

Abstract
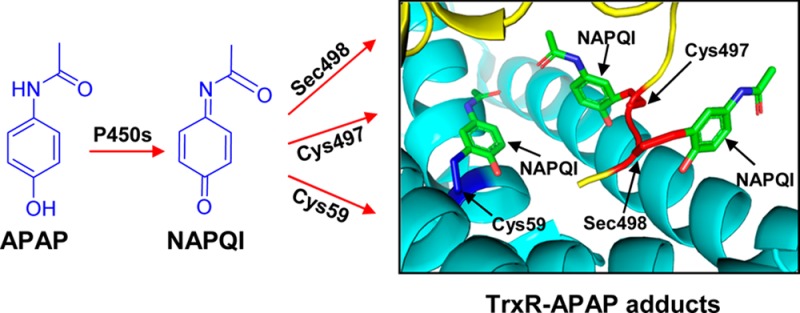
Acetaminophen (APAP) is metabolized in the liver to N-acetyl-p-benzoquinone imine (NAPQI), an electrophilic metabolite known to bind liver proteins resulting in hepatotoxicity. Mammalian thioredoxin reductase (TrxR) is a cellular antioxidant containing selenocysteine (Sec) in its C-terminal redox center, a highly accessible target for electrophilic modification. In the present study, we determined if NAPQI targets TrxR. Hepatotoxicity induced by APAP treatment of mice (300 mg/kg, i.p.) was associated with a marked inhibition of both cytosolic TrxR1 and mitochondrial TrxR2 activity. Maximal inhibition was detected at 1 and 6 h post-APAP for TrxR1 and TrxR2, respectively. In purified rat liver TrxR1, enzyme inactivation was correlated with the metabolic activation of APAP by cytochrome P450, indicating that enzyme inhibition was due to APAP-reactive metabolites. NAPQI was also found to inhibit TrxR1. NADPH-reduced TrxR1 was significantly more sensitive to NAPQI (IC50 = 0.023 μM) than the oxidized enzyme (IC50 = 1.0 μM) or a human TrxR1 Sec498Cys mutant enzyme (IC50 = 17 μM), indicating that cysteine and selenocysteine residues in the redox motifs of TrxR are critical for enzyme inactivation. This is supported by our findings that alkylation of reduced TrxR with biotin-conjugated iodoacetamide, which selectively reacts with selenol or thiol groups on proteins, was inhibited by NAPQI. LC-MS/MS analysis confirmed that NAPQI modified cysteine 59, cysteine 497, and selenocysteine 498 residues in the redox centers of TrxR, resulting in enzyme inhibition. In addition to disulfide reduction, TrxR is also known to mediate chemical redox cycling. We found that menadione redox cycling by TrxR was markedly less sensitive to NAPQI than disulfide reduction, suggesting that TrxR mediates these reactions via distinct mechanisms. These data demonstrate that APAP-reactive metabolites target TrxR, suggesting an additional mechanism by which APAP induces oxidative stress and hepatotoxicity.
Introduction
Acetaminophen (APAP) is a widely used analgesic and antipyretic agent considered safe at therapeutic doses; however, overdose can cause severe hepatotoxicity leading to acute liver failure.1,2 At therapeutic doses, APAP is largely eliminated via sulfation and glucuronidation reactions in the liver. APAP also undergoes cytochrome P450-mediated biotransformation to N-acetyl-p-benzoquinone imine (NAPQI), a highly reactive electrophile.3 The major P450 isoforms mediating NAPQI formation in humans are CYP2E1, CYP3A4, CYP1A2, and CYP2D6.4 APAP overdose saturates the conjugation pathways resulting in increased levels of NAPQI, which readily depletes hepatic glutathione (GSH). This results in covalent modification of nucleophilic sites on hepatic cellular proteins by NAPQI and cytotoxicity.5−7
It is well recognized that oxidative stress contributes to APAP toxicity.1,2 Following APAP intoxication, markers of oxidative stress including malondialdehyde, protein nitration, and 8-hydroxy-deoxyguanosine lesions in DNA are observed in the liver.8 Reactive oxygen and nitrogen species are also detected in the liver after APAP, along with posttranslational redox modifications of proteins.9−12 Concomitantly, antioxidants such as GSH, superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) are suppressed.9 Moreover, mice lacking antioxidant enzymes such as SOD2 or those treated with agents that reduce antioxidants rapidly succumb to APAP-induced hepatotoxicity.13−15 Conversely, animals overexpressing antioxidants including SOD1 and plasma GPx or enzymes important in GSH synthesis such as glutamate cysteine ligase are resistant to APAP toxicity.16,17 APAP poisoning is also mitigated by antioxidants such as N-acetylcysteine, a precursor of GSH, as well as liposomes containing SOD.18−20
The thioredoxin system is composed of NADPH, thioredoxin reductase (TrxR), and thioredoxin (Trx). It functions, at least in part, to protect cells against oxidative stress.21,22 Mammalian cells express three forms of TrxR, cytosolic TrxR1, mitochondrial TrxR2, and a testis-specific TrxR3. These are NADPH-dependent selenoproteins containing cysteine/selenocysteine and cysteine/cysteine redox motifs in their catalytic centers. These amino acids are essential for enzymatic activity and are preferential targets for covalent modification by electrophiles.23−25 TrxR reduces Trx, a disulfide reductase important in DNA synthesis and repair, antioxidant defense, and the regulation of thiol-containing proteins and redox-sensitive transcription factors.21 Inhibition of TrxR results in decreased activity of enzymes dependent on thioredoxin and reduced scavenging of reactive oxygen species (ROS). This can lead to oxidative stress, apoptosis, and necrosis.22 Since these processes are also involved in APAP-induced toxicity, we speculated that TrxR may be a molecular target for APAP-reactive metabolites, and this was investigated in the present studies.
Experimental Procedures
Chemicals and Enzymes
Purified rat liver TrxR1, recombinant human Trx1, and glutathione reductase (GR), recombinant E. coli Trx1, bovine glutathione peroxidase (GPx), insulin, NADPH, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), APAP, NAPQI, menadione (2-methyl-1,4-napthoquinone), reduced glutathione (GSH), oxidized glutathione (GSSG), phosphatase inhibitors (catalog no. P2850, which contains microcystinLR, cantharidin, and (−)-p-bromotetramisole), and protease inhibitor cocktail (catalog no. P2714, which contains 4-(2-aminoethyl)benzenesulfonyl fluoride, E-64, bestatin, leupeptin, aprotinin, and EDTA) were purchased from Sigma-Aldrich (St. Louis, MO). Sodium aurothiomalate hydrate was from Aldrich (Milwaukee, WI). Human recombinant TrxR mutant enzyme (Sec498Cys) was from AbFrontier (Seoul, Korea). Pooled human liver microsomes (HLM) and recombinant human P450s 1A2, 2E1, and 3A4 were purchased from BD Genetest (Woburn, MA). N-(Biotinoyl)-N′-(iodoacetyl) ethylenediamine (BIAM) was from Molecular Probes (Eugene, OR) and horseradish peroxidase (HRP)-conjugated streptavidin from GE Healthcare (Piscataway, NJ). Enhanced chemiluminescence (ECL) immunoblot detection reagents were from Thermo Scientific (Rockford, IL).
Animal Treatments
Male C57BL/6J mice (8–10 weeks) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were housed in microisolation cages and were provided with food and water ad libitum. All animals received humane care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Mice were fasted overnight prior to administration of a single dose of APAP (300 mg/kg; i.p.) or the phosphate-buffered saline (PBS) control. Food was returned 0.5 h following APAP treatment. Mice were euthanized 1–24 h following APAP administration. Blood samples were collected via cardiac puncture and livers harvested as previously described.10
Biochemical and Histological Assessment
Serum alanine (ALT) and aspartate transaminase (AST) activities were measured using diagnostic assay kits (ThermoElectron, Pittsburgh, PA) following the manufacturer’s instructions. Livers were fixed overnight at 4 °C in 3% paraformaldehyde in PBS containing 2% sucrose, washed 3 times with 2% sucrose/PBS, transferred to 50% ethanol, and then paraffin embedded. Six micrometer tissue sections were prepared and stained with hematoxylin and eosin. Images were acquired using an Olympus BX51 microscope equipped with a CCD camera.
Preparation of Subcellular Fractions from Liver
Livers were washed with 0.9% NaCl and homogenized using a Teflon homogenizer in homogenization buffer containing 20 mM Tris, 20 mM 3-(N-morpholino)propanesulfonic acid, 1 mM EGTA, 100 mM glucose, protease, and phosphatase inhibitors, pH 7.4. The homogenates were centrifuged at 800g for 10 min at 4 °C; supernatants were then centrifuged at 9000g for 20 min to sediment mitochondria. The resulting supernatants, designated as the S9 fraction, contained cytosolic proteins. The pellets were washed twice with homogenization buffer and then sonicated in homogenization buffer to obtain the mitochondrial fractions. Cytosolic and mitochondrial protein concentrations were determined using a BCA protein assay kit (Thermo Scientific) with bovine serum albumin as a standard.
TrxR Activity Assay
Liver TrxR activity was assayed using an insulin reduction assay by the method of Luthman and Holmgren26 with minor modifications. Reaction mixtures in a final volume of 50 μL contained 25 μg of cytosolic protein or 50 μg of mitochondrial protein, 50 mM HEPES, pH 7.6, 1 μM E. coli thioredoxin, 20 mM EDTA, 0.3 mM insulin, and 0.25 mM NADPH. After 30 min of incubation at 37 °C, the reactions were terminated by the addition of 200 μL of 8 M guanidine-HCl, 5 mM DTNB, and 200 μM Tris-HCl at pH 8.0 and absorbance at 412 nm recorded. Background TrxR-independent reduction of DTNB in subcellular fractions, determined in the absence of E. coli Trx, was subtracted from each value. TrxR activity was expressed as the percentage absorbance of APAP-treated samples relative to PBS-treated controls.
For NAPQI inhibition studies, TrxR activity was determined by the DTNB assay as described previously.26 Purified rat liver TrxR1 (50 nM) or human TrxR1 mutant enzyme (1 μM) was incubated in the absence or presence of NADPH (0.25 mM) at room temperature in 50 mM potassium phosphate buffer, pH 7.0, containing 1 mM EDTA and 50 mM KCl. After 5 min, NAPQI (1 nM–100 μM) or DMSO control was added, and the reaction mixture incubated for an additional 30 min. The reaction was initiated by the addition of DTNB (2.5 mM), and increases in absorbance at 412 nm were monitored. TrxR activity was defined as μmol of thionitrobenzoic acid formed per min per mg of protein, using a molar extinction coefficient for thionitrobenzoic acid of 13.6 mM–1min–1. For studies on the reversibility of TrxR inhibition, reaction mixtures containing NAPQI-modified TrxR were purified using Chroma Spin TE-10 columns (Clontech, Mountain View, CA) to remove unreacted NAPQI. Modified-TrxR was then analyzed for enzyme activity using the DTNB assay.
For some experiments, NADPH-reduced rat liver TrxR (50 nM), prepared as described above, was incubated at room temperature with increasing concentrations of NAPQI (0.001–100 μM) and 0.25 mM NADPH, in the presence or absence of glutathione (1 mM) in a final volume of 100 μL of TE buffer (50 mM Tris, pH 7.0, and 1 mM EDTA). After 30 min, 100 μL of a thioredoxin/insulin mixture (6 μM E. coli Trx, 250 μM NADPH, and 170 μM insulin in TE buffer) was added and changes in the absorbance at 340 nm analyzed. TrxR activity was calculated as the linear change in absorbance per min and expressed as the percentage of enzyme activity of DMSO-treated control samples.
Trx Activity Assay
Trx activity was assayed by an insulin reduction assay as described for the determination of liver TrxR activity using purified rat liver TrxR1 (50 nM) in place of E. coli Trx. For NAPQI inhibition studies, recombinant human Trx (1 mg/mL) was incubated in the absence or presence of DTT (10 mM) at 37 °C in TE buffer. After 15 min, Trx was purified using Chroma Spin TE-10 columns to remove DTT. Aliquots of purified Trx (final concentration, 1 μM) were then incubated with increasing concentrations of NAPQI (0.1–100 μM) or DMSO in a final volume of 100 μL of TE buffer at room temperature. After 30 min, 100 μL of a TrxR/insulin mixture (50 nM purified rat liver TrxR1, 0.5 mM NADPH, and 170 μM insulin in TE buffer) was added and changes in absorbance at 340 nm recorded. Trx activity was calculated as the linear change in absorbance per min and expressed as a percentage of the enzyme activity of DMSO-treated control samples.
Glutathione Reductase Assay
Glutathione reductase activity was measured by the NADPH reduction assay using oxidized glutathione disulfide as a substrate.26 Reaction mixtures, in a final volume of 0.5 mL, contained cytosolic fractions (0.2 mg/mL), oxidized glutathione (10 mM), and EDTA (1 mM) in phosphate buffer (100 mM, pH 7.5). The reaction was initiated by the addition of NADPH (250 μM) and monitored by decreases in absorbance at 340 nm at 25 °C. One unit of glutathione reductase activity was defined as the number of μmol of NADPH oxidized per min.
Studies on the Inhibition of TrxR by Metabolites of APAP Generated by Microsomes and Recombinant Cytochrome P450s
Human liver microsomes (0.4 mg/mL) or recombinant human P450s (60 pmol/mL) were incubated at 37 °C in 100 mM phosphate buffer, pH 7.4, with 0.2 or 1 mM APAP or PBS control, and purified rat liver TrxR1 (50 nM). Following a 3 min preincubation, an NADPH regenerating system (100 μM NADPH, 10 mM glucose-6-phosphate, and 0.5 unit/mL glucose-6-phosphate dehydrogenase) was added to initiate the reaction. After 30–60 min, TrxR activities were assayed using the DTNB assay. The concentrations of APAP were selected based on kinetic data published previously.4 APAP is known to have a high Km (Michaelis constant) for the individual P450s and liver microsomes (100–2000 μM), and only a small fraction is converted to NAPQI (less than 5%). In addition, once formed, NAPQI reacts with many targets in the microsomes that likely limit its interaction with TrxR.
BIAM Labeling of TrxR and Western Blotting
NAPQI-treated rat liver TrxR1, prepared as described in NAPQI inhibition studies, was incubated in the dark with 50 μM BIAM (dissolved in 50 mM Tris-HCl buffer at pH 6.5 or 8.5) at 37 °C for 30 min. BIAM-labeled protein was then separated by gel electrophoresis on Criterion 10.5–14% Tris-HCl gels (Bio-Rad, Hercules, CA) and electroblotted onto nitrocellulose membranes. The extent of BIAM labeling on TrxR was evaluated by the specific binding of streptavidin-HRP followed by the detection of labeled protein using ECL. After BIAM analysis, the blots were stripped and the membranes reprobed with antibody against TrxR (Santa Cruz) for analysis of total TrxR protein loaded in each well of the gel. Densitometric analysis of bands on the membranes was performed using a FluorChem Image System (Alpha Innotech, San Leandro, CA).
Analysis of TrxR-APAP Adducts by LC-MS/MS
NADPH-reduced rat liver TrxR1 (1 μM) was incubated with or without NAPQI (100 μM) at room temperature in a final volume of 100 μL in 50 mM potassium phosphate, pH 7.0, containing 1 mM EDTA and 50 mM KCl. After 1 h, the incubation mixture was desalted with Chroma Spin TE-10 columns to remove unreacted NAPQI. Five microliters of the filtered solution was analyzed for TrxR activity using the DTNB assay. Aliquots of this solution (40 μL) were then subjected to denaturing SDS–PAGE on 10.5–14% gels. After staining with Coomassie blue, bands containing TrxR were cut from the gels and reduced with 20 mM DTT. Samples were then alkylated with 40 mM iodoacetamide followed by in-gel trypsin digestion.27 Peptides were extracted from the gel, reconstituted in 0.1% trifluoroacetic acid, and analyzed by LC-MS/MS on a Dionex U3000 RSLC nano system (Dionex, Sunnyvale, CA) online with a Thermo LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The peptide mixtures were loaded onto an Acclaim PepMAP 100 nano trap column (5 μm particle, 100 Å pore size, 100 μm × 2 cm, Dionex) and washed for 5 min with solvent A (0.1% formic acid in water) at a flow rate of 10 μL/min. Separation was achieved with an Acclaim PepMAP RSLC nano column (2 μm particle, 100 Å pore size, 75 μm × 15 cm, Dionex) with a 30-min gradient from 2 to 45% of solvent B (0.1% formic acid in acetonitrile) at a flow rate of 300 nL/min. The effluent from the HPLC column was subsequently analyzed by electrospray ionization mass spectrometry. MS spectra were acquired in an Orbitrap with a resolution setting at 60000. MS/MS spectra were acquired in LTQ using a data-dependent acquisition procedure with a cyclic series of a full scan followed by MS/MS scans of 20 of the most intense ions with a repeat count of 2 and a dynamic exclusion duration of 30 s. The MS/MS data were searched against the IPI rat protein database with 10 ppm and 0.8 Da for MS and MS/MS tolerances, respectively, using the SEQUEST algorithm. S-Carbamidomethylation at cysteine (+57.02 Da), NAPQI-induced alkylation at cysteine (+149.15 Da), and oxidation at methionine (+15.60 Da) were set as dynamic modifications to identify the spectra of adducted peptides. Modifications on selenium-containing peptides were searched manually by distinctive isotope patterns and confirmed by the critical b and y ions in MS/MS. Because of the potential low recovery of adducted peptides following gel extraction, low efficiency in ionization of the adducted peptide, and limitations in the methods for MS/MS data collection, higher concentrations of protein and NAPQI were used for this experiment.
Assays for NADPH Oxidation and Redox Cycling Activity by TrxR
Oxidation of NADPH in reaction mixes was determined by measuring decreases in absorbance at 340 nm and quantified using an extinction coefficient of 6.2 mM–1 cm–1. Reaction mixes consisted of 100 nM TrxR, 0.25 mM NADPH, NAPQI (1 nM–100 μM), 1 mM EDTA, 50 mM KCl, and 50 mM potassium phosphate buffer (pH 7.0) in the presence and absence of 100 μM menadione, in a total volume of 100 μL. The same reaction mix with vehicle (DMSO) was used as a control. To analyze the effect of NAPQI-adducted TrxR on the production of ROS, reactions were run in the presence of 100 μM menadione, a quinone known to actively redox cycle with TrxR.28,29 Superoxide anion was measured using the superoxide dismutase-inhibitable acetylated cytochrome c reduction assay.29 Reactions were run in a total volume of 100 μL and contained 250 nM TrxR, 300 mM potassium phosphate buffer, pH 7.8, 0.1 mM EDTA, 0.25 mM NADPH, NAPQI (0.001–100 μM) or vehicle (DMSO), and 100 μM acetylated cytochrome c in the presence or absence of 100 μM menadione. Cytochrome c reduction was monitored at room temperature by increases in absorption at 550 nm and quantified using an extinction coefficient of 21.1 mM–1 cm–1. Reaction mixtures containing 2000 units/mL of SOD were used as controls.
H2O2 was measured either by using the Amplex Red assay or by its conversion to hydroxyl radicals in the presence of iron using the terephthalate assay.30 For Amplex Red assays, reactions were run in a total volume of 100 μL in 50 mM potassium phosphate buffer, pH 7.8, 0.25 mM NADPH, 1 unit/mL HRP, NAPQI (0.001–100 μM) or vehicle (DMSO), and 50 μM Amplex Red reagent in the presence or absence of 100 μM menadione. Reaction mixtures containing 2500 units/mL of catalase were used as controls. Product formation was assessed fluorometrically using excitation and emission wavelengths of 530 and 587 nm, respectively. To measure hydroxyl radicals, the reaction was supplemented with 1 mM terephthalate and the Fe3+ (100 μM)/EDTA (110 μM) complex in place of Amplex Red and HRP. For this assay, both NAPQI and menadione were dissolved in acetronitrile. Some samples also contained DMSO (1%) or catalase (2500 units/mL). After a 30 min incubation at 37 °C, the reaction was terminated by the addition of an equal volume of ice-cold methanol. After centrifugation at 20000g for 10 min, the supernatant was analyzed for 2-hydroxyterephthalate by reverse-phase HPLC as previously described.30
Molecular Modeling and Data Analysis
The 3D model of the rat liver TrxR1/NAPQI was generated using PyMOL software. The crystal structure of rat TrxR1 with a reduced C-terminal tail was obtained from the Protein Data Bank (PDI ID: 3EAN).31 DTNB reduction, cytochrome c reduction, NADPH oxidation, and the Amplex Red assay were monitored for increasing periods of time up to 30 min, and the initial velocities were analyzed using SoftMax Pro 6.3 software (Molecular Devices). IC50 values were determined by the nonlinear regression method of curve fitting using Prism 5 software (GraphPad Inc., San Diego, CA). Data were analyzed using Student’s t test. A value of p < 0.05 was considered significant.
Results
Reactive Intermediates of APAP Selectively Target TrxR in the Thioredoxin System
In initial experiments, we determined if the thioredoxin system is a target of APAP in vivo. Treatment of mice with APAP caused a time-related induction of hepatotoxicity as assessed histologically and by increases in serum transaminases (Figure 1). This was associated with marked decreases in hepatic TrxR1 and TrxR2 activities (Figure 2A and B). Whereas suppression of TrxR1 (<7% of control activity) was maximal within 1 h of APAP administration, decreases in TrxR2 (∼12% of control activity) were maximal after 6 h. By 24 h post-APAP, TrxR activities had returned to control or above control levels. In contrast, only small changes were observed in the activity of cytosolic glutathione reductase, a structurally related enzyme that is not a selenoprotein, or to Trx after APAP administration (Figure 2C and D). To determine if the inhibitory effects on TrxR were mediated by a reactive APAP metabolite, inhibition studies were performed using purified rat liver enzyme in the absence and presence of various cytochrome P450s. As shown in Figure 3, APAP only inhibited TrxR when incubation mixtures contained recombinant human CYP1A2, CYP2E1, CYP3A4, or human liver microsomes, which are all known to metabolize APAP.4 APAP alone had minimal effects of TrxR (<5% inhibition, data not shown). In the presence of the P450s, the inhibitory effects of APAP on purified TrxR were concentration- and time-dependent, and blocked by α-naphthoflavone and ketoconazole, inhibitors of CYP1A2 and CYP3A4, respectively.32 These results confirm that metabolic activation of APAP by the cytochrome P450 system is required for TrxR inhibition.
Figure 1.
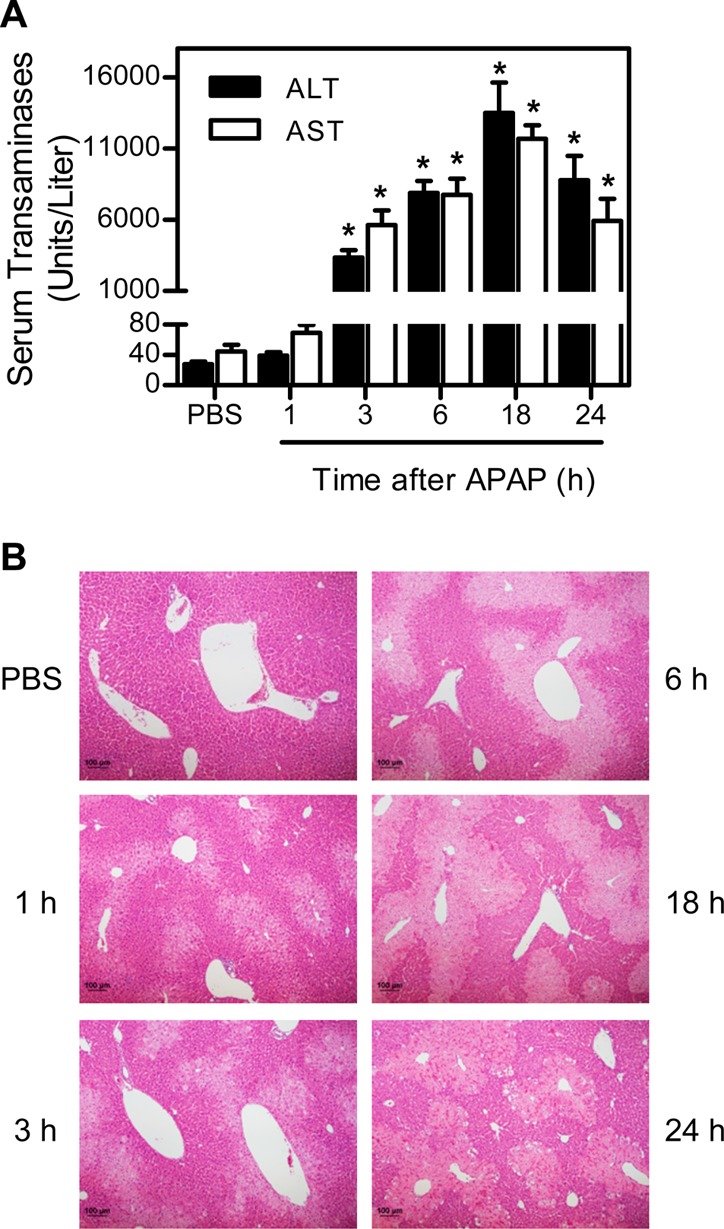
APAP-induced liver toxicity. Mice were treated i.p. with 300 mg/kg APAP or PBS control. The serum and liver were collected 1–24 h later. (A) Serum alanine transaminase (ALT) and aspartate aminotransferase (AST) activities. Data are the mean ± SE (n = 3 mice). *Significantly different (p < 0.05) from the PBS control. (B) Liver histology. Sections were stained with hematoxylin and eosin (original magnification × 100). One representative section from 3 mice/treatment group is shown.
Figure 2.

Effects of APAP on TrxR, Trx, and glutathione reductase (GR) activities in mouse liver. Subcellular fractions of the liver were prepared by differential centrifugation 1–24 h after the treatment of mice with APAP. TrxR1 (A), TrxR2 (B), and cytosolic Trx (C) activities were determined by the insulin reduction assay. Cytosolic GR activity was measured by the oxidation of NADPH using oxidized GSH as the substrate (D). Data are the mean ± SE (n = 3). *Significantly different (p < 0.05) from the PBS control.
Figure 3.

Effects of APAP and its metabolites on TrxR activity. (A) Inhibition of TrxR by APAP-reactive metabolites. Purified rat liver cytosolic TrxR (50 nM) was incubated at 37 °C with the indicated recombinant cytochrome P450s (60 pmol/mL) or human liver microsomes (HLM; 0.4 mg/mL), an NADPH-regeneration system (100 μM NADPH, 10 mM glucose-6-phosphate, and 0.5 unit/mL glucose-6-phosphate dehydrogenase), and APAP or control and 2.5 μM of α-naphthoflavone (α-NF) or ketoconazole (KTZ). After 30 min, TrxR activity was determined by the DTNB assay. Data are the mean ± SE (n = 3). *Significantly different (p < 0.05) from the PBS control. **Significantly different (p < 0.05) from the respective APAP-treated samples. (B) Time-dependent inhibition of TrxR by APAP-reactive metabolites in the presence of CYP3A4. TrxR activity in reaction mixes was assayed at the indicated times after the start of the reaction.
Redox- and Selenocysteine-Dependent Inhibition of TrxR by NAPQI
We next characterized the effects of the APAP metabolite, NAPQI, on TrxR using purified enzymes. NAPQI was found to readily inhibit purified rat liver TrxR1 in a redox-status-dependent manner (Figure 4A). Thus, the NADPH-reduced enzyme was more sensitive to NAPQI inhibition than the nonreduced enzyme (IC50 = 0.023 ± 0.001 μM vs 1.0 ± 0.070 μM; mean ± SE, n = 3). Additionally, a human mutant TrxR, in which the selenocysteine (residue 498) was replaced with cysteine, was significantly less sensitive to NAPQI (IC50 = 17 ± 2.7 μM), when compared to the wild type enzyme. We also found that inhibition of TrxR by NAPQI was time-dependent (Figure 4B), suggesting an irreversible process. To analyze this further, TrxR enzyme assays were used. We found that TrxR activity could not be recovered, even after the removal of unbound NAPQI from reaction mixtures using Chroma Spin TE-10 columns (Figure 4C). By comparison, neither oxidized nor reduced Trx was inhibited by NAPQI (Figure 4D). These data suggest that Trx is not a target for NAPQI, consistent with our in vivo findings that APAP administration had no significant effect on the activity of liver cytosolic Trx. GSH, a major intracellular scavenger of reactive metabolites including NAPQI, significantly suppressed NAPQI-mediated TrxR inactivation (Figure 4E). These results indicate that reduced TrxR is required for maximal sensitivity to NAPQI and that the selenocysteine-containing C-terminal redox center of TrxR is critical for enzyme inhibition.
Figure 4.

Effects of NAPQI on TrxR and Trx activities using purified enzyme proteins. (A) Effects of increasing concentrations of NAPQI on TrxR activity. Purified rat liver TrxR (rTrxR) or human TrxR Sec498Cys mutant (hTrxR mutant) enzyme was incubated with (reduced) or without (oxidized) NADPH (250 μM) at room temperature. After 5 min, NAPQI was added. Enzyme activity was assayed by the DTNB assay 30 min later. (B) Time course of TrxR inhibition by NAPQI. Reduced rat liver TrxR was incubated with NAPQI (5 or 20 nM) for increasing periods of time and TrxR activity analyzed. (C) Reversibility of NAPQI-induced TrxR inhibition. Reduced rat TrxR was incubated with NAPQI for 30 min at room temperature. TrxR was then purified using Chroma Spin TE-10 columns to remove free NAPQI and the remaining TrxR activity measured. (D) Effects of increasing concentrations of NAPQI on Trx activity. Recombinant human Trx1 was incubated with (reduced) or without (oxidized) DTT (10 mM) at 37 °C. After 15 min, Trx was purified using Chroma Spin TE-10 columns to remove DTT. Nonreduced or DTT-reduced Trx was then incubated with NAPQI at room temperature for 30 min. Trx activity was determined by the insulin reduction assay. (E) Effects of GSH on TrxR inhibition by NAPQI. Reduced rTrxR was treated with NAPQI in the presence or absence of GSH (1 mM) at room temperature for 30 min. Enzyme activity was determined by the insulin reduction assay. Data are expressed as the mean ± SE (n = 3). Note that experiments shown in panels A and B were the result of two different experiments done at different times.
We next examined the effects of NAPQI on free thiol groups in TrxR using a BIAM labeling technique. Previous studies have shown that BIAM alkylates thiol groups on proteins in a pH-dependent manner. Whereas at pH 6.5, BIAM preferentially binds the selenocysteine residue in TrxR, at pH 8.5 it binds both cysteine and selenocysteine residues in the enzyme.33 NAPQI treatment had minimal effects on BIAM labeling in nonreduced TrxR at both pH 6.5 and 8.5 (Figure 5). This suggests that catalytic residues in oxidized TrxR are not a target for the modification by NAPQI. Conversely, NAPQI caused a concentration-dependent decrease in BIAM labeling at both pH values in NADPH-treated TrxR, in which catalytic selenocysteine and cysteine residues in the redox centers of the enzyme were reduced (Figure 5). These results indicate that NAPQI selectively alkylates the redox centers in the reduced protein, a physiological active form of TrxR, resulting in enzyme inhibition.
Figure 5.

Effects of NAPQI on the labeling of TrxR by BIAM. Reduced (+ NADPH) and nonreduced (− NADPH) TrxR was treated with increasing concentrations of NAPQI or vehicle control. After 30 min, NAPQI-alkylated TrxR was purified using Chroma Spin TE-10 columns to remove free NAPQI and then labeled with BIAM at pH 6.5 or 8.5. Samples were analyzed by Western blotting 30 min after labeling. The extent of BIAM labeling on TrxR was evaluated by the specific binding of streptavidin-HRP followed by ECL detection. (A) Representative Western blots showing TrxR probed with streptavidin-HRP or antibody to TrxR. (B) Densitometric analysis of the labeling efficiency of TrxR by BIAM. The extent of BIAM labeling on TrxR was determined by the relative intensities of the streptavidin band to the TrxR band. Data are expressed as the mean ± SE (n = 3).
NAPQI Alkylated Catalytic Residues in the Redox Centers of TrxR
NAPQI alkylation sites in TrxR were next investigated by LC-MS/MS analysis of tryptic peptides prepared from the modified enzyme. NAPQI is known to preferentially react with nucleophilic sites in proteins resulting in the formation of APAP adducts; this process can lead to an increase in mass of 149.15 Da per molecule of NAPQI added to a modified peptide. Additionally, unmodified cysteine or selenocysteine residues on the protein are alkylated by iodoacetamide (addition of carbamidomethyl groups; mass increase of 57.02 Da), which is introduced during the in-gel digestion process to protect free thiol/selenol groups on the protein. Table 1 summarizes the observed NAPQI-modified peptides and their sequence assignments (isotopic spectra and tandem mass spectra of adducted peptides are shown in Figure 6 and in the Supporting Information Figures S1–S3). These adducted peptides were detected in NAPQI-treated samples but not in TrxR control samples. NAPQI was found to predominantly modify cysteine 59, cysteine 497, and selenocysteine 498 residues in redox centers containing trypic peptides, WGLGGTCVNVGCIPK and (R)SGGDILQSGCUG peptides.
Table 1. Modified Peptides in the Tryptic Digest of NAPQI-Treated TrxR.
| peptide sequence | position | m/z | charge | tR (min) | modificationa |
|---|---|---|---|---|---|
| WGLGGTCVNVGCIPK | 53–67 | 855.41 | 2 | 36.77 | C59-NAPQI |
| C64-CAMb | |||||
| SGGDILQSGCUG | 488–499 | 675.73 | 2 | 32.70 | C497-CAM |
| U498-NAPQI | |||||
| SGGDILQSGCUG | 488–499 | 722.25 | 2 | 34.80 | C497-NAPQI |
| U498-NAPQI | |||||
| RSGGDILQSGCUG | 487–499 | 753.78 | 2 | 31.10 | C497-NAPQI |
| U498-CAM | |||||
| RSGGDILQSGCUG | 487–499 | 799.79 | 2 | 33.22 | C497-NAPQI |
| U498-NAPQI |
Based on MS/MS results.
S-Carbamidomethylation (CAM) on cysteine results from the process of in-gel digestion using iodoacetamide to protect unreacted thiol/selenol groups.
Figure 6.

LC MS/MS analysis of NAPQI-adducted TrxR tryptic peptide. Reduced-rat liver TrxR (1 μM) was incubated with NAPQI (100 μM) at room temperature. After 1 h, the NAPQI-modified protein was purified by SDS–PAGE. The purified protein was reduced with DDT, reacted with iodoacetamide, and digested with trypsin in the gel. The resulting peptides were analyzed using LC-MS/MS. (A) MS/MS spectrum of CID product ions from the NAPQI-modified tryptic ion at m/z 721.62. This ion was identified as a doubly charged ion with one NAPQI adducted to the SGGDILQSGCUG peptide (residue 488–499) on cysteine 497 and one on selenocysteine 498. Matched b and y fragments are marked. Insets show the isotopic spectrum and sequence assignment of this doubly charged ion. (B) MS/MS spectrum of CID product ions from the NAPQI-modified tryptic ion at m/z 855.41. This adducted peptide was identified as a doubly charged ion of WGLGGTCVNVGCIPK (residues 53–67 in TrxR) with one NAPQI adducted to cysteine 59 and one carbamidomethylation on cysteine 64.
Examples of NAPQI-adducted peptides containing the N- and C-terminal redox motifs in TrxR are shown in Figure 6. An ion at m/z 721.62 displayed a distinctive isotope pattern of a selenium-containing peptide, suggesting that it is either a SGGDILQSGCUG or RSGGDILQSGCUG peptide. The isotopic spectrum revealed that it is a doubly charged ion (indicated by the mass difference of 0.5 Da between neighboring peaks) and corresponds to the [M + H]+ value of 1442.23. The mass of this parent ion is in agreement with the protonated SGGDILQSGCUG peptide (residue 488–499; [M + H]+ 1143.10 Da) plus 299.13 Da, corresponding to the mass of two NAPQI molecules (Figure 6A). The tandem mass spectrum confirmed that one NAPQI alkylated cysteine 497 and the other selenocysteine 498. This is supported by a series of normal b ions (b4–b9) and modified b10 ion with mass increases of 148.83 Da, indicating that cysteine 497 is modified by one NAPQI molecule. A mass increase of 150.02 Da on the y2 ion (376.02 Da) was also observed in this modified peptide when compared to the theoretical mass of the respective ion (226.05 Da) on the unmodified peptide, suggesting that selenocysteine 498 is modified by one NAPQI. We also found that fragment y5 through y8 ions increased by a mass of 299.09 Da, providing further evidence for the addition of two NAPQIs on these ions. Figure 6B shows an example of NAPQI modified to the N-terminal redox center of TrxR. This adducted peptide, a doubly charged ion at m/z 855.41 ([M + H]+ 1709.82 Da), corresponds to a mass addition of 206.08 Da to the protonated WGLGGTCVNVGCIPK peptide (residue 53–67; [M + H]+ 1503.74 Da). This mass increase is consistent with unmodified peptide plus one carbamidomethylation modification and one NAPQI modification. MS/MS sepctrum revealed a series of modified y ions (y4–y8) with a mass increase of 56.98 Da, suggesting that cysteine 64 is alkylated by an iodoacetamide. Moreover, fragment ions y9 through y14 increased by a mass of 205.94 Da, indicating the addition of one carbamidomethyl group and one NAPQI on these ions. These data also indicate that cysteine 59 is adducted by one molecule of NAPQI. These findings suggest that inhibition of TrxR by NAPQI resulted from specific alkylation on the catalytic cysteine and/or selenocysteine residues in the redox centers of the enzyme.
Effects of NAPQI on Chemical Redox Cycling by TrxR
In addition to reducing protein disulfides, TrxR catalyzes chemical redox cycling in an NADPH-dependent reaction, a process that generates ROS including superoxide anion, H2O2, and, in the presence of redox active metals, highly toxic hydroxyl radicals. We next determined if NAPQI altered the ability of TrxR to mediate chemical redox cycling. Using menadione as the redox-cycling chemical, TrxR was found to readily generate superoxide anion, H2O2, and hydroxyl radicals (Figure 7A–C). Consistent with the requirement for NADPH in redox-cycling reactions, an increase in NADPH utilization was evident during ROS formation. NAPQI and APAP by themselves did not redox cycle (Figure 7B,C and data not shown). The accumulation of superoxide anion and H2O2 in the redox-cycling assays was inhibited by superoxide dismutase and catalase, respectively (Figure 7B and data not shown). Hydroxyl radical production was inhibited by catalase, which degrades H2O2 formed in the assays, and DMSO, a hydroxyl radical trap (Figure 7C).
Figure 7.

Effects of NAPQI on TrxR-mediated menadione redox cycling. (A) Effects of NAPQI on menadione-generated superoxide anion, H2O2, hydroxyl radicals, and NADPH oxidation by purified rat liver TrxR. TrxR1 was incubated with the indicated concentrations of NAPQI or vehicle control in the presence or absence of menadione (MD, 100 μM) at 37 °C. Superoxide anion was measured by the reduction of acetylated cytochrome c and H2O2 production by the Amplex Red assay. Hydroxyl radicals were measured by the terephthalate assay in the presence of Fe3+/EDTA, and the product of the reaction, 2-hydroxyterephthalate (2-OH TPT), was quantitated by HPLC. NADPH oxidation was monitored by decreases in absorbance at 340 nm. Assays were run for increasing periods of time up to 30 min and the rate of ROS formation and NADPH oxidation calculated. Results are presented as the percentage of the rate observed in the absence of NAPQI. Data are the mean ± SE of triplicate measurements. *Significantly different (p < 0.05) from menadione-treated samples in the absence of NAPQI. (B) Time-dependent generation of H2O2 by purified rat liver TrxR. TrxR was incubated with menadione (100 μM), menadione plus NAPQI (0.1, 1, 10 μM), or NAPQI alone (10 μM). Inset shows TrxR incubated with 100 μM menadione or menadione plus 2500 units/mL calatase. Data are the mean ± SE (n = 3). (C) Representative HPLC chromatograms of menadione-generated hydroxyl radicals by purified rat liver TrxR, in the presence or absence of NAPQI. In some experiments, the enzyme was treated with NAPQI alone; DMSO was added as a hydroxyl radical trap, and catalase was used to inhibit H2O2 accumulation in the assays. (D) Effects of NAPQI on menadione-generated H2O2 by TrxR. Purified rat liver TrxR or human TrxR Sec498Cys mutant enzyme was incubated with menadione (100 μM) and increasing concentrations of NAPQI. H2O2 production was quantified by the Amplex Red assay. Results are presented as the percentage of the rate observed in the absence of NAPQI. Data are the mean ± SE (n = 3).
Menadione redox cycling by purified rat liver TrxR was found to be markedly less sensitive to NAPQI when compared to the disulfide reduction (IC50 = 0.980 ± 0.063 vs 0.023 ± 0.001 μM for the generation of H2O2 production during redox cycling and disulfide reduction, respectively) (compare Figures 4A and 7D). Differences in sensitivity of TrxR redox cycling were evident not only in the generation of H2O2 but also in superoxide anion and hydroxyl radicals. Thus, treatment with a concentration of NAPQI near the IC50 for disulfide reduction (20 nM) had no effect on the generation of these ROS. However, 100 μM NAPQI almost completely blocked the formation of the superoxide anion, H2O2 and hydroxyl radicals (Figure 7). Similarly, redox-cycling-mediated oxidation of NADPH was inhibited by 100 μM NAPQI but not 20 nM NAPQI. These data suggest that disulfide reduction and quinone redox cycling by TrxR occur by distinct mechanisms. The effects of selenocysteine on TrxR-mediated chemical redox cycling were also examined using the human TrxR Sec498Cys mutant enzyme. NAPQI caused a concentration-dependent inhibition of menadione-stimulated H2O2 production by this enzyme that was generally similar to that in the rat enzyme (IC50 = 1.2 ± 0.05 μM; Figure 7D).
Discussion
The present study demonstrates that APAP intoxication is associated with a rapid inhibition of both cytosolic TrxR1 and mitochondrial TrxR2 in the liver. Moreover, this is mediated by APAP-reactive metabolites. This is supported by our findings that the inhibition of purified TrxR1 was dependent on recombinant cytochrome P450s or P450s in human liver microsomes and that it was blocked by P450 inhibitors. Purified TrxR1 was also directly inhibited by the APAP metabolite NAPQI in a time-dependent and irreversible manner. These findings are in agreement with previous reports demonstrating that lethal doses of APAP (1000 mg/kg, 4 h) caused an inhibition of hepatic TrxR1 in wild type mice;34 in these studies, NAPQI was also found to inhibit recombinant TrxR1, although at significantly higher concentrations (200–1000 μM) than those that we observed. We also found that pretreatment of TrxR with NAPQI prevented the binding of iodoacetamide, a potent thiol and selenol alkylating agent, indicating covalent binding of NAPQI at specific target amino acid residues in TrxR. This is supported by LC-MS/MS analysis showing selective alkylation of cysteine 59, cysteine 497, and selenocysteine 498 by NAPQI in the N- and C-terminal redox centers of TrxR. These data are consistent with earlier reports showing that NAPQI, a soft electrophile, predominantly reacts with cysteine residues in liver proteins.5 Thus, it appears that by alkylating catalytic residues and taking the place of a disulfide and/or an essential selenenylsulfide structural element (see Figure 8 for the model of NAPQI binding to TrxR1), NAPQI inactivates TrxR. This process interferes with the formation of dithiol intermediates, resulting in the inhibition of catalytic reactions. It should be noted that cysteine 64 in TrxR, which is in proximity to cysteine 59 in the N-terminal redox center, is not alkylated by NAPQI. This is likely due to the fact that cysteine 64 is less solvent accessible than cysteine 59 as previously shown in the analysis of the crystal structure of TrxR.31,35 Both cysteine 59 and cysteine 497 are located in solvent-accessible regions of TrxR. Crystal structure analysis of TrxR also revealed the presence of several solvent-accessible noncatalytic cysteine residues including cysteine 177, 189, 382, and 383; however, none of these was modified by NAPQI. The reasons for selective modification of cysteine 59 and 497 by NAPQI are not known. TrxR forms dimers, tetramers, and oligomers under native conditions, and these protein–protein interactions may limit accessibility to other cysteine residues.36,37
Figure 8.
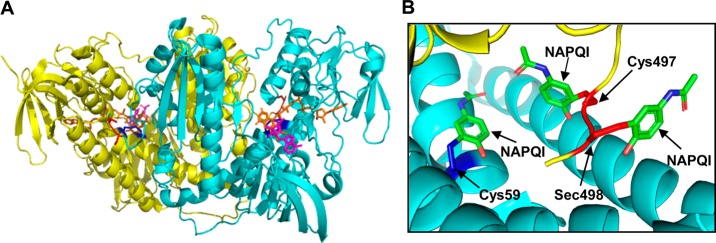
Molecular models for TrxR/NAPQI adducts based on the crystal structure of rat TrxR1. (A) Crystal structure of the rat TrxR1 dimer (PDB ID: 3EAN) with a reduced C-terminal redox center. One subunit is shown in yellow and the other in light blue. The N-terminal redox center (cysteine 59 and cysteine 64) is shown in the oxidized form in dark blue. The C-terminal redox center (cysteine 497 and selenocysteine 498) is shown in the reduced form in red. FAD is shown in orange and NADP+ in magenta. (B) Model of TrxR complexed with NAPQI. NAPQI modified to the cysteine 59, cysteine 497, and selenocysteine 498 in the redox centers of the enzyme, based on LC-MS/MS analysis.
The present study also shows that, in contrast to TrxR, only low level inhibition of Trx and glutathione reductase is noted after APAP administration to mice. NAPQI also had minimal effects on the activity of purified Trx in vitro. Glutathione reductase shares similar structural domains and catalytic mechanisms with TrxR but lacks the selenocysteine-containing C-terminal redox center.38 Both Trx and glutathione reductase contain cysteine residues in their redox centers, which are known to be targets for covalent modification by several electrophiles; it is possible that low levels of NAPQI bind to these proteins resulting in only limited inhibition of their activities.24,39−41 The ability of NAPQI to inhibit TrxR may be due to unique properties of the enzyme. In addition to having three cysteine residues in its redox centers, TrxR also contains a solvent accessible selenocysteine in the C-terminal redox center. This amino acid differs significantly in its pKa and nucleophilicity from cysteine (pKa (Sec) = 5.24–5.63; pKa (Cys) = 8.25);42,43 nucleophilic reactions of selenocysteine derivatives are approximately 2 to 3 orders of magnitude more rapid than those of cysteine derivatives.44 Moreover, selenoproteins typically display 100–1000-fold higher catalytic efficiency than Sec-Cys mutant enzymes.42,43 These data are consistent with the idea that selenocysteine in TrxR is a preferential target for covalent modification by NAPQI and that this results in inhibition of disulfide reduction by the enzyme.
Using purified rat liver TrxR1, we found that NAPQI was a more effective inhibitor when the enzyme was reduced by NADPH, which is similar to the activity of other TrxR inhibitors including, auranofin, curcumin, 2-chloroethyl ethyl sulfide, and mechlorethamine.27,29,45−47 NADPH is known to act on TrxR by reducing disulfide and selenenylsulfide in the redox centers of the enzyme to highly reactive dithiols and selenolthiol, two reaction centers required for enzyme activity.48 Conformational changes in the enzyme are triggered by the reduction of these thiols by NADPH. Crystallographic studies of rat TrxR1 have shown that reduction of selenenylsulfide exposes the selenocysteine residue to the enzyme surface, a process that facilitates substrate binding; this also exposes a reactive target for various electrophiles.31 Our finding that NAPQI is more efficient in inhibiting TrxR1 which contains selenocysteine than a mutant enzyme without selenocysteine (human Sec498Cys mutant TrxR) provides additional evidence for the idea that selenol in TrxR is a target for NAPQI. This is also supported by our iodoacetamide-binding experiments showing that NAPQI is effective in blocking the alkylation of TrxR by BIAM, which predominately reacts with selenolcysteine at acidic pH, and our LC-MS/MS analysis, which revealed the presence of NAPQI-selenocysteine adducts in the enzyme C-terminal redox center. Analogous findings of alkylation of TrxR on selenocysteine have been described for electrophiles such as 4-hydroxynonenal,33 curcumin,45 arsenic trioxide,49 2-chloroethyl ethyl sulfide,27 mechlorethamine,29 and nitroaromatic compounds,50 presumably due to the low pKa and high nucleophilicity of selenol and its location in the solvent-accessible C-terminal region of the enzyme.
NAPQI-induced inhibition of TrxR1 and TrxR2 can lead to a decrease in levels of reduced Trx in both cytosolic and mitochondrial compartments of cells. This would shut down the ability of the Trx system to scavenge ROS and TrxR/Trx-mediated dithiol/disulfide exchange reactions, processes regulating the activities of proteins important in antioxidant defense and cell growth control.22 Earlier studies have shown that inhibition of TrxR leads to an accumulation of oxidized Trx in cells, which can disrupt antioxidant balance, promoting oxidative stress. The mitochondrial Trx system is thought to play an essential role in detoxifying H2O2 in a substrate- and respiration-dependent manner.51,52 Inhibition of mitochondrial TrxR2 has been shown to initiate mitochondrial dysfunction, increase steady-state cellular levels of H2O2, and cause cell death.52,53 Our findings that APAP-reactive metabolites target TrxR in mitochondria are in accord with earlier reports that APAP, as well as NAPQI, can disrupt mitochondrial function; inhibition of mitochondrial TrxR may be an important mechanism of APAP-induced oxidative stress and cellular injury in hepatocytes.12,54,55 Reduced Trx is also known to bind to and inhibit apoptosis-signal-regulating kinase 1 (ASK1). Following the dissociation of oxidized Trx from ASK1, c-Jun N-terminal kinase (JNK) is activated resulting in cell death by either apoptosis or necrosis.22 It is possible that APAP causes Trx oxidation which subsequently activates ASK1/JNK signaling. The ASK1/JNK pathway has been reported to be activated following APAP overdose and to contribute to toxicity.56,57 Previous studies have also demonstrated that modification of TrxR on its Sec residue by electrophiles can induce SecTRAPs (selenium-compromised thioredoxin reductase-derived apoptotic proteins).58 These proteins can regulate both apoptosis and necrosis leading to rapid cell death.58 Modification of the Sec residue on TrxR by NAPQI and the generation of SecTRAPs may also contribute to APAP hepatotoxicity.
Of interest was our finding that following administration of APAP to mice, inhibition of TrxR2 was significantly slower than that of TrxR1. Thus, while the activity of hepatic TrxR1 was maximally inhibited (>90%) within 1 h of APAP treatment, maximum inhibition (>85%) of TrxR2 was evident at 6 h post-APAP. This may be the result of a differential distribution of NAPQI in the cytosol and mitochondria of hepatocytes and/or unique biochemical characteristics of the different TrxR isoforms. In this regard, earlier reports demonstrated that TrxR1 and TrxR2 are structurally distinct and have different substrate and inhibitor specificities.29,31,35,59−63 Analysis of the crystal structures of TrxR has revealed that the positioning of key residues around the redox centers of TrxR1 and TrxR2 are distinct.35,59,60 For example, a unique interaction via a salt bridge between the N- and C-terminal redox centers of the TrxR subunits is formed by amino acid residues lysine 56 and histidine 143 from one subunit and glutamate 503 and lysine 506 from the other subunit in mitochondrial TrxR2 but not in cytosolic TrxR1.59 Additionally, a guiding bar, amino acids 407–422, has been identified in cytosolic TrxR1 but not in mitochondrial TrxR2.60 These structural differences may contribute to their different inhibitor and substrate specificities, as well as their sensitivities to NAPQI. Also of interest were our findings that the activities of both TrxR1 and TrxR2 recovered to control levels at 24 h post-APAP treatment. Since inhibition of these enzymes by NAPQI in not reversible, it is likely that recovery is due to new synthesis of the enzyme during tissue repair. Modified TrxR is presumably eliminated by degradation of the enzyme.
In addition to functioning as a disulfide reductase, TrxR is also a prooxidant and mediates chemical redox cycling. Thus, the enzyme catalyzes the NADPH-dependent one-electron reduction of redox active chemicals including nitroaromatic compounds, quinones, and bipyridyl herbicides.28,50,64 Under aerobic conditions, radical ions formed from these chemicals rapidly react with oxygen, generating the superoxide anion and regenerating the parent compounds.64,65 Enzyme-mediated and spontaneous dismutation of superoxide anion leads to the production of H2O2. Highly toxic hydroxyl radicals are formed from H2O2 in the presence of trace metals.66 These ROS can damage cells and tissues resulting in toxicity.67 The present studies demonstrate that NAPQI can inhibit not only TrxR-mediated disulfide reduction but also quinone redox cycling. However, disulfide reduction is markedly more sensitive to inhibition by NAPQI. These findings are consistent with previous reports of selective inhibition of TrxR-mediated disulfide reduction over redox cycling by quinones,28 dinitrohalobenzenes,50 nobel metal compounds including cisplatin and auranofin,68 and mechlorethamine.29 Findings that NAPQI and these other chemicals have differential effects on TrxR-mediated disulfide reduction and redox cycling indicate that the enzyme mediates these reactions via distinct mechanisms. This is supported by docking studies showing that menadione, which redox cycles with TrxR, associates with hTrxR by interacting with glutamine 72 on both subunits of the dimer, a site distant from NAPQI alkylation sites on the enzyme identified by our LC-MS/MS analysis.69 Of note are our findings that a human TrxR Sec498Cys mutant enzyme, which displays low levels of disulfide reductase activity, continued to redox cycle in a process that was inhibited by NAPQI but only at very high concentrations (1–100 μM). These data further support the idea that the selenocysteine residue in the C-terminal redox center of TrxR is not required for quinone redox cycling. This is in agreement with findings that the prooxidant NADPH oxidase activity of TrxR is selenocysteine-independent.70
Since TrxR is a target for NAPQI, it might be expected that mice deficient in TrxR would show increased sensitivity to APAP. However, during the preparation of this article, two studies were published demonstrating that hepatocyte-specific TrxR1-null mice were resistant to acute APAP intoxication, a response which appeared to be due to compensatory increases in enzymes mediating APAP metabolism, glutathione biosynthesis, and other antioxidants and enhanced extracellular transport of conjugated xenobiotics.34,72 Many of the genes mediating the expression of the enzyme involved in APAP metabolism and detoxification of NAPQI are controlled by the transcription factor nuclear factor, erythroid 2-related factor 2 (Nrf2), whose activity was enhanced in the livers of TrxR-null mice.71,72 Taken together, these data suggest that resistance to APAP in the livers of TrxR1-null mice is multifactorial and independent of the levels of expression of TrxR.
In summary, our studies suggest a novel mechanistic basis for APAP-induced oxidative stress and hepatotoxicity, which involves NAPQI-mediated inhibition of reduced TrxR. This inhibition appears to be due to the modification of catalytic residues in both N- and C-terminal redox centers of TrxR. As the blocking of catalytic sites on TrxR prevents substrate reduction, this can disrupt antioxidant balance and other metabolic processes that require reduced Trx. Modified TrxR can also result in the formation of SecTRAPs which contribute to cytotoxicity. We speculate that inhibition of hepatic TrxR plays an important role in mediating APAP-induced liver injury. Strategies aimed at preventing or reversing damage to TrxR or upregulating its synthesis may be beneficial in ameliorating APAP toxicity.73,74
Glossary
Abbreviations
- APAP
acetaminophen
- NAPQI
N-acetyl-p-benzoquinone imine
- TrxR
thioredoxin reductase
- Sec
selenocysteine
- CYP
cytochrome P450
- GSH
glutathione
- SOD
superoxide dismutase
- GPx
glutathione peroxidase
- Trx
thioredoxin
- ROS
reactive oxygen species
- GR
glutathione reductase
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- HLM
human liver microsomes
- BIAM
N-(biotinoyl)-N′-(iodoacetyl) ethylenediamine
- HRP
horseradish peroxidase
- ECL
chemiluminescence
- ASK1
apoptosis signaling-regulating kinase 1
- JNK
c-Jun N-terminal kinase
- SecTRAPs
selenium-compromised thioredoxin reductase-derived apoptotic proteins
- Nrf2
nuclear factor erythroid 2-related factor 2
Supporting Information Available
MS/MS spectra and sequence assignments of NAPQI-adduced peptides at m/z 675.73, 753.78, and 799.79. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported in part by National Institutes of Health grants ES005022 (to J.D.L. and D.L.L.), ES004738 (to D.L.L. and J.D.L.), CA132624 (to D.L.L. and J.D.L.), AR055073 (to J.D.L., D.L.L., and D.E.H.), and GM034310 (to D.L.L. and J.D.L.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- James L. P.; Mayeux P. R.; Hinson J. A. (2003) Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 31, 1499–1506. [DOI] [PubMed] [Google Scholar]
- Hinson J. A.; Roberts D. W.; James L. P. (2010) Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 196, 369–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin D. C.; Miwa G. T.; Lu A. Y.; Nelson S. D. (1984) N-acetyl-p-benzoquinone imine: A cytochrome P-450-mediated oxidation product of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 81, 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine J. E.; Auriola S.; Pasanen M.; Juvonen R. O. (2009) Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica 39, 11–21. [DOI] [PubMed] [Google Scholar]
- Streeter A. J.; Dahlin D. C.; Nelson S. D.; Baillie T. A. (1984) The covalent binding of acetaminophen to protein. Evidence for cysteine residues as major sites of arylation in vitro. Chem.-Biol. Interact. 48, 349–366. [DOI] [PubMed] [Google Scholar]
- Hart S. G.; Cartun R. W.; Wyand D. S.; Khairallah E. A.; Cohen S. D. (1995) Immunohistochemical localization of acetaminophen in target tissues of the CD-1 mouse: correspondence of covalent binding with toxicity. Fundam. Appl. Toxicol. 24, 260–274. [DOI] [PubMed] [Google Scholar]
- James L. P.; Letzig L.; Simpson P. M.; Capparelli E.; Roberts D. W.; Hinson J. A.; Davern T. J.; Lee W. M. (2009) Pharmacokinetics of acetaminophen-protein adducts in adults with acetaminophen overdose and acute liver failure. Drug Metab. Dispos. 37, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C. L.; Kosyk O.; Ross P. K.; Schoonhoven R.; Boysen G.; Swenberg J. A.; Heinloth A. N.; Boorman G. A.; Cunningham M. L.; Paules R. S.; Rusyn I. (2006) Phenotypic anchoring of acetaminophen-induced oxidative stress with gene expression profiles in rat liver. Toxicol. Sci. 93, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa N.; Shinohara M.; Saberi B.; Gaarde W. A.; Han D.; Kaplowitz N. (2008) Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 283, 13565–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner C. R.; Heck D. E.; Yang C. S.; Thomas P. E.; Zhang X. J.; DeGeorge G. L.; Laskin J. D.; Laskin D. L. (1998) Role of nitric oxide in acetaminophen-induced hepatotoxicity in the rat. Hepatology 27, 748–754. [DOI] [PubMed] [Google Scholar]
- Ito Y.; Abril E. R.; Bethea N. W.; McCuskey R. S. (2004) Role of nitric oxide in hepatic microvascular injury elicited by acetaminophen in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G60–67. [DOI] [PubMed] [Google Scholar]
- Han D.; Shinohara M.; Ybanez M. D.; Saberi B.; Kaplowitz N. (2010) Signal transduction pathways involved in drug-induced liver injury. Handb. Exp. Pharmacol. 13, 267–310. [DOI] [PubMed] [Google Scholar]
- Fujimoto K.; Kumagai K.; Ito K.; Arakawa S.; Ando Y.; Oda S.; Yamoto T.; Manabe S. (2009) Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. Toxicol. Pathol. 37, 193–200. [DOI] [PubMed] [Google Scholar]
- Yoshikawa Y.; Morita M.; Hosomi H.; Tsuneyama K.; Fukami T.; Nakajima M.; Yokoi T. (2009) Knockdown of superoxide dismutase 2 enhances acetaminophen-induced hepatotoxicity in rat. Toxicology 264, 89–95. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Sagisaka H.; Arakawa S.; Shibaya Y.; Watanabe M.; Igarashi I.; Tanaka K.; Totsuka S.; Takasaki W.; Manabe S. (2003) A novel model of continuous depletion of glutathione in mice treated with L-buthionine (S,R)-sulfoximine. J. Toxicol. Sci. 28, 455–469. [DOI] [PubMed] [Google Scholar]
- Mirochnitchenko O.; Weisbrot-Lefkowitz M.; Reuhl K.; Chen L.; Yang C.; Inouye M. (1999) Acetaminophen toxicity. Opposite effects of two forms of glutathione peroxidase. J. Biol. Chem. 274, 10349–10355. [DOI] [PubMed] [Google Scholar]
- Botta D.; Shi S.; White C. C.; Dabrowski M. J.; Keener C. L.; Srinouanprachanh S. L.; Farin F. M.; Ware C. B.; Ladiges W. C.; Pierce R. H.; Fausto N.; Kavanagh T. J. (2006) Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J. Biol. Chem. 281, 28865–28875. [DOI] [PubMed] [Google Scholar]
- Prescott L. F.; Park J.; Ballantyne A.; Adriaenssens P.; Proudfoot A. T. (1977) Treatment of paracetamol (acetaminophen) poisoning with N-acetylcysteine. Lancet 2, 432–434. [DOI] [PubMed] [Google Scholar]
- Corcoran G. B.; Wong B. K. (1986) Role of glutathione in prevention of acetaminophen-induced hepatotoxicity by N-acetyl-L-cysteine in vivo: studies with N-acetyl-D-cysteine in mice. J. Pharmacol. Exp. Ther. 238, 54–61. [PubMed] [Google Scholar]
- Nakae D.; Yamamoto K.; Yoshiji H.; Kinugasa T.; Maruyama H.; Farber J. L.; Konishi Y. (1990) Liposome-encapsulated superoxide dismutase prevents liver necrosis induced by acetaminophen. Am. J. Pathol. 136, 787–795. [PMC free article] [PubMed] [Google Scholar]
- Gromer S.; Urig S.; Becker K. (2004) The thioredoxin system--from science to clinic. Med. Res. Rev. 24, 40–89. [DOI] [PubMed] [Google Scholar]
- Lu J.; Holmgren A. (2014) The thioredoxin antioxidant system. Free Radical Biol. Med. 66, 75–87. [DOI] [PubMed] [Google Scholar]
- Arner E. S. (2009) Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim. Biophys. Acta 1790, 495–526. [DOI] [PubMed] [Google Scholar]
- Tonissen K. F.; Di Trapani G. (2009) Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 53, 87–103. [DOI] [PubMed] [Google Scholar]
- Cai W.; Zhang L.; Song Y.; Wang B.; Zhang B.; Cui X.; Hu G.; Liu Y.; Wu J.; Fang J. (2012) Small molecule inhibitors of mammalian thioredoxin reductase. Free Radical Biol. Med. 52, 257–265. [DOI] [PubMed] [Google Scholar]
- Luthman M.; Holmgren A. (1982) Rat liver thioredoxin and thioredoxin reductase: Purification and characterization. Biochemistry 21, 6628–6633. [DOI] [PubMed] [Google Scholar]
- Jan Y. H.; Heck D. E.; Gray J. P.; Zheng H.; Casillas R. P.; Laskin D. L.; Laskin J. D. (2010) Selective targeting of selenocysteine in thioredoxin reductase by the half mustard 2-chloroethyl ethyl sulfide in lung epithelial cells. Chem. Res. Toxicol. 23, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenas N.; Nivinskas H.; Anusevicius Z.; Sarlauskas J.; Lederer F.; Arner E. S. (2004) Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein. J. Biol. Chem. 279, 2583–2592. [DOI] [PubMed] [Google Scholar]
- Jan Y. H.; Heck D. E.; Malaviya R.; Casillas R. P.; Laskin D. L.; Laskin J. D. (2014) Cross-linking of thioredoxin reductase by the sulfur mustard analogue mechlorethamine (methylbis(2-chloroethyl)amine) in human lung epithelial cells and rat lung: Selective inhibition of disulfide reduction but not redox cycling. Chem. Res. Toxicol. 27, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishin V. M.; Thomas P. E. (2004) Characterization of hydroxyl radical formation by microsomal enzymes using a water-soluble trap, terephthalate. Biochem. Pharmacol. 68, 747–752. [DOI] [PubMed] [Google Scholar]
- Cheng Q.; Sandalova T.; Lindqvist Y.; Arner E. S. (2009) Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 284, 3998–4008. [DOI] [PubMed] [Google Scholar]
- Khojasteh S. C.; Prabhu S.; Kenny J. R.; Halladay J. S.; Lu A. Y. (2011) Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: A re-evaluation of P450 isoform selectivity. Eur. J. Drug Metab. Pharmacokinetics 36, 1–16. [DOI] [PubMed] [Google Scholar]
- Fang J.; Holmgren A. (2006) Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J. Am. Chem. Soc. 128, 1879–1885. [DOI] [PubMed] [Google Scholar]
- Iverson S. V.; Eriksson S.; Xu J.; Prigge J. R.; Talago E. A.; Meade T. A.; Meade E. S.; Capecchi M. R.; Arner E. S.; Schmidt E. E. (2013) A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radical Biol. Med. 63, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandalova T.; Zhong L.; Lindqvist Y.; Holmgren A.; Schneider G. (2001) Three-dimensional structure of a mammalian thioredoxin reductase: Implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc. Natl. Acad. Sci. U.S.A. 98, 9533–9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev V. N.; Jeang K. T.; Stadtman T. C. (1996) Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. U.S.A. 93, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengby O.; Cheng Q.; Vahter M.; Jornvall H.; Arner E. S. (2009) Highly active dimeric and low-activity tetrameric forms of selenium-containing rat thioredoxin reductase 1. Free Radical Biol. Med. 46, 893–904. [DOI] [PubMed] [Google Scholar]
- Nordman T.; Xia L.; Bjorkhem-Bergman L.; Damdimopoulos A.; Nalvarte I.; Arner E. S.; Spyrou G.; Eriksson L. C.; Bjornstedt M.; Olsson J. M. (2003) Regeneration of the antioxidant ubiquinol by lipoamide dehydrogenase, thioredoxin reductase and glutathione reductase. Biofactors 18, 45–50. [DOI] [PubMed] [Google Scholar]
- Becker K.; Gui M.; Schirmer R. H. (1995) Inhibition of human glutathione reductase by S-nitrosoglutathione. Eur. J. Biochem. 234, 472–478. [DOI] [PubMed] [Google Scholar]
- Bauer H.; Fritz-Wolf K.; Winzer A.; Kuhner S.; Little S.; Yardley V.; Vezin H.; Palfey B.; Schirmer R. H.; Davioud-Charvet E. (2006) A fluoro analogue of the menadione derivative 6-[2′-(3′-methyl)-1′,4′-naphthoquinolyl]hexanoic acid is a suicide substrate of glutathione reductase. Crystal structure of the alkylated human enzyme. J. Am. Chem. Soc. 128, 10784–10794. [DOI] [PubMed] [Google Scholar]
- Picaud T.; Desbois A. (2006) Interaction of glutathione reductase with heavy metal: the binding of Hg(II) or Cd(II) to the reduced enzyme affects both the redox dithiol pair and the flavin. Biochemistry 45, 15829–15837. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; Alewood P. F. (2008) Selenopeptide chemistry. J. Pept. Sci. 14, 1223–1239. [DOI] [PubMed] [Google Scholar]
- Arner E. S. (2010) Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine?. Exp. Cell Res. 316, 1296–1303. [DOI] [PubMed] [Google Scholar]
- Nauser T.; Steinmann D.; Koppenol W. H. (2012) Why do proteins use selenocysteine instead of cysteine?. Amino Acids 42, 39–44. [DOI] [PubMed] [Google Scholar]
- Fang J.; Lu J.; Holmgren A. (2005) Thioredoxin reductase is irreversibly modified by curcumin: A novel molecular mechanism for its anticancer activity. J. Biol. Chem. 280, 25284–25290. [DOI] [PubMed] [Google Scholar]
- Lu J.; Papp L. V.; Fang J.; Rodriguez-Nieto S.; Zhivotovsky B.; Holmgren A. (2006) Inhibition of mammalian thioredoxin reductase by some flavonoids: Implications for myricetin and quercetin anticancer activity. Cancer Res. 66, 4410–4418. [DOI] [PubMed] [Google Scholar]
- Chew E. H.; Lu J.; Bradshaw T. D.; Holmgren A. (2008) Thioredoxin reductase inhibition by antitumor quinols: A quinol pharmacophore effect correlating to antiproliferative activity. FASEB J. 22, 2072–2083. [DOI] [PubMed] [Google Scholar]
- Zhong L.; Arner E. S.; Holmgren A. (2000) Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. U.S.A. 97, 5854–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Chew E. H.; Holmgren A. (2007) Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U.S.A. 104, 12288–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg J.; Zhong L.; Holmgren A.; Arner E. S. (1998) Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J. Biol. Chem. 273, 10835–10842. [DOI] [PubMed] [Google Scholar]
- Drechsel D. A.; Patel M. (2010) Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 285, 27850–27858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley B. A.; Sivakumaran V.; Shi S.; McDonald I.; Lloyd D.; Watson W. H.; Aon M. A.; Paolocci N. (2011) Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J. Biol. Chem. 286, 33669–33677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopert P.; Day B. J.; Patel M. (2012) Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS One 7, e50683-1–e50683-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis M.; Kass G. E.; Orrenius S.; Moldeus P. (1992) N-acetyl-p-benzoquinone imine induces Ca2+ release from mitochondria by stimulating pyridine nucleotide hydrolysis. J. Biol. Chem. 267, 804–809. [PubMed] [Google Scholar]
- Han D.; Dara L.; Win S.; Than T. A.; Yuan L.; Abbasi S. Q.; Liu Z. X.; Kaplowitz N. (2013) Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol. Sci. 34, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawan B. K.; Liu Z. X.; Han D.; Hanawa N.; Gaarde W. A.; Kaplowitz N. (2006) c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131, 165–178. [DOI] [PubMed] [Google Scholar]
- Henderson N. C.; Pollock K. J.; Frew J.; Mackinnon A. C.; Flavell R. A.; Davis R. J.; Sethi T.; Simpson K. J. (2007) Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut 56, 982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anestal K.; Prast-Nielsen S.; Cenas N.; Arner E. S. (2008) Cell death by SecTRAPs: Thioredoxin reductase as a prooxidant killer of cells. PLoS One 3, e1846-1–e1846-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biterova E. I.; Turanov A. A.; Gladyshev V. N.; Barycki J. J. (2005) Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 15018–15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Wolf K.; Urig S.; Becker K. (2007) The structure of human thioredoxin reductase 1 provides insights into C-terminal rearrangements during catalysis. J. Mol. Biol. 370, 116–127. [DOI] [PubMed] [Google Scholar]
- Rigobello M. P.; Vianello F.; Folda A.; Roman C.; Scutari G.; Bindoli A. (2006) Differential effect of calcium ions on the cytosolic and mitochondrial thioredoxin reductase. Biochem. Biophys. Res. Commun. 343, 873–878. [DOI] [PubMed] [Google Scholar]
- Rackham O.; Shearwood A. M.; Thyer R.; McNamara E.; Davies S. M.; Callus B. A.; Miranda-Vizuete A.; Berners-Price S. J.; Cheng Q.; Arner E. S.; Filipovska A. (2011) Substrate and inhibitor specificities differ between human cytosolic and mitochondrial thioredoxin reductases: Implications for development of specific inhibitors. Free Radical Biol. Med. 50, 689–699. [DOI] [PubMed] [Google Scholar]
- Lothrop A. P.; Snider G. W.; Ruggles E. L.; Hondal R. J. (2014) Why is mammalian thioredoxin reductase 1 so dependent upon the use of selenium?. Biochemistry 53, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. P.; Heck D. E.; Mishin V.; Smith P. J.; Hong J. Y.; Thiruchelvam M.; Cory-Slechta D. A.; Laskin D. L.; Laskin J. D. (2007) Paraquat increases cyanide-insensitive respiration in murine lung epithelial cells by activating an NAD(P)H:paraquat oxidoreductase: Identification of the enzyme as thioredoxin reductase. J. Biol. Chem. 282, 7939–7949. [DOI] [PubMed] [Google Scholar]
- Criddle D. N.; Gillies S.; Baumgartner-Wilson H. K.; Jaffar M.; Chinje E. C.; Passmore S.; Chvanov M.; Barrow S.; Gerasimenko O. V.; Tepikin A. V.; Sutton R.; Petersen O. H. (2006) Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 281, 40485–40492. [DOI] [PubMed] [Google Scholar]
- Dicker E.; Cederbaum A. I. (1991) NADH-dependent generation of reactive oxygen species by microsomes in the presence of iron and redox cycling agents. Biochem. Pharmacol. 42, 529–535. [DOI] [PubMed] [Google Scholar]
- Auten R. L.; Davis J. M. (2009) Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 66, 121–127. [DOI] [PubMed] [Google Scholar]
- Prast-Nielsen S.; Cebula M.; Pader I.; Arner E. S. (2010) Noble metal targeting of thioredoxin reductase--covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radical Biol. Med. 49, 1765–1778. [DOI] [PubMed] [Google Scholar]
- Munigunti R.; Gathiaka S.; Acevedo O.; Sahu R.; Tekwani B.; Calderon A. I. (2013) Characterization of PfTrxR inhibitors using antimalarial assays and in silico techniques. Chem. Cent. J. 7, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Antholine W. E.; Myers J. M.; Kalyanaraman B.; Arner E. S.; Myers C. R. (2010) The selenium-independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. J. Biol. Chem. 285, 21708–21723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorova E. S.; Lucas O.; Weisend C. M.; Rollins M. F.; Merrill G. F.; Capecchi M. R.; Schmidt E. E. (2009) Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One 4, e6158–1–e6158–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson A. D.; Carlson B. A.; Li F.; Bonzo J. A.; Yoo M. H.; Krausz K. W.; Conrad M.; Chen C.; Gonzalez F. J.; Hatfield D. L. (2013) Disruption of thioredoxin reductase 1 protects mice from acute acetaminophen-induced hepatotoxicity through enhanced NRF2 activity. Chem. Res. Toxicol. 26, 1088–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos A.; Berggren M.; Gasdaska J. R.; Powis G. (1997) Mechanisms of the regulation of thioredoxin reductase activity in cancer cells by the chemopreventive agent selenium. Cancer Res. 57, 4965–4970. [PubMed] [Google Scholar]
- Berggren M. M.; Mangin J. F.; Gasdaka J. R.; Powis G. (1999) Effect of selenium on rat thioredoxin reductase activity: Increase by supranutritional selenium and decrease by selenium deficiency. Biochem. Pharmacol. 57, 187–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.