

Abstract
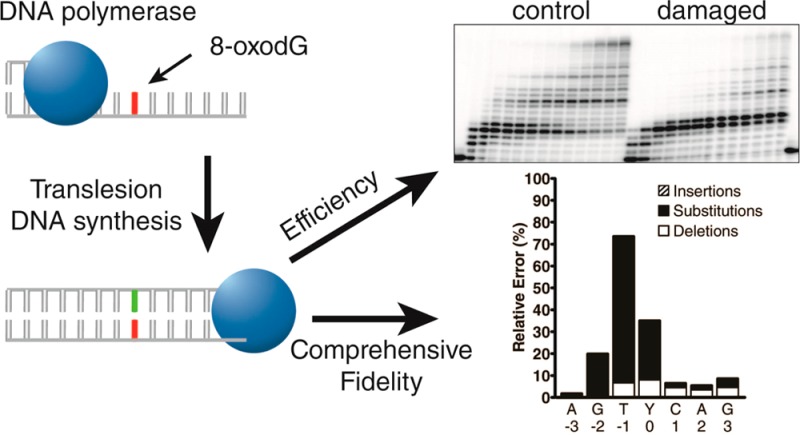
One of the most common lesions induced by oxidative DNA damage is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG). Replicative DNA polymerases poorly traverse this highly mutagenic lesion, suggesting that the replication fork may switch to a polymerase specialized for translesion DNA synthesis (TLS) to catalyze 8-oxodG bypass in vivo. Here, we systematically compared the 8-oxodG bypass efficiencies and fidelities of the TLS-specialized, human Y-family DNA polymerases eta (hPolη), iota (hPolι), kappa (hPolκ), and Rev1 (hRev1) either alone or in combination. Primer extension assays revealed that the times required for hPolη, hRev1, hPolκ, and hPolι to bypass 50% of the 8-oxodG lesions encountered (t50bypass) were 0.58, 0.86, 108, and 670 s, respectively. Although hRev1 bypassed 8-oxodG efficiently, hRev1 failed to catalyze the extension step of TLS, and a second polymerase was required to extend the lesion bypass products. A high-throughput short oligonucleotide sequencing assay (HT-SOSA) was used to quantify the types and frequencies of incorporation errors produced by the human Y-family DNA polymerases at and near the 8-oxodG site. Although hPolη bypassed 8-oxodG most efficiently, hPolη correctly incorporated dCTP opposite 8-oxodG within only 54.5% of the sequences analyzed. In contrast, hPolι bypassed the lesion least efficiently but correctly incorporated dCTP at a frequency of 65.8% opposite the lesion. The combination of hRev1 and hPolκ was most accurate opposite 8-oxodG (92.3%), whereas hPolκ alone was the least accurate (18.5%). The t50bypass value and correct dCTP incorporation frequency in the presence of an equal molar concentration of all four Y-family enzymes were 0.60 s and 43.5%, respectively. These values are most similar to those of hPolη alone, suggesting that hPolη outcompetes the other three Y-family polymerases to catalyze 8-oxodG bypass in vitro and possibly in vivo.
Introduction
Cellular genomes are constantly damaged by endogenous agents, such as oxygen radicals formed during aerobic respiration. This oxidative DNA damage is proposed to be a major contributor to carcinogenesis and aging.1,2 One of the most common oxidative DNA lesions generated within cells is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG). Although structural studies indicate that 8-oxodG induces only minor distortions to the helical structure of DNA that are localized to the modified base,3−5 8-oxodG is highly mutagenic due to its dual coding potential.6 During DNA replication, 8-oxodG adopts either an anti or syn conformation within a polymerase active site. While in the syn conformation, a templating 8-oxodG lesion utilizes the Hoogsteen edge of the damaged base to preferentially form base pairs with incoming dATP.7 If left unrepaired, these 8-oxodG:dA mispairs will lead to G → T transversion mutations, which have been linked to cancer induction.8
Although 8-oxodG does not completely block DNA synthesis catalyzed by replicative DNA polymerases,9−13 this lesion does induce the pausing of DNA polymerases alpha (Polα), delta (Polδ), and epsilon (Polε). Such a stalling event may provide an opportunity for a cell to switch to a DNA polymerase that is specialized for the bypass of DNA lesions, a process known as translesion DNA synthesis (TLS). The majority of lesion bypass DNA polymerases belong to the Y-family. Notably, humans encode four Y-family enzymes: DNA polymerases eta (hPolη), iota (hPolι), kappa (hPolκ), and Rev1 (hRev1). These four low-fidelity DNA polymerases are characterized by a lack of proofreading 3′ → 5′ exonuclease activity, low processivity, and a relatively spacious, solvent-exposed active site. Importantly, the human Y-family enzymes demonstrate a large degree of functional divergence due to the fact that during TLS, the nucleotide incorporation fidelity and efficiency of each Y-family polymerase is lesion specific.14 Therefore, it is likely that each Y-family polymerase has evolved to catalyze TLS of a specific set of lesions.
Investigation of the lesion bypass efficiencies and fidelities of the Y-family polymerases may help illuminate the in vivo roles of these polymerases for the TLS of specific lesions. To this end, earlier reports have examined the 8-oxodG bypass abilities of hPolη,10 hPolκ,15−17 hPolι,18 and hRev119 separately by using various enzymatic assays. However, these studies have utilized different reaction conditions and DNA substrates to individually investigate each enzyme, complicating direct comparisons between the activities of all four human Y-family enzymes. Furthermore, the kinetic methods previously utilized were unable to accurately measure deletion or insertion events induced by TLS. Therefore, we sought to systematically compare the 8-oxodG bypass efficiency of each Y-family enzyme under the same reaction conditions and to determine the type and frequency of errors induced by TLS of 8-oxodG through a high-throughput short oligonucleotide sequencing assay (HT-SOSA) that was recently developed in our laboratory.20 The HT-SOSA method is advantageous for a comprehensive analysis of replication errors induced by TLS of a damaged site as this method (1) provides a complete mutagenic profile of DNA damage-induced mutations by accounting for all mutagenic events, such as base deletions, insertions, and substitutions; (2) supplies sequencing information for incorporation events opposite, upstream, and downstream from the lesion site; and (3) allows for analysis of multibase mutations from a single full-length product. Here, we use a combination of primer extension assays and HT-SOSA to investigate for the first time the efficiency and accuracy of 8-oxodG bypass catalyzed by the human Y-family DNA polymerases both alone and in combination.
Experimental Procedures
Materials
OptiKinase was purchased from USB Corporation, dNTPs were purchased from GE Healthcare, and [γ-32P]ATP was purchased from PerkinElmer. Human Polη with a C-terminal His6 tag, human Polκ with an N-terminal His6 tag, and human Rev1 with an N-terminal His6 tag were expressed in Escherichia coli and purified as previously described.21 Human Polι with an N-terminal GST tag was expressed in Escherichia coli and purified, and the N-terminal GST tag was subsequently removed by proteolytic cleavage with tobacco etch viral protease as previously described.21
DNA Substrates
The synthetic 40-mer DNA template containing 8-oxodG (40-mer-8oxodG) was purchased from Midland Certified Reagent Company (Table 1). All other synthetic DNA oligomers were purchased from Integrated DNA Technologies (Table 1 and Table S1, Supporting Information). All DNA oligomers were gel purified by denaturing polyacrylamide gel electrophoresis (PAGE, 10% polyacrylamide, 8 M urea). Primers were 5′-[32P]-radiolabeled by incubating each primer with OptiKinase and [γ-32P]ATP for 3 h at 37 °C. DNA substrates were generated by annealing a 5′-[32P]-radiolabeled primer with either the control 40-mer template or damaged 40-mer-8oxodG template at a molar ratio of 1.00:1.15. Annealing solutions were heat denatured by incubation at 75 °C for 5 min, followed by slow cooling to 25 °C over several hours.
Table 1. DNA Primers and Templates.

Y represents the 8-oxodG lesion site.
Reaction Buffer
All kinetic and HT-SOSA reactions were performed in optimized reaction buffer R (50 mM HEPES, pH 7.5 at 37 °C, 50 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, 10% glycerol, and 0.1 mg/mL BSA) at 37 °C.22 All reported concentrations are final after mixing.
Running Start Assays
A preincubated solution containing either a single polymerase (100 nM) or all four human Y-family polymerases (25 nM each) and 100 nM of a 5′-[32P]-radiolabeled DNA substrate (17-mer/40-mer or 17-mer/40-mer-8oxodG) was rapidly mixed with a solution of all four dNTPs (200 μM each). The reaction mixtures were quenched at various times by the addition of 0.37 M EDTA. A rapid chemical quench flow apparatus (KinTek) was used for kinetic experiments with reaction times ranging from milliseconds to several minutes. The reaction products were resolved by using denaturing PAGE and quantified by using a Typhoon Trio (GE Healthcare).
Analysis of the running start assays was performed as previously described21,23 by calculating the relative lesion bypass efficiencies (8-oxodG bypass%) of each polymerase as a function of reaction time. For each time point t, 8-oxodG bypass% was calculated as the ratio of the bypass events to the encounter events (eq 1):
| 1 |
where the total 8-oxodG bypass events (B) was the sum of the concentrations of all products with sizes greater than or equal to the 22-mer, and the total 8-oxodG “encounter” events (E) was the summation of the 21-mer product concentration and the total 8-oxodG bypass events (B). The 8-oxodG bypass% as calculated by eq 1 was then plotted as a function of reaction time. To compare the 8-oxodG bypass efficiency of each polymerase, the t50bypass was defined as the time required for each Y-family polymerase to bypass 50% of the total 8-oxodG lesions encountered. This t50bypass value was calculated from the 8-oxodG bypass% plots assuming a constant rate of lesion bypass between the time point immediately before and the time point immediately after the bypass of 50% of the lesion sites encountered.
hRev1 Standing Start Assays
A preincubated solution of 100 nM or 400 nM of hRev1 and 100 nM of a 5′-[32P]-radiolabeled DNA substrate (21-mer/40-mer or 21-mer/40-mer-8oxodG) was rapidly mixed with a solution containing all four dNTPs (200 μM each). Where indicated, hPolκ (300 nM) was added to the reactions 30 s after the addition of the dNTP solution. The reaction mixtures were quenched and resolved by denaturing PAGE as described above.
High-Throughput Short Oligonucleotide Sequencing Assay
HT-SOSA was performed as previously described20 with the following modifications. Lesion bypass products were generated by mixing either the radiolabeled 13-mer/40-mer substrate or the 13-mer/40-mer-8oxodG substrate (100 nM) into a solution containing a single enzyme (100 nM) or all four enzymes (25 nM each). This solution was incubated briefly and subsequently mixed with a solution of all four dNTPs (200 μM each). The reactions containing only hPolη or only hPolκ were incubated at 37 °C for 1 h, and the reactions containing only hPolι or all 4 human Y-family polymerases were incubated at 37 °C for 4 h. To produce lesion bypass products with the combination of hRev1 and hPolκ, either the 16-mer/40-mer or 16-mer/40-mer-8oxodG DNA substrate (100 nM) was briefly preincubated with hRev1 (100 nM) and subsequently mixed with a solution containing all four dNTPs (200 μM each). After 10 or 60 s for the control or damaged DNA substrate reactions, respectively, hPolκ (300 nM) was added, and the reactions were incubated for an additional 1 h at 37 °C. After generation of the lesion bypass products, the full-length, newly synthesized DNA strands were effectively separated from the template strands by using denaturing PAGE, as the 13-mer and 16-mer primers annealed 5 bases upstream of the 3′-end of the 40-mer and 40-mer-8oxodG templates (Scheme 1).
Scheme 1.
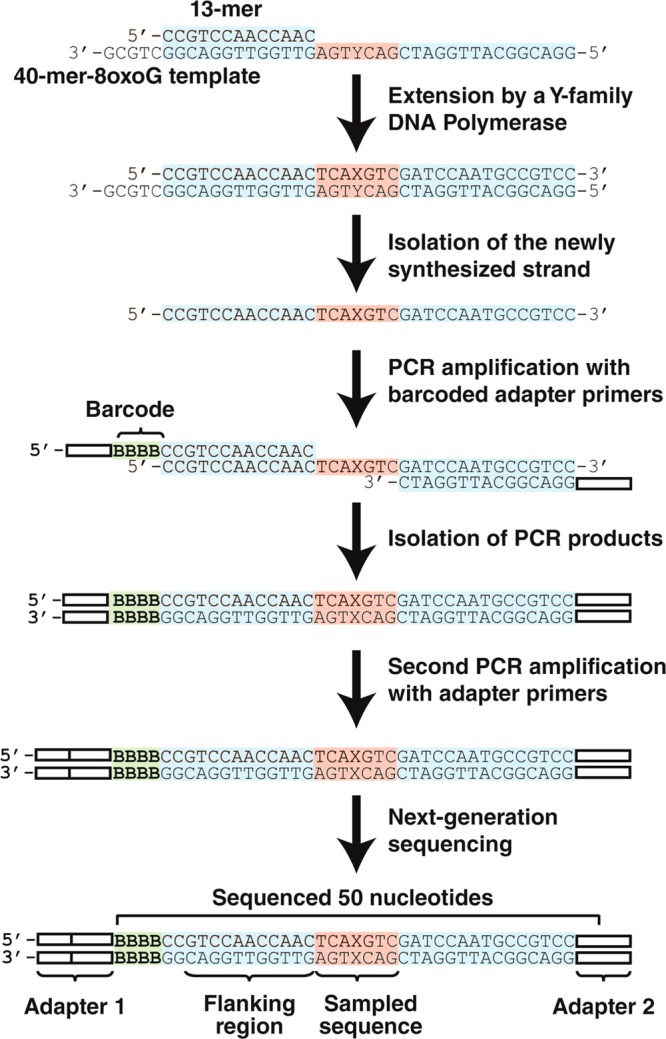
Creation of Sequencing Libraries, Next-Generation Sequencing, and DNA Sequence Analysis
Next-generation sequencing libraries were generated by first amplifying each purified, single-stranded DNA lesion bypass product by using 1 of 10 unique barcoded primers (Table S1, Supporting Information) and a common reverse primer. The PCR products were then gel purified, and the remaining adapter sequences required for next-generation sequencing were added in a second round of PCR by using Illumina PCR primers 1 and 2 (Table S1, Supporting Information). The completed sequencing libraries were then gel purified and mixed in equal molar ratios. The sequencing libraries generated from the lesion bypass products were then mixed with an equal molar amount of bacteriophage ΦX sequencing libraries to increase sequence diversity. Finally, the sequencing library solution was sequenced by using a HiSeq 2000 DNA sequencer (Illumina). This HT-SOSA method is described in Scheme 1. DNA sequencing reads were then sorted and analyzed as previously described.20
Results
Running Start Assays
To compare the ability of each human Y-family DNA polymerase to bypass 8-oxodG under identical in vitro conditions, we carried out running start primer extension assays. To this end, DNA substrates were generated by annealing a 5′-[32P]-radiolabeled 17-mer primer to either the undamaged 40-mer template or the damaged 40-mer-8oxodG template, which contained a site-specifically placed 8-oxodG lesion (Table 1 and Figure 1K). These DNA substrates were then extended by the action of a Y-family DNA polymerase. The 8-oxodG lesion did not induce any noticeable stalling of hPolη when compared to the undamaged control (Figure 1A and B). Thus, the efficiency of hPolη nucleotide incorporation was not inhibited by the lesion during the bypass or extension step of 8-oxodG TLS. In contrast, hPolκ paused during incorporations both opposite 8-oxodG and during the following extension step as indicated by the transient accumulation of the 22-mer and 23-mer products (Figure 1C and D). This result is consistent with previous steady-state kinetic studies indicating that the efficiency of hPolκ nucleotide incorporation is reduced opposite this lesion and that during the extension step of TLS, hPolκ more efficiently extends the 8-oxodG:dA mismatch compared to the correct 8-oxodG:dC base pair.17 The 8-oxodG lesion significantly blocked nucleotide incorporation by hPolι both opposite the lesion and for the extension step as indicated by the accumulation of the 21-mer and 22-mer products, respectively (Figure 1E and F). These two consecutive pause sites were also observed with the control substrate but to a lesser extent, as hPolι is known to preferentially misincorporate dG opposite template dT and inefficiently extend the mismatch. This combination of events reduces hPolι activity opposite template dTs, a phenomenon known as the T stop.24 Thus, the relative efficiency of nucleotide incorporation catalyzed by hPolι opposite 8-oxodG and during the extension step of TLS is predicted to be sequence-dependent. Polymerase hRev1 failed to extend past the lesion site within 20 h (Figure 1H). This failure to produce full-length DNA products was expected as hRev1 preferentially functions as a dCTP transferase by predominantly incorporating dCTP opposite DNA lesions or undamaged bases.19 Nevertheless, the running start assay indicated that the 8-oxodG lesion reduced the activity of this polymerase as hRev1 extended the control DNA substrate to a 23-mer but was only capable of extending the damaged DNA substrate to a 21-mer product (Figure 1G and H).
Figure 1.

Running start assays for the human Y-family DNA polymerases on undamaged and damaged DNA templates. A preincubated solution containing 100 nM of 5′-[32P]-radiolabeled (A, C, E, G, and I) 17-mer/40-mer or (B, D, F, H, and J) 17-mer/40-mer-8oxodG and either 100 nM of the indicated DNA polymerase or 25 nM of each Y-family polymerase was rapidly mixed with a solution containing all 4 dNTPs (200 μM each). The reaction mixtures were quenched at the indicated times with 0.37 M EDTA and resolved by using denaturing PAGE. The sizes of important products are indicated, and the 22nd position is denoted with an asterisk (*) to indicate an incorporation opposite the 8-oxodG lesion site. (K) The damaged 17-mer/40-mer-8oxodG substrate. The position of the 8-oxodG lesion within the template strand is indicated by a “Y.”
To determine the effect of 8-oxodG bypass in the presence of all four human Y-family DNA polymerases in vitro, a running start assay was performed with an equal molar concentration of all four human Y-family enzymes (Figure 1I and J). The time required for all of the human Y-family enzymes to bypass the 8-oxodG lesion was similar to that of hPolη alone, suggesting that in the absence of any auxiliary factors, hPolη dominates the replication of both undamaged and damaged DNA. Furthermore, as with hPolη alone, no strong pause sites were observed opposite the lesion (Figure 1J). Interestingly, the only strong pause site observed in the presence of all four Y-family enzymes was an accumulation of the 18-mer product. The only Y-family enzyme that produced this strong 18-mer product was hRev1 (Figure 1G and H), suggesting that the accumulation of the 18-mer was due to a single extension by hRev1, followed by sequestration of this 18-mer product by bound hRev1. Running start assays performed with the combination of hPolη, hPolκ, and hPolι lack this 18-mer product (Figure S1, Supporting Information), indicating that the accumulation of the 18-mer product in the presence of all four enzymes is indeed due to hRev1. We concluded that when the human Y-family polymerases were tested in combination, hPolη catalyzed the majority of the nucleotide incorporation events.
To define the relative lesion bypass efficiency of each Y-family polymerase by using established methods,21,23 the percent of lesions bypassed by each polymerase with respect to the total number of lesions encountered (8-oxodG bypass%) within the autoradiograms shown in Figure 1 was plotted as a function of reaction time (Figure S2, Supporting Information). To compare the relative lesion bypass efficiency of each enzyme, the time required for each polymerase to bypass 50% of the 8-oxodG sites encountered (t50bypass) relative to the replication of 50% of the corresponding dG sites (t50) was then calculated from these plots (Table 2). This qualitative analysis confirmed that the activity of hPolη was not reduced by the 8-oxodG lesion. In contrast, the lesion reduced the nucleotide incorporation rate of hPolκ and hPolι by 2.2- and 4.5-fold, respectively. The calculated t50bypass values also indicated that hPolη bypassed the 8-oxodG lesion 186-fold and 1160-fold faster than hPolκ and hPolι, respectively. Consistently, the efficiency of 8-oxodG bypass by all four human Y-family enzymes combined was nearly identical to that of hPolη alone. Taken together, these data indicate that in the absence of auxiliary factors, hPolη bypassed 8-oxodG more efficiently than either hPolκ or hPolι.
Table 2. 8-OxodG Bypass Efficiencies of the Human Y-Family DNA Polymerases.
| enzyme | t50bypass (s)a | t50 (s)b | t50bypass/t50 |
|---|---|---|---|
| hPolηc | 0.58 | 0.61 | 0.95 |
| hPolκc | 108 | 49 | 2.2 |
| hPolιc | 670 | 150 | 4.5 |
| hRev1c | >72 000 | 5300 | >14 |
| hRev1d | 0.86 | 0.13 | 6.8 |
| all 4 polsc | 0.60 | 0.62 | 0.98 |
Calculated as the time required to bypass 50% of the 8-oxodG sites.
Calculated as the time required to bypass 50% of the undamaged dG sites.
Determined by using the 17-mer/40-mer and 17-mer/40-mer-8oxodG substrates.
Determined by using the 21-mer/40-mer and 21-mer/40-mer-8oxodG substrates.
Standing Start hRev1 Assays
Because hRev1 failed to extend the 17-mer primer past the lesion site within 20 h (Figure 1 H), we chose to evaluate the ability of hRev1 to bypass the 8-oxodG lesion by using a standing start primer extension assay. To this end, an undamaged 21-mer/40-mer or damaged 21-mer/40-mer-8oxodG DNA substrate (Figure 2A) was extended by the action of hRev1 such that the first nucleotide incorporation by hRev1 was opposite either undamaged dG or the 8-oxodG lesion site (Figure 2B and C). Interestingly, the 8-oxodG lesion reduced the rate of nucleotide incorporation catalyzed by hRev1 opposite the lesion and eliminated the ability of hRev1 to extend the lesion bypass product as indicated by the lack of 23-mer product formation in the presence of 8-oxodG (Figure 2C). The plot of dG and 8-oxodG bypass (Figure 2D) revealed that 15% of the 8-oxodG lesions were not bypassed within 30 s, compared to only 6% of the undamaged dG sites that were not bypassed within the same time. This result suggests that approximately 9% of the hRev1 that encountered 8-oxodG formed nonproductive complexes due to interactions with the lesion. Analyses of the standing-start t50bypass values indicated that the 8-oxodG lesion reduced the efficiency of hRev1 nucleotide incorporation by 6.8-fold (Table 2). Thus, of the four human Y-family enzymes, hRev1 was most significantly inhibited by the 8-oxodG lesion relative to undamaged dG. However, due to the relatively fast rate of nucleotide incorporation of this enzyme, hRev1 was the second most efficient enzyme to bypass 8-oxodG overall and only 1.5-fold less efficient than hPolη (Table 2). To examine the ability of hRev1-generated lesion bypass products to be extended, the undamaged 21-mer/40-mer or damaged 21-mer/40-mer-8oxodG DNA substrate was extended by the action of either hRev1 alone or in combination with hPolκ, the Y-family enzyme purported to be specialized for the extension step of TLS.25−27 Within 30 s, hRev1 traversed the majority of the dG or 8-oxodG sites as demonstrated by the accumulation of the 22-mer product (Figure 2E and F). As previously observed in the running start primer extension assays, these 22-mer products were also not extended to full-length products by the action of hRev1 alone. However, if the control or damaged substrates were reacted with hRev1 for 30 s to allow for the bypass of the lesion or undamaged dG, followed by the addition of hPolκ to facilitate the extension of the hRev1-generated lesion bypass products, these 22-mer products were extended to full-length within 1 h. Thus, although hRev1 is capable of bypassing the 8-oxodG lesion efficiently, a second polymerase is required for the extension of the lesion bypass products.
Figure 2.

Standing start assays for hRev1 and hPolκ on undamaged and damaged DNA templates. (A) The damaged 21-mer/40-mer-8oxodG substrate. The position of the 8-oxodG lesion within the template strand is indicated by a “Y.” A preincubated solution containing 400 nM of hRev1 and 100 nM of 5′-[32P]-radiolabeled (B) 21-mer/40-mer or (C) 21-mer/40-mer-8oxodG was rapidly mixed with a solution of all 4 dNTPs (200 μM each). The reaction mixtures were quenched at the indicated times with 0.37 M EDTA and resolved by using denaturing PAGE. (D) Time-dependent lesion bypass by hRev1. The control dG or damaged 8-oxodG bypass% was plotted as a function of reaction time. A preincubated solution containing 100 nM of hRev1 and 100 nM of 5′-[32P]-radiolabeled (E) 21-mer/40-mer or (F) 21-mer/40-mer-8oxodG was rapidly mixed with a solution of all 4 dNTPs (200 μM each). Where indicated, hPolκ (300 nM) was added to the reaction 30 s after the addition of the dNTP solution. The reaction mixtures were quenched and resolved as described above.
HT-SOSA to Determine the Mutagenic Consequences of 8-oxodG Translesion DNA Synthesis
To examine the patterns and frequencies of mutations induced by TLS of 8-oxodG catalyzed by the Y-family DNA polymerases, we used HT-SOSA as summarized in Scheme 1. By using this high-throughput method,20 greater than two million nucleotide sequences generated from lesion bypass products were analyzed to provide a statistically robust data set (Table S2, Supporting Information). As a control for errors induced by the high-throughput sequencing approach, we calculated the average error rates within a control flanking region (positions −14 to −4 relative to the lesion site) of each sequence, which was initially derived from the 13-mer or 16-mer primers (Table 1 and Scheme 1). The average base insertion, deletion, and substitution frequencies within this flanking region were found to be 0.014%, 0.055%, and 0.080%, respectively. These rates combined for an overall background error rate of 0.15 ± 0.01% at each template position. This background error rate was at least 51-fold below the error rate of each enzyme opposite the lesion site and more than 10-fold below the background error rate at virtually every other position analyzed within the undamaged or damaged templates. As an additional control, using HT-SOSA we calculated the average base substitution error rates for hPolη, hPolκ, and hPolι replicating the undamaged, control template to be 1.8 × 10–2, 1.5 × 10–2, and 1.5 × 10–1, respectively. These values are similar to the nucleotide misincorporation fidelities of hPolη (2.0 × 10–3 to 2.1 × 10–2), hPolκ (3.5 × 10–3 to 2.9 × 10–2), and hPolι (9.3 × 10–3 to 1.1 × 10–1) as previously determined by presteady-state kinetic assays with undamaged DNA substrates,23 and as previously measured by HT-SOSA.20 Therefore, HT-SOSA is a viable technique for the study of the mutagenic profiles induced by TLS of 8-oxodG catalyzed by the error-prone Y-family polymerases.
hPolη
We found that hPolη correctly incorporated dCTP opposite 8-oxodG in 54.5% of the lesion bypass sequences analyzed (Figure 3). Therefore, hPolη was 15.7-fold more error prone opposite the lesion than opposite template dG. Opposite 8-oxodG, hPolη misincorporated dATP (41.7%) over misincorporating dTTP (1.3%) or dGTP (1.2%) and rarely generated a base insertion (0.1%) or deletion (1.3%) mutation. Interestingly, this result indicates that even though the nucleotide incorporation rate of hPolη opposite undamaged dG and 8-oxodG was almost identical (Figure 1A and B, and Table 2), hPolη was the second most error prone Y-family enzyme opposite the lesion site. To compare the hPolη error frequency opposite the 8-oxodG lesion to other template positions, we examined the errors generated by hPolη both three template positions upstream and downstream from the lesion site (Figure 4A and B). This analysis demonstrated that the error rates of hPolη were nearly unchanged (within 1.6-fold) at these template positions in the presence or absence of 8-oxodG (Tables S3, S4 and S5, Supporting Information). Therefore, the lesion only influenced the error rate of hPolη during incorporations opposite from 8-oxodG.
Figure 3.

Comparison of the preferred actions of the human Y-family polymerases opposite 8-oxodG or dG. The relative number of nucleotide incorporations, insertion mutations, or deletion mutations produced by each polymerase or polymerase combination opposite template (A) dG or (B) 8-oxodG are indicated.
Figure 4.

Histogram of the relative errors generated by the human Y-family DNA polymerases as a function of template position. Lesion bypass analysis for (A and B) hPolη, (C and D) hPolκ, (E and F) hPolι, and (G and H) a combination of all 4 Y-family polymerases by using either the undamaged 17-mer/40-mer substrate or the damaged 17-mer/40-mer-8oxodG substrate. The relative number of base insertions (striped bar), substitutions (black bar), and deletions (white bar) as a percentage of the total dNTP incorporations are indicated at each template position. The template bases are denoted, and the 8-oxodG lesion is represented as “Y.” The indicated template positions are relative to the 8-oxodG lesion site within the 40-mer-8oxodG template.
hPolκ
Of the four human Y-family enzymes, hPolκ was the most error prone opposite 8-oxodG (Figure 3B), preferentially misincorporating dATP (78.0%) over correct dCTP (18.5%). Overall, we found that hPolκ was 14-fold more error prone opposite the lesion when compared to undamaged dG. Interestingly, the hPolκ error rates at positions downstream from the lesion site were nearly unchanged between the damaged or control templates (Figure 4C and D), even though the nucleotide incorporation rate of hPolκ was reduced at these positions within the primer extension assays (Figure 1D). In contrast, at positions upstream from the lesion site, the frequency of base insertion mutations increased by an average of 3.6-fold in the presence of 8-oxodG (Tables S3, S6, and S7, Supporting Information). However, these insertion mutation events were rare, occurring at an average frequency of only 0.16%, indicating that 8-oxodG seldom altered the fidelity of hPolκ before the lesion entered the polymerase active site.
hPolι
HT-SOSA analysis indicated that hPolι correctly incorporated dCTP and incorrectly incorporated dA opposite 8-oxodG in 65.8% and 21.7% of the sequences analyzed, respectively (Figure 3). Thus, even though 8-oxodG reduced the nucleotide incorporation efficiency of hPolι to a greater extent than either hPolη or hPolκ (Table 2), hPolι was less error-prone than hPolη or hPolκ opposite the lesion. Overall, hPolι generated more errors than either hPolη or hPolκ at nearly every template position (Figure 4E and F). The fidelity of hPolι has been shown to be lower opposite template pyrimidines than template purines and to violate normal Watson–Crick base pairing opposite template dT.24,28,29 Consistently, we observed the fidelity of hPolι to be the lowest opposite template base dT followed by dC, dG, and finally dA when replicating the undamaged template (Figure 4E) and found hPolι to preferentially incorporate dG opposite template dT (Figure S3, Supporting Information). Although hPolι produced a significant number of frameshift mutations while replicating the undamaged control template, the average hPolι base deletion rate at template positions downstream from the 8-oxodG site increased by an average of 3.0-fold (Tables S3, S8, and S9, Supporting Information). This increase in the average base deletion error rate of hPolι downstream from 8-oxodG indicates that the lesion perturbed nucleotide incorporation by hPolι after the lesion exited the polymerase active site.
Combination of hRev1 and hPolκ
To determine the frequencies and types of mutations induced by 8-oxodG bypass catalyzed by hRev1, the control 16-mer/40-mer substrate or damaged 16-mer/40-mer-8oxodG substrate were first reacted with hRev1 to allow for hRev1-mediated incorporation opposite undamaged dG or the 8-oxodG lesion, respectively. Then, because hRev1 is incapable of completely extending similar DNA substrates (Figure 2E and F), hPolκ was subsequently added to extend the DNA products to full-length for HT-SOSA analysis (Figure 5). Of all the Y-family polymerases investigated individually or in combination, we found the combination of hRev1 and hPolκ was the least error prone opposite 8-oxodG by inserting correct dCTP (92.3%) over incorrect dATP (6.9%). Importantly, the standing start primer extension assays indicated that approximately 15% of hRev1 formed unproductive complexes with the damaged 21-mer/40-mer-8oxodG DNA substrate (Figure 2D). Therefore, we hypothesize that hPolκ catalyzed a portion of these observed dATP incorporation events after the liberation of the 21-mer/40-mer-8oxodG substrate from unproductively bound hRev1. This finding suggests that the error rate of hRev1 opposite 8-oxodG may be even lower than the error frequency observed here. Nevertheless, this result is consistent with the demonstrated activity of hRev1 as a specialized dCTP transferase, preferentially inserting dCTP opposite damaged or undamaged templating bases.19 Thus, we conclude that hRev1 is the best suited Y-family polymerase to catalyze error-free TLS of 8-oxodG. Interestingly, the combination of hRev1 and hPolκ produced a significant number of deletion mutations at template positions +1 and +2 (Tables S10 and S11, Supporting Information). The majority of these deletion mutations are double base deletion mutations, suggesting that these double base deletion mutations arose after hRev1 incorporated two dC nucleotides into the primer strand, followed by realignment of the primer strand to loop out the +1 and +2 template positions, and subsequent extension by hPolκ. This proposed loop out mechanism is consistent with the previously reported ability of hPolκ to extend primers that contain limited sequence complementarity at the 3′ primer terminus.17
Figure 5.

Histogram of the relative errors generated by the combination of hRev1 and hPolκ as a function of template position. Sequence analysis of the lesion bypass products generated by the combination of hRev1 and hPolκ with (A) the undamaged 21-mer/40-mer substrate or (B) the 21-mer/40-mer-8oxodG substrate. The relative number of base insertions (striped bar), substitutions (black bar), and deletions (white bar) as a percentage of the total dNTP incorporations are shown at each template position. The template bases are denoted, and the 8-oxodG lesion is represented as “Y.” The indicated template positions are relative to the 8-oxodG lesion site within the 40-mer-8oxodG template.
Combination of All Four Human Y-Family Polymerases
All four human Y-family enzymes are present within the nucleus during S-phase. However, the question of how an individual Y-family polymerase is recruited to a stalled replication complex in order to bypass a particular lesion in vivo remains unanswered. We hypothesize that the inherent properties of each polymerase, such as affinity for the lesion site and lesion bypass efficiency, are predictive of the potential for each enzyme to bypass a particular lesion in vivo. In order to establish a direct competition between the human Y-family enzymes for 8-oxodG bypass, HT-SOSA was performed with an equal molar concentration of all four DNA polymerases (Figure 4G and H). This combination of all four enzymes misincorporated dATP (50.0%) over correctly incorporating dCTP (43.5%), misincorporating dGTP (1.2%) or dTTP (1.3%), or generating a deletion mutation (4.0%) or an insertion mutation (0.6%). Therefore, the fidelity of lesion bypass by the combination of all four human enzymes most closely resembled the 8-oxodG bypass fidelity of hPolη alone (Tables S4, S5, S12 and S13, Supporting Information). Consistent with the running start primer extension assays (Figure 1I and J), we concluded that in the presence of all four human Y-family polymerases, hPolη catalyzed the majority of nucleotide incorporation events, including opposite the lesion site. Therefore, even though hPolη is not the best suited human Y-family member to catalyze error-free TLS of 8-oxodG, the properties hPolη allow this polymerase to outcompete hPolκ, hPolι, and hRev1 for bypass of 8-oxodG.
Discussion
The highly mutagenic 8-oxodG lesion is one of the most prevalent lesions induced by oxidative DNA damage. In this article, we systematically compared the 8-oxodG bypass efficiencies and fidelities of all four human Y-family DNA polymerases for the first time in order to predict the enzyme(s) that have evolved to catalyze TLS of 8-oxodG in vivo. We conclude that (1) hPolη outcompetes hPolκ, hPolι, and hRev1 for nucleotide incorporations opposite 8-oxodG in vitro; (2) hPolη is the only Y-family member to traverse 8-oxodG with the same efficiency as undamaged dG; (3) the efficiency of nucleotide incorporation by hRev1 is most significantly reduced opposite the lesion when compared to incorporations opposite undamaged dG; (4) hRev1 fails to extend 8-oxodG lesion bypass products; and (5) the combination of hRev1 to bypass 8-oxodG and a second polymerase to extend the lesion bypass product is the most accurate polymerase combination to perform TLS of 8-oxodG, followed by hPolι, hPolη, and hPolκ individually.
The relative 8-oxodG bypass efficiencies based upon the calculated t50bypass values of the human Y-family polymerases are hPolη > hRev1 ≫ hPolκ ≫ hPolι (Table 2). These findings agree with a previous report investigating 8-oxodG bypass by hPolη, hPolκ, and hPolι by using a gapped DNA substrate strand displacement assay,30 although hRev1 was not included in that study. Importantly, we found that the ability of hRev1 to perform the extension step of TLS was almost completely eliminated by 8-oxodG (Figure 2C and F). Therefore, even though 8-oxodG is efficiently bypassed by hRev1, a switch to a second polymerase is required for the extension step. In fact, this inherent inability of hRev1 to extend the lesion bypass products may promote polymerase switching in vivo. If the switching does not occur, the extension of any lesion bypass product by hRev1 would result in one or more dCTP incorporations into DNA due to the function of hRev1 as a dCTP transferase, likely leading to the substitution or frameshift mutations. Thus, we hypothesize that the mostly error-free extension of lesion bypass products generated by hRev1 would be carried out in vivo by a second polymerase, such as hPolκ17,25 or DNA polymerase ζ, a B-family polymerase that is also suitable for the extension step of TLS.31
By using HT-SOSA, we found that the most common mutation induced by 8-oxodG bypass catalyzed by all four human Y-family polymerases either alone or in combination was the incorporation of dA opposite 8-oxodG (Figure 3B). These results are consistent with previous kinetic studies indicating the predominant substitution catalyzed by hPolη,10 hPolκ,16 and hPolι18 opposite 8-oxodG is the misincorporation of dATP. This miscoding potential of 8-oxodG is strongly influenced by the ratio of the anti and syn conformations of 8-oxodG within a polymerase active site. Interestingly, this dual coding potential of lesions such as 8-oxodG is irrelevant to nucleotide incorporations by hRev1, as structural studies indicate this enzyme uses a novel mechanism whereby incoming dCTP interacts and forms two hydrogen bonds with the side chain of Arg357 within the N-digit domain of hRev1, rather than the templating base.32 This dCTP transferase activity is also supported by kinetic analysis demonstrating hRev1 preferentially incorporates dCTP opposite any templating base, damaged or undamaged.19,33 Here, we found that the combination of hRev1 and hPolκ was nearly error-free opposite 8-oxodG (Figure 3B). Indeed the low frequency of dATP incorporation (6.9%) detected by HT-SOSA may be due to the displacement of unproductively bound hRev1 by active hPolκ, which in turn bypassed the lesion and predominantly incorporated dATP opposite 8-oxodG. Therefore, hRev1 may be almost completely error free when bypassing 8-oxodG.
In this study, we demonstrate that when all four human Y-family polymerases are tested in combination, hPolη outcompetes hPolκ, hPolι, and hRev1 for TLS of 8-oxodG (Table 2). Notably, previous studies indicate that among the human Y-family polymerases, hPolη possesses the highest efficiency of nucleotide incorporation into undamaged DNA substrates23 and a 2- to 5-fold greater affinity for undamaged DNA substrates when compared to that of hPolι, hRev1, or hPolκ.23,34,35 Furthermore, it has been shown that the presence of 8-oxodG as a templating base does not reduce the binding affinity of either yeast Polη36 or the model Y-family polymerase Dpo437 for a DNA substrate, suggesting that the Y-family polymerases are well suited to accommodate this small lesion within their relatively large active sites, without a reduction in DNA binding capacity. Thus, the superior DNA binding affinity of hPolη likely contributes to its dominance within the four enzyme combination assays (Figures 1I and J). However, differences in DNA binding affinity alone are not sufficient to explain the 100- to 100,000- fold difference in the t50bypass values between hPolη and the other three Y-family DNA polymerases (Table 2). Instead, we conclude that a superior nucleotide incorporation efficiency of hPolη is the primary contributor to the dominance of hPolη for 8-oxodG bypass. Importantly, we chose to test the abilities of the human Y-family polymerases to bypass 8-oxodG at equal molar concentrations in order to gain insight into the intrinsic abilities of these polymerases to compete for TLS of 8-oxodG. However, the relative in vivo expression profiles of the human Y-family polymerases under conditions of oxidative DNA damage are not known. Indeed the expression of each of the Y-family polymerases may not be similar between cell types or may be dynamic during the cell cycle. Further studies are especially needed to elucidate the regulation of the expression and activities of the Y-family polymerases in response to cellular DNA damage in vivo, which remains an important, unanswered question.
Eukaryotes utilize a number of mechanisms to reduce the mutagenic consequences of 8-oxodG, including (1) excision of the damaged base from 8-oxodG·dC pairs by 8-oxoguanine DNA glycosylase 1 (OGG1) to initiate base excision repair (BER), (2) excision of the dA base from 8-oxodG:dA mispairs by MYH adenine glycosylase to facilitate a new incorporation event opposite the lesion, and (3) hydrolysis of 8-oxodGTP to 8-oxodGMP by MTH1 to prevent incorporation of the damaged dNTP into replicating DNA. Despite these important repair mechanisms, 8-oxodG lesions do persist during S-phase and are encountered by the replication fork. The majority of mutations induced by 8-oxodG in vivo are G → T transversion mutations,38,39 indicating that the error-prone TLS of 8-oxodG principally results in dATP misincorporation. Our finding that hPolη outcompetes hPolκ, hPolι, and hRev1 for nucleotide incorporation opposite 8-oxodG is most consistent with a model wherein hPolη is the predominant Y-family member to catalyze the error-prone bypass of 8-oxodG in vivo. However, in the absence of functional Polη, another Y-family polymerase may catalyze the bypass 8-oxodG. Consistently, plasmid-based mutation assays conducted within wild-type or hPolη-deficient human cell lines demonstrated that in the absence of hPolη, the action of an unknown polymerase increased the total 8-oxodG-induced mutation frequency by 1.2- to 2-fold,40,41 primarily due to an increase in G → T transversion mutations. Our data suggest that hPolκ is the unknown polymerase since it is the only Y-family enzyme which misincorporates dATP opposite 8-oxodG with a higher frequency than hPolη (Figure 3B).
The elucidation of which TLS-specialized polymerases are responsible for the error-free or error-prone bypass of specific types of DNA lesion sites in vivo remains an unanswered and challenging question. Here, we used primer extension assays paired with the recently established HT-SOSA approach to directly compare the efficiency and fidelity of 8-oxodG bypass catalyzed by each of the human Y-family polymerases individually and in combination. Overall, our data indicate that even though hPolη is error-prone opposite 8-oxodG, the superior efficiency of hPolη for TLS of 8-oxodG suggests that this enzyme is the primary Y-family enzyme to catalyze the bypass of 8-oxodG in vivo. However, we cannot completely rule out a role for the other three Y-family members for TLS of 8-oxodG in vivo. Indeed, many DNA polymerases may catalyze the bypass of 8-oxodG lesions in vivo to different extents. The comprehensive biochemical studies reported here will provide a basis for further evaluation of these possibilities by in vivo studies.
Acknowledgments
We thank Jason D. Fowler, Sean Newmister, and Shanen M. Sherrer for the purification of hPolη, hPolκ, and hPolι, respectively. In addition, we thank John Curfman and Dr. Pearly Yan at The Ohio State Nucleic Acid Shared Resource for their aid in performing the next-generation sequencing.
Glossary
Abbreviations
- 8-oxodG
8-oxo-7,8-dihydro-2′-deoxyguanosine
- DTT
dithiothreitol
- OGG1
8-oxoguanine glycosylase DNA glycosylase 1
- BER
base excision repair
- BSA
bovine serum albumin
- CPD
cyclobutane pyrimidine dimer
- dNTP
2′-deoxynucleoside 5′-triphosphate
- EDTA
ethylenediaminetetraacetic acid
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- HT-SOSA
high-throughput short oligonucleotide sequencing assay
- hPolα
human DNA polymerase alpha
- hPolη
human DNA polymerase eta
- hPolκ
human DNA polymerase kappa
- hPolι
human DNA polymerase iota
- hRev1
human DNA polymerase Rev1
- PAGE
polyacrylamide gel electrophoresis
- PCNA
proliferating cell nuclear antigen
- Polα
DNA polymerase alpha
- Polδ
DNA polymerase delta
- Polε
DNA polymerase epsilon
- TLS
translesion DNA synthesis
Supporting Information Available
Primers sequences, number of sequences analyzed by HT-SOSA, calculated error rates, lesion bypass graphs, and lesion bypass analysis of the combination of hPolη, hPolκ, and hPolι. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by grants from the National Science Foundation [MCB-0960961] and the National Institutes of Health [GM079403 and ES009127] to Z.S.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fraga C. G.; Shigenaga M. K.; Park J. W.; Degan P.; Ames B. N. (1990) Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. U.S.A. 87, 4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames B. N.; Shigenaga M. K.; Hagen T. M. (1993) Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U.S.A. 90, 7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y.; Uesugi S.; Ikehara M.; Nishimura S.; Kawase Y.; Ishikawa H.; Inoue H.; Ohtsuka E. (1991) NMR studies of a DNA containing 8-hydroxydeoxyguanosine. Nucleic Acids Res. 19, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb L. A.; Peek M. E.; Morningstar M. L.; Verghis S. M.; Miller E. M.; Rich A.; Essigmann J. M.; Williams L. D. (1995) X-ray structure of a DNA decamer containing 7,8-dihydro-8-oxoguanine. Proc. Natl. Acad. Sci. U.S.A. 92, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley-Hecht K. E.; Leonard G. A.; Gibson N. J.; Thomson J. B.; Watson W. P.; Hunter W. N.; Brown T. (1994) Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry 33, 10266–10270. [DOI] [PubMed] [Google Scholar]
- Marnett L. J. (2000) Oxyradicals and DNA damage. Carcinogenesis 21, 361–370. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Batra V. K.; Wilson S. H. (2010) DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat. Res. 703, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Tassan N.; Chmiel N. H.; Maynard J.; Fleming N.; Livingston A. L.; Williams G. T.; Hodges A. K.; Davies D. R.; David S. S.; Sampson J. R.; Cheadle J. P. (2002) Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232. [DOI] [PubMed] [Google Scholar]
- Einolf H. J.; Guengerich F. P. (2001) Fidelity of nucleotide insertion at 8-oxo-7,8-dihydroguanine by mammalian DNA polymerase delta. Steady-state and pre-steady-state kinetic analysis. J. Biol. Chem. 276, 3764–3771. [DOI] [PubMed] [Google Scholar]
- McCulloch S. D.; Kokoska R. J.; Garg P.; Burgers P. M.; Kunkel T. A. (2009) The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases delta and eta. Nucleic Acids Res. 37, 2830–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markkanen E.; Castrec B.; Villani G.; Hubscher U. (2012) A switch between DNA polymerases delta and lambda promotes error-free bypass of 8-oxo-G lesions. Proc. Natl. Acad. Sci. U.S.A. 109, 20401–20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maga G.; Villani G.; Crespan E.; Wimmer U.; Ferrari E.; Bertocci B.; Hubscher U. (2007) 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature 447, 606–608. [DOI] [PubMed] [Google Scholar]
- Sabouri N.; Viberg J.; Goyal D. K.; Johansson E.; Chabes A. (2008) Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 36, 5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch S. D.; Kunkel T. A. (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A.; Eoff R. L.; Guengerich F. P.; Egli M. (2009) Structural and functional elucidation of the mechanism promoting error-prone synthesis by human DNA polymerase kappa opposite the 7,8-dihydro-8-oxo-2′-deoxyguanosine adduct. J. Biol. Chem. 284, 22467–22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yuan F.; Wu X.; Wang M.; Rechkoblit O.; Taylor J. S.; Geacintov N. E.; Wang Z. (2000) Error-free and error-prone lesion bypass by human DNA polymerase kappa in vitro. Nucleic Acids Res. 28, 4138–4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L.; Prakash L.; Prakash S. (2002) Role of human DNA polymerase kappa as an extender in translesion synthesis. Proc. Natl. Acad. Sci. U.S.A. 99, 16000–16005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yuan F.; Wu X.; Taylor J. S.; Wang Z. (2001) Response of human DNA polymerase iota to DNA lesions. Nucleic Acids Res. 29, 928–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Wu X.; Rechkoblit O.; Geacintov N. E.; Taylor J. S.; Wang Z. (2002) Response of human REV1 to different DNA damage: preferential dCMP insertion opposite the lesion. Nucleic Acids Res. 30, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart D. J.; Camerlengo T. L.; Harrison J. K.; Sherrer S. M.; Kshetry A. K.; Taylor J. S.; Huang K.; Suo Z. (2013) A high-throughput and quantitative method to assess the mutagenic potential of translesion DNA synthesis. Nucleic Acids Res. 41, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrer S. M.; Fiala K. A.; Fowler J. D.; Newmister S. A.; Pryor J. M.; Suo Z. (2011) Quantitative analysis of the efficiency and mutagenic spectra of abasic lesion bypass catalyzed by human Y-family DNA polymerases. Nucleic Acids Res. 39, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala K. A.; Suo Z. (2004) Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry 43, 2106–2115. [DOI] [PubMed] [Google Scholar]
- Sherrer S. M.; Sanman L. E.; Xia C. X.; Bolin E. R.; Malik C. K.; Efthimiopoulos G.; Basu A. K.; Suo Z. (2012) Kinetic analysis of the bypass of a bulky DNA lesion catalyzed by human Y-family DNA polymerases. Chem. Res. Toxicol. 25, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yuan F.; Wu X.; Wang Z. (2000) Preferential incorporation of G opposite template T by the low-fidelity human DNA polymerase iota. Mol. Cell. Biol. 20, 7099–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington M. T.; Johnson R. E.; Prakash L.; Prakash S. (2002) Human DINB1-encoded DNA polymerase kappa is a promiscuous extender of mispaired primer termini. Proc. Natl. Acad. Sci. U.S.A. 99, 1910–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L.; Unk I.; Johnson R. E.; Phillips B. B.; Hurwitz J.; Prakash L.; Prakash S. (2002) Stimulation of DNA synthesis activity of human DNA polymerase kappa by PCNA. Mol. Cell. Biol. 22, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez-Del Carpio R.; Silverstein T. D.; Lone S.; Johnson R. E.; Prakash L.; Prakash S.; Aggarwal A. K. (2011) Role of human DNA polymerase kappa in extension opposite from a cis-syn thymine dimer. J. Mol. Biol. 408, 252–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D. T.; Johnson R. E.; Prakash S.; Prakash L.; Aggarwal A. K. (2004) Replication by human DNA polymerase-iota occurs by Hoogsteen base-pairing. Nature 430, 377–380. [DOI] [PubMed] [Google Scholar]
- Tissier A.; McDonald J. P.; Frank E. G.; Woodgate R. (2000) poliota, a remarkably error-prone human DNA polymerase. Genes Dev. 14, 1642–1650. [PMC free article] [PubMed] [Google Scholar]
- Dorjsuren D.; Wilson D. M. 3rd; Beard W. A.; McDonald J. P.; Austin C. P.; Woodgate R.; Wilson S. H.; Simeonov A. (2009) A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases. Nucleic Acids Res. 37, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. E.; Washington M. T.; Haracska L.; Prakash S.; Prakash L. (2000) Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406, 1015–1019. [DOI] [PubMed] [Google Scholar]
- Swan M. K.; Johnson R. E.; Prakash L.; Prakash S.; Aggarwal A. K. (2009) Structure of the human Rev1-DNA-dNTP ternary complex. J. Mol. Biol. 390, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. A.; Fowler J. D.; Suo Z. (2010) Kinetic basis of nucleotide selection employed by a protein template-dependent DNA polymerase. Biochemistry 49, 5504–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone S.; Townson S. A.; Uljon S. N.; Johnson R. E.; Brahma A.; Nair D. T.; Prakash S.; Prakash L.; Aggarwal A. K. (2007) Human DNA polymerase kappa encircles DNA: implications for mismatch extension and lesion bypass. Mol. Cell 25, 601–614. [DOI] [PubMed] [Google Scholar]
- Washington M. T.; Johnson R. E.; Prakash L.; Prakash S. (2004) Human DNA polymerase iota utilizes different nucleotide incorporation mechanisms dependent upon the template base. Mol. Cell. Biol. 24, 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson K. D.; Washington M. T. (2005) Mechanism of efficient and accurate nucleotide incorporation opposite 7,8-dihydro-8-oxoguanine by Saccharomyces cerevisiae DNA polymerase eta. Mol. Cell. Biol. 25, 2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell B. A.; Suo Z. (2012) Kinetic basis for the differing response to an oxidative lesion by a replicative and a lesion bypass DNA polymerase from Sulfolobus solfataricus. Biochemistry 51, 3485–3496. [DOI] [PubMed] [Google Scholar]
- Kalam M. A.; Basu A. K. (2005) Mutagenesis of 8-oxoguanine adjacent to an abasic site in simian kidney cells: tandem mutations and enhancement of G-->T transversions. Chem. Res. Toxicol. 18, 1187–1192. [DOI] [PubMed] [Google Scholar]
- Moriya M. (1993) Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G.C-->T.A transversions in simian kidney cells. Proc. Natl. Acad. Sci. U.S.A. 90, 1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avkin S.; Livneh Z. (2002) Efficiency, specificity and DNA polymerase-dependence of translesion replication across the oxidative DNA lesion 8-oxoguanine in human cells. Mutat. Res. 510, 81–90. [DOI] [PubMed] [Google Scholar]
- Lee D. H.; Pfeifer G. P. (2008) Translesion synthesis of 7,8-dihydro-8-oxo-2′-deoxyguanosine by DNA polymerase eta in vivo. Mutat. Res. 641, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.