

Abstract
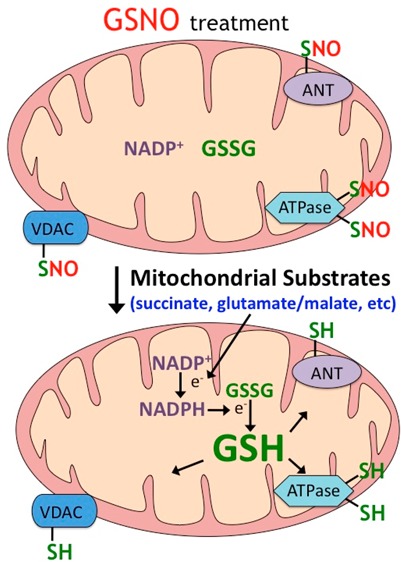
S-Nitrosylation is a reversible post-translational modification on cysteinyl thiols that can modulate the function of redox-sensitive proteins. The S-nitrosylation of mitochondrial proteins has been shown to regulate various mitochondrial activities involved in energy-transducing systems and mitochondrion-driven apoptosis. In isolated rat brain mitochondria, we demonstrate that mitochondrial protein S-nitrosylation is regulated by respiratory substrates (glutamate/malate) through a thiol-dependent pathway. Mitochondrial proteins become susceptible to S-nitrosoglutathione (GSNO)-induced S-nitrosylation in mitochondria with an oxidized environment (low glutathione (GSH), NADH, and NADPH, and high GSSG, NAD+, and NADP+) caused by isolation of mitochondria using a discontinuous Percoll gradient. Activation of mitochondrial respiration by respiratory substrates leads to increased NAD(P)H and GSH levels, which in turn reduces mitochondrial S-nitrosylated proteins. 1-Chloro-2,4-dinitrobenzene (CDNB), which depletes mitochondrial GSH and inhibits the thioredoxin–thioredoxin reductase system, prevented the denitrosylation of mitochondrial proteins caused by respiratory substrate treatment. Using biotin-switch coupled with LC-MS/MS, several mitochondrial proteins were identified as targets of S-nitrosylation including adenine nucleotide translocase (ANT) and voltage-dependent anion channel (VDAC), important components of the mitochondria permeability transition pore (MPTP), as well as ATP synthase. The S-nitrosylation of ATP synthase by GSNO was found to inhibit its activity. These findings emphasize the importance of respiratory substrates in regulating S-nitrosylation through a thiol-dependent (GSH and/or thioredoxin) pathway, with implications for mitochondrial bioenergetics and mitochondrion-driven apoptosis.
Introduction
Nitric oxide (•NO), generated by the constitutive and inducible isoforms of nitric oxide synthase, is a signaling molecule involved in a wide variety of physiological and pathophysiological events, ranging from vascular regulation and neuronal plasticity to neurodegeneration, diabetes, and chronic inflammation, thus accounting for the beneficial and toxic effects of this gaseous free radical. The biochemistry of •NO has been discussed in terms of both concentration and spatial considerations that determine activation of signaling through the soluble guanylate cyclase, regulation of mitochondrial respiration upon reversible binding to cytochrome oxidase, and reactivity with nonheme iron.1 Diverse •NO signaling mechanisms and their cross-talk have been recently reviewed.2
Protein S-nitrosylation is a post-translational modification of cysteinyl thiols via oxidative reaction with a nitric oxide derivative (e.g., nitrosonium cation) to form the structure R–S-N=O, and the determinants of specificity for S-nitrosylation in a cellular setting have been extensively documented.3S-Nitrosothiols (e.g., S-nitrosoglutathione, GSNO) and •NO regulate thiol-dependent pathways by several different mechanisms.4 At a cellular level, S-nitrosylation is involved in the regulation of some signaling pathways as well as of other protein post-translational modifications, including acetylation, ubiquitylation, and phosphorylation.5,6 S-nitrosylation of mitochondrial targets is associated with a degree of variability, inasmuch as this post-translational modification affects mostly tissues with high energy metabolism and can lead to protection or inactivation of enzymes.7 For example, incubation of rat heart mitochondrial protein lysate with a nitrosothiol, GSNO, identified 60 proteins that were S-nitrosylated and that were involved in various mitochondrial functions, such as solute transport, electron transfer, TCA cycle, oxidative stress defense, and fatty acid and amino acid metabolic pathways.8 The reversible inhibition of complex I in heart mitochondria through S-nitrosylation of the 75 kDa subunit, as it occurs in the presence of S-nitrosothiols or during ischemic-preconditioning, results in the protection of mitochondria against ischemia-reperfusion.9−11 The S-nitrosylation of complex I in heart mitochondria depends on its structural conformation and has implications for hypoxic conditions.12,13 In addition to complex I, a subproteome of heart mitochondrial S-nitrosylated proteins during ischemic preconditioning identified enzymes involved in energetics and calcium homeostasis.14 More recently, it was shown that myocardial caveolae mediate eNOS/NO/S-nitrosylation cardioprotective signaling to mitochondria.15 S-Nitrosylation of complex I was also found in brain mitochondria and exacerbated following the dopaminergic depletion of GSH.16 S-Nitrosylation has also been implicated in neuropathogenic roles inherent in Alzheimer’s and Parkinson’s disease; these roles entail alterations of mitochondrial dynamics (by affecting via S-nitrosylation of the dynamin-related protein 1 (DRP1)) as well as S-nitrosylation of protein-disulfide isomerase that mediates protein misfolding.17 Liver injury following ischemia-reperfusion and in alcoholic fatty liver was associated with S-nitrosylation of mitochondrial proteins and the decreased energy supply associated with mitochondrial dysfunction.18,19
We have previously shown that respiratory substrates (e.g., glutamate/malate and succinate) regulate GSH redox status and protein S-glutathionylation in mitochondria by modulating NADPH levels.20 Although S-nitrosylation of mitochondrial proteins plays an important role in physiology and pathophysiology, the regulatory mechanisms of mitochondrial protein S-nitrosylation remain unclear. This study is aimed at establishing the influence of the mitochondrial metabolic state on protein S-nitrosylation in the organelles with an experimental model consisting of brain mitochondria isolated by the discontinuous Percoll gradient and supplemented with GSNO, a nitrosating agent. GSNO catalyzes the S-nitrosylation of proteins by a transnitrosation pathway; brain mitochondria isolated by a discontinuous Percoll gradient are in an oxidized state as indicated by the high levels of GSSG, protein mixed disulfides, and oxidized pyridine nucleotides.20
Experimental Procedures
Animals and Chemicals
Male Wistar rats at 4 months of age were purchased from Simonsen Laboratories, Inc. (Gilroy, CA, USA). Micro Bio-Spin 6 Chromatography Columns were obtained from Bio-Rad (Hercules, CA, USA). Blocker casein in TBS and EZ-Link Biotin-HPDP were from Pierce Biotechnologies (Rockford, IL, USA). Sypro Ruby protein gel stain was obtained from Molecular Probes (Eugene, OR, USA). S-Nitrosoglutathione (GSNO), protease inhibitor cocktail, Percoll, MMTS (methylmethanethiosulfonate), pyruvate kinase/lactate dehydrogenase enzyme mixture, and all other chemicals were from Sigma (St. Louis, MO, USA). Rabbit polyclonal antibody against COX IV was from ABcam (Cambridge, UK).
Isolation of Mitochondria and Proteinase K Treatment
Rat brain mitochondria were isolated by a discontinuous Percoll gradient.21 All procedures were subjected to the regulations of the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California (Los Angeles, CA). Male Wistar rats at 4 months of age, housed with food and water ad libitum in a facility (USC Department of Animal Resources) with automated light/dark (12 h/12 h) cycle, were fasted overnight, anesthetized by CO, and sacrificed. Brain was excised, washed twice, and minced to fine pieces with a pair of scissors in ice-cold mitochondria isolation buffer (250 mM sucrose, 20 mM HEPES, 1 mM EDTA, 1 mM EGTA, 0.5% (m/v) BSA, and 0.1% (v/v) protease inhibitor cocktail, pH 7.4). Brain tissue was homogenized by a Potter-Elvehjem tissue grinder in the same ice-cold isolation buffer. The homogenate was centrifuged briefly at 1,330g for 5 min at 4 °C in an Eppendorf 5810R centrifuge to remove cell debris and nuclei. The supernatant was transferred to a clean tube and centrifuged at 21,200g for 10 min at 4 °C in a Beckman J2-21 centrifuge with a JA17 rotor. Pellet was resuspended in 15% Percoll (prepared in mitochondria isolation buffer) and centrifuged again at 21,200g for 10 min at 4 °C. The pellet, which appeared loose and fluffy, was obtained by a plastic transfer pipet and carefully layered on top of a discontinuous 23% on 40% Percoll gradient, followed by centrifugation at 30,700g for 5 min at 4 °C. The material that resided at the interface between 40% and 23% was highly enriched with mitochondria and was taken and diluted at 1:4 ratio with isolation buffer, followed by centrifugation at 16,700g for 10 min at 4 °C. The pellet was diluted again at a 1:4 ratio with isolation buffer and centrifuged at 6,900g for 10 min at 4 °C. The pellet was then resuspended in mitochondria isolation buffer containing 0.1% fatty acid-free BSA and centrifuged at 6,900g for 10 min at 4 °C. Finally, the pellet of the last centrifugation was resuspended in BSA-free mitochondria isolation buffer and kept on ice until use. Respiratory control ratios of ∼4–5 were obtained from these isolated mitochondria using glutamate/malate or succinate as electron donors. GSH and protein thiols become stronger nucleophiles when deprotonated at basic pH values; thus, all experiments were performed at pH 7.4. Broken mitochondria were prepared by freeze/thaw 3 times in liquid nitrogen, followed by brief sonication. CDNB (1-chloro-2,4-dinitrobenzene) treatment was used to deplete key thiols (GSH and thioredoxin–thioredoxin reductase) in mitochondria.22
Proteinase K Treatment
Stock solution of proteinase K was prepared at 5 mg/mL in double-distilled water and kept frozen at −80 °C until use. Mitochondria were treated with 50 μg/mL proteinase K for 25 min at 0 °C. PMSF was then added to a final concentration of 2 mM. Mitochondria were incubated for an additional 10 min at 0 °C, followed by centrifugation at 9,000 rpm for 10 min at 4 °C, and resuspended in mitochondria isolation buffer.
Mitochondrial Swelling
Mitochondrial swelling was monitored in an Agilent 8453 spectrophotometer (Palo Alto, CA, USA) at 520 nm.23 The experiment was performed on freshly isolated intact rat brain mitochondria. Isotonic swelling buffer contained 150 mM KCl, 20 mM MOPS, 10 mM Trizma base, pH 7.3, 0.5 μM rotenone, 0.5 μM antimycin A, and 2 μM calcium ionophore A23187. The experiment was initiated by the addition of 50 μg of mitochondria in the volume of 10 μL, preincubated with 1 μL of vehicle or 1 μL of 10 mM GSNO (final concentration 1 mM) at room temperature for 10 min in the dark, into the glass cuvette containing 990 μL of swelling buffer; 1 min after the addition of mitochondria, 1 μL of CaCl2 of various concentrations was added to reach final concentrations ranging from 100 μM to 1 mM. Five minutes after the addition of CaCl2, 40 μM of alamethicin was added to induce maximal mitochondrial swelling, and the recording of absorbance was continued for 4 more minutes. The A520 recorded right before the addition of CaCl2 was designated as A520a; that right before the addition of alamethicin was designated as A520b; and that following the addition of alamethicin was designated as A520c. Ca2+-induced swelling was calculated as A520a – A520b. Maximal mitochondrial swelling was calculated as A520a – A520c. Therefore, the percentage of Ca2+-induced swelling was [A520a – A520b]/[A520a – A520c] × 100%. Basal swelling of mitochondria, which were pretreated with vehicle or GSNO, was obtained by adding 50 μg of mitochondria (10 μL) to 990 μL of swelling buffer, and the A520 was recorded for 6 min. Immediately, alamethicin was added, and the recording was continued for 4 more minutes.
Biotin-Switch Method and Western Blot
The biotin-switch method was followed as described with slight modifications.24,25 Intact rat brain mitochondria, as a suspension in the mitochondria isolation buffer (1 mg/mL), were incubated with GSNO in the dark at room temperature for 30 min. GSNO solutions were freshly made before each experiment since aged GSNO solutions form various degradation products.26 HEN buffer (250 mM HEPES, 1 mM EDTA, and 0.1 mM neocuproine, pH 7.7) was prepared freshly before experiments. Mitochondria were centrifuged at 6,900g for 10 min at 4 °C, and mitochondrial proteins were extracted by resuspending the mitochondria pellet in HEN/2% CHAPS, followed by 3 freeze/thaw cycles in liquid nitrogen and vigorous vortexing. Undissolved lipid fragments were spun down at 10,000g for 10 min at 4 °C. Protein concentration was determined by the Bradford dye-binding method using the Bio-Rad protein assay reagent (Hercules, CA, USA). Then, 75 μg of proteins was taken, and the volume was adjusted with HEN buffer to 1 μg/μL, to which 7 μL of 25% SDS and 1.5 μL of 20% MMTS (freshly prepared in DMF) were added and incubated for 20 min at 50 °C in the dark to thiol-methylate the free protein sulfhydryl groups. MMTS was cleaned up from the protein solution by passing the samples through Bio-Rad Micro Bio-Spin 6 chromatography columns 3 times with three fresh columns. The columns were pre-equilibrated with HEN buffer before use; 68 μL of the final flow-through was taken, to which were added 1.36 μL of 50 mM biotin-HPDP (final concentration 1 mM) and 3.4 μL of 100 mM ascorbate (final concentration 5 mM), and incubated for 1 h at room temperature. An extra sample was treated with DMSO as a vehicle control for biotin-HPDP. This would reveal any endogenous protein biotinylation or nonspecific binding by the antibiotin antibody. At the end of incubation, a nonreducing sample loading buffer from Pierce (Rockford, IL, USA) was added at 1:4 dilution. Proteins were separated by SDS–PAGE and transferred to the nitrocellulose membrane. The membrane was then blocked in Pierce Blocker casein for 1 h at room temperature, followed by incubation in TBST (Tris buffered saline with 1% Tween 20) containing 1:1,000 diluted Pierce polyclonal goat anti-biotin antibody overnight at 4 °C with constant agitation. The membrane was washed twice each time for 10 min in TBST, followed by incubation in TBST containing 1:10,000 diluted mouse anti-goat HRP secondary antibody for 1 h at room temperature. Then, the membrane was washed 6 times each for 10 min in TBST. Chemiluminescence was carried out according to the manufacturer’s instructions on a Kodak BioMax Light film.
Purification of S-Nitrosylated Proteins
Intact rat brain mitochondria (1 mg/mL) were incubated with 1 mM GSNO in the mitochondrial isolation buffer for 1 h at room temperature in the dark. Mitochondria were centrifuged at 6,900g for 10 min at 4 °C, and mitochondrial proteins were extracted by resuspending the mitochondrial pellet in HEN/2% CHAPS, followed by 3 freeze/thaw cycles in liquid nitrogen and vigorous vortexing. Undissolved lipid fragments were spun down by centrifugation at 10,000g for 10 min at 4 °C. The supernatant was collected and adjusted with HEN buffer to 1 mg/mL, to which 2% of SDS and 0.4% of MMTS (freshly prepared as 20% stock in DMF) were added to block the free protein sulfhydryl groups for 20 min at 50 °C in the dark. The blocking reagent was cleaned up by protein acetone precipitation twice: 10 volumes of acetone prechilled in −20 °C was added to the protein solution and incubated in −20 °C for 20 min to facilitate protein precipitation. Proteins were centrifuged at 2,000g for 10 min at 4 °C. The protein pellet and the inner surface of the centrifuge tube were carefully rinsed with prechilled acetone to wash out residual organic solvent. The protein pellet was then dissolved in HENS buffer (1% SDS in HEN buffer; 0.1 mL of HENS buffer per mg of protein in the initial protein sample). Biotin-HPDP 1 mM and ascorbate 5 mM were added to the protein solution and incubated for 1 h at room temperature. Proteins were again cleaned up twice by acetone precipitation (2 volumes were used instead of 10 volumes); 2 volumes of neutralization buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100, pH 7.7) and streptavidin-agarose beads equilibrated in neutralization buffer were added to the protein solution and incubated with constant agitation for 1 h at room temperature. The beads were washed in wash buffer (20 mM HEPES, 600 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100, pH 7.7). Proteins, eluted by incubating in elution buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, 100 mM 2-mercaptoethanol, and pH 7.7-NaOH) for 20 min, were separated by SDS–PAGE and finally stained by SYPRO Ruby. Proteins were visualized on a standard UV-transilluminator, and protein bands were sliced out for LC-MS/MS analysis.
Measurement of Mitochondrial Thiol Content, GSH, GSSG, and Pyridine Nucleotides
Thiol levels in mitochondria were measured using DTNB as previously described.27 GSH and GSSG concentrations were analyzed using HPLC electrochemical detection as described previously.28 NAD+, NADH, NADP+, and NADPH levels were measured by HPLC as previously described.29 Briefly, brain homogenate and isolated mitochondria were homogenized in buffer (0.06 M KOH, 0.2 M KCN, and 1 mM bathophenanthroline disulfonic acid) followed by chloroform extraction. Chloroform extraction was carried out by centrifugation at 22,000g in a microcentrifuge at 4 °C; the resulting aqueous supernatant with soluble pyridine nucleotides was collected and extracted twice to remove lipids and proteins. Finally, it was filtered with a 0.45 μm positively charged filter (Pall Life Sciences) to remove the RNA and DNA in the microcentrifuge at 4 °C. The mobile phase consisted of 0.2 M ammonium acetate (buffer A) at pH 5.5 and HPLC-grade methanol (buffer B). A gradient program with initial conditions as 100% buffer A and 0% buffer B was set; from 0 to 4 min, 0 to 3% B and from 4 to 23 min, 3–6.8% B, followed by washing the column with 50% A and 50% B and re-equilibrating to initial conditions for the next run. Quantitation of pyridine nucleotides was performed by integrating the peaks and adding the cyanide adducts as detected by the fluorescence spectrophotometer (λexc = 330 nm; λem = 460 nm).
LC-MS/MS
In-Gel Tryptic Digest
Protein bands from SDS–PAGE were excised from the gels using biopsy punches (Acuderm, Lauderdale, FL, USA). In-gel tryptic digest was carried out using trypsin that was reductively methylated to reduce autolysis (Promega, Madison, WI, USA). Prior to digestion, samples were neither reduced with DTT nor alkylated with iodoacetamide in order to keep potential cysteine modifications stable. The digestion reaction was carried out overnight at 37 °C. Digestion products were extracted from the gel with a 5% formic acid/50% acetonitrile solution (2×) and one acetonitrile extraction followed by evaporation using an APD SpeedVac (ThermoSavant, Milford, MA, USA).
Analysis of Tryptic Peptide Sequence Tags by Tandem Mass Spectrometry
The dried tryptic digest samples were cleaned with ZipTip and resuspended in 10 μL of 60% formic acid. Chromatographic separation of the tryptic peptides was achieved using a ThermoFinnigan Surveyor MS-Pump in conjunction with a BioBasic-18 100 mm × 0.18 mm reverse phase capillary column (ThermoFinnigan, San Jose, CA). Mass analysis was done using a ThermoFinnigan LCQ Deca XP Plus ion trap mass spectrometer equipped with a nanospray ion source (ThermoFinnigan) employing a 4.5 cm long metal needle (Hamilton, 950–00954) using data-dependent acquisition mode. Electrical contact and voltage application to the probe tip took place via the nanoprobe assembly. Spray voltage of the mass spectrometer was set to 2.9 kV and a heated capillary temperature at 190 °C. The column was equilibrated for 5 min at 1.5 μL/min with 95% solution A and 5% solution B (A, 0.1% formic acid in water; B, 0.1%formic acid in acetonitrile) prior to sample injection. A linear gradient was initiated 5 min after sample injection ramping to 35% A, 65% B after 50 min, and 20% A, 80% B after 60 min. Mass spectra were acquired in the m/z 400–1800 range.
Protein Identification
Protein identification was carried out with the MS/MS search software Mascot 1.9 (Matrix Science) with confirmatory or complementary analyses with TurboSequest as implemented in the Bioworks Browser 3.2, build 41 (ThermoFinnigan). NCBI Sus scrofa protein sequences were used as the primary search database, and searches were complemented with the NCBI nonredundant protein database.
Results
GSNO Causes S-Nitrosylation of Brain Mitochondria Isolated by Discontinuous Percoll Gradient
Mitochondria isolated by a discontinuous Percoll gradient are in an oxidized state with extensive protein glutathionylation, but metabolic functionality is recovered upon addition of respiratory substrates.20 The integrity of the isolated mitochondria was also demonstrated by the resistance of inner membrane cytochrome c against proteinase K treatment while the outer membrane protein monoamine oxidase A (MAO-A) was efficiently digested (Figure 1A). Treatment of the thus isolated brain mitochondria with GSNO induced protein S-nitrosylation in a dose-dependent manner (Figure 1B, lanes 3 and 4). The susceptibility to GSNO was increased when the mitochondrial membrane integrity was compromised by freeze/thaw and sonication (Figure 1B, lanes 5 and 6), thus suggesting that the extent of GSNO-mediated S-nitrosylation of mitochondrial proteins is limited by transport of GSNO across the mitochondrial membranes. Without GSNO treatment, a low basal level of S-nitrosylation was detected (Figure 1B, lane 2). Transnitrosation (GSNO + Pr-SH ↔ GSH + Pr–S–NO) is the likely mechanism that accounts for the post-translational modification of protein cysteinyl residues.4
Figure 1.

GSNO-induced S-nitrosylation of mitochondrial proteins. (A) Isolated rat brain mitochondria were treated with proteinase K as described in the Experimental Procedures section. Western blot was performed to detect cytochrome c (cyt c) and monoamine oxidase A (MAO-A). Lane 1, purified mitochondria; lane 2, proteinase K-treated mitochondria. (B) Intact (underline a) and broken (underline b) mitochondria were incubated with GSNO at 0.1 and 1 mM concentrations for 30 min at room temperature in the dark. S-Nitrosylated proteins were detected by the biotin-switch method as described in the Experimental Procedures section. Lane 1 reveals endogenous biotinylated proteins, while lane 2 with biotin-HPDP treatment reveals endogenous S-nitrosylated proteins. COX IV was used as the loading control.
Respiratory Substrates and DTT Attenuate GSNO-Induced S-Nitrosylation of Mitochondrial Proteins
Supplementation of brain mitochondria with complex I substrates (glutamate/malate) increased the levels of NADH, NADPH, and GSH, consistent with a reduced mitochondrial environment (Figure 2A).20 The 6-fold increase in GSH (Figure 2A, lower panel and Figure 2B, column 4) resulted in a decreased intensity of the bands corresponding to the S-nitrosylated proteins (Figure 2B, lane 4 versus 3). Some proteins remained S-nitrosylated even in the presence of respiratory substrates, thus suggesting that GSNO-mediated S-nitrosylation can occur even in a reducing environment and lead to stable S-nitrosylated proteins. DTT also extensively reduced GSNO-induced S-nitrosylation (Figure 2B, lane 5 versus 3) and was more effective than respiratory substrate treatment in reducing the levels of S-nitrosylated proteins (Figure 2B, lane 4 versus 5). DTT treatment was also effective in increasing GSH levels (Figure 2B, column 5) through the reduction of GSSG and deglutathionylation of proteins (data not shown) (as observed with respiratory substrates).20 The bands that appear on lanes 1 and 2 of Figure 2B as well as similar Western blots on subsequent figures represent endogenous biotinylated and endogenous S-nitrosylated proteins, respectively.
Figure 2.

Respiratory substrates replenish mitochondrial GSH and protect mitochondrial proteins from GSNO-induced S-nitrosylation. (A) Respiratory substrate (glutamate/malate 7.5 mM) treatment to isolated brain mitochondria regenerate NADH, NADPH, and GSH levels. (B) Intact rat brain mitochondria were treated with 30 μM GSNO for 30 min at room temperature in the absence or presence of respiratory substrates or 0.5 mM DTT. S-Nitrosylated proteins were detected by the biotin-switch method as described in the Experimental Procedures section. NADH, NADPH, and GSH were detected using HPLC. A representative gel from 3 experiments is shown. COX IV was used as a loading control.
Respiratory Substrates and DTT Reverse GSNO-Induced S-Nitrosylation through a Thiol-Dependent Pathway
CDNB was used to deplete thiols in mitochondria to assess their importance in mitochondrial substrate driven denitrosylation. CDNB depletes primarily mitochondrial GSH through a GSH transferase-dependent pathway.30,31 Since CDNB conjugation is mediated by GSH transferase, protein thiols do not appear to react with CDNB, with thioredoxin reductase being a noted exception.30,32 The addition of respiratory substrates increased mitochondrial GSH, which was dose-dependently depleted by CDNB treatment (Figure 3A). Treatment with respiratory substrates did not decrease GSNO-induced S-nitrosylation in mitochondria treated with CDNB (Figure 3B, left panel). A S-nitrosylated protein band that was responsive to CDNB (indicated by an arrowhead) was quantified and plotted against CDNB concentration. CDNB decreased GSH levels and promoted protein S-nitrosylation even in the presence of respiratory substrates (Figure 3B, right panel). Similarly, when mitochondrial thiols were depleted by CDNB, DTT no longer reduced S-nitrosylation induced by GSNO. These findings confirm that intramitochondrial thiols, likely GSH and/or thioredoxin, are needed in denitrosylation induced by respiratory substrates or DTT (Figure 3C).
Figure 3.

Depletion of mitochondrial GSH by CDNB diminished the protection, afforded by respiratory substrates and DTT, against GSNO-induced S-nitrosylation. (A) Intact mitochondria were treated with or without respiratory substrates (2.4 mM glutamate, 2.4 mM malate, and 0.7 mM ADP) in the absence or presence of increasing concentrations of CDNB for 30 min at room temperature. One hundred micrograms of treated mitochondria were obtained from each sample and analyzed by HPLC for reduced GSH. Sample 2, which was treated with respiratory substrate alone, was taken as 100%. Error bar represents the standard deviation obtained from two separate experiments. (B) Left panel: Intact mitochondria were treated with 30 μM GSNO for 30 min at room temperature in the absence or presence of respiratory substrates (7 mM glutamate, 7 mM malate, and 2 mM ADP). In samples 5–7, increasing concentrations of CDNB were coincubated with GSNO and respiratory substrates. At the end of the incubation, S-nitrosated proteins were detected by the biotin-switch method. A representative band near 75 kDa, indicated by an arrowhead, was selected for densitometry measurement. Right panel: 100 μg of the treated mitochondria, obtained from the experiment in the left panel, was analyzed by HPLC for the detection of GSH. Intensities of the representative band (open circle) and GSH levels (solid circle) from samples 4–7 were plotted against the CDNB concentrations. (C) The experiment was essentially the same as in that B except that mitochondria were treated with 0.5 mM DTT instead of respiratory substrates.
The notion that mitochondrial GSH is important in denitrosylation is further strengthened by the observation that exogenous GSH reduced GSNO-induced S-nitrosylation in a dose-dependent manner (Figure 4A). The levels of GSH, GSSG, and GSNO in the experimental model in Figure 4A are shown in Figure 4B. GSH levels began to rise with increasing addition of exogenous GSH to Percoll-isolated brain mitochondria. GSNO was undetectable in isolated mitochondria, but detected upon the addition of exogenous GSNO. Its level was not altered by exogenous GSH. This suggests that the inhibition of GSNO-induced protein S-nitrosylation is not due to a depletion of GSNO; alternatively, this fits with the hypothesis that exogenously added GSH directly reduces S-nitrosylated proteins via trans-nitrosylation (protein-S-NO + GSH → protein-S-H + GSNO). The newly formed GSNO did not have significant impact on the detectable GSNO level due to the low concentration of protein-S-NO. The direct reduction of S-nitrosylated proteins by exogenous GSH was supported by the observation that adding GSH 10 min after GSNO incubation could effectively reduce the S-nitrosylation of mitochondrial proteins (Figure 4C, lane 5). The denitrosylation of mitochondrial proteins by GSH addition 30 min following GSNO treatment was similar to the denitrosylation observed by coincubation of GSNO and GSH (Figure 4C, lane 4). Taken together, these data suggest that mitochondrial GSH plays a central role in the denitrosylation of mitochondrial proteins.
Figure 4.

Exogenous GSH protected mitochondrial proteins from GSNO-induced S-nitrosylation. (A) Intact mitochondria were incubated with 0.1 mM GSNO for 30 min at room temperature in the dark in the absence or presence of increasing concentrations of GSH. COX IV was used as a loading control. (B) One hundred micrograms of mitochondria was obtained from the samples in A and analyzed by HPLC for GSH, GSSG, and GSNO detection. (C) Intact rat brain mitochondria were treated with 100 μM GSNO for 30 min at room temperature in the absence or presence of 5 mM GSH. In sample 5, after mitochondria were treated with 100 μM GSNO for 30 min, 5 mM GSH was added and further incubated for 10 min. In lane 4, 5 mM GSH was added at the same time as 100 μM GSNO and incubated for 30 min. COX IV was used as a loading control.
Identification of S-Nitrosylated Mitochondrial Proteins
The biotin-switch method was employed to label S-nitrosylated mitochondrial proteins for identification by LC-MS/MS. Identification of three prominent bands near 25, 40, and 50 kDa (Figure 5A) were revealed as mitochondrial nitrosylation targets ATPase and glutamate dehydrogenase (bands near 50 kDa), pyruvate dehydrogenase, creatine kinase, and succinyl-CoA ligase (bands near 40 kDa), and adenine nucleotide translocase (ANT), NADH-ubiquinone oxidoreductase, VDAC, prohibitin, and 2-oxoglutarate/GSH carrier (bands near 25 kDa). The identified proteins had sequence coverage greater than 15% and Mascot scores greater than 200. To reconfirm the identities of some of the mitochondrial proteins found by LC-MS/MS, these pulled-down proteins were separated by SDS–PAGE and transferred to a nitrocellulose membrane for Western blot analysis. Adenine nucleotide translocase (ANT) and voltage-dependent anion channel (VDAC-1) were detected by respective antibodies (Figure 5B), thus supporting mass spectrometry identification.
Figure 5.

Identification of S-nitrosylated proteins in isolated rat brain mitochondria. (A) Intact rat brain mitochondria were treated with 1 mM GSH or GSNO for 1 h at room temperature in the dark. Mitochondrial proteins were treated with the biotin-switch method. Biotinylated proteins were pulled down by streptavidin-agarose beads, eluted, and separated by SDS–PAGE. Proteins were stained by SYPRO-Ruby (Bio-Rad) and visualized by a VersaDoc 1000 imaging system (Bio-Rad). Proteins were identified by LC-MS/MS. (B) Intact rat brain mitochondria were incubated with 1 mM GSH or GSNO for 1 h at room temperature in the dark. Proteins were treated with the biotin-switch method. Biotinylated proteins were pulled down by streptavidin-agarose beads, eluted, and separated by SDS–PAGE, followed by transfer to the nitrocellulose membrane, and Western blot analysis with anti-ANT and anti-VDAC1 antibodies.
Effect of S-Nitrosylation on Mitochondrial Functions
ANT and VDAC are major components in the mitochondrial permeability transition pore (MPTP) complex.33−35 The functional consequences of S-nitrosylation on calcium-induced MPTP are shown in Figure 6: GSNO consistently exacerbated the swelling caused by Ca2+ in a dose-dependent manner. Hence, S-nitrosylation of ANT and VDAC correlated with enhanced calcium-induced MPTP opening. The ATP synthase F1α subunit isoform 1 is inhibited by S-glutathionylation in brain mitochondria isolated with a discontinuous Percoll gradient.20 Interestingly, in this study ATPase was again identified as a target of GSNO-induced S-nitrosylation (Figure 7). Pretreatment of isolated mitochondria with respiratory substrates increases ATPase activity due to deglutathionylation. Addition of GSNO to mitochondria (respiratory substrate pretreated mitochondria broken by freezeing/thawing in liquid nitrogen, then treated with GSNO 1 mM) strongly inhibited ATPase activity; hence, similar to S-glutathionylation, S-nitrosylation of ATPase results in inhibition of its activity.
Figure 6.

GSNO potentiated Ca2+-induced mitochondrial swelling. Freshly isolated intact rat brain mitochondria were incubated with either vehicle (gray bar) or 1 mM GSNO (black bar) for 10 min at room temperature in the dark. The pretreated mitochondria were then added to a quartz cuvette containing swelling buffer, and mitochondrial swelling was induced by various concentrations of CaCl2 and monitored at 520 nm in a spectrophotometer. Basal swelling of mitochondria, treated with vehicle or GSNO, was subtracted from Ca2+-induced swelling. Unpaired Student’s t-test was performed. (*P < 0.05, ** P < 0.01, and n = 3).
Figure 7.
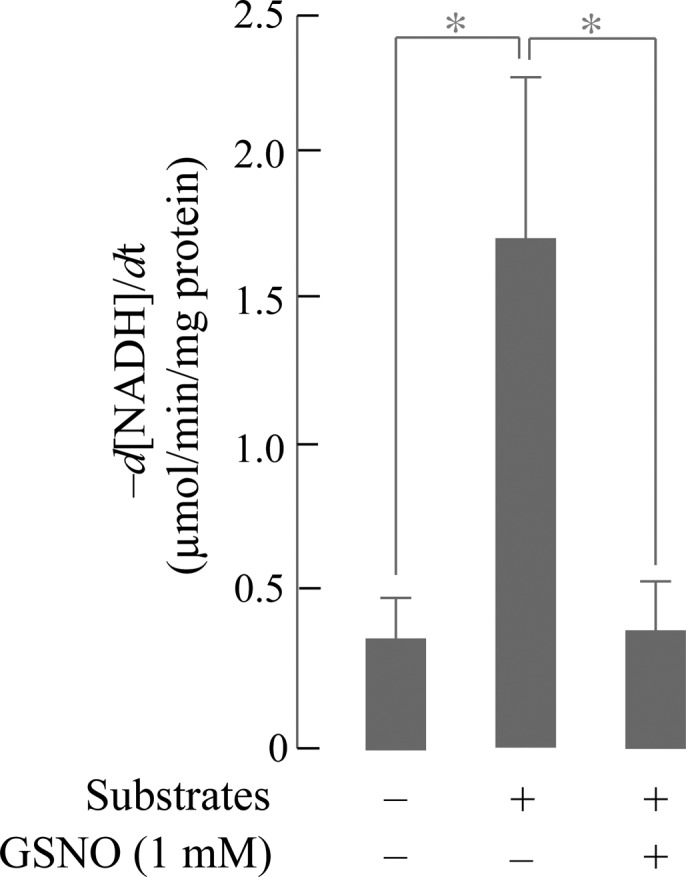
GSNO inhibited ATP synthase activity. Freshly isolated intact rat brain mitochondria were treated with or without respiratory substrates (2.4 mM glutamate, 2.4 mM malate, and 0.7 mM ADP) for 20 min at room temperature. Mitochondria were spun down and resuspended in mitochondrial isolation buffer, followed by three cycles of freeze/thaw in liquid nitrogen to obtain broken mitochondria. Fifty micrograms of respiratory substrate-treated broken mitochondria, in the volume of 10 μL, were incubated with 1 μL of 10 mM GSNO (final concentration 1 mM) for 10 min at room temperature in the dark. The ATPase activities of control, respiratory substrate-treated, respiratory substrate-, and GSNO-treated mitochondria were monitored as detailed in the Experimental Procedures section. Unpaired Student’s t test was performed (*P < 0.05, n = 3).
Discussion
We have previously described that S-glutathionylation of mitochondrial proteins was a major consequence of oxidative stress and that respiratory substrates were key regulators of the mitochondrial redox status, viewed by thiol/disulfide exchange, by maintaining mitochondrial NADPH levels.20 Brain mitochondria isolated by a discontinuous Percoll gradient have a very oxidizing state, which was reduced by supplementation of mitochondria with complex I or complex II respiratory substrates. LC-MS/MS analyses identified the S-glutathionylated proteins as succinyl-CoA transferase and ATPase (F1 complex, α subunit), and likely the NADP+-dependent isocitrate dehydrogenase-2.20 Exposure of primary cortical neurons or astrocytes to increasing flow rates of •NO resulted in a decrease of cellular GSH accompanied by the formation of GSSG and GSNO and protein S-glutathionylation; it was suggested that the latter followed S-nitrosylation; neurons being far more sensitive than astrocytes to •NO exposure.36
In this study, protein S-nitrosylation observed upon incubation of brain mitochondria with GSNO can be partially reversed by energizing mitochondria with complex I or complex II substrates, thus emphasizing their significance in the regulation of mitochondrial redox status. The intracellular stability of GSNO is governed by many factors, including chemically driven degradation reactions4 and enzymatically driven reactions. The main enzyme-dependent degradation described thus far is the reduction of GSNO to GSSG by glutathione-dependent formaldehyde dehydrogenase (or alcohol dehydrogenase III), later renamed GSNO reductase37,38 due to its high specificity and affinity for GSNO and a reflection of the increasingly important role of GSNO in redox chemistry. In this study, proteins from across all mitochondrial compartments have been identified as targets of GSNO-induced S-nitrosylation. The mechanism of S-nitrosylation of mitochondrial matrix proteins by extra-mitochondrial GSNO remains to be determined. However, GSH has been found to be imported into mitochondria via 2-oxoglutarate and dicarboxylate transporters on the mitochondrial inner membrane, and extensive thiol derivatization of GSH did not significantly affect the transport,39,40 suggesting that GSNO may enter the mitochondrial matrix via the same carrier system. This cannot be generalized to the cellular transport of GSNO that requires transmembrane transport of S-nitrosocysteine by the amino acid transporter system L, its intracellular conversion to GSNO, and its metabolism to GSSG.41−43 Regardless of the transport mechanism, extra-mitochondrial GSNO affected various aspects of the mitochondrial functions via S-nitrosylation (probably by transnitrosation reactions), including solute transport across the inner membrane (2-oxoglutarate/GSH transporter); electron transfer (NADH-ubiquinone oxidoreductase); permeability transition and apoptosis (ANT, VDAC, and creatine kinase); and energy metabolism (glutamate dehydrogenase, pyruvate dehydrogenase, succinyl Co-A ligase, and ATP synthase). Among the mitochondrial proteins which are identified as targets of GSNO-induced S-nitrosylation in this study, five of them (ATP synthase, creatine kinase, pyruvate dehydrogenase, ANT, and VDAC) have been previously reported in heart and liver mitochondria and human pulmonary arterial endothelia cells.8,44 The remaining five (glutamate dehydrogenase 1, succinyl Co-A ligase β-subunit, NADH ubiquinone oxidoreducase 30 kDa subunit, 2-oxoglutarate/GSH carrier, and prohibitin) were identified in this study.
Addition of respiratory substrates to brain mitochondria proceeds with significant increases of GSH, NADH, and NADPH; the latter provides the reducing equivalents that support thiol-dependent systems such as thioredoxin and subsequent denitrosylation. The substantial increase in mitochondrial GSH was associated with denitrosylation, gain in activity (in the case of ATPase), and GSSG formation. Of note, the formation of GSSG may be ascribed to a GSH-dependent denitrosylating activity. Studies with cultured endothelial cells have shown that S-nitrosylation by S-nitrosocysteine (CysNO) results in the decline in mitochondrial bioenergetics and at high doses cell death.45,46 Our data suggests that mitochondrial substrate flux is an important regularatory mechanism against S-nitrosylation of mitochondrial proteins. It would be interesting to determine what the cytotoxicity of CysNO would be if flux of mitochondrial substrates in cells were perturbed. Cellular systems would be the next logical step to study the regulation of S-nitrosylation by flux of mitochondrial substrates. The system utilized in this study (isolated mitochondria treated with GSNO) does not take into account localization of GSNO, flux of oxidants, and other factors that may occur in cells.
ATPase was one of the mitochondrial targets for S-nitrosylation (this study) as well as S-glutathionylation.20 Both S-nitrosylation and S-glutathionylation cause a decrease in ATPase activity and are reversible post-translational modifications; thus, the S-nitrosylation of complex I can function as a protective mechanism.10 The amino acid sequences of the subunits of rat ATP synthase F1 complex show that only α and γ contain cysteines: two in α and one in γ; the cysteine at the interface between α and β subunits is close to the glycine-rich loop47 and is a potential residue for GSNO-induced S-nitrosylation. Modification of these cysteines may affect subunit interaction and/or cause conformational changes that modulate ATPase catalytic activity. Also, ATP synthase F1 complex α subunit isoform 1 was found to be both S-glutathionylated20 and S-nitrosylated (this study). Furthermore, glutathionylation also inhibits the ATP synthase activity, similar to nitrosylation. Our findings suggest that ATP generation during oxidative stress will be limited due to these oxidative modifications on complex V; conversely, reversible S-nitrosylation and S-glutathionylation of ATPase may temporarily protect the complex against irreversible oxidative modifications.
Mitochondrial permeability transition (MPT) refers to a transient increase of permeability across mitochondrial inner membranes to solutes ≤1500 Da.48 Transient MPT has been suggested to be a regulatory mechanism for mitochondria to rapidly release Ca2+ into the cytosol, while prolonged MPT can lead to cytochrome c release and apoptosis.48−51 The identification of ANT and VDAC as targets of extra-mitochondrial GSNO imply that MPT may be regulated by S-nitrosylation of critical cysteines on these two proteins. Pretreatment of isolated mitochondria with GSNO slightly increase Ca2+-induced swelling. Similarly, NO has been reported to cause permeabilization of ANT-reconstituted liposomes,52 and GSNO enhanced Ca2+-dependent swelling of isolated rat heart mitochondria53 and induced cyclosporine A-sensitive (i) cytochrome c release in perfused hearts.54 ANT contains cysteine residues on the three matrix-facing loops,55 which are potential candidates for S-nitrosylation. Collectively, these findings support a regulatory role of S-nitrosylation on MPT. S-Nitrosylation represents just one of several modifications to protein thiols (i.e., oxidation, lipid electrophiles such as 15-deoxy-d12,14-prostaglandin J2 (15d-PGJ2)) in mitochondria to regulate MPT.56 Although the exact assembly of the mitochondrial permeability transition pore (mPTP) is unclear, redox-sensitive cyclophilin D in the mitochondrial matrix has been confirmed as a regulator of the mPTP: S-nitrosylation of cyclophilin D at Cys203 seems critical for the activation of the mPTP.57
It may be surmised that the S-nitrosylation of mitochondrial proteins is in a dynamic equilibrium with respiratory activity and the redox environment (GSH, thioredoxin). S-Nitrosylation may represent a protection of protein thiols in response to the changing mitochondrial respiration and redox environment. While respiratory substrates reversed most GSNO-mediated S-nitrosylation of brain mitochondrial proteins, some S-nitrosylation can occur even when the mitochondrial redox environment is reduced, i.e., high levels of NAD(P)H and GSH, thus suggesting different reactivities of S-nitrosylated mitochondrial proteins. Several studies have identified a set of proteins that remain S-nitrosylated under physiological conditions: brain cytosolic lysates exposed to GSNO show a subset of stable S-nitrosylated proteins, some of them related to metabolism (glyceraldehyde-3P-dehydrogenase and pyruvate kinase) and oxidative stress (peroxiredoxin-6).58 Two enzyme systems catalyzed protein denitrosylation: GSNO reductase (GSNOR) and the thioredoxin systems.59 The former, also known as alcohol dehydrogenase-3, is ubiquitously distributed and strongly influenced by the cellular redox potential as determined by GSH and NADH levels.60 GSNOR activity is influenced by GSH redox status but does not require GSH for activity. The fact that DTT treatment could not reverse S-nitrosylation when thiols (GSH and/or thioredoxin) were depleted by CDNB suggests that a certain level of thioredoxin and/or GSH is essential for denitrosylation of proteins in mitochondria. This suggests that thioredoxin and/or GSH-dependent enzymes such as glutaredoxin play an important role in the denitrosylation of mitochondrial proteins, either acting directly to denitrosylate proteins or working in conjunction with GSNOR and/or thioredoxin to mediate denitrosylation of proteins in mitochondria. Further studies are needed to understand enzyme contribution for denitrosylation of proteins in mitochondria following the addition of respiratory substrates. Overall, our findings emphasize the importance of respiratory substrates in regulating S-nitrosylation through a thiol-dependent pathway, with implications for mitochondrial bioenergetics and mitochondrion-driven apoptosis.
Glossary
Abbreviations
- ANT
adenine nucleotide translocase
- biotin-HPDP
N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide
- BSA
bovine serum albumin
- CDNB
1-chloro-2,4-dinitrobenzene
- CHAPS
3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate
- DMF
dimethylformamide
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycoltetraacetic acid
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- GSSG
glutathione disulfide
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- HRP
horse radish peroxidase
- HPLC
high prerformance liquid chromatography
- MMTS
methylmethanethiosulfonate
- MOPS
3-morpholinopropane-1-sulfonic acid
- MPT
mitochondrial permeability transition
- MPTP
mitochondria permeability transition pore
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- PMSF
phenylmethylsulfonyl fluoride
- SDS
sodium dodecyl sulfate
- VDAC
voltage-dependent anion channel
Supported by NIH grant RO1AG016718 to E.C. and PO1AG026572 (to Roberta Díaz Brinton; E.C. as coinvestigator in Project 1).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Hill B. G.; Dranka B. P.; Bailey S. M.; Lancaster J. R. J.; Darley-Usmar V. M. (2010) What part of NO don’t you understand? Some answers to the cardinal questions in nitric oxide biology. J. Biol. Chem. 285, 19699–19704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Ruiz A.; Cadenas S.; Lamas S. (2011) Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radical Biol. Med. 51, 17–29. [DOI] [PubMed] [Google Scholar]
- Hess D. T.; Matsumoto A.; Kim S. O.; Marshall H. E.; Stamler J. S. (2005) Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166. [DOI] [PubMed] [Google Scholar]
- Hogg N. (2002) The biochemistry and physiology of S-nitrosothiols. Annu. Rev. Pharmacol. Toxicol. 42, 585–600. [DOI] [PubMed] [Google Scholar]
- Stamler J. S.; Lamas S.; Fang F. C. (2001) Nitrosylation: the prototypic redox-based signaling mechanism. Cell 106, 675–683. [DOI] [PubMed] [Google Scholar]
- Hess D. T.; Stamler J. S. (2012) Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 287, 4411–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi C. A. (2012) Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 1820, 712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. I.; Uhrigshardt H.; O’Meally R. N.; Cole R. N.; Van Eyk J. E. (2012) Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol. Cell. Proteomics 11, M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell L. S.; Nadtochiy S. M.; Tompkins A. J.; Young S.; Brookes P. S. (2006) Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 394, 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prime T. A.; Blaikie F. H.; Evans C.; Nadtochly S. M.; James A. M.; Dahm C. C.; Vitturi D. A.; Patel R. P.; Hiley C. R.; Akbakumova I.; requejo R.; Chouchani E. T.; Hurd T. R.; Garvey J. F.; taylor C. T.; Brookes P. S.; Smith R. A.; Murphy M. P. (2009) A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 106, 10764–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochly S. M.; Burwell L. S.; Brookes P. S. (2007) Cardioprotection and mitochondrial S-nitrosation: effects on S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 42, 821–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi E.; Brown G. C.; Feelish M.; Moncada S. (1998) Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. U.S.A. 95, 7631–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin A.; Moncada S. (2007) S-Nitrosation of mitochondrial complex I depends on its structural conformation. J. Biol. Chem. 282, 37448–37453. [DOI] [PubMed] [Google Scholar]
- Sun J.; Morgan M.; Shen R. F.; Steenbergen C.; Murphy E. (2007) Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ. Res. 101, 1155–1163. [DOI] [PubMed] [Google Scholar]
- Sun J.; Kohr M. J.; Nguyen T.; Aponte A. M.; Connelly P. S.; Esfahani S. G.; Gucek M.; Daniels M. P.; Steenbergen C.; Murphy E. (2012) Disruption of caveolae blocks ischemic preconditioning-mediated S-nitrosylation of mitochondrial proteins. Antioxid. Redox Signaling 16, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta S. J.; Andersen J. K. (2006) Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radical Biol. Med. 41, 1442–1448. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Lipton S. A. (2011) Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 18, 1478–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon K.-H.; Hood B. L.; Kim B. J.; Hardwick J. P.; Conrads T. P.; Veenstra T. D.; Song B. J. (2006) Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology 44, 1218–1230. [DOI] [PubMed] [Google Scholar]
- Moon K.-H.; Hood B. L.; Mukhopadhyay P.; Rajesh M.; Abdelmegeed M. A.; Kwon Y.-I.; Conrads T. P.; Veenstra T. D.; Song B.-J.; Pacher P. (2008) Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology 135, 1344–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J.; Han D.; Sancheti H.; Yap L. P.; Kaplowitz N.; Cadenas E. (2010) Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M. F.; Sims N. R. (2000) Improved recovery of highly enriched mitochondrial fractions from small brain tissue samples. Brain Res. Protoc. 5, 95–101. [DOI] [PubMed] [Google Scholar]
- Han D.; Canali R.; Rettori D.; Kaplowitz N. (2003) Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol. Pharmacol. 64, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Hansson M. J.; Persson T.; Friberg H.; Keep M. F.; Rees A.; Wieloch T.; Elmer E. (2003) Powerful cyclosporin inhibition of calcium-induced permeability transition in brain mitochondria. Brain Res. 960, 99–111. [DOI] [PubMed] [Google Scholar]
- Foster M. W.; Stamler J. S. (2004) New insights into protein S-nitrosylation. Mitochondria as a model system. J. Biol. Chem. 279, 25891–25897. [DOI] [PubMed] [Google Scholar]
- Jaffrey S. R.; Snyder S. H. (2001) The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, PL1. [DOI] [PubMed] [Google Scholar]
- Tao L.; English A. M. (2004) Protein S-glutathiolation triggered by decomposed S-nitrosoglutathione. Biochemistry 43, 4028–4038. [DOI] [PubMed] [Google Scholar]
- Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77. [DOI] [PubMed] [Google Scholar]
- Yap L. P.; Sancheti H.; Ybanez M. D.; Garcia J. V.; Cadenas E.; Han D. (2010) Determination of GSH, GSSG, and GSNO using HPLC with electrochemical detection. Methods Enzymol. 473, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaidman L. K.; Leung A. C.; Adams J. D. Jr. (1995) High-performance liquid chromatography analysis of oxidized and reduced pyridine dinucleotides in specific brain regions. Anal. Biochem. 228, 312–317. [DOI] [PubMed] [Google Scholar]
- Awasthi Y. C.; Garg H. S.; Dao D. D.; Partridge C. A.; Srivastava S. K. (1981) Enzymatic conjugation of erythrocyte glutathione with 1-chloro-2,4-dinitrobenzene: the fate of glutathione conjugate in erythrocytes and the effect of glutathione depletion on hemoglobin. Blood 58, 733–738. [PubMed] [Google Scholar]
- Jocelyn P. C. (1967) The standard redox potential of cysteine-cystine from the thiol-disulphide exchange reaction with glutathione and lipoic acid. Eur. J. Biochem. 2, 327–331. [DOI] [PubMed] [Google Scholar]
- Arner E. S.; Bjornstedt M.; Holmgren A. (1995) 1-Chloro-2,4-dinitrobenzene is an irreversible inhibitor of human thioredoxin reductase. Loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity. J. Biol. Chem. 270, 3479–3482. [DOI] [PubMed] [Google Scholar]
- Beutner G.; Rück A.; Riede B.; Brdiczka D. (1997) Complexes between hexokinase, mitochondrial porin and adenylate translocator in brain: regulation of hexokinase, oxidative phosphorylation and permeability transition pore. Biochem. Soc. Trans. 25, 151–157. [DOI] [PubMed] [Google Scholar]
- Crompton M.; Virji S.; Ward J. M. (1998) Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 258, 729–735. [DOI] [PubMed] [Google Scholar]
- Rück A.; Dolder M.; Wallimann T.; Brdiczka D. (1998) Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Lett. 426, 97–101. [DOI] [PubMed] [Google Scholar]
- Yap L. P.; Garcia J. V.; Han D.; Cadenas E. (2010) Role of nitric oxide-mediated glutathionylation in neuronal function. Potential regulation of energy utilization. Biochem. J. 428, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg J. J.; Griffiths W. J.; Nilsson S. J.; Hoog J. O. (2003) Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analysed by electron spray mass spectrometry. Eur. J. Biochem. 270, 1249–1256. [DOI] [PubMed] [Google Scholar]
- Liu L.; Hausladen A.; Zeng M.; Que L.; Heitman J.; Stamler J. S. (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410, 490–404. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Putt D. A.; Lash L. H. (2000) Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria: further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch. Biochem. Biophys. 373, 193–202. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Lash L. H. (1998) Evicdence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J. Pharmacol. Exp. Ther. 285, 608–618. [PubMed] [Google Scholar]
- Zhang D.; Lu C.; Whiteman M.; Chance B.; Armstrong J. S. (2008) The mitochondrial permeability transition regulates cytochrome c release for apoptosis during endoplasmic reticulum stress by remodeling the cristae junction. J. Biol. Chem. 283, 3476–3486. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Hogg N. (2004) The mechanism of transmembrane S-nitrosothiol transport. Proc. Natl. Acad. Sci. U.S.A. 101, 7891–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Hogg N. (2005) S-Nitrosothiols: cellular formation and transport. Free Radical Biol. Med. 38, 831–838. [DOI] [PubMed] [Google Scholar]
- Chouchani E. T.; Hurd T. R.; Nadtochiy S. M.; Brookes P. S.; Fearnley I. M.; Lilley K.; Smith R. A.; Murphy M. P. (2010) Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 430, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers A. R.; Broniowska K. A.; Darley-Usmar V. M.; Hogg N. (2011) Differential regulation of metabolism by nitric oxide and S-nitrosothiols in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 301, H803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers A. R.; Broniowska K. A.; Hogg N. (2013) Nitrosative stress and redox-cycling agents synergize to cause mitochondrial dysfunction and cell death in endothelial cells. Redox Biol. 1, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet M.; Ysern X.; Hullihen J.; Pedersen P. L.; Amzel L. M. (1991) Mitochondrial ATP synthase. Quaternary structure of the F1 moiety at 3.6 A determined by x-ray diffraction analysis. J. Biol. Chem. 266, 21197–211201. [PubMed] [Google Scholar]
- Gunter T. E.; Pfeiffer D. R. (1990) Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258, C755–C786. [DOI] [PubMed] [Google Scholar]
- Petronilli V.; Miotto G.; Canton M.; Brinni M.; Colonna R.; Bernardi P.; Di Lisa F. (1999) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 76, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V.; Penzo D.; Scorrano L.; Bernardi P.; Di Lisa F. (2001) The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 276, 12030–12034. [DOI] [PubMed] [Google Scholar]
- Zamzami N.; Kroemer G. (2001) The mitochondrion in apoptosis: how Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2, 67–71. [DOI] [PubMed] [Google Scholar]
- Vieira H. L.; Belzacq A. S.; Haouzi D.; Bernassola F.; Cohen I.; Jacotot E.; Ferri K. F.; El Hamel C.; Bartle L. M.; Melino G.; Brenner C.; Goldmach V.; Kromer G. (2001) The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene 20, 4305–4316. [DOI] [PubMed] [Google Scholar]
- Borutaite V.; Morkuniene R.; Brown G. C. (2000) Nitric oxide donors, nitrosothiols and mitochondrial respiration inhibitors induce caspase activation by different mechanisms. FEBS Lett. 467, 155–159. [DOI] [PubMed] [Google Scholar]
- Jekabsone A.; Kapkunas Z.; Brown G. C.; Borutaite V. (2003) S-nitrosothiol-induced rapid cytochrome c release, caspase activation and mitochondrial permeability transition in perfused heart. Biochem. Pharmacol. 66, 1513–1519. [DOI] [PubMed] [Google Scholar]
- Majima E.; Yamaguchi N.; Chuman H.; Shinohara Y.; Ishida M.; Goto S.; Terada H. (1998) Binding of the fluorescein derivative eosin Y to the mitochondrial ADP/ATP carrier: characterization of the adenine nucleotide binding site. Biochemistry 37, 424–432. [DOI] [PubMed] [Google Scholar]
- Landar A.; Shiva S.; Levonen A. L.; Oh J. Y.; Zaragoza C.; Johnson M. S.; Darley-Usmar V. M. (2006) Induction of the permeability transition and cytochrome c release by 15-deoxy-Delta12,14-prostaglandin J2 in mitochondria. Biochem. J. 394, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T.; Steven M. V.; Kohr M.; Steenbergen C.; Sack M. N.; Murphy E. (2011) Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 286, 40184–40192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige J. S.; Xu G.; Stancevic B.; Jaffrey S. R. (2008) Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem. Biol. 15, 1307–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M.; Forrester M. T.; Stamler J. S. (2009) Protein denitrosylatioon: enzymatic mechanisms and cellular functions. Nat. Cell Biol. 10, 721–732. [DOI] [PubMed] [Google Scholar]
- Staab C. A.; Hellgren M.; Höög J.-O. (2008) Dual functions of alcohol dehydrogenase 3: implications with focus on formaldehyde dehydrogenase and S-nitrosoglutathione reductase activities. Cell. Mol. Life Sci. 65, 3950–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]