

Abstract
Shogaols, the major constituents of thermally processed ginger, have been proven to be highly effective anticancer agents. Our group has identified cysteine-conjugated shogaols (M2, M2′, and M2″) as the major metabolites of [6]-, [8]-, and [10]-shogaol in human and found that M2 is a carrier of its parent molecule [6]-shogaol in cancer cells and in mice, while being less toxic to normal colon fibroblast cells. The objectives of this study are to determine whether M2′ and M2″ behave in a similar manner to M2, in both metabolism and efficacy as anticancer agents, and to further explore the biological pro-apoptotic mechanisms of the cysteine-conjugated shogaols against human colon cancer cells HCT-116 and HT-29. Our results show that [8]- and [10]-shogaol have similar metabolic profiles to [6]-shogaol and exhibit similar toxicity toward human colon cancer cells. M2′ and M2″ both show low toxicity against normal colon cells but retain potency against colon cancer cells, suggesting that they have similar activity to M2. We further demonstrate that the cysteine-conjugated shogaols can cause cancer cell death through the activation of the mitochondrial apoptotic pathway. Our results show that oxidative stress activates a p53 pathway that ultimately leads to p53 up-regulated modulator of apoptosis (PUMA) induction and down-regulation of B-cell lymphoma 2 (Bcl-2), followed by cytochrome c release, perturbation of inhibitory interactions of X-linked inhibitor of apoptosis protein (XIAP) with caspases, and finally caspase 9 and 3 activation and cleavage. A brief screen of the markers attenuated by the proapoptotic activity of M2 revealed similar results for [8]- and [10]-shogaol and their respective cysteine-conjugated metabolites M2′ and M2″. This study highlights the cysteine-conjugated metabolites of shogaols as novel dietary colon cancer preventive agents.
Keywords: ginger, cysteine-conjugated metabolites, shogaols, colon cancer, apoptosis
Introduction
Ginger, the rhizome of Zingiber officinale, has been cultivated as both a ubiquitous spice and a traditional medicinal food for thousands of years.1 Its many curative properties include treatment of nausea and dyspepsia, ameliorating symptoms of arthritis via anti-inflammation, and therapeutic activity against asthma, respiratory disorders, and rheumatic ailments.2−5 Most recently, pharmacologically active components of ginger have been implicated in chemoprevention via numerous mechanisms. Gingerols and shogaols have been identified as the most active ingredients in ginger. Shogaols, the products of ginger thermal processing, consistently show greater anticarcinogenic activity than gingerols, with [6]-shogaol (6S) being the most abundant one followed by [10]- and [8]-shogaol (10S and 8S) (Figure 1A).6−10 In particular, 6S has repeatedly shown strong chemopreventive activity against colorectal cancer, the third most common cancer diagnosed in the United States.11−15
Figure 1.

M2′ and M2″ serve as a carrier to their parent compounds [8]- and [10]-shogaol (8S and 10S), respectively. Chemical structures of shogaols (A) and their cysteine-conjugated metabolites (B); HPLC-ECD chromatograms representing the metabolic profiles of 8S, M2′, 10S, and M2″ in human colon normal cells, CCD-18Co (C, F), and human colon cancer cells, HCT-116 (D, G) and HT-29 (E, H).
Therapeutics with high anticancer potency often come under scrutiny for their accompanying toxicity against noncancerous cells in the viable organism. Consequently, there is escalating interest in employing dietary components and their metabolic constituents as less noxious treatments toward chemoprevention.16−18 Our group has recently established that the reactive ginger component 6S is extensively metabolized in mice and in cancer cells19,20 and that redox transformation and a phase II mercapturic acid pathway (MAP) are its major metabolic routes.19,20 As previously reported, an initial reaction between the α,β-unsaturated ketone functional group of 6S and the cysteine sulfhydryl component of GSH takes place in the MAP, giving rise to the corresponding conjugates. The conjugates then undergo series of enzymatic modifications on the GSH moiety, forming cysteinylglycine, cysteine, and finally N-acetylcysteine conjugates. Additional studies show that both 8S and 10S are also metabolized in humans through the MAP and the cysteine-conjugated metabolites, M2′ and M2″, respectively (Figure 1B), were identified as their major metabolites in human urine.21 Phase II metabolism products such as thiol conjugates are more water-soluble and often less toxic and pungent than their parent compounds, making them preferable for chemoprevention trials.22 Our group recently investigated the bioactivity of the cysteine-conjugated metabolite of [6]-shogaol (M2) and its putative mode of action as a hydrophilic carrier of the parent molecule to its site of efficacy. We established that cysteine-conjugation reduced the toxicity of 6S against normal human colon and lung fibroblast cells while still retaining efficacy against human colon and lung cancer cells.13 Through a series of chemical stability and biological assays, we demonstrated the function of M2 as a carrier of 6S to its active site, wherein the parent compound is rendered less toxic but regains potency and similar bioactivity upon deconjugation.23 It is still unknown whether M2′ and M2″ behave in a similar manner to M2, in both efficacy as anticancer agents and metabolically. As an expansion of our proposed chemical mechanism for cysteine conjugates as carriers of their parent molecule shogaols, we investigated whether M2′ and M2″ are the respective carriers of 8S and 10S in cancer cells and can induce apoptosis through a mechanism similar to M2.
Although we have established the functionality and importance of M2, its mechanism of action against colon cancer cells has not been completely elucidated. There have been studies implicating 6S as a pro-apoptotic agent, via reactive oxygen species (ROS) production and subsequent growth arrest and DNA damage (GADD)-inducible transcription factor 153 (GADD153) expression in COLO 205 cells,15 an inducer of G2/M arrest and aberrant mitotic cell death associated with tubulin aggregation in HCT-116 colon carcinoma cells,14 and an inhibitor of colon cancer cell proliferation through activation of peroxisomal proliferator activated receptor γ (PPARγ).12 Given the significance of M2 as a potential therapeutic with selective toxicity toward cancerous cells, the impetus of this study is also to determine the mechanistic role M2 plays in promoting apoptosis in human colon cancer cells HCT-116 and HT-29. In particular, examining the impact of M2 in colon cancer cells containing wild-type p53 (HCT-116) and mutant p53 (HT-29) will be emphasized. The progressive loss or inactivation of p53 is well associated with the corresponding advancement of cancer to more aggressive stages, and is one of the most prevalent genetic defects in human malignancies that often correlates to chemotherapeutic resistance.24−26 Thus, determining whether the bioactivity of M2, M2′, or M2″ is dependent upon p53 status is of critical concern in terms of cancer prevention, since usable compounds must be able to exert their activity through the early mutations stages of cancer.
Materials and Methods
Cell Culture
HCT-116 and HT-29 human colon cancer cells, CCD-18Co human fibroblast cells derived from colon, and Eagle’s minimum essential media (EMEM) were obtained from American Type Tissue Culture (Manassas, VA). McCoy’s 5A medium was purchased from Thermo Fisher Scientific (Waltham, MA). Supplements of fetal bovine serum (FBS) and penicillin/streptomycin were purchased from Gemini Bio-Products (West Sacramento, CA).
Reagents
6S, 8S, and 10S were purified from ginger extract in our laboratory.10 M2 was synthesized in our laboratory, as previously reported.13 HPLC-grade solvents and other reagents were obtained from VWR International (South Plainfield, NJ). LC/MS grade solvents and other reagents were obtained from Thermo Fisher Scientific (Rockford, IL). Glutathione was obtained from Sigma-Aldrich (St. Louis, MN). Crystal violet, glutaraldehyde, MTT [3-(4,5-dimethylthiaxol-2-yl)-2,5-diphenyltetrazolium bromide], and propidium iodide were procured from Thermo Fisher Scientific (Waltham, MA). Primary antibodies against β-actin, B-cell lymphoma 2 (Bcl-2), caspase 9, caspase 3, cytochrome c, PUMA (p53 up-regulated modulator of apoptosis), p53, XIAP (X-linked inhibitor of apoptosis protein), as well as secondary antibodies conjugated to HRP (horseradish peroxidase) against mouse and rabbit were purchased from Cell Signaling Technology (Beverly, MA).
HPLC-ECD Analysis
A high-performance liquid chromatrography/ESA electrochemical detector (HPLC-ECD) (ESA, Chelmsford, MA) consisting of an ESA model 584 HPLC pump, an ESA model 542 autosampler, an ESA organizer, and an ESA electrochemical detector (ECD) coupled with two ESA model 6210 four sensor cells was used. A Gemini C18 column (150 mm × 4.6 mm, 5 μm; Phenomenex, Torrance, CA) was used for chromatographic analysis at a flow rate of 1.0 mL/min. The mobile phases consisted of solvent A (30 mM sodium phosphate buffer containing 1.75% ACN and 0.125% THF, pH 3.35) and solvent B (15 mM sodium phosphate buffer containing 58.5% ACN and 12.5% THF, pH 3.45). The gradient elution had the following profile: 20% B from 0 to 3 min; 20–55% B from 3 to 11 min; 55–60% B from 11 to 12 min; 60–65% B from 12 to 13 min; 65–100% B from 13 to 40 min; 100% B from 40 to 45 min; then 20% B from 45.1 to 50 min. The cells were then cleaned at a potential of 1000 mV for 1 min. The injection volume of the sample was 10 μL. The eluent was monitored by the Coulochem electrode array system (CEAS) with potential settings at 200, 250, 300, 350, 400, 450, and 500 mV. Data for Figure 1 were from the channel set at 350 mV of the CEAS.
LC/MS Analysis
LC/MS analysis was carried out with a Thermo-Finnigan Spectra System, which consisted of an Accela high-speed MS pump, an Accela refrigerated autosampler, and an LTQ Velos ion trap mass detector (Thermo Electron, San Jose, CA) incorporated with heated electrospray ionization (H-ESI) interfaces. A Gemini C18 column (50 mm × 2.0 mm i.d., 3 μm; Phenomenex, Torrance, CA) was used for separation at a flow rate of 0.2 mL/min. The column was eluted from 100% solvent A (5% aqueous methanol with 0.2% acetic acid) for 3 min, followed by linear increases in B (95% aqueous methanol with 0.2% acetic acid) to 40% from 3 to 15 min, to 91% from 15 to 49 min, and to 100% from 49 to 50 min, and then with 100% B from 50 to 55 min. The column was then re-equilibrated with 100% A for 5 min. The LC eluent was introduced into the H-ESI interface. The positive ion polarity mode was set for the H-ESI source with the voltage on the H-ESI interface maintained at approximately 4.5 kV. Nitrogen gas was used as the sheath gas and auxiliary gas. Optimized source parameters, including ESI capillary temperature (300 °C), capillary voltage (50 V), ion spray voltage (3.6 kV), sheath gas flow rate (30 units), auxiliary gas flow rate (5 units), and tube lens (120 V), were tuned using authentic 6S. The collision-induced dissociation (CID) was conducted with an isolation width of 2 Da and normalized collision energy of 35 for MS2 and MS3. Default automated gain control target ion values were used for MS–MS3 analyses. The mass range was measured from 50 to 1000 m/z. Data acquisition was performed with Xcalibur 2.0 version (Thermo Electron, San Jose, CA).
Chemical Synthesis of M2′ and M2″
The experimental procedure to synthesize M2′ and M2″ was similar to that of M2.13 In brief, a catalytic amount of NaHCO3 (1.3 mg, 0.015 mmol) was added to a mixture of 8S (91.2 mg, 0.3 mmol) and cysteine (54 mg, 0.45 mmol) in methanol/water (6 mL, 1:1, v/v). The mixture was stirred at room temperature for 24 h, and adjusted to pH 6 with a diluted acetic acid solution (0.1 M). The mixture was then purified by preparative HPLC to give M2′ as a white solid (70 mg, yield 55%). Shogaol 10S (100 mg, 0.3 mmol) instead of 8S was used in the above reaction to give M2″ (73 mg, yield 54%).
Purification of M2′ or M2″ Using Preparative HPLC
Waters preparative HPLC systems with 2545 binary gradient module, Waters 2767 sample manager, Waters 2487 autopurification flow cell, Waters fraction collector III, dual injector module, and 2489 UV/Visible detector were used to separate M2′ or M2″ from the reaction mixture. A Phenomenex Gemini-NX C18 column (250 mm × 30.0 mm i.d., 5 μm) was used with a flow rate of 20.0 mL/min. The wavelength of the UV detector was set at 280 nm. The injection volume was 1.0 mL for each run. The mobile phase consisted of solvent A (H2O + 0.1% formic acid) and solvent B (MeOH + 0.1% formic acid). The reaction mixture of M2′ was injected to the preparative column and eluted with a gradient solvent system (75% to 87% B from 0 to 12 min; to 75% B from 12 to 12.5 min; then 75% B from 12.5 to 15 min). A total of six runs resulted in 70 mg of M2′ (tR 9.45 min). Similarly, reaction mixture of M2″ was injected to the preparative column and eluted with a gradient solvent system (85% to 100% B from 0 to 15 min; then 100% B from 15 to 16 min; to 85% B from 16 to 16.5 min; then 85% B from 16.5 to 20 min). A total of seven runs resulted in 73 mg of M2″ (tR 8.13 min).
Nuclear Magnetic Resonance (NMR)
1H (600 MHz), and 13C (150 MHz) NMR spectra of M2′ and M2″ in CD3OD were acquired on a Bruker AVANCE 600 MHz NMR spectrometer (Bruker, Inc., Silberstreifen, Rheinstetten, Germany). The 1H and 13C NMR data of M2′ and M2″ are listed in Table 1.
Table 1. δH (600 MHz) and δC (150 MHz) NMR Spectra Data of M2′ and M2″a.
| M2′ |
M2″ |
|||
|---|---|---|---|---|
| δH, multi (J) | δC | δH, multi (J) | δC | |
| 1 | 2.81, m | 31.6 | 2.81, m | 31.8 |
| 2 | 2.81, m | 48.1 | 2.81, m | 48.1 |
| 3 | 210.4 | 210.4 | ||
| 4 | 2.88, m | 53.8 | 2.89, m | 53.8 |
| 2.81, m | 2.81, m | |||
| 5 | 3.11, m | 40.9 | 3.11, m | 40.9 |
| 6 | 1.56, m | 34.9 | 1.56, m | 34.9 |
| 7 | 1.42, m | 27.4 | 1.42, m | |
| 8 | 1.31, m | 31.8 | 1.31, m | 31.8 |
| 9 | 1.31, m | 29.2b | 1.31, m | 29.3b |
| 10 | 1.31, m | 28.9b | 1.31, m | 29.3b |
| 11 | 1.31, m | 28.9b | 1.31, m | 29.2b |
| 12 | 1.31, m | 26.4b | 1.31, m | 29.1b |
| 13 | 0.92, t (6.9) | 22.3 | 1.31, m | 28.9b |
| 14 | 13.0 | 1.31, m | 26.4b | |
| 15 | 1.31, m | 22.3 | ||
| 16 | 0.92, t (6.9) | 13.0 | ||
| 1′ | 132.4 | 132.4 | ||
| 2′ | 6.80, d (2.0) | 111.7 | 6.80, d (2.0) | 111.8 |
| 3′ | 147.8 | 147.7 | ||
| 4′ | 144.4 | 144.4 | ||
| 5′ | 6.71, d (7.98) | 115.0 | 6.71, d (7.98) | 115.0 |
| 6′ | 6.65, dd (7.98, 2.0) | 120.4 | 6.65, dd (7.98, 2.0) | 120.5 |
| 1″ | (a) 3.21, m | 32.0 | (a) 3.21, m | 32.1 |
| (b) 2.81, m | (b) 2.81, m | |||
| 2″ | 3.66, m | 54.9 | 3.66, m | 54.9 |
| 3″ | 171.1 | 171.2 | ||
| OMe | 3.85, s | 55.1 | 3.85, s | 55.0 |
CD3OD; δ in ppm and J in Hz.
Assignments interchangeable.
Metabolism of 8S, 10S, M2′, and M2″ in Human Colon Cancer Cells
Cells (1.0 × 106) were plated in six-well culture plates and were allowed to attach for 24 h at 37 °C in 5% CO2 incubator. Shogaol 8S or 10S in DMSO or the corresponding cysteine-conjugated metabolites M2′ and M2″ were diluted in McCoy’s 5A medium (containing 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% glutamine) to reach a final concentration of 10 μM and were incubated with different colon cancer cell lines (HCT-116 or HT-29). At different time points (0, 2, 4, 8, 24, and 48 h), 190 μL samples of supernatant were taken and transferred to vials containing 10 μL of 2% acetic acid to stabilize these compounds and their respective metabolites. An equal volume of acetonitrile was added to the samples before centrifugation. The supernatant was harvested, and the samples were then analyzed by HPLC-ECD.
Evaluation of Toxicity in Human Colon Cancer and Normal Colon Cells
Cell viability was determined by an MTT colorimetric assay as described previously.27 Briefly, human colon fibroblast cells, CCD-18Co, or human colon cancer cells, HCT-116 or HT-29, were plated in 96-well microtiter plates with 3000 cells/well and allowed to attach for 24 h at 37 °C and 5% CO2. The test compounds (in DMSO) were added to cell culture medium to desired final concentrations (final DMSO concentrations for control and treatments were 0.1%). After the cells were cultured for 24 h, the medium was aspirated, and cells were treated with 200 μL of fresh medium containing 2.41 mmol/L MTT. After incubation for 3 h at 37 °C, the medium containing MTT was aspirated, 100 μL of DMSO was added to solubilize the formazan precipitate, and the plates were shaken gently for an hour at room temperature. Absorbance values were derived from the plate reading at 550 nm on a Biotek (Winooski, VT) microtiter plate reader. The reading reflected the number of viable cells and was expressed as a percentage of viable cells in the control. CCD-18Co cells were grown in EMEM. Both HCT-116 and HT-29 cells were cultured in McCoy’s 5A medium. All of the above media were supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% glutamine, and the cells were kept in a 37 °C incubator with 95% humidity and 5% CO2.
Apoptosis Analysis
Apoptosis was determined by FACS analysis of propidium iodide (PI)-stained cells. In brief, cells were trypsinized, washed with cold phosphate-buffered saline (PBS), fixed in ice-cold 70% ethanol for 1 h, and then resuspended in 2 mL of PBS supplemented with 10 μL of RNase (100 mg/mL) and incubated at 37 °C for 30 min. After incubation, DNA was stained with 1 mg/mL PI in PBS. Cell staining was analyzed using a Cell Lab QuantaTM SC flow cytometer (Beckman Coulter, Danvers, MA), and data were processed using FCS Express software (DeNovo Software, Los Angeles, CA). The percentage of apoptotic cells in each sample was determined based on the sub-G0 peaks detected in monoparametric histograms.
Measurement of Reactive Oxygen Species
This assay employed the cell-permeable fluorogenic probe 2′,7′-dichlorodihydrofluorescein diacetate [DCFH-DA] (Sigma-Aldrich, St. Louis, MO) to measure the relative changes in O2– and H2O2 levels in HCT-116 or HT-29 cells after treatment with 5, 10, and 20 μM 6S or M2 (or DMSO) over 0, 2, 4, 8, and 24 h. In brief, DCFH-DA is diffused into cells and is deacetylated by cellular esterases to nonfluorescent 2′,7′-dichlorodihydrofluorescein (DCFH), which is rapidly oxidized to highly fluorescent 2′,7′-dichlorofluorescein (DCF) by intracellular hydrogen peroxide or other low molecular weight peroxides.13,28 Measured fluorescence intensity is thus proportional to the amount of such peroxides in the cell at a given time. Human colon cancer cells HCT-116 or HT-29 were seeded in 96-well black-sided, clear-bottomed culture plates, with 5000 cells/well and were allowed to adhere for 24 h in a 37 °C incubator with 5% CO2. Media was aspirated, and 5, 10, or 20 μM M2, 6S, or DMSO diluted in media was added to designated wells, which were run in triplicate. After desired incubation times of 0, 2, 4, 8, or 24 h, media and test compounds were aspirated. Cells were washed three times with 200 μL of PBS before addition of 100 μL of 1 mM DCFH-DA. The fluorogenic probe permeated cell membranes and was processed to DCF for 1 h at 37 °C. After incubation, plates were immediately placed in a Biotek microplate reader to measure fluorescence at wavelengths of 485 (excitation) and 528 (emission). Raw values were normalized to DMSO control for each time point and are presented as fold induction versus 0 h time point (n = 3).
Western Blot Analysis
Cell lysates were prepared in ice-cold RIPA lysis buffer [25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, Thermo Fisher Scientific] supplemented with a protease inhibitor cocktail (AEBSF, aprotinin, bestatin, E-64, leupeptin, and pepstatin A in DMSO with EDTA, Thermo Fisher Scientific). Protein content was measured by a Pierce BCA Assay Kit (Thermo Fisher Scientific). Protein contents of cell lysates (30 μg of protein/lane) were resolved by SDS-PAGE. Proteins were then electro-transferred onto PVDF membranes, and blots were blocked for 1 h at room temperature in 1 × TBS with 1% casein (Bio-Rad Laboratories, Berkeley, CA). Blots were then incubated overnight at 4 °C with the desired primary antibody diluted in TBS with 0.5% Tween-20. Blots were then washed with TBS-Tween 20 and probed for 1 h with the appropriate secondary antibody (1:1000). Protein bands were visualized with chemiluminescence using West Femto maximum detection substrate (Thermo Fisher Scientific). To confirm equal protein loading in each lane, immunoblots were stripped and reprobed for β-actin. Protein fold-induction was calculated by normalizing the intensity of the band of interest to β-actin first and then to DMSO control lanes.
Colony Formation Assay
Human colon cancer cells HCT-116 or HT-29 (1000 cells per well) were seeded in 6-well culture plates for 24 h and then incubated with M2 (0, 1, 5, 10, 20, or 40 μM) in DMSO in a 37 °C incubator with 5% CO2. After 2 weeks, colonies were washed with phosphate-buffered saline (PBS), then stained with a mixture of 6.0% glutaradehyde and 0.5% crystal violet for 30 min at room temperature, rinsed in water, air-dried, and then photographed.
Statistical Analysis
Student’s t-test or two-way analysis of variance (ANOVA) with the Bonferroni post-test were used to determine the statistical significance of data, which was performed on GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA).
Results
Synthesis and Structure Elucidation of M2′ and M2″
A similar experimental procedure was used for the syntheses of M2′ and M2″ as the previous protocol used for M2.13 M2′ showed the molecular formula C22H35NO5S on the basis of positive ESI-MS at m/z 426 [M + H]+ and its 1H and 13C NMR data. The molecular weight of M2′ was 121 mass units more than that of 8S (MW 304), indicating that M2′ was the cysteine-conjugated 8S, which is an expected result from the reaction between 8S and l-cysteine. This was also supported by the observation of the absence of a double bond in the 1H and 13C NMR spectra of M2′. Therefore, M2′ was confirmed to be 5-cysteinyl-8S. In the same way, M2″ was identified as 5-cysteinyl-10S, based on its positive ESI-MS at m/z 454 [M + H]+ and its 1H and 13C NMR data (Table 1).
M2′ and M2″ Give Similar Metabolic Profiles as Their Parent Compounds 8S and 10S
Recent work in our group has focused on the biotransformation of the cysteine-conjugated metabolite of 6S, M2. The metabolic profiles of M2 in cancer cells HCT-116 and H-1299 resembled those of 6S, indicating that its biotransformation route was initiated by deconjugation.23 Evidence supporting the identification of M2′ and M2″ as carriers of their respective shogaols is presented in Figure 1, in which the metabolic profiles of 8S and 10S in human colon fibroblast cells CCD18Co (Figure 1C,F) or human colon cancer cells HCT-116 (Figure 1D,G) and HT-29 (Figure 1E,H) are compared with the profiles of M2′ and M2″ in the same respective cell lines. Upon removal of the cysteine residue, which occurs after less than 2 h of treatment, M2′ and M2″ are metabolized in an almost identical fashion as their parent shogaols. We have reported that M9 and M11 are the major metabolites of 6S in cancer cells; M11 is the double bond reduced metabolite of 6S, and M9 is the ketone group reduced metabolite of M11.19 In the present study, reduced products were also identified as the major metabolites of 8S and 10S in human colon fibroblast cells and cancer cells, M9′ and M11′ for 8S and M9″ and M11″ for 10S. Their structures were confirmed by comparing their MS/MS spectra with those of M9 and M11 (Figure 2).
Figure 2.

Chemical structures and MS/MS spectra of M9, M9′, and M9″ (A) and M11, M11′, and M11″ (B).
M2′ and M2″ Exert Similar Bioactivities as Their Parent Compounds 8S and 10S
We have reported that M2 has low toxicity in normal human fibroblast colon and lung cells while still remaining toxic against colon cancer and lung cancer cells.13 As the structure of the cysteine-conjugated metabolites of 8S and 10S only differ from 6S by their respective increases in side chain length, we hypothesized that M2′ and M2″ should have similar activities compared with their parent molecules as M2 has compared with 6S.
Our results from measurement of cell viability by MTT assay show that 8S and 10S and their respective cysteine-conjugated metabolites M2′ and M2″ have low toxicity in normal colon cells CCD-18Co (Figure 3A), with IC50 values of 104.66 and 135.53 μM for 8S and 10S, respectively, and IC50 values greater than 200 μM for M2′ and M2″. All compounds were highly potent against human colon cancer cells HCT-116 (Figure 3B) and HT-29 (Figure 3C), with slightly different efficacy profiles between the two types of cells. M2′ and M2″ show higher activity against HCT-116 than their parent molecules, with IC50 values of 15.21 and 13.28 μM, respectively, versus values of 22.8 and 25.09 μM for 8S and 10S, respectively. Similar results were observed in the p53 mutant HT-29 cell line, albeit slightly more resistant to treatment from all compounds. The IC50 values for 8S and 10S, 27.88 and 23.92 μM, were about 10–16% lower than those for M2′ and M2″, at 31.15 and 28.75 μM, respectively.
Figure 3.

M2′ and M2″ remain bioactive to human colon cancer cells but less toxic to human normal colon cells than 8S and 10S and M2 induces apoptosis on HCT-116 and HT-29 human colon cancer cells. Growth inhibitory effects of 8S, M2′, 10S, and M2″ against human normal colon cells CCD-18Co (A) and human colon cancer cells HCT-116 (B) and HT-29 (C) treated with 8S, M2′, 10S, or M2″ for 24 h at different doses (n = 6) (measured by MTT assay). (D) Effect of M2 on apoptosis in human colon cancer cells HCT-116 and HT-29 treated with M2 (0, 10, 20, and 40 μM) for 24 h measured by PI staining; data represent mean ± SD. (E) Western blot analysis of HCT-116 and HT-29 cell extracts treated with DMSO, 10, 20, and 40 μM of M2 for 24 h. Fold induction for each marker compared with DMSO is indicated under the corresponding line.
M2 Induces Apoptosis in Human Colon Cancer Cells HCT-116 and HT-29
In order to compare the biological similarities of M2, M2′, and M2″, we first needed to acquire a more accurate view of the mechanisms of action of M2. To study the impact of M2 on induction of apoptosis in both HCT-116 and HT-29 colon cancer cells, the percent of apoptotic cells were quantified after 24 h treatment of increasing doses of M2 (Figure 3D). A dose-dependent effect of M2 was observed with HCT-116 cells being notably more sensitive to M2 treatment than HT-29 cells. Treatment with 40 μM M2 gave the greatest induction of apoptosis in HCT-116 and HT-29 cells, with 26.34% or 14.38% apoptotic cells, respectively. The 10 and 20 μM treatments also yielded twice as many apoptotic HCT-116 cells compared with HT-29 cells (Figure 3D).
Induction of apoptosis was further confirmed by Western blot analysis of markers of the intrinsic mitochondrial apoptosis pathway (Figure 3E). Increasing doses of M2 led to PUMA induction (up to ∼17-fold increase in both cell lines when treated with 40 μM M2) and progressive reduction of Bcl-2 levels (undetectable in HCT-116 and ∼0.1-fold in HT-29 cells when treated with 40 μM M2). We detected a progressive increase in cytochrome c release with increasing doses of M2 (up to ∼2.2-fold increase in both cell lines when treated with 40 μM M2). We also observed a clear, progressive reduction in XIAP expression with increasing doses of M2 (undetectable in HCT-116 and ∼0.3-fold in HT-29 cells when treated with 40 μM M2). Finally, increasing concentration of the cleaved forms of caspase 9 (up to ∼4.6-fold increase in both cell lines when treated with 40 μM M2) and caspase 3 (∼3–4-fold increase in both cell lines when treated with 40 μM M2) were detected with increasing doses of M2. Overall, these results show that markers of the mitochondrial pathway (PUMA, Bcl-2) of apoptosis were activated upon exposure to M2 and ultimately led to the release of the corresponding apoptosis markers (cytochrome c, cleaved caspases 3 and 9).
8S, 10S, M2′, and M2″ Activate Apoptosis
Our results on M2′ and M2″ are consistent with our previous observation that M2 has low toxicity in normal colon cells while still retaining strikingly similar potency to 6S in cancer cell growth inhibition.13 Thus, we could tentatively propose that M2′ and M2″ have similar biological roles as the function we previously proposed for M2, as low toxicity carriers of parent molecular 8S and 10S to active sites, where function is restored upon removal of the cysteine group.23 To verify this hypothesis, we studied the apoptotic markers modulated by M2 (described Figure 3E) in HCT-116 and HT-29 cells treated with 8S, M2′, 10S, or M2″ for 24 h. We chose a dose of 20 μM since it is close to the IC50 values of all compounds.
Screening of the markers modulated by the pro-apoptotic activity of M2 gave similar results for 8S and 10S and their respective cysteine-conjugated metabolites M2′ and M2″(Figure 4A). In other words, PUMA and cleaved caspase-3 were up-regulated, with concomitant down-regulation of Bcl-2. The changes in marker expression was observed in both cell lines in a nearly identical amplitude between the parent compound and its corresponding metabolite.
Figure 4.

Cysteine conjugated shogaols induce apoptosis via oxidative stress-mediated p53 pathway. Western blot analysis of HCT-116 or HT-29 cell extracts treated with DMSO or 20 μM of 8S, M2′, 10S, or M2″ for 24 h on apoptosis-related protein (A) or with 0, 10, 20, and 40 μM of M2 or 6S on p53 expression with or without the presence of GSH (5 mM) (B and C). Protein levels were relatively quantitated by densitometric analysis using β-actin as a loading control. Fold induction for each marker compared with DMSO is indicated under the corresponding line. (D, E) Induction of ROS by M2 or 6S. HCT-116 and HT-29 cells were treated with indicated concentrations of 6S or M2 for different time periods, and the intracellular ROS levels were determined as described under Materials and Methods. (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001).
Shogaols and Their Cysteine-Conjugated Metabolites Affect Wild-Type and Mutant p53 Expression in Human Colon Cancer Cells HCT-116 and HT-29
Since we established that the cysteine-conjugated shogaols could activate apoptosis through the same pathway, we wanted to further study their mechanisms of action using 6S and its metabolite M2 as a model. We established that PUMA, a transcriptional target of p53, accumulated almost identically in p53 wild-type HCT-116 and p53 mutant HT-29 cell lines, with both showing greater than 17-fold induction of PUMA after 24 h treatment with 40 μM M2 (Figure 3E). To study the impact of M2 on p53 regulation in human colon cancer cells and its dependency on p53 integrity, p53 wild-type HCT-116 or p53 mutant HT-29 cells were cultured with M2 or 6S for 24 h at concentrations of 10, 20, or 40 μM. The p53 response in colon cancer cells to 6S treatment (10, 20, or 40 μM for 24 h) was observed in HCT-116 and HT-29. After M2 or 6S treatment, a dose-dependent up-regulation of p53 is noted in both wild-type and mutant cancer cell lines (Figure 4B,C), indicating that M2 or 6S regulation of p53 does not require a wild-type gene, although induction of p53 expression by M2 or 6S is dramatic in HCT-116 cells and slightly less striking (but still significant) in HT-29 cells (Figure 4B,C). Consistent with our premise, we then studied p53 expression in HCT-116 and HT-29 cells after exposure to 20 μM of 8S, M2′, 10S, or M2″. We observed an increase in p53 accumulation for all compounds (Figure 4A). The cysteine-conjugated metabolites were able to increase p53 accumulation in a similar manner to their parent compound.
6S and Its Cysteine-Conjugated Metabolite M2 Affect Reactive Oxygen Species Generation in Human Colon Cancer Cells HCT-116 and HT-29
Our observations up to this point indicated that M2 induces apoptosis in colon cancer cells through modulation of p53 expression and subsequent activation of the mitochondrial apoptotic pathway through PUMA and Bcl-2. However, the mechanism triggering the p53 accumulation was still unclear. Because overabundance of reactive oxygen species has been implicated as a genotoxic stressor and trigger for p53 activation,29−33 we observed the production of ROS by M2 (compared with parent compound 6S for reference) in HCT-116 and HT-29 cells. As shown in Figure 4D,E, the trends of ROS induction by both M2 and 6S are strikingly similar, with the greatest peak at 2 h after treatment and a steady decline thereafter, which is consistent with the changes of glutathione levels in cancer cells that we reported previously.23 In this case, treatment of HCT-116 cells with 40 μM M2 produced the greatest ROS activity after 2 h, with greater than 2-fold induction (with statistical significance, p < 0.0001). The scales of induction of ROS by M2 or 6S are parallel to the p53 induction response in the two respective cell lines. That is, in HCT-116 cells, ROS activity is induced 2-fold or greater by M2 or 6S treatment, while in HT-29 cells, ROS activity is induced less than 1.5-fold.
To demonstrate that ROS induction was indeed the trigger to p53 accumulation, we treated HCT-116 and HT-29 cells for 24 h with M2 or 6S (10, 20, or 40 μM) and supplemented with 5 mM GSH in order to suppress ROS accumulation in the cells. As shown on Figure 4B,C, addition of GSH suppressed p53 induction in both cell lines for all concentrations of M2 or 6S, establishing that p53 does not accumulate if there is no ROS generation.
M2 Inhibits Colony Formation in Human Colon Cancer Cells HCT-116 and HT-29
While we established that the cysteine conjugate of 6S can activate apoptosis, it is possible that other mechanisms leading to cancer cell death are triggered, especially in light of the specific activation of the multiacting p53. Since p53 is one of the major regulators of the cell cycle, we further investigated the potential of our cysteine conjugated metabolites to inhibit cancer cell proliferation, using a colony assay and M2 as a model. Human colon cancer cells HCT-116 (Figure 5A) and HT-29 (Figure 5B) were treated with M2 with doses ranging from 0 to 40 μM for 2 weeks. Inhibition of colony formation was observed in a dose-dependent manner in both cell lines, with 50% inhibition between 5 and 10 μM treatments. Following a trend underlined previously, HCT-116 cells are slightly more sensitive to M2 than HT-29 cells.
Figure 5.

Dose dependent inhibition of human colon cancer cell colony formation by M2 in HCT-116 (A) and HT-29 (B) cells. HCT-116 (A) and HT-29 (B) cells were treated with M2 (0, 1, 5, 10, 20, and 40 μM) and incubated in 6-well plates for 2 weeks, and the cells were then stained with crystal violet and counted for colony formation. Each column represents a mean ± SD (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001).
Discussion
There is currently considerable interest in developing cancer therapeutics and preventive agents from dietary sources, with extensive research over the past few decades focusing on plant derivatives.34−38 Ginger has been comprehensively studied in recent years for its chemopreventive properties, particularly against colorectal cancer.39−42 Our group and others have identified active constituents of ginger, with our most recent focus on shogaols, especially 6S.12,13,19,20,23,39,43,44 The metabolism and biotransformation of 6S was investigated thoroughly, providing further insight into its means of exerting bioactivity.19 Upon testing the efficacy of the major metabolites of 6S, we found that 5-cysteinyl-conjugated 6S (M2) exerted strong antiproliferative activity against human colon cancer cells, while having significantly lessened toxicity in normal human colon cells.13 Further, M2 was shown to act as a carrier of parent molecule 6S.23 The discovery of M2 was via correlation of 6S metabolism as a highly electrophilic xenobiotic through the detoxifying mercapturic acid pathway (MAP). We also found that increased chain length of shogaols does not cause a departure from the MAP. M2′ and M2″ were identified as the major metabolites of 8S and 10S, respectively, from humans upon consumption of ginger tea.21 The current study shows that M2′ and M2″ are the carriers of 8S and 10S, respectively, and have similar antiproliferative activity against human colon cancer cells and less toxicity in normal human colon cells to their respective parent compounds. Our results clearly indicate that this portion of phase II metabolism transforms electrophiles to less reactive and more water-soluble intermediates, thus aiding in their mobility and decreasing their toxicity en route.45
It has already been established through many studies that 6S induces apoptosis in cancer cells via several pathways,12−15 and thus our focus was to identify the pathway that encompasses the mode of activity of M2. We demonstrated that M2 treatment could induce ROS generation, which in turn up-regulated p53 expression and induced apoptosis through the mitochondrial pathway. This proposed mechanism is summarized in Figure 6. Since tumor suppressor gene p53 is one of the most frequent mutations in cancers, leading to resistance to pharmacological interventions and more aggressive progression,46−49 we were interested in testing the efficacy of M2 in wild-type p53 HCT-116 human colon cancer cells as well as mutant p53 HT-29 human colon cancer cells. In what could be a fortunate outcome for the putative roles of ginger constituents and metabolites toward chemoprevention, M2 induced apoptosis in both cell lines. It is notable that although the p53 pro-apoptotic pathway was exploited for at least some of M2’s bioactivity and that we observed a general trend of superior bioactivity in HCT-116 cells, the metabolite’s efficacy was ultimately not compromised by p53 mutation. This is particularly attractive in terms of cancer chemoprevention, since it suggests that the cysteine-conjugated metabolite of 6S would still be able to activate a p53 apoptotic response even in cancer cells containing mutations of the p53 gene.
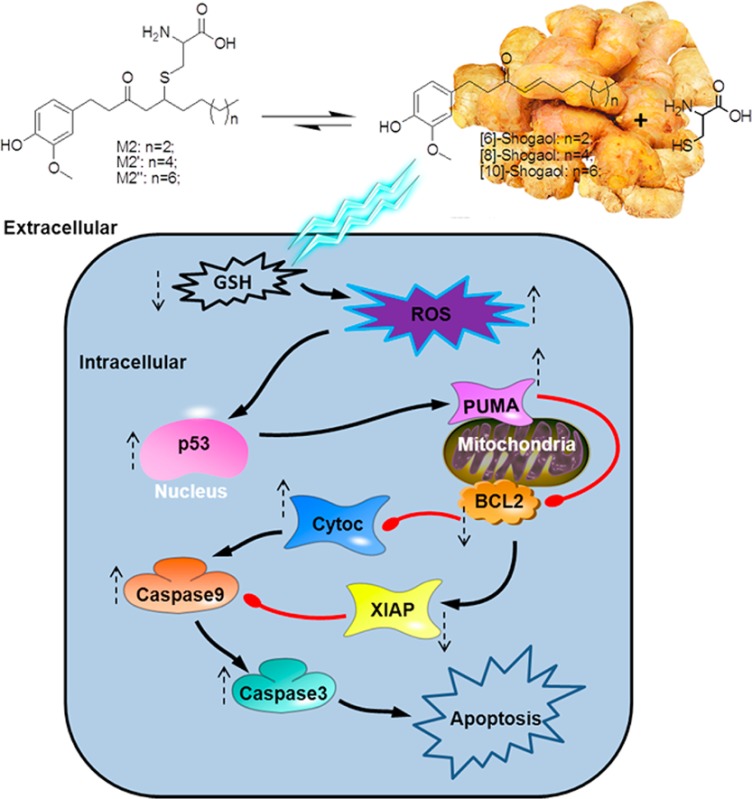
Figure 6.

Schematic representation of the mechanisms of action of shogaols and their cysteine-conjugated metabolites in regulating apoptosis by p53 induction, activation of PUMA via p53 translocation to the mitochrondrial surface, Bcl-2 down-regulation, cytochrome c release, XIAP inhibition, and finally caspases 9 and 3 activation and cleavage.
A commonly proposed p53-induced apoptosis mechanism involves direct interaction with Bcl-2 family members and down-regulation of antiapoptotic genes, such as Bcl-2, which was also noted in the current study.47,50−53 Both HCT-116 and HT-29 human colon cancer cells experienced a dramatic down-regulation of Bcl-2 after M2 treatment. Interestingly, PUMA, a transcriptional target of p53,54 was also up-regulated in both colon cancer cell lines after treatment with M2. Upon a transcriptionally induced signal from p53, PUMA assists in promoting apoptosis by disrupting the association restraints Bcl-xL exerts on p53, thus liberating the molecule to exert pro-apoptotic activity, but binding to Bcl-xL in the process.55 This evidence supports the strong role of M2 as a chemopreventive agent against colon cancer cells that induces p53 expression and downstream regulation.
Treatment of colon cancer cells HCT-116 and HT-29 with M2 in this study lead to apoptosis, through early production of reactive oxygen species. Overabundance of ROS combined with a cancer cell’s reduced detoxification ability can often lead to oxidative stress sufficient to induce programmed cell death.56 Apoptosis induced by p53 is at least partially dependent upon accumulation of ROS,57 and we demonstrated this to be also true in our model. Therefore, we can conclude that M2 causes p53 induction of apoptosis via ROS production in both HCT-116 and HT-29 human colon cancer cells. Since we demonstrated that the other metabolites M2′ and M2″ activate similar markers of apoptosis (Figure 4A) and contain the same chemical reactivity as M2, it is reasonable to conclude that these metabolites would behave in a similar way and could also activate cancer cell apoptosis through ROS induction and the subsequent p53 accumulation. In addition, we also demonstrated that M2 does not exclusively induce cancer cell death through apoptosis and can also influence other major mechanisms such as cell proliferation (Figure 5); it follows that other cysteine conjugated metabolites originating from the same MAP have that potential as well. These compounds are worthy of further investigation, and further studies could examine commonly consumed ginger mixtures containing these compounds for more direct translation of these data to in vivo dietary models.
Ginger components are highly lauded for their vast and varied chemopreventive properties. Because shogaols have been proven as highly effective anticancer agents, the continued study of their mechanisms of activity can allow us to discover novel anticancer targets. Further, investigation of the metabolic products of shogaols can provide new therapeutic routes that are less toxic, while retaining high efficacy. This study has shown that cysteine-conjugated shogaols are novel compounds with a putative role as natural pharmaceuticals with low-toxicity, high-potency, and at least partial indifference to p53 integrity in colon cancer cells.
Acknowledgments
This work was supported by NIH Grants CA138277 and CA138277S1 to S. Sang.
Glossary
Abbreviations
- 6S
[6]-shogaol
- 8S
[8]-shogaol
- 10S
[10]-shogaol
- GSH
glutathione
- HPLC
high-performance liquid chromatography
- LC/MS
liquid chromatrography/mass spectrometry
- M2
5-cysteinyl-6S
- M2′
5-cysteinyl-8S
- M2″
5-cysteinyl-10S
- p53
tumor protein 53
- ROS
reactive oxygen species
Author Contributions
§ H.C. and D.N.S. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Park E. J.; Pezzuto J. M. Botanicals in cancer chemoprevention. Cancer Metastasis Rev. 2002, 21, 231–255. [DOI] [PubMed] [Google Scholar]
- Kubra I. R.; Rao L. J. An impression on current developments in the technology, chemistry, and biological activities of ginger (Zingiber officinale Roscoe). Crit. Rev. Food Sci. Nutr 2011, 52, 651–688. [DOI] [PubMed] [Google Scholar]
- Ali B. H.; Blunden G.; Tanira M. O.; Nemmar A. Some phytochemical, pharmacological and toxicological properties of ginger (Zingiber officinale Roscoe): A review of recent research. Food Chem. Toxicol. 2008, 46, 409–420. [DOI] [PubMed] [Google Scholar]
- Altman R. D.; Marcussen K. C. Effects of a ginger extract on knee pain in patients with osteoarthritis. Arthritis Rheum. 2001, 44, 2531–2538. [DOI] [PubMed] [Google Scholar]
- Lantz R. C.; Chen G. J.; Sarihan M.; Solyom A. M.; Jolad S. D.; Timmermann B. N. The effect of extracts from ginger rhizome on inflammatory mediator production. Phytomedicine 2007, 14, 123–128. [DOI] [PubMed] [Google Scholar]
- Kim J. S.; Lee S. I.; Park H. W.; Yang J. H.; Shin T. Y.; Kim Y. C.; Baek N. I.; Kim S. H.; Choi S. U.; Kwon B. M.; Leem K. H.; Jung M. Y.; Kim D. K. Cytotoxic components from the dried rhizomes of Zingiber officinale Roscoe. Arch Pharm. Res. 2008, 31, 415–418. [DOI] [PubMed] [Google Scholar]
- Bak M. J.; Ok S.; Jun M.; Jeong W. S. 6-Shogaol-rich extract from ginger up-regulates the antioxidant defense systems in cells and mice. Molecules 2012, 17, 8037–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugasani S.; Pichika M. R.; Nadarajah V. D.; Balijepalli M. K.; Tandra S.; Korlakunta J. N. Comparative antioxidant and anti-inflammatory effects of [6]-gingerol, [8]-gingerol, [10]-gingerol and [6]-shogaol. J. Ethnopharmacol. 2010, 127, 515–520. [DOI] [PubMed] [Google Scholar]
- Wu H.; Hsieh M. C.; Lo C. Y.; Liu C. B.; Sang S.; Ho C. T.; Pan M. H. 6-Shogaol is more effective than 6-gingerol and curcumin in inhibiting 12-O-tetradecanoylphorbol 13-acetate-induced tumor promotion in mice. Mol. Nutr. Food Res. 2010, 54, 1296–1306. [DOI] [PubMed] [Google Scholar]
- Sang S.; Hong J.; Wu H.; Liu J.; Yang C. S.; Pan M. H.; Badmaev V.; Ho C. T. Increased growth inhibitory effects on human cancer cells and anti-inflammatory potency of shogaols from Zingiber officinale relative to gingerols. J. Agric. Food Chem. 2009, 57, 10645–10650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.; Ward E.; Brawley O.; Jemal A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. Ca—Cancer J. Clin. 2011, 61, 212–36. [DOI] [PubMed] [Google Scholar]
- Tan B. S.; Kang O.; Mai C. W.; Tiong K. H.; Khoo A. S.; Pichika M. R.; Bradshaw T. D.; Leong C. O. 6-Shogaol inhibits breast and colon cancer cell proliferation through activation of peroxisomal proliferator activated receptor gamma (PPARgamma). Cancer Lett. 2013, 336, 127–39. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Warin R. F.; Soroka D. N.; Chen H.; Sang S. Metabolites of ginger component [6]-shogaol remain bioactive in cancer cells and have low toxicity in normal cells: chemical synthesis and biological evaluation. PloS One 2013, 8, e54677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan F. F.; Nagle A. A.; Ang X.; Ho O. H.; Tan S. H.; Yang H.; Chui W. K.; Chew E. H. Shogaols at proapoptotic concentrations induce G(2)/M arrest and aberrant mitotic cell death associated with tubulin aggregation. Apoptosis 2011, 16, 856–867. [DOI] [PubMed] [Google Scholar]
- Pan M. H.; Hsieh M. C.; Kuo J. M.; Lai C. S.; Wu H.; Sang S.; Ho C. T. 6-Shogaol induces apoptosis in human colorectal carcinoma cells via ROS production, caspase activation, and GADD 153 expression. Mol. Nutr. Food Res. 2008, 52, 527–537. [DOI] [PubMed] [Google Scholar]
- Bansal P.; Khoobchandani M.; Kumar V.; Srivastava M. M., Cruciferous Vegetables: Novel Cancer Killer and Guardians of Our Health. In Chemistry of Phytopotentials: Health, Energy and Environmental Perspectives, Khemani L. D., Srivastava M. M., Srivastava S., Eds.; Springer: Berlin Heidelberg, 2012; pp 3–7. [Google Scholar]
- Tan X.-L.; Spivack S. D. Dietary chemoprevention strategies for induction of phase II xenobiotic-metabolizing enzymes in lung carcinogenesis: A review. Lung Cancer 2009, 65, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Allyl isothiocyanate as a cancer chemopreventive phytochemical. Mol. Nutr. Food Res. 2010, 54, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Lv L.; Soroka D.; Warin R. F.; Parks T. A.; Hu Y.; Zhu Y.; Chen X.; Sang S. Metabolism of [6]-shogaol in mice and in cancer cells. Drug Metab. Dispos. 2012, 40, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Sang S. Identification of phase II metabolites of thiol-conjugated [6]-shogaol in mouse urine using high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 907, 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Soroka D. N.; Hu Y.; Chen X.; Sang S. Characterization of thiol-conjugated metabolites of ginger components shogaols in mouse and human urine and modulation of the glutathione levels in cancer cells by [6]-shogaol. Molecular Nutr. Food Res. 2013, 57, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway C. C.; Krzeminski J.; Amin S.; Chung F. L. Decomposition rates of isothiocyanate conjugates determine their activity as inhibitors of cytochrome p450 enzymes. Chem. Res. Toxicol. 2001, 14, 1170–1176. [DOI] [PubMed] [Google Scholar]
- Chen H.; Soroka D. N.; Zhu Y.; Hu Y.; Chen X.; Sang S. Cysteine-conjugated metabolite of ginger component [6]-shogaol serves as a carrier of [6]-shogaol in cancer cells and in mice. Chem. Res. Toxicol. 2013, 26, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussi T.; Lozano G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [DOI] [PubMed] [Google Scholar]
- Toledo F.; Wahl G. M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [DOI] [PubMed] [Google Scholar]
- Seemann S.; Maurici D.; Olivier M.; Caron de Fromentel C.; Hainaut P. The tumor suppressor gene TP53: Implications for cancer management and therapy. Crit. Rev. Clin. Lab. Sci. 2004, 41, 551–583. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
- Brandt R.; Keston A. S. Synthesis of diacetyldichlorofluorescin: A stable reagent for fluorometric analysis. Anal. Biochem. 1965, 11, 6–9. [DOI] [PubMed] [Google Scholar]
- Qin H.; Yu T.; Qing T.; Liu Y.; Zhao Y.; Cai J.; Li J.; Song Z.; Qu X.; Zhou P.; Wu J.; Ding M.; Deng H. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J. Biol. Chem. 2007, 282, 5842–5852. [DOI] [PubMed] [Google Scholar]
- Speidel D.; Helmbold H.; Deppert W. Dissection of transcriptional and non-transcriptional p53 activities in the response to genotoxic stress. Oncogene 2006, 25, 940–953. [DOI] [PubMed] [Google Scholar]
- Tao Y.; Li W.; Liang W.; Van Breemen R. B. Identification and quantification of gingerols and related compounds in ginger dietary supplements using high-performance liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2009, 57, 10014–10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waster P. K.; Ollinger K. M. Redox-dependent translocation of p53 to mitochondria or nucleus in human melanocytes after UVA- and UVB-induced apoptosis. J. Invest. Dermatol. 2009, 129, 1769–1781. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Chaiswing L.; Velez J. M.; Batinic-Haberle I.; Colburn N. H.; Oberley T. D.; St Clair D. K. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005, 65, 3745–3750. [DOI] [PubMed] [Google Scholar]
- Cerella C.; Radogna F.; Dicato M.; Diederich M. Natural compounds as regulators of the cancer cell metabolism. Int. J. Cell Biol. 2013, 2013, 639401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S.; Flescher E. Methyl jasmonate: A plant stress hormone as an anti-cancer drug. Phytochemistry 2009, 70, 1600–1609. [DOI] [PubMed] [Google Scholar]
- Elkady A. I.; Abuzinadah O. A.; Baeshen N. A.; Rahmy T. R. Differential control of growth, apoptotic activity, and gene expression in human breast cancer cells by extracts derived from medicinal herbs Zingiber officinale. J, Biomed. Biotechnol. 2012, 2012, 614356. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Karna P.; Chagani S.; Gundala S. R.; Rida P. C.; Asif G.; Sharma V.; Gupta M. V.; Aneja R. Benefits of whole ginger extract in prostate cancer. Br. J. Nutr. 2012, 107, 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlikova B.; Diederich M. Power from the garden: Plant compounds as inhibitors of the hallmarks of cancer. Curr. Med. Chem. 2012, 19, 2061–2087. [DOI] [PubMed] [Google Scholar]
- Stoner G. D. Ginger: Is it Ready for Prime Time?. Cancer Prev. Res. 2013, 6, 257–262. [DOI] [PubMed] [Google Scholar]
- Citronberg J.; Bostick R.; Ahearn T.; Turgeon D. K.; Ruffin M. T.; Djuric Z.; Sen A.; Brenner D. E.; Zick S. M. Effects of Ginger Supplementation on Cell-Cycle Biomarkers in the Normal-Appearing Colonic Mucosa of Patients at Increased Risk for Colorectal Cancer: Results from a Pilot, Randomized, and Controlled Trial. Cancer Prev. Res. 2013, 6, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogosawa S.; Yamada Y.; Yasuda S.; Sun Q.; Takizawa K.; Sakai T. Dehydrozingerone, a structural analogue of curcumin, induces cell-cycle arrest at the G2/M phase and accumulates intracellular ROS in HT-29 human colon cancer cells. J. Nat. Prod. 2012, 75, 2088–2093. [DOI] [PubMed] [Google Scholar]
- Ramos S. Cancer chemoprevention and chemotherapy: Dietary polyphenols and signalling pathways. Mol. Nutr Food Res. 2008, 52, 507–526. [DOI] [PubMed] [Google Scholar]
- Park G.; Kim H. G.; Ju M. S.; Ha S. K.; Park Y.; Kim S. Y.; Oh M. S. 6-Shogaol, an active compound of ginger, protects dopaminergic neurons in Parkinson’s disease models via anti-neuroinflammation. Acta Pharmacol. Sin. 2013, 34, 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Soroka D.; Zhu Y.; Sang S. Metabolism of ginger component [6]-shogaol in liver microsomes from mouse, rat, dog, monkey, and human. Molecular Nutr. Food Res. 2013, 57, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Li C.-T.; Kong A.-N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharmacal Res. 2005, 28, 249–268. [DOI] [PubMed] [Google Scholar]
- Wang B.; Xiao Z.; Ko H. L.; Ren E. C. The p53 response element and transcriptional repression. Cell Cycle (Georgetown, Tex.) 2010, 9, 870–879. [DOI] [PubMed] [Google Scholar]
- Vousden K. H.; Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [DOI] [PubMed] [Google Scholar]
- Lane D.; Levine A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harbor Perspect. Biol. 2010, 2, a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garritano S.; Inga A.; Gemignani F.; Landi S. More targets, more pathways and more clues for mutant p53. Oncogenesis 2013, 2, e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O.; Prives C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [DOI] [PubMed] [Google Scholar]
- Vazquez A.; Bond E. E.; Levine A. J.; Bond G. L. The genetics of the p53 pathway, apoptosis and cancer therapy. . Nat. Rev. Drug Discovery 2008, 7, 979–987. [DOI] [PubMed] [Google Scholar]
- Youle R. J.; Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. . Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [DOI] [PubMed] [Google Scholar]
- Nakano K.; Vousden K. H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [DOI] [PubMed] [Google Scholar]
- Speidel D. Transcription-independent p53 apoptosis: An alternative route to death. Trends Cell Biol. 2010, 20, 14–24. [DOI] [PubMed] [Google Scholar]
- Ozben T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [DOI] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [DOI] [PubMed] [Google Scholar]