

Abstract
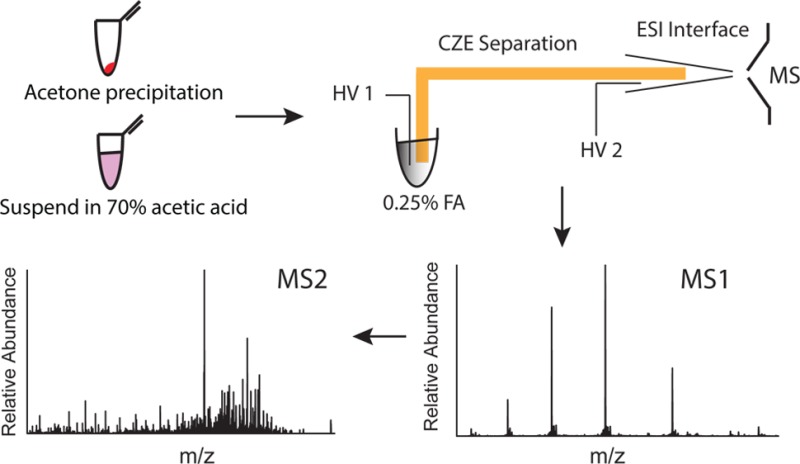
Capillary zone electrophoresis (CZE) with an electrokinetically pumped sheath-flow nanospray interface was coupled with a high-resolution Q-Exactive mass spectrometer for the analysis of culture filtrates from Mycobacterium marinum. We confidently identified 22 gene products from the wildtype M. marinum secretome in a single CZE–tandem mass spectrometry (MS/MS) run. A total of 58 proteoforms were observed with post-translational modifications including signal peptide removal, N-terminal methionine excision, and acetylation. The conductivities of aqueous acetic acid and formic acid solutions were measured from 0.1% to 100% concentration (v/v). Acetic acid (70%) provided lower conductivity than 0.25% formic acid and was evaluated as low ionic-strength and a CZE–MS compatible sample buffer with good protein solubility.
Mass spectrometry-based proteomics is an effective tool for protein identification, characterization, and quantitation.1−3 Most proteomic studies employ a bottom-up approach where proteins are enzymatically digested, and the resulting peptides are then analyzed by tandem mass spectrometry to infer the identity of proteins in the sample. While fast and efficient, this analysis seldom generates complete protein coverage. The resulting gaps can hide both post-translational modifications and alternative splice forms.
In contrast, top-down proteomics employs tandem mass spectrometry to analyze intact proteins. When successful, this analysis generates outstanding sequence coverage and aids in the identification and localization of post-translational modifications.4−6 However, top-down proteomics requires sophisticated front-end separation and extremely high-resolution mass spectrometers. High-resolution Fourier transform ion cyclotron resonance (FTICR) mass spectrometry was first employed in top-down protein analysis by McLafferty’s group.6−8 That group later demonstrated the successful characterization of proteins with masses greater than 200 kDa.9 One of the most impressive demonstrations of top-down proteomics for complex sample was reported by Tran et al.,10 wherein 1 043 gene products and over 3 000 protein species were identified from a human cell lysate with a three-stage separation system; that analysis required roughly 45 h of analysis time using a FTICR mass spectrometer and generated ∼20 protein IDs and ∼60 proteoform IDs per hour of mass spectrometer time. In another study, Ansong and colleagues employed a 4 h UPLC separation of intact proteins from Salmonella typhimurium. Top-down analysis identified 563 unique proteins and 1 665 proteoforms.11
Reverse phase liquid chromatography (RPLC) is the most commonly used separation method for both peptides and proteins.12−16 However, while RPLC is efficient for the separation of peptides, protein separations suffer from strong retention on the stationary phase, which can result in broad peaks and poor peak capacity, time-consuming washing steps, and short column lifetime.
Capillary electrophoresis (CE) is an alternative to reverse phase liquid chromatography that can provide efficient protein separation.17−21 For example, capillary isoelectric focusing (cIEF) coupled with FTICR mass spectrometry was applied to analysis of the Escherichia coli proteome by Smith’s group; that study generated parent ion mass information for 400–1 000 putative proteins in a single run.22 Capillary zone electrophoresis (CZE) is an alternative separation mode that is much easier to automate than cIEF. Up to 74 glycoforms have been identified and characterized from a single pharmaceutical glycoprotein using CZE coupled with time-of-flight MS.19 That work employed a sheathless electrospray ionization (ESI) interface.
Our group has developed an electrokinetically pumped sheath-flow nanospray CE–MS interface that employs electro-osmosis to generate very low sheath flow rates.23 This sheath-flow nanospray interface has been applied to a number of bottom-up proteomics analyses.24−29 We recently demonstrated that this sheath-flow interface could also be applied for top-down protein analysis.29 Model proteins and several impurities were separated and analyzed by that system in 12 min. After database searching of the tandem spectra, three proteins, their post-translational modifications, and one impurity were identified. Kelleher’s group has very recently reported the use of the electrokinetically pumped nanospray interface and a Q-Exactive mass spectrometer to analyze intact proteins from Pseudomonas aeruginosa.30 A total of 30 proteins were identified in the mass range of 30–80 kDa during a 25 min CZE separation.
In this work, we coupled CZE to a high resolution Q-Exactive mass spectrometer via the electrokinetically pumped sheath-flow electrospray interface. The Mycobacterium marinum secretome was separated and analyzed using this platform. We first evaluated the compatibility of high concentration (70%) acetic acid as sample preparation buffer with the CZE-MS/MS system using bovine heart cytochrome c as a model protein. We then applied this system to the analysis secretome from M. marinum. This experiment requires minimal sample preparation. We identified 22 gene products and 58 proteoforms in a single run from the wildtype secretome.
Experimental Section
Materials and Reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless stated otherwise. Formic acid (FA) and glacial acetic acid were purchased from Fisher Scientific (Pittsburgh, PA). Methanol was purchased from Honeywell Burdick & Jackson (Wicklow, Ireland). Water was deionized by a NanoPure system from Thermo Scientific (Marietta, OH). Linear polyacrylamide (LPA)-coated fused capillary (50 μm i.d. × 150 μm o.d.) was purchased from Polymicro Technologies (Phoenix, AZ).
Sample Preparation
The culturing of M. marinum and generation of short-term culture filtrates have been described elsewhere.31 A secreted protein fraction containing approximately 200 μg of protein, as determined by the bicinchoninic acid assay, was purified by ice-cold acetone precipitation and resuspension in 50 μL of 70% acetic acid, followed by sonication for 5 min. The suspension was then centrifuged and the supernatant was taken for CZE–ESI-MS/MS analysis.
CZE–ESI-MS/MS Analysis
CZE was coupled to a Q Exactive mass spectrometer for secretome characterization. Electrospray was generated using an electrokinetically pumped sheath flow through a nanospray emitter.24 The borosilicate glass emitter (1.0 mm o.d. × 0.75 mm i.d., 10 cm length) was pulled with a Sutter instrument P-1000 flaming/brown micropipet puller. The emitter inner diameter was 7–12 μm. Separation was performed in a 50 cm long, 50 μm i.d., 150 μm o.d. LPA-coated fused capillary. The separation buffer was 0.25% (v/v) FA. The electrospray sheath liquid was 10% (v/v) methanol and 0.1% (v/v) FA. A ∼500 ng protein aliquot (∼6 cm in length) was injected into the separation capillary by pressure. The separation voltage was 15 kV, and the electrospray voltage was 1.2 kV.
Mass Spectrometer Operating Parameters
A Q Exactive mass spectrometer (Thermo Fisher Scientific) was operated with the S-lens rf level set at 50% and the ion transfer tube temperature at 280 °C. Full MS scans were acquired in the Orbitrap over the m/z 600–2000 range with resolution of 140 000 at m/z 200. The three most intense peaks with charge state ≥2 were selected for fragmentation in the higher energy collisional dissociation (HCD) cell and detection in the Orbitrap with resolution of 70 000 at m/z 200. The target value for MS and MS/MS acquisition were 3.00 × 106 and 1.00 × 106, respectively. One microscan was used. The maximum injection times for MS and MS/MS were both 500 ms. Dynamic exclusion was 60 s.
Data Analysis
The tandem spectra were decharged and deisotoped by MS-Deconv (version 0.8.0.7370), followed by database searching with MS-Align+ software (version 0.7.1.7143).32 Raw files from Q Exactive were first converted to mzXML files with ReAdW (version 4.3.1). Then, MS-Deconv (v 0.8.0.7370) was used to generate msalign files with mzXML files as the input. Finally, the MS-Align+ software (http://bix.ucsd.edu/projects/msalign/) was used for database searching with msalign files as the input. NCBI protein database for M. marinum including common contaminates (5 583 protein sequences) was used for database searching. The parameters for database searching included a maximum number of modifications (shift number) as 2, mass error tolerance as 10 ppm, “doOneDaltonCorrection” and “doChargeCorrection” as false, “cutoffType” as EVALUE, and cutoff as 0.01. For protein identification, results were filtered with an E-value better than 0.001.
Results and Discussion
Sample
This study employed the proteins derived from short-term culture filtrates of M. marinum. This bacterium is closely related to the causative agent of tuberculosis (M. tuberculosis) and is often used as a model system for the study of some aspects of that disease,31 specifically ESX-1 protein secretion. We have previously reported the comparison of both CZE and UPLC for the bottom-up analysis of this secretome; CZE identified 140 proteins and UPLC identified 134 proteins.25 In both cases, analysis required roughly 3 h of mass spectrometer time.
Conductivity of Acetic and Formic Acids
Despite the success of CZE in bottom-up proteomics and the top-down analysis of standard proteins, there has been limited work on extension of CZE–ESI-MS/MS for the top-down characterization of proteins from a complex sample. One challenge hindering the application of CZE to top-down proteomics is protein solubilization. A clue to enhanced protein solubilization comes from reports that employ organic acids to solubilize membrane proteins.33 As an example, Catherman employed a high concentration of formic acid to solubilize intact proteins for LC–MS analysis.12 Unfortunately, high concentrations of formic acid are not compatible with CZE because of the high conductivity of formic acid results in high current and band broadening.
Intriguingly, there is a dramatic difference in conductivity between acetic and formic acid solutions at concentrations up to 50% in concentration.34 Published data cover a limited concentration range. To extend data to higher concentrations, we determined the conductivity of aqueous acetic acid and formic acid solutions by applying 6 kV across a 60 cm capillary filled with acetic acid and formic acid in water at concentrations ranging from 0.1% to 100% and measuring current. Ohm’s law and the capillary geometry were used to calculate conductivity, Figure 1 and Table S1 in the Supporting Information. Across all concentration ranges studied, acetic acid solutions have much lower conductivity than formic acid. Furthermore, this data suggests that very high concentrations of acetic acid (>50%) will have lower conductivity than the 0.25% formic acid running buffer that is commonly used in CZE analysis of proteins.
Figure 1.
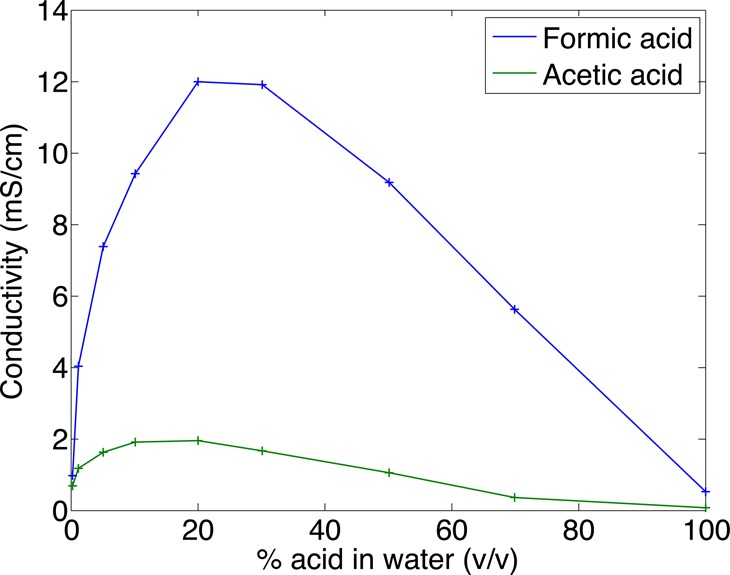
Conductivity of aqueous solutions of acetic and formic acids at 25 °C. Conductivity was determined from the current generated when applying 6 kV voltage across a 60 cm long, 20 μm i.d. capillary. Both capillary ends were immersed in 0.1% FA during electrophoresis. To produce a stable reading, current was recorded 10 s after applying the voltage. Uncertainties in data are ∼5%. Data points are connected by straight lines.
We also examined the current in a capillary filled with plugs of 70% acetic acid in a capillary filled with 0.25% formic acid running buffer. Plugs of acetic acid between 0 and 27 cm in length were injected into a 40 cm LPA coated capillary by pressure. The resistance of the capillary increased linearly with plug length, Figure 2. The resistance across the 40 cm long capillary was 1.4 GΩ when the capillary was filled with formic acid, and the resistance increased at a rate of 96 MΩ per centimeter of injected acetic acid. These resistance values correspond to a conductivity of 1.5 mS/cm for 0.25% formic acid and 0.5 mS/cm for 70% acetic acid; the conductivity of 70% acetic acid is roughly 3 times lower than the 0.25% formic acid separation buffer. These results suggest that a modest stacking effect can be expected for samples prepared in 70% acetic acid used with a 0.25% formic acid separation buffer, due to the lower conductivity of the 70% acetic acid sample buffer.
Figure 2.
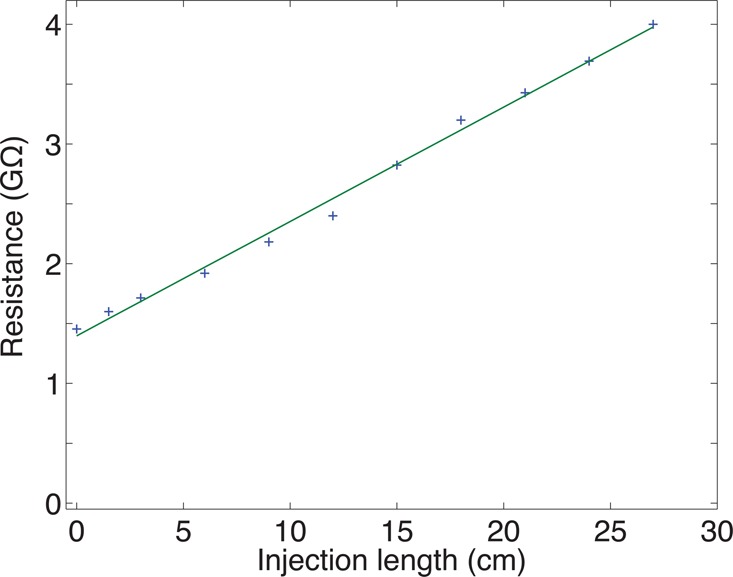
Electrical resistance across a 40 cm long, 50 μm i.d. capillary filled with plugs of 70% acetic acid. The running buffer was 0.25% formic acid. Both capillary ends were immersed in 0.25% FA during electrophoresis after the acetic acid solution was injected. To produce a stable reading, current was measured 10 s after applying a 16 kV across the capillary.
Next, to evaluate the compatibility of 70% acetic acid as sample buffer with a CZE–MS system, we dissolved about 30 ng of cytochrome c in 0.25% FA and in 70% acetic acid solutions and analyzed the samples by CZE–ESI-MS under the same conditions. Triplicate runs were performed for both sample solutions with an LTQ-XL mass spectrometer. On average the peak height and widths were the same for the two buffers, although the variance for both peak height and width were larger in 70% acetic acid. The migration time was consistently 20% longer for the sample prepared in 70% acetic acid (p < 0.025) (Table S2 in the Supporting Information). Longer migration time in 70% acetic acid was likely due to the higher viscosity of the acetic acid solution compared with water.35
Analysis of Secretome from Mycobacterium marinum
Normalized collision energy (NCE) was first varied to optimize the number of protein identifications with a M. marinum WT secreted protein sample. The number of identifications maximized with NCE near 30%.
Examples of fragmentation for 10 kDa culture filtrate antigen EsxB (CFP-10) with these three NCEs are provided in the Supporting Information (Figure S1). Lower NCE resulted in poor fragmentation of the selected precursor ion, so fewer product ions were generated, causing poor tandem mass spectra matching. Higher NCE generated tandem spectra that were too complex for identification. It is worth mentioning that all mass spectrometry parameters used here were generic, and there was no modification made to the commercially available Q Exactive mass spectrometer.
We characterized the M. marinum WT secreted protein sample. A ∼500 ng protein aliquot was injected. As shown in Figure 3, the separation window was about 35 min, and the peak widths were less than 1 min. A total of 22 proteins were identified in a single run with NCE was set to 28% (Table 1). The protein identification efficiency (the number of protein IDs per hour instrument time) is similar to those reported by Tran et al.,10 who identified 1 043 proteins in 45 h-long LC–MS runs. The size of identified proteins ranged from several kDa to over 20 kDa. The high-resolution mass spectrometer resolved isotopic peaks for these relatively low molecular weight proteins (Figure 3). Most of these proteins were also identified in our bottom-up study of this secretome. Five of the detected proteins were not present in our earlier bottom-up proteomics study of M. marinum’s secretome; those proteins were all hypothetical proteins.
Figure 3.

Base peak electropherogram of the secreted proteins analyzed by the CZE–ESI-MS/MS system. Selected peaks were labeled with identified protein spectra. Superscript numbers indicate the protein rank in Table 1. The voltage applied was 15 kV for CE separation and 1.2 kV for electrospray. Inserts show parent ion spectra for proteins centered at the indicated m/z values.
Table 1. Identified Proteins in a Single Top-down CZE Analysis of the M. marinum Secretome.
| ranka | accession | name | size (kDa) | species | bottom-up data setb |
|---|---|---|---|---|---|
| 1 | gi|183 980 221 | 10 kDa culture filtrate antigen EsxB | 10.6 | M. marinum M | CE, LC |
| 2 | gi|183 985 424 | hypothetical protein MMAR_5453 | 5.7 | M. marinum M | CE, LC |
| 3 | gi|183 980 745 | hypothetical protein MMAR_0722 | 15.0 | M. marinum M | CE, LC |
| 4 | gi|183 985 379 | immunogenic protein Mpt64 | 22.7 | M. marinum M | CE, LC |
| 5 | gi|183 983 668 | low molecular weight antigen Cfp2 | 12.2 | M. marinum M | CE, LC |
| 6 | gi|183 984 660 | hypothetical protein MMAR_4692 | 12.3 | M. marinum M | |
| 7 | gi|183 985 108 | cold shock protein A CspA_1 | 7.2 | M. marinum M | LC |
| 8 | gi|183 982 932 | hypothetical protein MMAR_2929 | 8.3 | M. marinum M | CE, LC |
| 9 | gi|183 985 378 | hypothetical protein MMAR_5548 | 4.2 | M. marinum M | |
| 10 | gi|183 982 679 | hypothetical protein MMAR_2672 | 8.9 | M. marinum M | |
| 11 | gi|183 985 410 | hypothetical protein MMAR_5439 | 3.7 | M. marinum M | CE, LC |
| 12 | gi|183 982 898 | PE family protein | 4.5 | M. marinum M | CE, LC |
| 13 | gi|183 984 791 | cold shock protein a, CspA | 7.2 | M. marinum M | CE |
| 14 | gi|183 981 569 | hypothetical protein MMAR_1553 | 14.5 | M. marinum M | CE, LC |
| 15 | gi|183 983 350 | transmembrane protein, MmpS5_2 | 9.1 | M. marinum M | LC |
| 16 | gi|183 985 421 | 6 kDa early secretory antigenic target EsxA (EsaT-6) | 10.0 | M. marinum M | CE, LC |
| 17 | gi|183 980 929 | hypothetical protein MMAR_0908 | 9.5 | M. marinum M | |
| 18 | gi|183 985 025 | lipoprotein DsbF | 14.6 | M. marinum M | LC |
| 19 | gi|183 980 785 | PPE family protein, PPE10 | 8.6 | M. marinum M | CE, LC |
| 20 | gi|183 982 895 | hypothetical protein MMAR_2891 | 10.2 | M. marinum M | |
| 21 | gi|183 982 952 | hypothetical protein MMAR_2949 | 15.3 | M. marinum M | CE, LC |
| 22 | gi|183 983 815 | hypothetical protein MMAR_3840 | 9.8 | M. marinum M | CE, LC |
Rank is based on E-value (E < 9 × 10–4).
CE = present in bottom-up data set of secretome using CZE; LC = present in bottom-up data set using LC.24
All of the identified proteins had molecular weights less than 25 kDa. The Q-Exactive mass spectrometer has a resolution of 140 000 (m/z 200), which limits our ability to identify larger proteins; a mass spectrometer with higher resolving power will be required to extend our top-down analysis to higher molecular weight proteins. This low-molecular weight bias likely accounts for the decreased number of protein identifications compared with our bottom-up analysis of the M. marinum secretome. Moreover, the nature of this secretome suggests that large proteins are present in low abundance, which makes their identification difficult. Also, there are several small proteins with extremely high abundance, which can induce ionization suppression of comigrating proteins. On the basis of the number and size of identified proteins, our system still has limited separation and identification ability compared to the LC–MS system.11 This limitation is caused by the small sample injection amount and the narrow separation window of capillary electrophoresis compared with HPLC. Protein prefractionation should improve the results, which will be addressed in future studies.
Top-down proteomics has a distinct advantage in exploring protein complexity by generating information on proteoforms. We observed 58 proteoforms from 22 gene products, including 16 proteoforms components of the TypeVII ESX-1 protein secretion system (CFP-10 and ESAT-6), which is essential for virulence in pathogenic mycobacteria and conserved in several Gram-positive pathogens. The proteoforms details are listed in the Supporting Information (Table S3). For CFP-10, protein isomers were also separated and observed from the base peak electropherogram showing as small peaks (Figure 3), from which 15 proteoforms were identified. Post-translational modifications include signal peptide removal, N-terminal methionine excision, and acetylation. Only the N-terminal acetylation form of ESAT-6 was found in our database search. However, we confirmed the existence of its unacetylated form by manually checking the spectrum (Figure S2 in the Supporting Information).
Quality tandem spectra were obtained with the optimized collision energy. An example is shown in Figure 4A, the best matching spectrum for 10 kDa culture filtrate antigen EsxB (CFP-10) generated 85 matched fragment ions, and 80 of them were of less than 5 ppm mass error. Also, an N-terminal methionine excision was observed from the tandem mass spectrum.
Figure 4.

HCD fragmentation of the 10-kDa culture filtrate antigen EsxB. (A) Fragmentation spectra of the [M + 7H] 7+ charge state with HCD (normalized collision energy was 28%). (B) Sequence of this protein and the fragmentation patterns observed with HCD.
Acknowledgments
We thank Dr. Patricia A. Champion (ND Biology) for the kind donation of M. marinum culture filtrates. We also thank Dr. William Boggess in the Notre Dame Mass Spectrometry and Proteomics Facility for his help with this project. This project was supported by a grant from the National Institutes of Health (Grant R01GM096767).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Aebersold R.; Goodlett D. R. Chem. Rev. 2001, 101, 269–295. [DOI] [PubMed] [Google Scholar]
- Angel T. E.; Aryal U. K.; Hengel S. M.; Baker E. S.; Kelly R. T.; Robinson E. W.; Smith R. D. Chem. Soc. Rev. 2012, 41, 3912–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Aslanian A.; Yates J. R. 3rd. Curr. Opin. Chem. Biol. 2008, 12, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chait B. T. Science 2006, 314, 65–66. [DOI] [PubMed] [Google Scholar]
- Kelleher N. L. Anal. Chem. 2004, 76, 197A–203A. [PubMed] [Google Scholar]
- Kelleher N. L.; Lin H. Y.; Valaskovic G. A.; Aaserud D. J.; Fridriksson E. K.; McLafferty F. W. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar]
- Ge Y.; Lawhorn B. G.; ElNaggar M.; Strauss E.; Park J. H.; Begley T. P.; McLafferty F. W. J. Am. Chem. Soc. 2002, 124, 672–678. [DOI] [PubMed] [Google Scholar]
- McLafferty F. W.; Fridriksson E. K.; Horn D. M.; Lewis M. A.; Zubarev R. A. Science 1999, 284, 1289–1290. [DOI] [PubMed] [Google Scholar]
- Han X. M.; Jin M.; Breuker K.; McLafferty F. W. Science 2006, 314, 109–112. [DOI] [PubMed] [Google Scholar]
- Tran J. C.; Zamdborg L.; Ahlf D. R.; Lee J. E.; Catherman A. D.; Durbin K. R.; Tipton J. D.; Vellaichamy A.; Kellie J. F.; Li M. X.; Wu C.; Sweet S. M. M.; Early B. P.; Siuti N.; LeDuc R. D.; Compton P. D.; Thomas P. M.; Kelleher N. L. Nature 2011, 480, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansong C.; Wu S.; Meng D.; Liu X. W.; Brewer H. M.; Kaiser B. L. D.; Nakayasu E. S.; Cort J. R.; Pevzner P.; Smith R. D.; Heffron F.; Adkins J. N.; Pasa-Tolic L. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10153–10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman A. D.; Li M.; Tran J. C.; Durbin K. R.; Compton P. D.; Early B. P.; Thomas P. M.; Kelleher N. L. Anal. Chem. 2013, 85, 1880–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton J. D.; Tran J. C.; Catherman A. D.; Ahlf D. R.; Durbin K. R.; Lee J. E.; Kellie J. F.; Kelleher N. L.; Hendrickson C. L.; Marshall A. G. Anal. Chem. 2012, 84, 2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellaichamy A.; Tran J. C.; Catherman A. D.; Lee J. E.; Kellie J. F.; Sweet S. M.; Zamdborg L.; Thomas P. M.; Ahlf D. R.; Durbin K. R.; Valaskovic G. A.; Kelleher N. L. Anal. Chem. 2010, 82, 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi A.; Bai D. L.; Geer L. Y.; Shabanowitz J.; Hunt D. F. Int. J. Mass Spectrom. 2007, 259, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman A. D.; Durbin K. R.; Ahlf D. R.; Early B. P.; Fellers R. T.; Tran J. C.; Thomas P. M.; Kelleher N. L. Mol. Cell. Proteomics 2013, 12, 3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simó C.; Herrero M.; Neusüss C.; Pelzing M.; Kenndler E.; Barbas C.; Ibáñez E.; Cifuentes A. Electrophoresis 2005, 26, 2674–2683. [DOI] [PubMed] [Google Scholar]
- Jorgenson J. W.; Lukacs K. D. Science 1983, 222, 266–272. [DOI] [PubMed] [Google Scholar]
- Haselberg R.; de Jong G. J.; Somsen G. W. Anal. Chem. 2013, 85, 2289–2296. [DOI] [PubMed] [Google Scholar]
- Haselberg R.; Ratnayake C. K.; de Jong G. J.; Somsen G. W. J. Chromatogr., A 2010, 1217, 7605–7611. [DOI] [PubMed] [Google Scholar]
- Valaskovic G. A.; Kelleher N. L.; McLafferty F. W. Science 1996, 273, 1199–1202. [DOI] [PubMed] [Google Scholar]
- Jensen P. K.; Pasa-Tolic L.; Anderson G. A.; Horner J. A.; Lipton M. S.; Bruce J. E.; Smith R. D. Anal. Chem. 1999, 71, 2076–2084. [DOI] [PubMed] [Google Scholar]
- Wojcik R.; Dada O. O.; Sadilek M.; Dovichi N. J. Rapid Commun. Mass Spectrom. 2010, 24, 2554–2560. [DOI] [PubMed] [Google Scholar]
- Li Y.; Champion M. M.; Sun L.; Champion P. A.; Wojcik R.; Dovichi N. J. Anal. Chem. 2012, 84, 1617–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Zhu G.; Li Y.; Wojcik R.; Yang P.; Dovichi N. J. Proteomics 2012, 12, 3013–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wojcik R.; Dovichi N. J.; Champion M. M. Anal. Chem. 2012, 84, 6116–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G.; Sun L.; Yan X.; Dovichi N. J. Anal. Chem. 2013, 85, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Essaka D. C.; Sun L.; Zhu G.; Dovichi N. J. Proteomics 2013, 13, 2546–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Knierman M. D.; Zhu G.; Dovichi N. J. Anal. Chem. 2013, 85, 5989–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Compton P. D.; Tran J. C.; Ntai I.; Kelleher N. L. Proteomics 2014, 10.1002/pmic.201300381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion P. A.; Champion M. M.; Manzanillo P.; Cox J. S. Mol. Microbiol. 2009, 73, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Inbar Y.; Dorrestein P. C.; Wynne C.; Edwards N.; Souda P.; Whitelegge J. P.; Bafna V.; Pevzner P. A. Mol. Cell. Proteomics 2010, 9, 2772–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers A. E.; Wu C. C. Chem. Rev. 2007, 107, 3687–3714. [DOI] [PubMed] [Google Scholar]
- Weast R. C., Ed. CRC Handbook of Chemistry and Physics, 70th ed.; CRC Press: Boca Raton, FL, 1989; p D-221. [Google Scholar]
- Qiao Y.; Di Z.; Ma Y.; Ma P.; Xia S. Chin. J. Chem. Eng. 2010, 18, 446–454. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.