI] [PubMed] [Google Scholar]
- Gilar M.; Olivova P.; Daly A. E.; Gebler J. C. Anal. Chem. 2005, 77, 6426–6434. [DOI] [PubMed] [Google Scholar]
- Kalili K. M.; Vestner J.; Stander M. A.; de Villiers A. Anal. Chem. 2013, 85, 9107–9115. [DOI] [PubMed] [Google Scholar]
- Czarnecki C. M. Comp. Biochem. Physiol., Part C 1984, 79, 9–14. [DOI] [PubMed] [Google Scholar]
- Powell S. R.; McCay P. B. Free Radical Biol. Med. 1995, 18, 159–168. [DOI] [PubMed] [Google Scholar]
- Han X. Z.; Gao S.; Cheng Y. N.; Sun Y. Z.; Liu W.; Tang L. L.; Ren D. M. Biosci. Trends 2012, 6, 19–25. [DOI] [PubMed] [Google Scholar]
- Riad A.; Bien S.; Gratz M.; Escher F.; Westermann D.; Heimesaat M. M.; Bereswill S.; Krieg T.; Felix S. B.; Schultheiss H. P.; Kroemer H. K.; Tschope C. Eur. J. Heart Failure 2008, 10, 233–243. [DOI] [PubMed] [Google Scholar]
- Liao S. Y.; Tse H. F.; Chan Y. C.; Mei-Chu Yip P.; Zhang Y.; Liu Y.; Li R. A. Heart Rhythm 2013, 10, 273–282. [DOI] [PubMed] [Google Scholar]
- Tan G.; Lou Z.; Liao W.; Zhu Z.; Dong X.; Zhang W.; Li W.; Chai Y. PLoS One 2011, 6, e27683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G.; Liao W.; Dong X.; Yang G.; Zhu Z.; Li W.; Chai Y.; Lou Z. PLoS One 2012, 10.1371/journal.pone.0034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G.; Lou Z.; Liao W.; Dong X.; Zhu Z.; Li W.; Chai Y. Mol. BioSyst. 2012, 8, 548–556. [DOI] [PubMed] [Google Scholar]
- Tan G.; Lou Z.; Jing J.; Li W.; Zhu Z.; Zhao L.; Zhang G.; Chai Y. Biomed. Chromatogr. 2011, 25, 1343–1351. [DOI] [PubMed] [Google Scholar]
- Takaseya T.; Ishimatsu M.; Tayama E.; Nishi A.; Akasu T.; Aoyagi S. J. Am. Coll. Cardiol. 2004, 44, 2239–2246. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Gao Y.; Tan G.; Wu S.; Dong X.; Lou Z.; Zhu Z.; Chai Y. J. Ethnopharmacol. 2013, 147, 349–356. [DOI] [PubMed] [Google Scholar]
- Jain A. N. J. Med. Chem. 2003, 46, 499–511. [DOI] [PubMed] [Google Scholar]
- Chen J. Z.; Wang J.; Xie X. Q. J. Chem. Inf. Model. 2007, 47, 1626–1637. [DOI] [PubMed] [Google Scholar]
- Xie X. Q.; Chen J. Z.; Billings E. M. Proteins 2003, 53, 307–319. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Wang J.; Cui Y.; Wu X. J. Tradit. Chin. Med. 2012, 32, 308–313. [DOI] [PubMed] [Google Scholar]
- Dodd D. A.; Atkinson J. B.; Olson R. D.; Buck S.; Cusack B. J.; Fleischer S.; Boucek R. J. Jr. J. Clin. Invest. 1993, 91, 1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenfuss G. Cardiovasc. Res. 1998, 37, 279–289. [DOI] [PubMed] [Google Scholar]
- Jensen B. C.; Swigart P. M.; De Marco T.; Hoopes C.; Simpson P. C. Circ.: Heart Failure 2009, 2, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer H. G.; Pinedo H. M.; Schuurhuis G. J.; Joenje H. Pharmacol. Ther. 1990, 47, 219–231. [DOI] [PubMed] [Google Scholar]
- qXin H.; Liu X. H.; Zhu Y. Z. Eur. J. Pharmacol. 2009, 612, 75–79. [DOI] [PubMed] [Google Scholar]
- Bartha E.; Solti I.; Szabo A.; Olah G.; Magyar K.; Szabados E.; Kalai T.; Hideg K.; Toth K.; Gero D.; Szabo C.; Sumegi B.; Halmosi R. J. Cardiovasc. Pharmacol. 2011, 58, 380–391. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Song M.; Hou L.; Yu Z.; Chen H. Neurosci. Lett. 2012, 518, 122–127. [DOI] [PubMed] [Google Scholar]
- Song M. K.; Liu H.; Jiang H. L.; Yue J. M.; Hu G. Y.; Chen H. Z. Neuroscience 2008, 155, 469–475. [DOI] [PubMed] [Google Scholar]
- Caballero N. A.; Melendez F. J.; Nino A.; Munoz-Caro C. J. Mol. Model. 2007, 13, 579–586. [DOI] [PubMed] [Google Scholar]
- Doyle D. A.; Morais Cabral J.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. Science 1998, 280, 69–77. [DOI] [PubMed] [Google Scholar]
- Bortner C. D.; Cidlowski J. A. Arch. Biochem. Biophys. 2007, 462, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P. C.; Turker I.; Lopshire J. C.; Masroor S.; Nguyen B. L.; Tao W.; Rubart M.; Chen P. S.; Chen Z.; Ai T. J. Am. Heart Assoc. 2013, 2, e004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krick S.; Platoshyn O.; Sweeney M.; McDaniel S. S.; Zhang S.; Rubin L. J.; Yuan J. X. Am. J. Physiol.: Heart Circ. Physiol. 2002, 282, H184–193. [DOI] [PubMed] [Google Scholar]
- Wang L.; Xu D.; Dai W.; Lu L. J. Biol. Chem. 1999, 274, 3678–3685. [DOI] [PubMed] [Google Scholar]
- Krick S.; Platoshyn O.; McDaniel S. S.; Rubin L. J.; Yuan J. X. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2001, 281, L887–894. [DOI] [PubMed] [Google Scholar]
- Hu C. L.; Liu Z.; Zeng X. M.; Liu Z. Q.; Chen X. H.; Zhang Z. H.; Mei Y. A. Neuropharmacology 2006, 51, 737–746. [DOI] [PubMed] [Google Scholar]
- Ekhterae D.; Platoshyn O.; Zhang S.; Remillard C. V.; Yuan J. X. Am. J. Physiol.: Cell Physiol. 2003, 284, C1405–1410. [DOI] [PubMed] [Google Scholar]
- Kirsch G. E.; Shieh C. C.; Drewe J. A.; Vener D. F.; Brown A. M. Neuron 1993, 11, 503–512. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.