

Abstract
Mcl-1, an antiapoptotic member of the Bcl-2 family of proteins, is a validated and attractive target for cancer therapy. Overexpression of Mcl-1 in many cancers results in disease progression and resistance to current chemotherapeutics. Utilizing high-throughput screening, compound 1 was identified as a selective Mcl-1 inhibitor and its binding to the BH3 binding groove of Mcl-1 was confirmed by several different, but complementary, biochemical and biophysical assays. Guided by structure-based drug design and supported by NMR experiments, comprehensive SAR studies were undertaken and a potent and selective inhibitor, compound 21, was designed which binds to Mcl-1 with a Ki of 180 nM. Biological characterization of 21 showed that it disrupts the interaction of endogenous Mcl-1 and biotinylated Noxa-BH3 peptide, causes cell death through a Bak/Bax-dependent mechanism, and selectively sensitizes Eμ-myc lymphomas overexpressing Mcl-1, but not Eμ-myc lymphoma cells overexpressing Bcl-2. Treatment of human leukemic cell lines with compound 21 resulted in cell death through activation of caspase-3 and induction of apoptosis.
Introduction
Evasion of apoptosis or programmed cell death, a key regulator of physiological growth control and regulation of tissue homeostasis, is a hallmark of cancer and a contributor to the emergence of resistance to current therapies.1−3 The B-cell lymphoma-2 (Bcl-2) family of proteins regulate the intrinsic (mitochondrial) pathway of apoptosis through a network of protein–protein interactions between pro- and antiapoptotic members.4 Twenty-five known members of the family can be grouped functionally according to their pro- and antiapoptotic effects as well as structurally according to their Bcl-2 homology (BH) regions. The Bcl-2 antiapoptotic proteins, consisting of Bcl-2, Bcl-XL, Bcl-b, Bcl-w, Mcl-1, and A1, share up to four BH domains which form the hydrophobic BH3-binding groove for binding their cognate partners. The pro-apoptotic proteins are divided into two groups: (a) multidomain proteins including Bax and Bak with BH1–BH4 domains and (b) BH3-only proteins including Bad, Bid, Bim, Noxa, and Puma, which share homology only in the BH3 α-helical domain. The BH3 domain possesses four conserved hydrophobic residues that are involved in the interaction with BH3-binding groove of the pro-survival Bcl-2 family members, which results in the sequestering and blocking of the function of pro-death members.
Overexpression of Bcl-2 survival members is observed in different types of human tumor samples and cancer cell lines, and much effort has been focused on developing therapeutics against this family of proteins for the treatment of cancers.5−7 Selective and potent small-molecule inhibitors have been successfully developed against Bcl-2,8 Bcl-XL,9 and Bcl-2/Bcl-XL10−13 with Navitoclax (ABT-263), currently in phase I/II clinical trials. The efficacy of ABT-263 as a single agent has been demonstrated in tumors with low levels of the pro-survival Mcl-1. Several studies14−18 have shown that resistance to ABT-263 and its analogue ABT-737 is linked to high expression levels of Mcl-1, and in many instances this resistance can be overcome by treatment with agents that downregulate, destabilize, or inactivate Mcl-1. The biological significance of Mcl-1 protein expression in support of cell survival has been well documented in a number of cell systems, including human myeloblastic leukemia,19 myeloma,20−22 B-lymphoma,23 nonsmall cell lung,24 melanoma,25 pancreatic,26,27 and prostate.28 Furthermore, Mcl-1 is overexpressed in many human tumor specimens26,29−32 and metastatic tissue.33 This overexpression contributes to chemoresistance and disease relapse.34−36 It has been shown that Mcl-1 down-regulation is important toward making multiple myeloma cells susceptible to BH3-only proteins and therefore to mitochondrial disruption.37,38 Such down-regulation can increase the sensitivity to rituximab-mediated killing of chronic and acute lymphoid leukemia (CLL and ALL).39 Antisense strategies targeting Mcl-1 in vitro and in vivo have provided promising results in sensitizing human melanoma and pancreatic cancer.40−42 These data suggest that therapies specifically targeting Mcl-1, either as a single agent or in combination, can be effective in the treatment of different human cancers.
Recently, several groups including us have reported on small molecules43−50 and stapled peptides51,52 as Mcl-1 inhibitors. Herein we disclose a novel series of small-molecule Mcl-1 inhibitors discovered through high-throughput screening (HTS) followed by the utilization of structure-based design to develop structure–activity relationships (SAR) around lead compound 1 (Figure 1A). Docking studies and two-dimensional 1H– 15N heteronuclear single quantum coherence spectroscopy (HSQC) NMR studies were employed to provide information about its binding mode which was used for the design and synthesis of additional analogues. A SAR was developed which resulted in the identification of several selective small-molecule Mcl-1 inhibitors with improved binding. Selected analogues were then tested in a series of complementary biochemical, biophysical, functional, and cellular assays to evaluate their potency, specificity, and mechanism of action.
Figure 1.

(A) Structure of the HTS lead compound 1. (B) Putative binding mode of 1 to Mcl-1(PDB ID: 2NLA). Side chains residues of mNoxa peptide are shown in blue sticks. The surface of Mcl-1 protein is colored according to the chemical shift intensity. Significant shift (>0.09 ppm) is represented with purple, moderate shift (≥0.03 and ≤0.09 ppm) represented with pink. (C) Overlaid 15N–1H HSQC spectra of Mcl-1 (red) and in the presence of 1 (Mcl-1:1 ratio of 1:2) (black), (Mcl-1:1 ratio of 1:1) (purple). Arrows show the direction of chemical shift changes upon binding of 1. (D) Plot of chemical shift changes of Mcl-1 amide upon addition of 1 (Mcl-1:1 ratio of 1:2) as a function of Mcl-1 residue numbers.
Results and Discussion
Lead Discovery
HTS of a 53.3K small-molecule library was performed using a fluorescence polarization (FP) binding assay based on the interaction between recombinant human Mcl-1 and fluorescently labeled Bid BH3 peptide (Flu-Bid). Compound 1 (Figure 1A) was one of the validated hits, which was also previously identified as a proteasome inhibitor.53 Compound 1 was resynthesized and its binding to Mcl-1 was confirmed with a Ki of 1.55 ± 0.18 μM. Compound 1 exhibits similar potency as MIM1 (IC50 = 4.72 μM), Mcl-1 small-molecule inhibitor with a thiazolyl substituted core, which was also discovered with high-throughput competitive FP screen approach.47 It is known that the binding efficiency index (BEI) is important in identifying leads that exhibit good potency relative to their size.54 The BEI of compound 1, calculated as a ratio between pKi and molecular weight, is 14.7, which encouraged us to further pursue with modifications of this scaffold. To explore the binding mode of 1 with Mcl-1, in silico induced fit docking (IFD)55 studies were performed using the crystal structure of Mcl-1 in complex with mNOXA BH3 peptide (PDB ID: 2NLA).56 The predicted binding model of 1 in complex with Mcl-1 revealed that the thiophene and the naphthalene rings of 1 occupy two hydrophobic pockets, h2 and h3, in the Mcl-1 protein, mimicking two conserved hydrophobic residues in the BH3 binding motif represented by Leu 78 and Ile 81, respectively, in mNoxaB (Figure 1B). The importance of the two conserved hydrophobic residues, Leu and Ile, in the BH3 binding motif for the high affinity interaction and selective binding to Mcl-1 has been demonstrated with structural studies of Bims2A,57 a highly selective peptide derived from Bim BH3-only protein, as well as from recently reported selective small-molecule inhibitor in complex with Mcl-1.45 The carboxylic acid group of 1 forms a network of hydrogen bonds with Arg 263 and Asn 260 of Mcl-1, mimicking the conserved Asp in BH3 peptides (Asp 83 in mNoxa). The predicted binding model also suggests that the phenolic group forms a hydrogen bond with His 224, which is one of the residues composing the h3 pocket of Mcl-1. A recent report58 on the conformational flexibility of Mcl-1 and its binding hotspots identified His 224 as an acidic hotspot in the h3 site of Mcl-1, supporting the predicted hydrogen bonding in this region of Mcl-1.
To validate the computationally predicted binding site and confirm the binding of 1 to the BH3 groove of Mcl-1 protein, HSQC NMR spectroscopy studies were performed. For this purpose, the assignment of the backbone amides of apo human Mcl-1 was based on the work by Liu et al.,59 and a series of 1H,15N-HSQC studies were carried out. The HSQC spectra were of good quality with well-dispersed peaks, and concentration-dependent perturbations of residues were observed (Figure 1C), indicating that 1 binds Mcl-1 specifically and causes dose-dependent perturbations of backbone amides. The chemical shift changes in the presence of a 2-fold excess of 1 were mapped and plotted against Mcl-1 residues (Figure 1D). Compound 1 caused moderate to significant chemical shift perturbations for residues of Met 231, Met 250, Leu 267, and Phe 270, which constitute the h2 and h3 pockets and which are predicted to be occupied by the thiophene and naphthalene rings of 1, respectively. Moderate chemical shift perturbations of Arg 263 and His 224 were also observed, which are predicted to form hydrogen bonds with the thioacidic acid and phenolic moieties of 1, respectively. Additionally, the residues in the vicinity of the predicted binding pose (Leu 232, Val 243, Arg 248) and the ones located on an unstructured loop connecting α-helix 3 to α-helix 4 (Lys 234, Leu 235, Lys 238, Asn 239) were also perturbed. Overall analysis of the chemical shift changes of the compound 1 in complex with Mcl-1 showed that 1 affects the residues forming the BH3-binding groove and provided conclusive evidence that 1 binds Mcl-1 protein at the same site that the conserved BH3 peptides interact with Mcl-1 protein. Therefore, on the basis of our modeling and NMR results, the substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamide represents a promising class for further optimization. A structure-based design approach was undertaken, and a focused library of analogues of 1 was designed and synthesized to improve the potency of this series.
Synthesis
Except for analogues 4–8, 15, and 39, which were commercially available, all analogues were synthesized through a novel, modular route (Scheme 1), which differs from that recently reported by Ge et al.53 Our route allows for facile access to a variety of analogues with variations at R1, R2, and R3 and the linker region (X). It starts with an electrophilic aromatic substitution of 1-methoxy-4-nitronaphthalene with N-iodosuccinamide to provide aryl iodide (47).60 This was subjected to Pd-catalyzed C–S or C–C cross-coupling using conditions previously reported,61,62 or developed in our lab based on recent literature,63−65 to provide several desired intermediates (48). The nitro function was reduced with iron66 or via catalytic hydrogenation67 to provide the corresponding amines. Reaction of the amines with appropriate sulfonyl or acyl chlorides or 3-methyl-1-((4-phenylpiperazin-1-yl)sulfonyl)-1H-imidazol-3-ium68 provided the penultimate compounds (49), which were demethylated with BBr3 followed by purification by trituration or reverse-phase HPLC to afford the target compounds (1–3, 9–14, 16–23, 25–26, 28–29, 31–38) of >95% purity (Tables 1–3). In the case of analogues with an ester side chain, the BBr3 step provided a convenient way to concomitantly hydrolyze the ester in a single pot. To preserve the methyl ester of analogue 32 (Table 3), analogue 41 (Table 4) was subjected to BBr3 conditions followed by a quench with MeOH. For the synthesis of 49o, intermediate 49k underwent Suzuki–Miyaura coupling69,70 with phenyl boronic acid to provide the desired compound which was subjected to BBr3 to provide 17 (Table 1). Aminolysis of 41 and 49s with NH4OH71 provided carboxamides 42 (Table 4) and 49aa, respectively. 42 was then subjected to BBr3 to give 33 (Table 3). The terminal amide of 42 and 49aa were converted by a known procedure72 to tetrazoles 49cc and 49dd, which after BBr3 demethylation provided 36 and 37 (Table 3), respectively. The thioacidic acid analogues with a methoxy at R3 (40, 43–44) (Table 4) were obtained via LiOH hydrolysis of esters 41, 49n, and 49o, respectively. Acetylation of 10 (Table 1) with acetyl chloride73 provided 46 (Table 4). Analogue 24 (Table 1) with a phenyl scaffold in place of naphthalene was synthesized using conditions similar to those described for 10 starting from 2-iodoanisole (Supporting Information Scheme S1). The syntheses of compounds 27 (Table 2) and 30 (Table 3) were completed starting from 4-methoxy-1-naphthaldehyde and 1-nitronaphthalene, respectively (Supporting Information Schemes S2–S3).
Scheme 1. Synthetic Route for 1 and Analogues.

Reagents and conditions: (a)
NIS, TFA, reflux, 24 h; (b) HS(CH2)nCOOCH3 (n = 1, 2), Pd(OAc)2, Xantphos, Cs2CO3, LiI, ZnCl2, THF, 60 °C, overnight, or HS(CH2)3CH3, Pd2(dba)3, Dppf, Et3N,
NMP, 80 °C, 2 h, or HCC(CH2)nOH (n = 1, 2), Pd(PPh3)2Cl2, CuI, Et3N/THF, 60 °C, 2 h (4:1),
60 °C, 2 h; (c) Fe, AcOH, 70 °C, 1 h, or Pd/C, H2 30 psi, EtOH/EtOAc (6:1), rt, overnight; (d) RSO2Cl,
pyridine, CH2Cl2, rt, overnight, or RCOCl, Et3N, CH2Cl2, rt, overnight, or 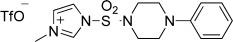 , CH3CN, 80 °C, 15
h; (e) BBr3, CH2Cl2, 0 °C to
rt, 1 h, or BBr3, CH2Cl2, 0 °C
to rt, 1 h, quench with MeOH at 0 °C; (g) phenyl boronic acid,
Pd(PPh3)4, Na2CO3, THF/H2O, 60 °C, 2 h; (h) NH4OH, rt, 1 h; (i) NaN3, SiCl4, CH3CN, 80 °C, 15 h; (j)
LiOH, THF, rt, 1 h; (k) H3CCOCl, Et3N, 0 °C,
rt, 30 min.
, CH3CN, 80 °C, 15
h; (e) BBr3, CH2Cl2, 0 °C to
rt, 1 h, or BBr3, CH2Cl2, 0 °C
to rt, 1 h, quench with MeOH at 0 °C; (g) phenyl boronic acid,
Pd(PPh3)4, Na2CO3, THF/H2O, 60 °C, 2 h; (h) NH4OH, rt, 1 h; (i) NaN3, SiCl4, CH3CN, 80 °C, 15 h; (j)
LiOH, THF, rt, 1 h; (k) H3CCOCl, Et3N, 0 °C,
rt, 30 min.
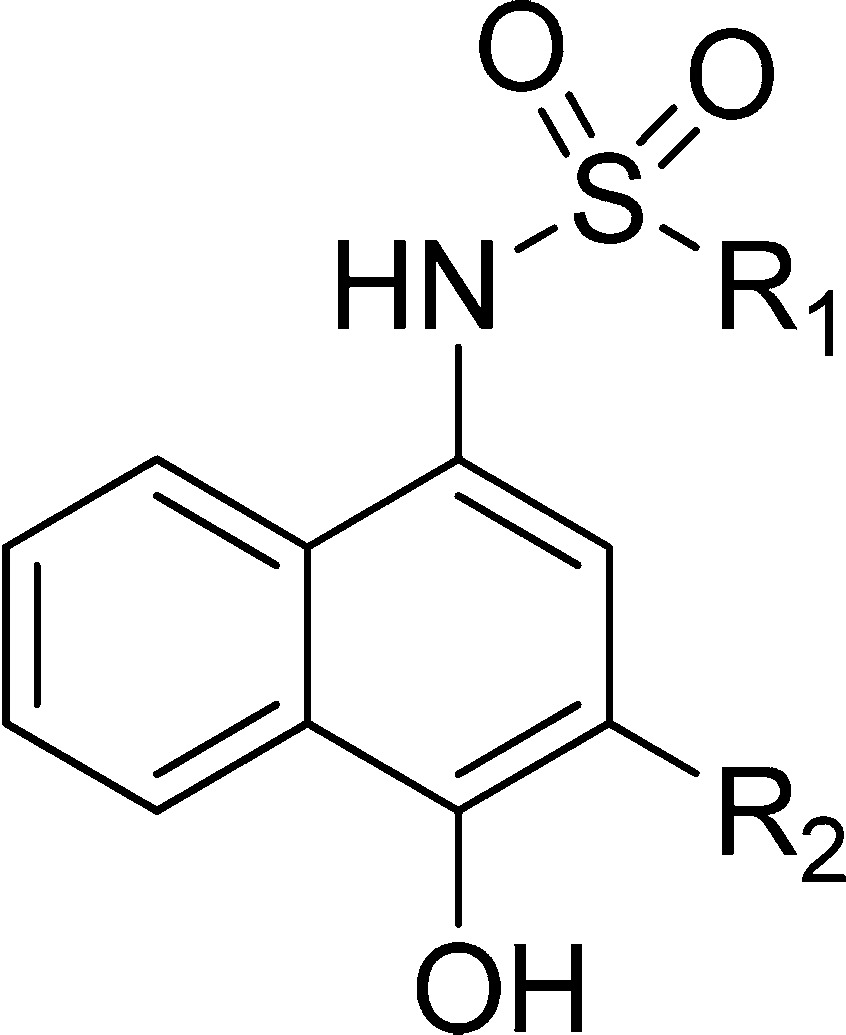
Table 1. Binding Affinities of Sulfonamide Analogues with Variations at R1.
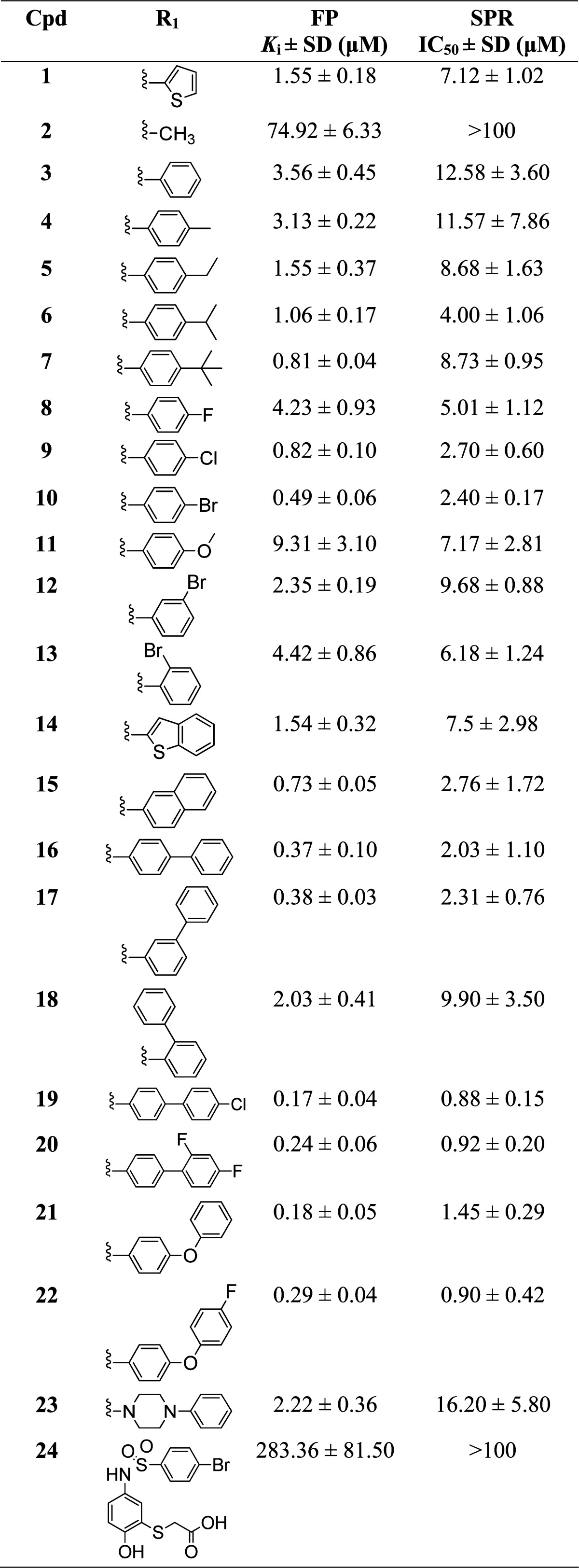
Table 3. Binding Affinities of Sulfonamide Analogues with Variations at R1 and R2a.
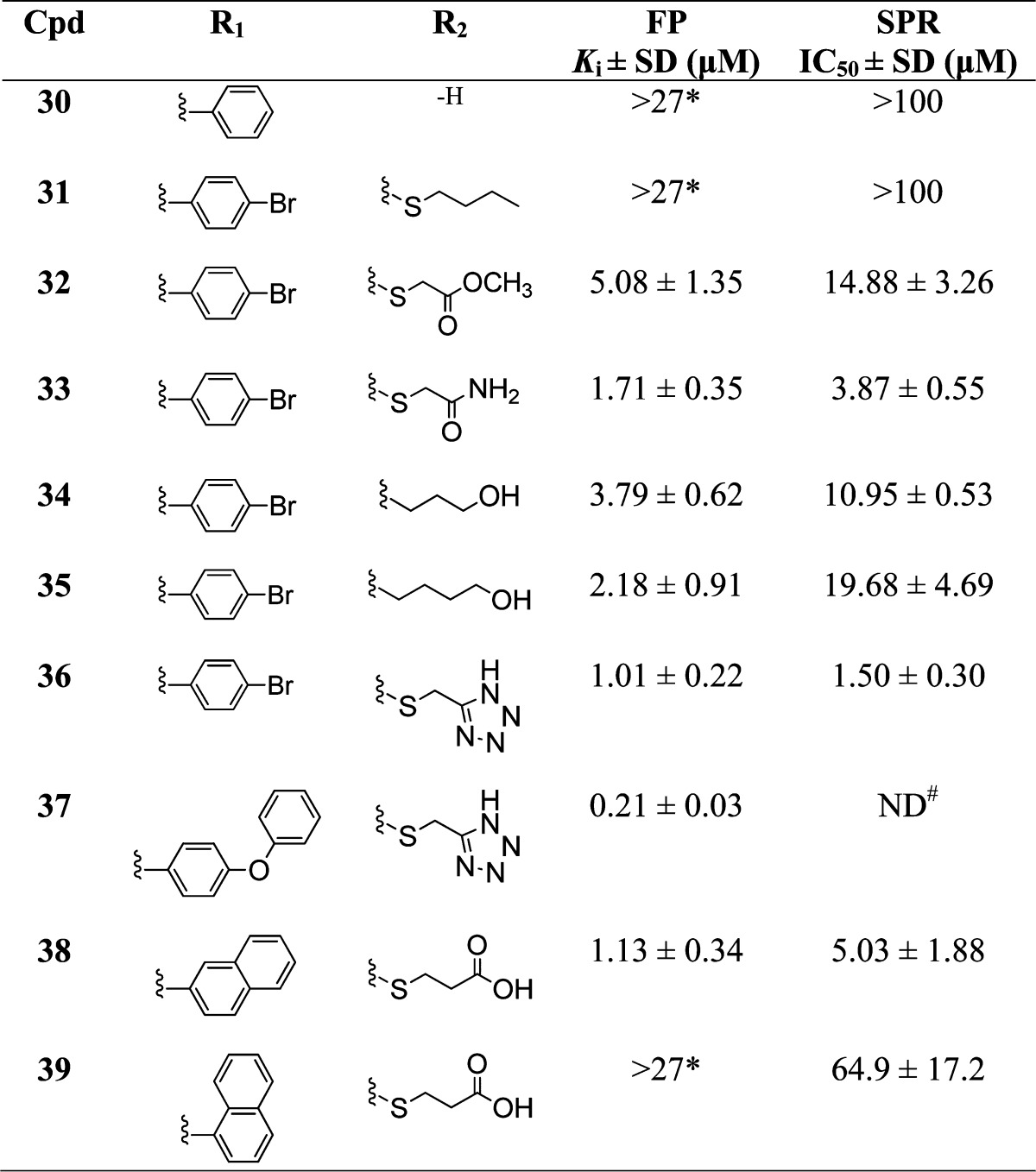
#ND: The IC50 was not determined because the compound showed nonspecific binding to the control surface; *Compounds were tested up to 100 μM.
Table 4. Binding Affinities of Sulfonamide Analogues with Variations at R1, R2, and R3a.
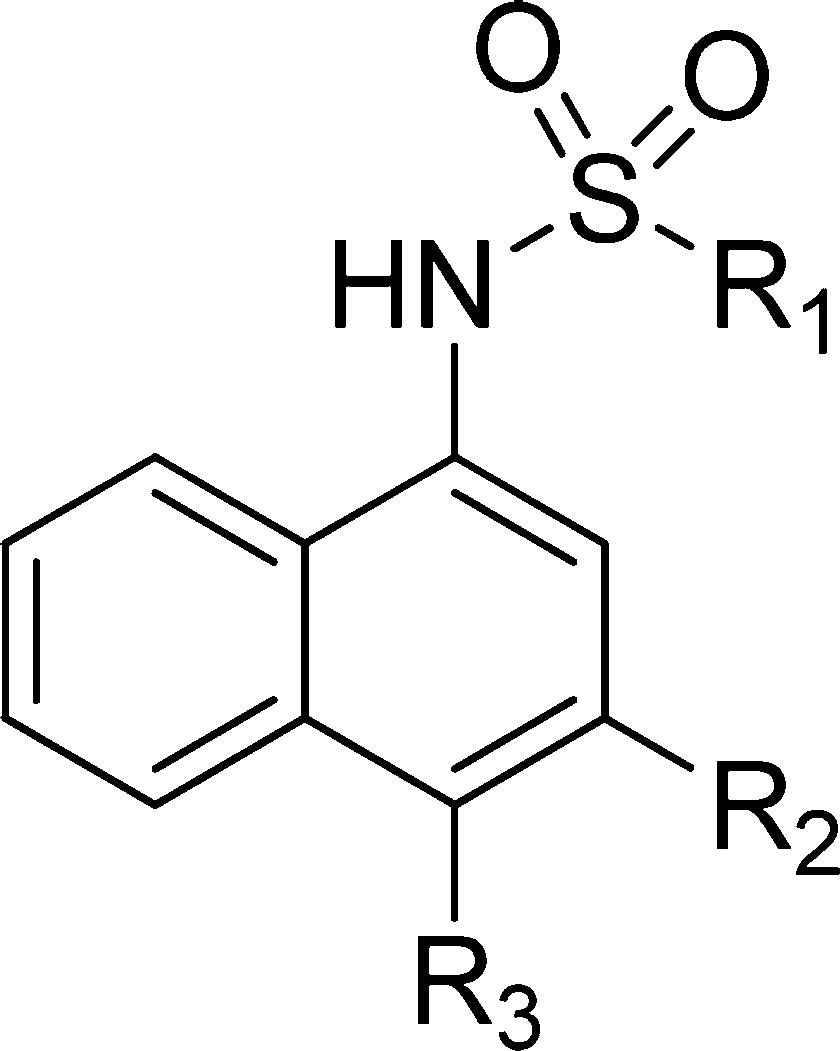
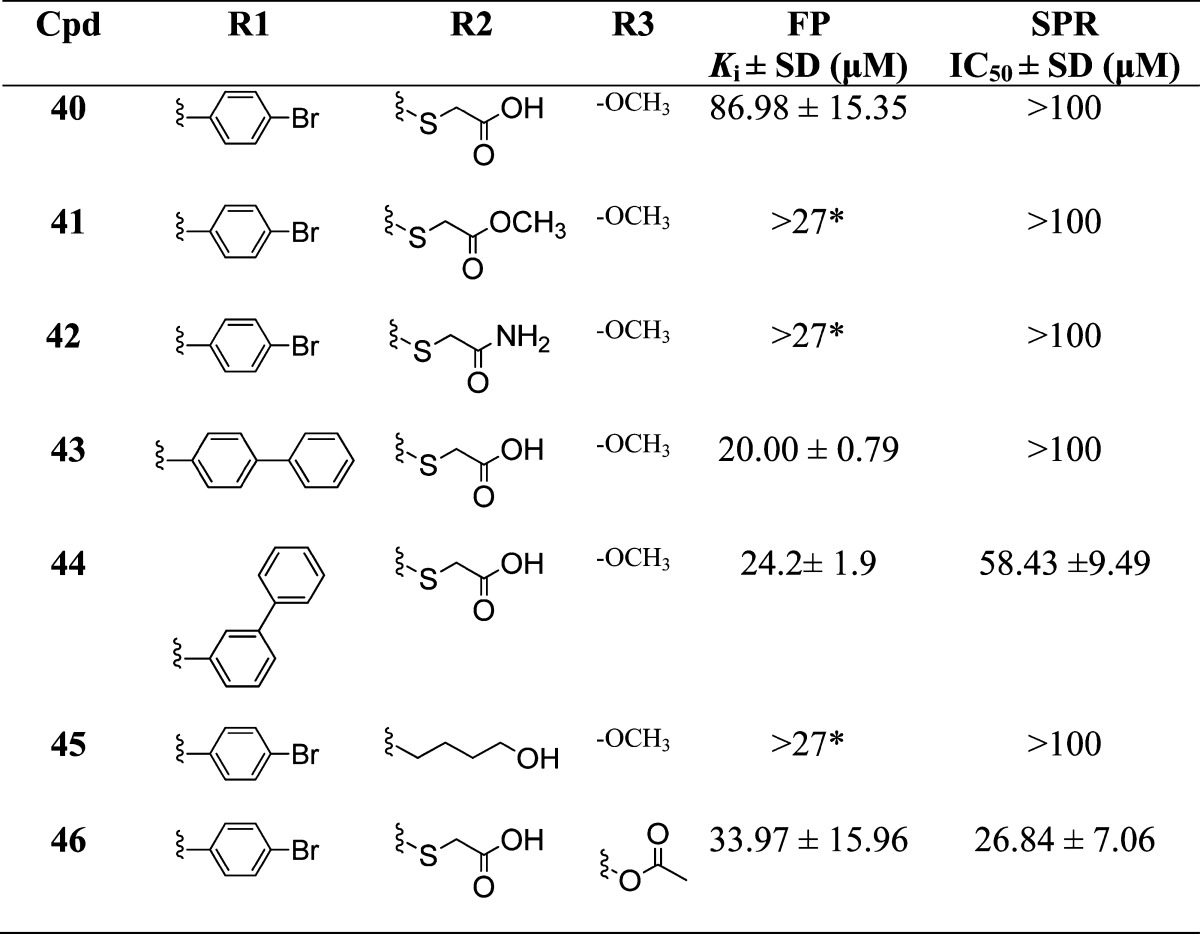
*Compounds were tested up to 100 μM.
Table 2. Binding Affinities of Analogues with Linker (X) Variations.
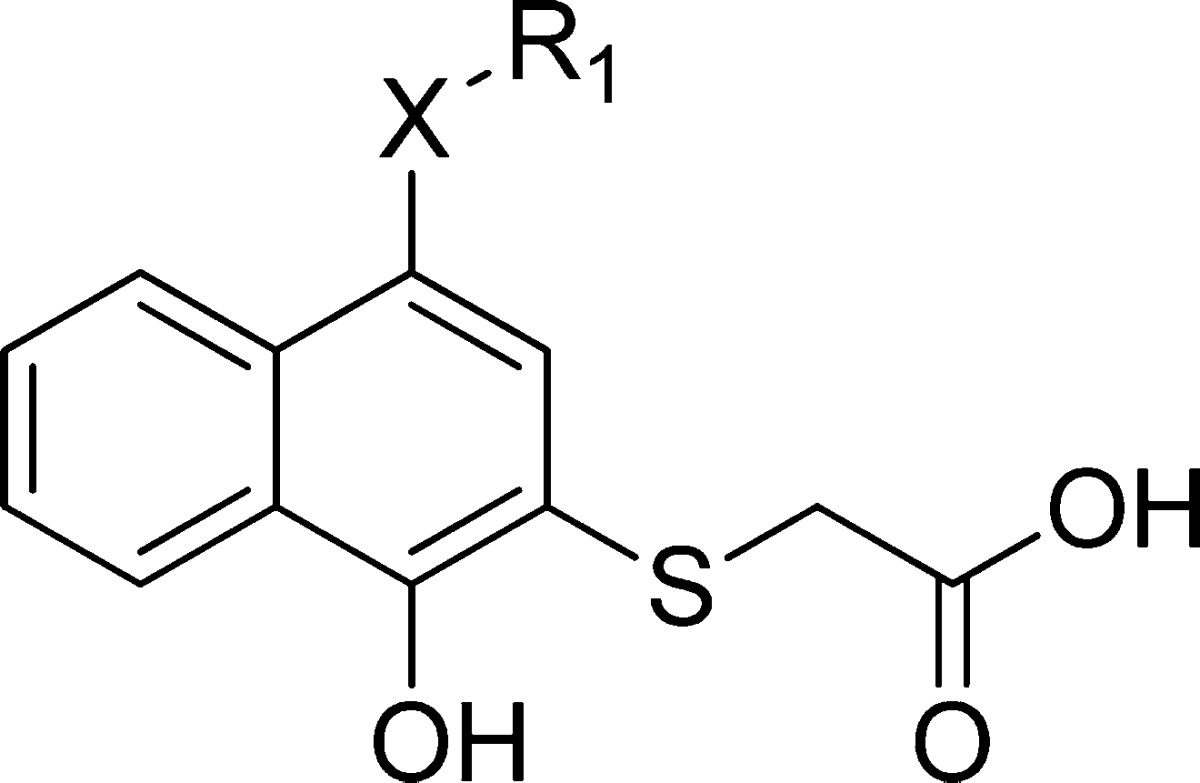
Structure–Activity Relationships
Structure-based design of analogues based on 1 yielded a focused library of compounds leading to clear SAR for this series. The binding affinities of our Mcl-1 inhibitors were determined by using competitive fluorescence polarization (FP) and surface plasmon resonance (SPR) binding assays, which test the ability of inhibitors to disrupt interaction between Mcl-1 and two different BH3 peptides, fluorescently labeled Bid and biotin-labeled Bim, respectively. Concurrently, HSQC NMR experiments were performed to provide structural insights of protein-bound ligand and experimental validation for the modeling studies.
The predicted binding model showed that the thiophene ring at R1 of 1 projects into the h2 pocket (Figure 1B), which is the biggest and deepest pocket among the four hydrophobic pockets of Mcl-1.56 To investigate the importance of hydrophobic interaction at this site and increase the binding affinity of 1, a series of analogues with variation at R1 was synthesized and evaluated (Table 1). When R1 is changed to a methyl group in 2, the binding affinity is significantly reduced, confirmed by SPR (IC50 > 100 μM) and NMR experiments which showed lack of chemical shift perturbation of backbone residues in the Mcl-1 BH3 binding site after adding 2 (Supporting Information Figure S1). As was expected, isosteric replacement of the thiophene in 1 to a phenyl in 3 maintained binding affinity with Ki of 3.56 ± 0.45 μM. To probe the hydrophobic interactions in the h2 pocket, analogues with alkyl and halogen substituents of various sizes at the para-position of the phenyl ring were prepared. From this set of compounds, a trend emerged showing improved binding with increasing size and hydrophobicity of substituents. tert-Butyl phenyl (7) exhibited 4.5-fold enhancement in FP assay (Ki = 0.81 ± 0.04 μM) and 1.5-fold in SPR assay (IC50 = 8.73 ± 0.95 μM), and bromo phenyl (10) 7-fold in FP assay (Ki = 0.49 ± 0.06 μM) and 5-fold in SPR assay (IC50 = 2.40 ± 0.17 μM) improved binding over 3. Introduction of a more polar methoxy group in 11 was accommodated but resulted in a 3-fold decreased binding (Ki = 9.31 ± 3.1 μM) compared to 3. Bromine substitution at the meta-(12) and ortho-positions (13) of the phenyl ring was also explored, and the obtained binding results from both assays, FP (Ki of 2.35 ± 0.19 μM and 4.42 ± 0.86 μM, respectively) and SPR (IC50 = 9.68 ± 0.88 μM and IC50 = 6.18 ± 1.24 μM, respectively), indicated that para-bromo substitution in 10 is the best, exhibiting the highest binding affinity to Mcl-1 among these three isomers. Because of peak overlap and possibility of ambiguous assignment of the peaks in the presence of a ligand and to improve the quality and accuracy of ligand-induced Mcl-1 chemical shift perturbation, resonance assignments were also determined for 10 in complex with Mcl-1 using 13C,15N double-labeled Mcl-1 protein in the presence of a 2-fold excess of 10. The HSQC spectrum of 10 showed a similar pattern of chemical shift perturbation as with 1, which we attribute to the structural similarity of the two analogues. Consistent with the 7-fold improved binding of 10 in comparison with 1, the chemical shift perturbations were larger for 10 (Supporting Information Figure S2A). In particular, significant perturbation of residues Ser 247, Arg 248, and Val 253 on α-helix 4, which forms the upper rim of the h2 pocket, and residues Val 216, Val 220, and Gln 221 located toward the C-terminus of α-helix 2, which are at the border between the h3 and h4 pockets, were observed for 10, suggesting that the Mcl-1 conformational flexibility accommodates larger R1 substituent into the h2 pocket. Chemical shift perturbation plots derived from 1H,15N-HSQC experiments of the other two bromophenyl isomers showed a similar perturbation pattern as 10 with chemical shift changes that correlate well with the binding data in which the strongest perturbation is observed for 10, followed by 12 and 13 (Supporting Information Figure S2A–C).
To further extend into the h2 pocket and gain additional interaction, analogues with biphenyl substituents were synthesized because biphenyl is considered a privileged moiety in targeting protein–protein interactions.74 Analogues 16 and 17, with para- and meta-biphenyl substituents, respectively, showed a similar 10-fold improvement in FP-based binding assay compared to 3 with Ki values of 0.37 ± 0.10 and 0.38 ± 0.03 μM, respectively, further confirmed with SPR assay showing 6-fold improvement. On the other hand, the ortho-biphenyl analogue 18 showed almost the same binding affinity as 3 with Ki of 2.03 ± 0.41 μM and IC50 = 9.90 ± 3.50 μM, in FP and SPR assays, respectively. The predicted binding models of these compounds showed that the distal phenyl ring of 16 and 17 inserts deeper into the h2 pocket (parts A and B of Figure 2, respectively). For 18, this phenyl ring is partially solvent exposed (Figure 2C), which might explain its lower affinity compared to 16 and 17. In agreement with their binding affinities, chemical shift perturbation plots of 16 and 17 show stronger perturbation of residues involved in the binding site compared to 18 (Figure 2D–F).
Figure 2.

Putative binding modes of (A) 16, (B) 17, (C) 18 to Mcl-1 (PDB ID: 2NLA). Surface of Mcl-1 protein is colored according to the chemical shift intensity. Significant shift (>0.09 ppm) is represented with purple, moderate shift (≥0.03 and ≤0.09 ppm) represented with pink. Plots of chemical shift changes of Mcl-1 amide upon addition of (D) 16 (Mcl-1:16 ratio of 1:2), (E) 17 (Mcl-1:17 ratio of 1:2), (F), and 18 (Mcl-1:18 ratio of 1:2) as a function of Mcl-1 residue numbers.
Analysis of the HSQC spectra of analogues 16–18 resulted in an important finding that strongly supports their predicted binding mode. One long-standing question for us was the possibility of a flipped binding mode for this class of analogues where R1 would occupy the h4 pocket instead of h2. The possibility of a flipped conformation has been previously documented for a different class of dual Mcl-1 and Bcl-xL inhibitors.49 The differential chemical shift mapping method75,76 was applied, using the three biphenyl analogues (16, 17, and 18) differing only in the orientation of distal phenyl ring. Comparison of the spectra of the Mcl-1 bound to these three analogues shows distinct changes in chemical shifts exclusively for the resonances of the residues that are in direct contact with the modified structure of the inhibitors, facilitating binding site mapping. Careful analysis of the HSQC spectra of these analogues reveals a different direction of the chemical shift perturbations of the residues forming the h2 pocket (Met 250) or in its vicinity (Phe 228) as well as residues located on α-helix 4 (Val 249, Val 253) (Figures 3A–C). On the other hand, residues which are not part of the h2 pocket, such as Leu267, Val265, His224, and Arg263, show very similar chemical shift perturbations in the presence of the biphenyl analogues (Supporting Information Figure S6). This finding provides conclusive evidence for biphenyl occupation of the h2 pocket and further supports that α-helix 4 of Mcl-1 is flexible.58
Figure 3.

Overlaid 15N–1H HSQC spectra of Mcl-1 (red) and in the presence of 16 (Mcl-1:16 ratio of 1:2) (purple), 17 (Mcl-1:17 ratio of 1:2) (blue), 18 (Mcl-1:18 ratio of 1:2) (green) for (A) Phe 228, (B) Met 250, and (C) Val 249 and Val 253. Arrows show the direction of chemical shift changes upon binding of compounds. (D) Overlay of putative binding modes of 16 (purple), 17 (blue), and 18 (green) to Mcl-1 (PDB ID: 2NLA) highlighting in red Val 249, Met 250, and Val 253 on helix 4, Phe 228 on helix 3 of Mcl-1.
Because of encouraging binding data with 16, analogues 19 and 20 with halogenated biphenyl rings at R1 were synthesized to further increase potency. Satisfyingly, 19 with para-chlorobiphenyl showed a 21-fold improved binding compared to 3, becoming the most potent analogue in our series, with a Ki = 0.17 ± 0.04 μM determined by the FP binding assay using fluorescent labeled Bid BH3 peptide and IC50 of 0.88 ± 0.15 μM in displacement of biotin labeled Bim BH3 peptide in the SPR-based assay (14-fold improvement). The chemical shift perturbation plot of 19 (Supporting Information Figure S3) showed a similar pattern of perturbations to 16, suggesting a similar binding conformation but with a lower perturbation magnitude, likely due to the lower solubility of 19 in our NMR studies. To improve the physicochemical properties of the biphenyl analogues, we synthesized and evaluated analogues 21 with para-phenoxyphenyl at R1 and its fluorine substituted congener, 22. Analogue 21 showed similar binding affinity as 19 (Ki = 0.18 ± 0.05 μM in FP, and IC50 = 1.45 ± 0.29 μM in SPR) but stronger chemical shift perturbation (Figure 4A–C), probably due to its improved solubility. Computational docking of 21 predicted a π–π stacking of the distal phenyl of the para-phenoxyphenyl moiety with the phenyl of Phe270 of Mc-1 (Figure 4D), which could account for its improved affinity. Compounds 19 and 21 became the most potent analogues of our series and exhibited BIEs of 13.5 and 14.0, respectively, maintaining 1’s BIE. In comparison with compound 53 (Ki of 0.055 μM), recently reported Mcl-1 selective inhbitor with an indole core structure, discovered and optimized by fragment-based screening strategy,4519 and 21 have binding affinity in a similar nanomolar range, being 3-fold less potent than 53. To further increase the aqueous solubility of this class of analogues, a para-phenylpiperazine group at R1 was introduced in 23 which led to a 6-fold decrease in binding based on FP assay (Ki = 2.22 ± 0.36 μM) and 8-fold decrease based on SPR assay (IC50 = 16.20 ± 5.80 μM) in comparison with the biphenyl substituent in 16. This can be attributed to a relatively polar substitutent projecting into the hydrophobic h2 pocket and to a less optimal conformational preference of phenylpiperazine versus biphenyl for the h2 pocket.
Figure 4.

Putative binding mode of 21. (A) Surface of the Mcl-1 protein (PDB ID: 2NLA) is colored according to the chemical shift intensity. Significant shift (>0.09 ppm) is represented with purple, moderate shift (≥0.03 and ≤0.09 ppm) represented with pink. (B) Plot of chemical shift changes of Mcl-1 amide upon addition of 21 (Mcl-1:21 ratio of 1:2) as a function of Mcl-1 residue numbers. (C) Overlaid 15N–1H HSQC spectra of Mcl-1 (red) and in the presence of 21 (Mcl-1:21 ratio of 1:2) (black), (Mcl-1:21 ratio of 1:1) (purple). Arrows show the direction of chemical shift changes upon binding of 21. (D) Mcl-1 residues shown to be perturbed in HSQC NMR in the presence of 21 (Mcl-1:21 ratio of 1:2) (green).
To briefly explore the importance of the naphthalene core to the binding potency, which occupies the h3 hydrophobic pocket, analogue 24 with a phenyl core was synthesized. Binding studies showed a significant drop in potency (Ki = 283.36 ± 81.50 μM in FP and IC50 > 100 μM in SPR) compared to the corresponding compound 10 with the naphthalene core, supporting the importance of hydrophobic interactions at h3 to the overall Mcl-1 binding.
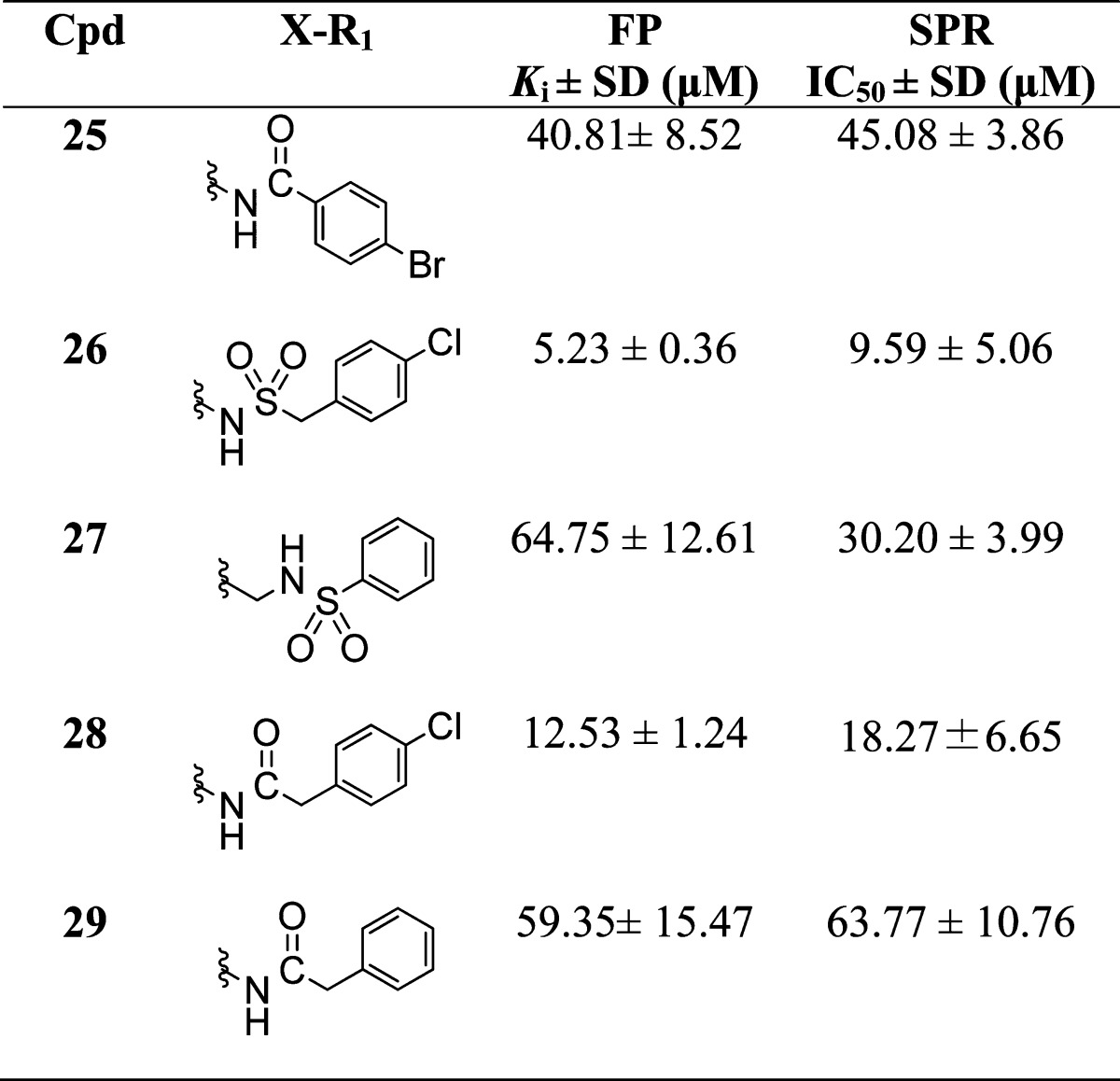
The next focus of our SAR studies was the sulfonamide linker for which docking results suggested lack of any specific interactions with Mcl-1. To investigate the importance of the sulfonamide linker, several analogues were synthesized (Table 2). Replacement of the sulfonamide with a carboxamide in 25 led to significant 83-fold decrease of the binding affinity with Ki value of 40.8 ± 8.50 μM compared to 10, possibly due to the unfavorable orientation of R1 by the carboxamide linker. The decreased binding potency was confirmed by SPR and NMR (Supporting Information Figure S4) binding studies. To explore the impact of the flexibility of the linker, a methylene linker distal (26) or proximal (27) to the naphthalene core was inserted and both analogues showed decreased binding affinity with Ki = 5.23 ± 0.36 μM and Ki = 64.75 ± 12.61 μM, relative to 9 (6-fold) and 3 (18-fold), respectively. As expected, change of the sulfonamide linker in 26 to a carboxamide in 28 decreased the potency, but only by 2-fold (Ki = 12.53 ± 1.24 μM), suggesting that the insertion of the methylene linker between the carboxamide and the pendant aryl ring provids a degree of freedom to better orient R1 into h2 pocket of Mcl-1.
Modeling studies showed that the thioacetic acid moiety at R2 mimics the conserved Asp of BH3-only peptides and that the carboxylate is involved in electrostatic interaction with Asn 260 and Arg 263 of Mcl-1. Therefore, to further explore this site, analogues with different substituents at R2 were synthesized (Table 3). Removal of the acid side chain (30) or its replacement with thiobutyl (31) did not show binding up to 100 μM in both FP and SPR assays. Introducing a methyl ester (32) or a primary carboxamide (33) resulted in decreased binding by 10-and 3-fold in FP assay respectively compared to 10 (6- and 2-fold decrease in SPR assay respectively), consistent with each of these functional groups, forming a weaker interaction with Arg 263 relative to the carboxylic acid moiety. When the carboxylic acid was changed to an alcohol and the sulfur to methylene (34) or ethylene (35), both analogues showed decreased binding compared to the parent congener 10 with Ki values of 3.79 ± 0.62 and 2.18 ± 0.91 μM, respectively. As was expected, bioisosteric replacement of the carboxylic acid group with a tetrazole in analogues 36 and 37 maintained the binding affinity in comparison with their parent congeners, 10 and 21. Homologation of the thioacetic acid in 38 had no detrimental effect on binding and showed a similar Ki of 1.13 ± 0.34 μM in FP assay and IC50 = 5.03 ± 1.88 μM in SPR assay as 15, which can be explained with the flexibility of both the thiopropanoic acid moiety and Arg 263 side chain. However, changing the point of fusion of the naphthalene ring in 38 substantially affected the binding and 39 with 1-naphthyl substituent showed a significant reduction in binding to Mcl-1. Our docking studies suggest that this might be attributed to a clash with the residues in the h2 pocket of Mcl-1.
Modeling showed that the phenolic group of the core naphthalene scaffold forms a hydrogen bond with His 224 of Mcl-1, consistent with the reported acidic hotspot in the h3 site of Mcl-1 close to His 224.58 Several analogues were synthesized to probe the contribution of the phenolic group to binding to Mcl-1 (Table 4). When the hydroxyl group is changed to a methoxy in analogue 40, the potency decreased by 170 fold (Ki = 86.98 ± 15.35 μM) compared to the phenolic congener 10. The analogue 41, where R2 is methyl thioacetate and R3 is methoxy, did not show binding up to 100 μM, and the loss of the binding was confirmed with SPR and HSQC experiment (Supporting Information Figure S5). Similar significant loss of binding is also apparent in compounds 42 to 45 compared to their corresponding phenolic analogues, clearly indicating the importance of the phenolic group to the overall binding to Mcl-1. Compound 46, where the hydroxyl group was acetylated exhibited a Ki of 33.97 ± 15.96 μM, which is a 69-fold decrease in binding compared to 10. However, 46 showed improved binding relative to 40, which might be attributed to the formation of a hydrogen bond between the carbonyl of the acetyl group and the protonated His 224, supported by computational prediction.
We determined the selectivity of this class of compounds against four other Bcl-2 antiapoptotic proteins (Bcl-2, Bcl-xL, Bcl-w, Bfl-1/A1). The most potent analogues were tested in competitive FP-based assays that were optimized for each protein, and Ki values were calculated using equations developed previously77 (Table 5). In general, all the analogues inhibit Mcl-1 most potently with the following order of selectively: Bfl-1/A1 > Bcl-w > Bcl-2 > Bcl-xL. As the BH3 domain binding profile of Bfl-1/A1, as well as its BH3 binding groove, is most similar to that of Mcl-1,78 it is not surprising that the tested compounds showed less selective inhibition of A1. The most potent analogues in this series, 19 and 21, show a profile for selectively inhibiting Mcl-1 with 7-and 19-fold versus Bfl-1/A1, 8-and 9-fold versus Bcl-w, 36- and 42-fold versus Bcl-2, and 56- and 59-fold versus Bcl-xL, respectively. Other reported selective Mcl-1 inhibitors, MIM1 and 53, also show high selectivity against Bcl-xL (IC50 > 50 μM and Ki > 15 μM, respectively), while 19 and 21 show better selectivity against Bcl-2 in comparison with 53 which has 16-fold selectivity with Ki value of 0.87 μM.
Table 5. Selectivity of Selected Analogues against Bcl-2 Antiapoptotic Proteins.
| compound | Mcl-1 Ki ± SD (μM) | A1/Bfl-1 Ki ± SD (μM) | Bcl-2 Ki ± SD (μM) | Bcl-w Ki ± SD (μM) | Bcl-XLKi ± SD (μM) |
|---|---|---|---|---|---|
| 1 | 1.55 ± 0.18 | 6.14 ± 1.0 | 54.65 ± 9.56 | 37.53 ± 7.96 | 99.00 ± 22.63 |
| 10 | 0.49 ± 0.06 | 5.33 ± 1.01 | 23.83 ± 1.81 | 8.19 ± 1.91 | 32.99 ± 4.33 |
| 16 | 0.37 ± 0.10 | 2.34 ± 0.37 | 8.82 ± 0.65 | 1.92 ± 0.37 | 8.05 ± 0.50 |
| 17 | 0.38 ± 0.03 | 3.18 ± 0.41 | 7.85 ± 0.65 | 2.19 ± 0.45 | 15.14 ± 1.11 |
| 18 | 2.03 ± 0.41 | 17.26 ± 0.56 | 23.84 ± 3.16 | 4.41 ± 0.39 | 48.15 ± 3.28 |
| 19 | 0.17 ± 0.04 | 1.11 ± 0.19 | 6.11 ± 0.65 | 1.36 ± 0.51 | 9.59 ± 1.28 |
| 21 | 0.18 ± 0.05 | 3.36 ± 0.56 | 7.56 ± 1.08 | 1.58 ± 0.35 | 10.58 ± 1.53 |
Biological Characterization of Mcl-1 Inhibitors
To verify the specific binding of novel inhibitors to Mcl-1, we employed a pull-down assay using biotin-labeled Noxa (BL-Noxa) and whole cell lysate from the human breast cancer cell line 2LMP. As shown in Figure 5, Mcl-1 was pulled down by BL-Noxa and, as was expected, the Bim BH3 peptide disrupted the interaction between BL-Noxa and Mcl-1. Preincubation with several Mcl-1 inhibitors, 10, 19, and 21, completely blocked the binding of BL-Noxa to Mcl-1, similar to Bim peptide, demonstrating that these inhibitors can recognize and specifically bind to the BH3 binding groove of endogenous Mcl-1 protein.
Figure 5.
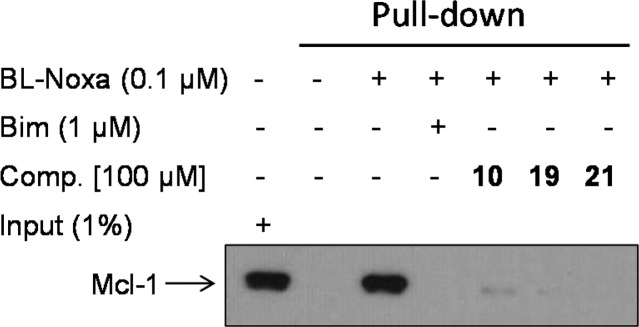
Interaction of Mcl-1 inhibitors with endogenous Mcl-1 protein and Noxa. Biotin-labeled Noxa (BL-Noxa, 0.1 μM) was incubated with whole cell lysates of 2LMP cells with or without tested Mcl-1 inhibitors and Bim BH3 peptide as a positive control, followed by incubation with precleared streptavidin agarose beads. Eluted beads were subjected to Western blot analysis with anti-Mcl-1 antibody.
It is well established that Bax and Bak are required for the initiation of intrinsic, mitochondrial, apoptotic cell death, and they are maintained in an inactive state through interaction with the antiapoptotic proteins.14,79 Therefore, cells subjected to inhibitors of antiapoptotic proteins are expected to undergo cell death in a Bax/Bak-dependent manner.80 To determine the contribution of Bak and Bax in the cell death induced by our Mcl-1 inhibitors, we employed murine embryonic fibroblasts (MEFs) wild-type (wt) and deficient in both Bax and Bak (double knock out, DKO). Exposure of the wt MEFs to 21 resulted in a concentration-dependent cell death (assessed by PI staining), while Bax/Bak deletion significantly rescued cells from 21 induced cell death (Figure 6). Although 21 initiates cell death also in DKO MEFs (23% positive PI at 16 μM), it is clear that 21 is more potent in wt MEFs (61% positive PI at 16 μM), indicating that Bax and/or Bak are involved in cell death induction. Taken together, our data suggest that 21 induces cell death in a Bax/Bak-dependent manner due to its Mcl-1 inhibitory function.
Figure 6.

Cell death induced by Mcl-1 inhibitor 21 is Bax/Bak-dependent. MEFs deficient in Bax and Bak (gray bars) along with their wild-type counterpart (black bars) were exposed for 15 h to different concentrations of 21, and the cell viability was assessed with PI staining. Error bars represent the mean ± SEM. The significance was calculated using unpaired t test, and the number of data is shown for each tested concentration with corresponding significance: (**) p < 0.01 and (***) p < 0.001.
To further confirm the specificity of our novel Mcl-1 inhibitors and to determine whether different prosurvival Bcl-2 proteins could suppress the apoptotic activities of novel Mcl-1 inhibitors, we used reported cell lines developed by retroviral transduction of lymphoma cells isolated from Eμ-myc transgenic mice which differ only in their expression of prosurvival Bcl-2 family proteins.81 Lymphoma cells overexpressing Mcl-1 and Bcl-2 were treated with varying concentrations of tested compounds for 15–18 h, and then cell viability was determined by flow cytometry using a fluorescent reactive dye (LIVE/DEAD fixable violet stain kit). ABT-263 (navitoclax), a selective inhibitor of Bcl-2, Bcl-xL, and Bcl-w, was used as a positive control. As predicted, lymphoma cells overexpressing Mcl-1 were significantly sensitive to 19 and 21 as assessed by an increased percentage of cell death in a concentration-dependent manner. In contrast, 19 and 21 were ineffective against Eμ-myc/Bcl-2 lymphomas (Figure 7). Importantly, 41 did not show any activity against both cell lines overexpressing Mcl-1 or Bcl-2, consistent with our binding studies which showed that 41 does not bind to Mcl-1. As expected, lymphoma cells overexpressing Bcl-2 were sensitive to cell death induced by ABT-263, while cells overexpressing Mcl-1 were insensitive to ABT-263, consistent with its binding specificity. Collectively, these results, demonstrate that 19 and 21 specifically bind and inhibit Mcl-1 and have no effect on Bcl-2, which is consistent with our biochemical data for their selectivity profiles. Furthermore, this supports the concept that tumor cells “addicted” to Mcl-1 protein will be the most sensitive target cell population for selective small-molecule Mcl-1 inhibitors.
Figure 7.

Sensitivity of Eμ-myc lymphoma cells overexpressing Mcl-1 and Bcl-2 antiapoptotic proteins to inhibitor-induced cell death. Eμ-myc/Mcl-1 and Eμ-myc/Bcl-2 lymphomas were treated for 15–18 h with increasing concentrations of 19, 21, 41, and ABT-263. Dead cells were assessed by LIVE/DEAD fixable dead cell stain kit (ViVID). The data shown represents means ± SEM from 3–7 independent experiments. The significance was calculated using unpaired t test, and the number of data is shown for each tested concentration with corresponding significance: (*) is p < 0.05, (**) p < 0.01, and (***) p < 0.001.
We next evaluated our most potent compounds for their ability to inhibit cell growth in the leukemia cell lines HL-60, MV4,11, and K-562 (Figure 8). It has been shown that AML-derived cell lines, HL-60 and MV4,11, are sensitive to inhibition of the antiapoptotic protein Mcl-1, while CML-derived K-562 cell line is less sensitive to Mcl-1 inhibition.82 Tested compounds from our series showed inhibition of the cell growth in a dose-dependent manner with similar potencies, IC50 values ranging from 2.06 to 11.72 μM against the HL-60 and MV4,11 cell lines (Figure 8A). Interestingly, these compounds showed decreased ability to inhibit the cell growth of K-562, with IC50 values of 11.76 to 29.89 μM. Compound 41, which does not bind to Mcl-1, did not show inhibition up to 50 μM. It is important to be pointed out that 36 and 37, where the carboxylic acid was replaced with a bioisostere tetrazole group, show similar cellular activity in all tested cell lines in comparison with their parent compounds, 10 and 21, respectively. These results demonstrate that the tetrazole group can effectively replace the acid group, achieving not only the same binding affinity to Mcl-1 but also comparable cellular activity. Furthermore, compound 32, in which the acid group has been replaced with methyl ester and showed 10-fold less binding to Mcl-1 as compared with 10, has also similar IC50 values in tested cell lines as 10. This is probably due to enzymatic hydrolysis of the methyl ester group in the cells and releasing the corresponding compound with free acid group, thus functioning as a pro-drug. These data also demonstrate that the compounds with free acidic group in this series are cell permeable, although the mechanism of their cell permeability need to be further studied. Compounds 21 and 37 were evaluated for their ability to induce apoptosis in the HL-60 cell line using annexin-V and propidium iodide (PI) double staining by flow cytometry (Figure 8B). Both compounds effectively induced apoptosis in a dose-dependent manner. Treatment of the HL-60 cells by 2.5, 5.0, and 10 μM of 21 and 37 for 20 h results in 28.9% and 23.5%, 37.8% and 51.6%, and 78.4% and 74.2% of apoptotic cells (early + late), respectively, as compared to 7.6% and 7.3% of apoptotic cells in the DMSO controls. These results further confirmed that these two analogues have similar cellular activity, and tetrazole can effectively replace the acid group. To determine if the induction of apoptosis by compound 37 in the HL-60 cell line depends upon caspases, we treated the cells with compound 37 alone or in the presence of Z-VED-FMK, a pan-caspase inhibitor (Figure 8B and Supporting Information Figure S7). The obtained results showed that the induction of apoptosis was significantly inhibited in the presence of Z-VAD-FMK, clearly indicating that the apoptotic activity of compound 37 is mediated by caspases. Of note, Z-VAD-FMK alone has no effect on cells (Supporting Information Figure S7). Compound 41 at 40 μM has no effect on apoptosis induction just like untreated control, which is consistent with its lack of binding to Mcl-1 and inhibition of cell growth. We further tested compounds 19 and 21 in HL-60 cell line for their ability to induce caspase-3 activity, one of the important biochemical markers of apoptosis (Figure 8C). Both compounds induce activation of caspase-3 activity in a dose-dependent manner, with 21 effectively inducing activation over a 24 h period starting at 2.5 μM. Importantly, these results correlate with the ability of 21 to induce apoposis and inhibit HL-60 cell growth.
Figure 8.

Cell death and apoptosis induction by Mcl-1 inhibitors in human leukemic cell lines. (A) Inhibition of cell growth by designed Mcl-1 inhibitors in the HL-60, MV4,11, and K-562 leukemia cell lines. Cells were treated for 3 days, and cell growth was determined using CellTiter Glo luminescent cell viability assay. (B) Analysis of apoptosis induced by 21 and 37 in the HL-60 leukemia cell line. Cells were treated with 21, 37, and 41 for 20 h using indicated concentrations, and apoptosis was analyzed with annexin-V and propidium iodide (PI) double staining by flow cytometry. Early apoptotic cells were defined as annexin-V positive/PI-negative, and late apoptotic cells as annexin-V/PI-double positive. Induction of the apoptosis by 37 was tested also in the presence of Z-VAD-FMK. (C) Induction of caspase-3 by 19 and 21 in the HL-60 cell line. Cells were treated for 20 h, and caspase-3 was detected with fluorometric-based assay. Results shown are the mean and SEM from at least three separate experiments.
Conclusions
Applying a HTS approach we have identified a novel class of small molecules as selective Mcl-1 inhibitors. Employing structure-based design supported by NMR studies, we synthesized a focused library of analogues and established a SAR for binding to Mcl-1. Careful HSQC analysis of analogues 16–18 provided strong evidence for the predicted binding model of these compounds and further confirmation that R1 substituent of this class of inhibitors binds to one well-defined pocket of Mcl-1 known as h2 pocket. Analogues 19 and 21, with Ki values of 170 and 180 nM, respectively, were developed as the most potent compounds in this series with an overall 9-fold increase in binding compared to 1. Binding studies showed that 19 and 21 maintained the selectivity profile of 1. Using wild-type and Bax/Bak double knockout MEFs cells, the contribution of these two multidomain pro-apoptotic proteins in 21-induced cell death was determined and demonstrated that 21 primarily causes cell death in wild-type MEFs in a Bax/Bak-dependent manner. Furthermore, 19 and 21 led to sensitization of Eμ-myc lymphomas overexpressing Mcl-1 but did not show effect on cells overexpressing Bcl-2 antiapoptotic protein, confirming their selective targeting of Mcl-1. Most potent developed 3-substituted-N-(4-Hydroxynaphthalen-1-yl)arylsulfonamide Mcl-1 inhibitors inhibited the cell growth of AML-derived cell lines, HL-60 and MV4,11. Compounds 19 and 21 induced activation of caspase-3 in HL-60 cell line and 21 effectively induced apoptosis starting at 2.5 μM. Future studies will be directed toward optimization of this class of compounds and in vivo evaluation.
Experimental Section
Chemistry Materials and Methods
All reactions were performed under anhydrous conditions. Reagents were used as supplied without further purification. Reactions were monitored by TLC using precoated silica gel 60 F254 plates. Silica gel chromatography was performed with silica gel (220–240 mesh) obtained from Silicycle. Purities of final compounds were assessed by analytical reverse-phase HPLC performed with one of the two methods. Method A: Agilent 1100 series with an Agilent Zorbax Eclipse Plus C18 (4.6 mm × 75 mm, 3.5 μm particle size) column with the gradient 10% ACN/water (1 min), 10–90% ACN/water (6 min), and 90% ACN/water (2 min) flow = 1 mL/min. Method B: Shimadzu system with a Restek Ultra C18 (4.6 mm × 150 mm, 5 μm particle size) column with the gradient 50% ACN/water (5 min), 50–70% ACN/water (2 min), and 70%–90% ACN/water (8 min) flow = 1 mL/min. Semipreparative reverse-phase HPLC was performed on a Shimadzu system with a Restek Ultra C18 (21.2 mm × 150 mm, 5 μm particle size) column. All NMR spectra were obtained in DMSO-d6 or CDCl3, and results were recorded at 400 or 500 MHz on a Varian 400 or 500 instrument. Mass spectra were recorded on a Micromass LCT time-of-flight instrument utilizing electrospray ionization operating in positive-ion (ESI+) or negative-ion (ESI) modes where indicated. High resolution mass spectrometry (HRMS) analysis was performed on an Agilent Q-TOF system. All final compounds were purified to >95% purity. Analogues 4–8, 15, and 39 were purchased from commercial vendors.
2-Iodo-1-methoxy-4-nitronaphthalene (47)
Synthesized using reported procedure with modification.60 A mixture of 1-methoxy-4-nitronaphthalene (2.1 g, 10.4 mmol) and N-iodosuccinimide (2.7 g, 12 mmol) in TFA (40 mL) was heated to reflux and stirred for 20 h under nitrogen. The reaction mixture was diluted with EtOAc (40 mL), washed with saturated aqueous Na2S2O3 solution (30 mL), saturated aqueous NaHCO3 (30 mL × 2), and brine (30 mL). The organic layer was dried (MgSO4), filtered, and silica was added to filtrate and the solvent was removed under reduced pressure. The adsorbed crude residue was purified by flash column chromatography (100% hexane) on silica gel to give 47 (2.4 g, 70%) as a light-yellow solid. Note: Rf of starting material and product were very close and a good separation was achieved with a relatively long silica gel column and 100% hexane gradient. >95% pure product was needed for pd-catalyzed coupling step. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.58 (s, 1H), 8.21 (d, J = 8.42 Hz, 1H), 7.74 (t, J = 7.53 Hz, 1H), 7.65 (t, J = 7.53 Hz, 1H), 4.03 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.69, 142.85, 134.07, 129.99, 128.55, 128.08, 126.55, 123.92, 123.02, 83.35, 62.20. ESI MS: m/z 330.0 (M + H)+.
Methyl 2-((1-Methoxy-4-nitronaphthalen-2-yl)thio)acetate (48a)
Synthesized using reported procedures with modification.63−65 To a solution of Cs2CO3 (1.5 g, 4.5 mmol) in dry THF (7 mL) under nitrogen was added methylthioglycolate (277 μL, 2.9 mmol). The mixture was stirred at room temperature for 10 min. At this time, a solution of ZnCl2 (288 mg, 2.1 mmol) in dry THF (3 mL) was added and the mixture was stirred at room temperature for an additional 10 min. Meanwhile, in a separate flask, Pd(OAc)2 (36 mg, 0.16 mmol) and xantphos (90 mg, 0.15 mmol) were premixed in dry THF (5 mL) under nitrogen and stirred at room temperature for about 20 min. To the solution of thiol, Cs2CO3, and ZnCl2 was added 47 (1.0 g, 3.1 mmol), LiI (200 mg, 1.5 mmol), and premixed solution of the catalyst and ligand. The mixture was stirred at 60 °C under nitrogen for 20 h. The reaction mixture was filtered to remove Cs2CO3 and silica was added to the mixture and the solvent was removed under reduced pressure. The adsorbed crude residue was purified by column chromatography (hexane/EtOAc 4:1) on silica gel to give 48a (606 mg, 66%) as a yellow oil which solidified. 1H NMR (400 MHz, CDCl3) δ 8.59 (d, J = 8.50 Hz, 1H), 8.37 (s, 1H), 8.19 (d, J = 8.50 Hz, 1H), 7.70 (t, J = 7.57 Hz, 1H), 7.64 (t, J = 7.57 Hz, 1H), 4.07 (s, 3H), 3.77 (s, 2H), 3.70 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.48, 159.95, 142.50, 129.73, 128.88, 127.85, 127.22, 125.94, 123.71, 122.93, 122.68, 61.92, 52.72, 35.10. ESI MS: m/z 308.1 (M + H)+.
Methyl 3-((1-Methoxy-4-nitronaphthalen-2-yl)thio)propanoate (48b)
Synthesized using a similar procedure used to prepare 48a except using methyl 3-mercaptopropionate. The mixture was stirred at 60 °C under nitrogen for 5 h. Crude was purified using flash column chromatography (hexane/EtOAc 4:1) on silica gel with dry loading to give 48b (194 mg, 66%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 8.48 Hz, 1H), 8.27 (s, 1H), 8.16 (d, J = 8.48 Hz, 1H), 7.70–7.64 (m, 1H), 7.64–7.58 (m, 1H), 4.03 (s, 3H), 3.65 (s, 3H), 3.28 (t, J = 7.24 Hz, 2H), 2.65 (t, J = 7.24 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 171.64, 159.79, 142.48, 129.51, 128.93, 127.81, 126.94, 125.60, 123.62, 123.45, 122.57, 61.59, 51.90, 34.11, 28.11. ESI MS: m/z 322.0 (M + H)+, 343.9 (M + Na)+.
Butyl(1-methoxy-4-nitronaphthalen-2-yl)sulfane (48c)
Synthesized using a reported procedure.61 A stirred mixture of 47 (300 mg, 0.91 mmol), Pd2(dba)3 (42 mg, 0.05 mmol), Dppf (104 mg, 0.18 mmol), and Et3N (0.2 mL) in dry NMP (7 mL) was flushed with nitrogen for 15 min at room temperature. Butanethiol (83 μL, 0.77 mmol) was then added, and the reaction mixture was heated to 80 °C and stirred for 2 h. The mixture was diluted with EtOAc (10 mL) and washed with H2O (10 mL × 4) and brine (10 mL). The organic layer was dried (MgSO4), filtered, and silica added to the filtrate, and the solvent was removed under reduced pressure. The adsorbed crude residue was purified by flash column chromatography (hexane to hexane/EtOAc 99:1) on silica gel to give 48c (189 mg, 71%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.58 (ddd, J = 0.72, 1.50, 8.36 Hz, 1H), 8.26 (s, 1H), 8.16 (ddd, J = 0.72, 1.50, 8.36 Hz, 1H), 7.68–7.59 (m, 2H), 4.03 (s, 3H), 3.03 (t, J = 7.36 Hz, 2H), 1.67 (p, J = 7.36 Hz, 2H), 1.48 (h, J = 7.36 Hz, 2H), 0.93 (t, J = 7.36 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 158.48, 142.53, 129.05, 128.81, 127.69, 125.69, 125.57, 125.03, 123.62, 122.34, 61.31, 32.23, 31.10, 21.92, 13.60. ESI MS: m/z 292.0 (M + H)+.
3-(1-Methoxy-4-nitronaphthalen-2-yl)prop-2-yn-1-ol (48d)
Synthesized using a reported procedure.62 A mixture of 47 (453 mg, 1.4 mmol), Pd(PPh3)2Cl2 (48 mg, 0.07 mmol), and CuI (28 mg, 0.15 mmol) in Et3N (8 mL) and dry THF (3 mL) was added dropwise to a solution of 2-propyn-1-ol (0.15 mL, 2.6 mmol) in Et3N (3 mL) under nitrogen at room temperature. Reaction mixture was heated to 60 °C and stirred for 2 h then diluted with EtOAc (10 mL) and washed with saturated aqueous NH4Cl (15 mL × 2) and brine (15 mL). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. The crude was purified by flash column chromatography (hexane/EtOAc 3:2) on silica gel to give 48d (342 mg, 97%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.59 (d, J = 8.66 Hz, 1H), 8.29 (s, 1H), 8.26 (d, J = 8.66 Hz, 1H), 7.74–7.66 (m, 1H), 7.64–7.55 (m, 1H), 4.58 (s, 2H), 4.29 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.40, 141.04, 130.49, 129.78, 128.26, 127.56, 126.29, 123.44, 123.24, 107.40, 93.96, 80.83, 61.95, 51.67. ESI MS: m/z 258.1 (M + H)+.
4-(1-Methoxy-4-nitronaphthalen-2-yl)but-3-yn-1-ol (48e)
Synthesized using a similar procedure used to prepare 48d except using 3-butyn-1-ol as the alkyne. The crude was purified by flash column chromatography (hexane/EtOAc 3:2) on silica gel to give 48e (338 mg, 83%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 8.56 Hz, 1H), 8.23 (s, 1H), 8.19 (d, J = 8.56 Hz, 1H), 7.63 (t, J = 7.59 Hz, 1H), 7.53 (t, J = 7.59 Hz, 1H), 4.23 (s, 3H), 3.87 (t, J = 6.14 Hz, 2H), 2.76 (t, J = 6.14 Hz, 2H), 2.45 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 162.29, 141.11, 130.19, 129.91, 128.36, 127.48, 126.03, 123.43, 123.10, 109.99, 108.73, 93.90, 61.77, 60.96, 24.07. ESI MS: m/z 272.1 (M + H)+.
A Representative Procedure for Iron Reduction of Nitro to Amine
Methyl 2-((4-Amino-1-methoxynaphthalen-2-yl)thio)acetate (49a)
Synthesized using a reported procedure.66 To a suspension of iron powder (538 mg, 9.6 mmol) in glacial acetic acid (5 mL) was added 48a (195 mg, 0.63 mmol) dissolved in glacial acetic acid (5 mL). The mixture was stirred at 70 °C under nitrogen for 1 h, when the mixture turned milky. The mixture was then diluted with EtOAc (15 mL) and washed with saturated aqueous NaHCO3 (20 mL × 2) and brine (20 mL). Organic layer was dried (MgSO4), filtered, and the solvent removed under reduced pressure to give 49a as a crude as a purple oil. The crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.29 Hz, 1H), 7.76 (d, J = 8.29 Hz, 1H), 7.50 (t, J = 7.58 Hz, 1H), 7.43 (t, J = 7.58 Hz, 1H), 6.78 (s, 1H), 3.91 (s, 3H), 3.73 (s, 2H), 3.68 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.48, 147.81, 138.68, 128.54, 126.54, 125.33, 124.24, 123.36, 122.45, 121.35, 111.08, 61.41, 52.53, 35.37. ESI MS: m/z 278.1 (M + H)+, 300.1 (M + Na)+.
Methyl 3-((4-Amino-1-methoxynaphthalen-2-yl)thio)propanoate (49b)
Synthesized using the procedure for 49a except using 48b as the starting material. Crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.36 Hz, 1H), 7.75 (d, J = 8.36 Hz, 1H), 7.49 (t, J = 7.56 Hz, 1H), 7.42 (t, J = 7.56 Hz, 1H), 6.72 (s, 1H), 6.26 (s, 2H), 3.89 (s, 3H), 3.65 (s, 3H), 3.22 (t, J = 7.46 Hz, 2H), 2.64 (t, J = 7.46 Hz, 2H);. 13C NMR (100 MHz, CDCl3) δ 172.38, 147.73, 138.77, 128.63, 126.51, 125.12, 123.92, 123.56, 122.36, 121.35, 110.85, 61.21, 51.78, 34.48, 27.90. ESI MS: m/z 292.0 (M + H)+, 314.0 (M + Na)+.
3-(Butylthio)-4-methoxynaphthalen-1-amine (49c)
Synthesized using the procedure for 49a except using 48c as the starting material. Crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.30 Hz, 1H), 7.75 (d, J = 8.30 Hz, 1H), 7.49 (t, J = 7.42 Hz, 1H), 7.40 (t, J = 7.42 Hz, 1H), 6.72 (s, 1H), 6.30 (s, 2H), 3.92 (s, 3H), 2.96 (t, J = 7.33 Hz, 2H), 1.65 (p, J = 7.33 Hz, 2H), 1.47 (h, J = 7.33 Hz, 2H), 0.92 (t, J = 7.33 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 146.85, 138.46, 128.54, 126.42, 125.50, 124.73, 123.45, 122.20, 121.30, 110.24, 61.02, 32.29, 31.50, 22.02, 13.67. ESI MS: m/z 262.0 (M + H)+.
A Representative Procedure for Hydrogenation Reaction
3-(4-Amino-1-methoxynaphthalen-2-yl)propan-1-ol (49d)
Syntheszied using a reported procedure.67 A stirred solution of 48d (325 mg, 1 mmol) in a mixture of EtOH (12 mL) and EtOAc (2 mL) was hydrogenated in the presence of 10% Pd/C (80 mg) at room temperature and under 30 psi of H2 overnight. The suspension was filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The crude was used in the next reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.35 Hz, 1H), 7.76 (d, J = 8.35 Hz, 1H), 7.51–7.43 (m, 1H), 7.43–7.35 (m, 1H), 6.54 (s, 1H), 3.85 (s, 3H), 3.54 (t, J = 6.08 Hz, 2H), 2.80 (t, J = 7.27 Hz, 2H), 1.85 (p, J = 6.69 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 146.20, 138.66, 129.80, 128.24, 126.01, 124.51, 123.79, 122.41, 121.33, 111.39, 62.18, 61.32, 33.18, 25.51. ESI MS: m/z 232.1 (M + H)+.
4-(4-Amino-1-methoxynaphthalen-2-yl)butan-1-ol (49e)
Synthesized using the procedure for 49d except using 48e as the starting material. Crude was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.39 Hz, 1H), 7.72 (d, J = 8.39 Hz, 1H), 7.43 (t, J = 7.33 Hz, 1H), 7.34 (t, J = 7.33 Hz, 1H), 6.52 (s, 1H), 3.79 (s, 3H), 3.56 (t, J = 6.43 Hz, 2H), 2.67 (t, J = 6.43 Hz, 2H), 1.65 (dt, J = 6.60, 14.36 Hz, 2H), 1.56 (dt, J = 6.60, 14.36 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 145.98, 138.36, 130.77, 128.39, 125.85, 124.33, 123.72, 122.34, 121.36, 111.52, 62.32, 61.97, 32.45, 29.21, 26.89.
A Representative Procedure for Sulfonamide/Amide Coupling Reaction of Aryl Amines with Sulfonyl/Acyl Chlorides
Methyl 2-((1-Methoxy-4-(thiophene-2-sulfonamido)naphthalen-2-yl)thio)acetate (49f)
A solution of the crude amine 49a dissolved in dry CH2Cl2 (4 mL) was added to 2-thiophenesulfonyl chloride (119 mg, 0.65 mmol). Addition of pyridine (0.08 mL, 0.99 mmol) was followed, and the mixture was stirred at room temperature under nitrogen overnight. The mixture was diluted with EtOAc (10 mL) and washed with H2O (10 mL × 3) and brine (10 mL). The organic layer was dried (MgSO4), filtered, and the solvent removed under reduced pressure. Crude was purified by flash column chromatography (hexane/EtOAc 7:3) on silica gel to give 49f (176 mg, 66% over two steps) as a purple oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.40 Hz, 1H), 7.82 (d, J = 8.40 Hz, 1H), 7.49–7.43 (m, 2H), 7.42–7.39 (m, 1H), 7.39–7.34 (m, 2H), 6.92–6.88 (m, 1H), 3.95 (s, 3H), 3.68 (s, 3H), 3.65 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.91, 154.12, 139.48, 133.09, 132.51, 130.06, 128.60, 127.74, 127.39, 126.96, 125.74, 123.39, 122.29, 61.49, 52.68, 35.12. ESI MS: m/z 423.9 (M + H)+, 445.8 (M + Na)+.
Methyl 2-((1-Methoxy-4-(methylsulfonamido)naphthalen-2-yl)thio)acetate (49g)
Synthesized using the procedure for 49f except using methanesulfonyl chloride, which afforded the title compound (69 mg, 39% over two steps) as a light-pink solid. 1H NMR (400 MHz, CDCl3) δ 8.13–8.08 (m, 1H), 8.05–8.00 (m, 1H), 7.65 (s, 1H), 7.60–7.54 (m, 2H), 6.80 (s, 1H), 3.99 (s, 3H), 3.75 (s, 2H), 3.69 (s, 3H), 3.04 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.98, 154.06, 129.61, 128.85, 127.84, 127.36, 127.19, 125.15, 123.67, 122.67, 122.07, 61.50, 52.64, 39.91, 35.00. ESI MS: m/z 355.9 (M + H)+, 377.9 (M + Na)+.
Methyl 2-((1-Methoxy-4-(phenylsulfonamido)naphthalen-2-yl)thio)acetate (49h)
Synthesized using the procedure for 49g except using benzenesulfonyl chloride, which afforded the title compound (119 mg, 64% over two steps) as a purple oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.40 Hz, 1H), 7.74 (t, J = 9.06 Hz, 3H), 7.47 (q, J = 8.19, 8.80 Hz, 2H), 7.36 (t, J = 7.61 Hz, 3H), 7.29 (s, 1H), 6.93 (s, 1H), 3.95 (s, 3H), 3.68 (s, 3H), 3.61 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.80, 154.02, 139.11, 132.95, 129.95, 128.98, 128.63, 127.82, 127.33, 126.95, 126.90, 125.67, 123.36, 122.31, 122.28, 61.49, 52.63, 35.06. ESI MS: m/z 417.9 (M + H)+, 439.9 (M + Na)+.
Methyl 2-((4-(4-Chlorophenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49i)
Synthesized using the procedure for 49f except using 4-chlorobenzenesulfonyl chloride, which afforded the title compound (280 mg, 67% over two steps) as a light-pink solid. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.40 Hz, 1H), 7.74 (d, J = 8.40 Hz, 1H), 7.66 (t, J = 1.96 Hz, 1H), 7.65 (t, J = 1.96 Hz, 1H), 7.47 (ddd, J = 1.17, 6.86, 8.24 Hz, 1H), 7.38 (ddd, J = 1.17, 6.86, 8.24 Hz, 1H), 7.35–7.33 (m, 2H), 7.32 (t, J = 1.94 Hz, 1H), 7.07 (s, 1H), 3.95 (s, 3H), 3.69 (s, 3H), 3.63 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.84, 154.18, 139.50, 137.67, 129.86, 129.25, 128.78, 128.66, 127.49, 127.05, 125.81, 123.42, 122.41, 122.14, 61.50, 52.64, 35.02. ESI MS: m/z 451.9 (M + H)+, 473.8 (M + Na)+.
Methyl 2-((4-(4-Bromophenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (41)
Synthesized using the procedure for 49f except using 4-bromobenzenesulfonyl chloride, which afforded the title compound (185 mg, 61% over two steps) as a pink/purple solid. HPLC (method A, tR = 7.66 min), purity 99%. 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.38 Hz, 1H), 7.72 (d, J = 8.38 Hz, 1H), 7.61–7.59 (m, 1H), 7.58 (t, J = 2.04 Hz, 1H), 7.54–7.47 (m, 3H), 7.44–7.38 (m, 1H), 7.34 (s, 1H), 6.76 (s, 1H), 3.97 (s, 3H), 3.71 (s, 3H), 3.64 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.83, 154.25, 138.21, 132.29, 129.91, 128.89, 128.71, 128.07, 127.41, 127.12, 125.92, 123.47, 122.48, 122.17, 122.09, 61.56, 52.70, 35.03. ESI HRMS: m/z 493.9724 (M – H)−.
Methyl 2-((1-Methoxy-4-(4-methoxyphenylsulfonamido)naphthalen-2-yl)thio)acetate (49j)
Synthesized using the procedure for 49f except using 4-methoxybenzenesulfonyl chloride, which afforded the title compound (190 mg, 77% over two steps) as a purple oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.42 Hz, 1H), 7.80 (d, J = 8.42 Hz, 1H), 7.69–7.63 (m, 2H), 7.48 (t, J = 7.64 Hz, 1H), 7.40 (t, J = 7.64 Hz, 1H), 7.29 (s, 1H), 6.86–6.80 (m, 3H), 3.95 (s, 3H), 3.79 (s, 3H), 3.70 (s, 3H), 3.63 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.82, 163.12, 153.79, 130.64, 129.85, 129.52, 128.61, 128.11, 126.95, 126.90, 125.20, 123.36, 122.31, 114.12, 61.50, 55.56, 52.65, 35.09. ESI MS: m/z 447.8 (M + H)+, 469.8 (M + Na)+.
Methyl 2-((4-(3-Bromophenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49k)
Synthesized using the procedure for 49f except using 3-bromobenzenesulfonyl chloride, which afforded the title compound (331 mg, 74%) as a purple/pink solid. 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.40 Hz, 1H), 7.88 (s, 1H), 7.73 (d, J = 8.40 Hz, 1H), 7.62 (t, J = 9.41 Hz, 2H), 7.49 (t, J = 7.59 Hz, 1H), 7.40 (t, J = 7.59 Hz, 1H), 7.34 (s, 1H), 7.26–7.21 (m, 1H), 6.82 (s, 1H), 3.98 (s, 3H), 3.70 (s, 3H), 3.65 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.72, 154.55, 141.06, 135.89, 130.41, 130.24, 130.03, 128.75, 127.31, 127.04, 127.02, 126.33, 125.89, 123.45, 122.90, 122.48, 122.09, 61.48, 52.55, 35.16. ESI MS: m/z 495.8, 497.8 (M + H)+, 517.8, 519.8 (M + Na)+.
Methyl 2-((4-(2-Bromophenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49l)
Synthesized using the procedure for 49f except using 2-bromobenzenesulfonyl chloride, which afforded the title compound (314 mg, 74% over two steps) as a purple oil. 1H NMR (400 MHz, CDCl3) δ 8.16–8.11 (m, 1H), 8.05–8.00 (m, 1H), 7.93 (d, J = 7.72 Hz, 1H), 7.78 (d, J = 7.72 Hz, 1H), 7.54–7.48 (m, 3H), 7.42–7.32 (m, 2H), 7.19 (s, 1H), 7.13 (s, 1H), 3.94 (s, 3H), 3.68 (s, 3H), 3.49 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.52, 154.35, 138.58, 135.17, 133.92, 132.04, 130.52, 128.75, 127.83, 127.55, 127.11, 127.07, 125.00, 123.16, 122.90, 122.26, 119.94, 61.40, 52.50, 35.13. ESI MS: m/z 495.8, 597.8 (M + H)+, 517.8, 519.8 (M + Na)+.
Methyl 2-((4-(Benzo[b]thiophene-2-sulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49m)
Synthesized using the procedure for 49f except using 1-benzothiophene-2-sulfonyl chloride, which afforded the title compound (132 mg, 57% over two steps) as a purple oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.40 Hz, 1H), 7.90 (d, J = 8.40 Hz, 1H), 7.79 (d, J = 7.99 Hz, 1H), 7.74 (d, J = 7.99 Hz, 1H), 7.69 (s, 1H), 7.48–7.42 (m, 2H), 7.41 (s, 1H), 7.40–7.35 (m, 2H), 7.01 (s, 1H), 3.96 (s, 3H), 3.64 (s, 3H), 3.51 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.77, 154.23, 141.85, 139.53, 137.38, 130.61, 130.05, 128.66, 127.47, 127.37, 127.06, 125.72, 125.47, 125.44, 123.47, 122.62, 122.34, 122.25, 61.51, 52.58, 34.95. ESI MS: m/z 473.9 (M + H)+, 495.8 (M + Na)+.
Methyl 2-((4-([1,1′-Biphenyl]-4-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49n)
Synthesized using the procedure for 49f except using 4-biphenylsulfonyl chloride, which afforded the title compound (152 mg, 71% over two steps) as a purple oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.34 Hz, 1H), 7.83–7.76 (m, 3H), 7.58 (d, J = 8.31 Hz, 2H), 7.51 (d, J = 7.14 Hz, 2H), 7.49–7.42 (m, 2H), 7.42–7.35 (m, 3H), 7.33 (s, 1H), 6.90 (s, 1H), 3.96 (s, 3H), 3.65 (s, 3H), 3.60 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.75, 153.99, 145.89, 139.09, 137.68, 129.97, 128.98, 128.66, 128.50, 127.90, 127.86, 127.56, 127.23, 126.98, 126.93, 125.58, 123.43, 122.35, 122.33, 61.49, 52.58, 35.02. ESI MS: m/z 494.1 (M + H)+, 516.1 (M + Na)+.
Methyl 2-((4-([1,1′-Biphenyl]-3-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49o)
Synthesized using reported procedures with modification.69,70 A stirred mixture of 49k (307 mg, 0.62 mmol), phenylboronic acid (113 mg, 0.91 mmol), 2 M aqueous Na2CO3 (0.93 mL), and Pd(PPh3)4 (72 mg, 0.06 mmol) in THF (6 mL) /H2O (1 mL) was heated at 60 °C under nitrogen for 2 h. Mixture was diluted with EtOAc (10 mL) and washed with H2O (15 mL × 2) and brine (15 mL). Organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. The crude was purified by flash column chromatography on silica to give 49o (284 mg, 92%) as a light-brown oil. 1H NMR (400 MHz, CDCl3/ couple of drops of D2O) δ 8.00 (d, J = 8.39 Hz, 1H), 7.83 (s, 1H), 7.79–7.73 (m, 2H), 7.69 (dd, J = 7.58, 14.09 Hz, 2H), 7.47–7.40 (m, 2H), 7.37–7.31 (m, 6H), 3.92 (s, 3H), 3.62 (s, 3H), 3.58 (s, 2H). 13C NMR (100 MHz, CDCl3/ couple of drops of D2O) δ 169.86, 154.00, 142.22, 139.49, 138.99, 133.60, 131.46, 131.02, 130.00, 129.44, 128.89, 128.63, 128.14, 127.89, 127.03, 126.98, 126.88, 125.92, 125.87, 125.80, 123.46, 122.38, 122.27, 61.43, 52.60, 34.97. ESI MS: m/z 494.1 (M + H)+.
Methyl 2-((4-([1,1′-Biphenyl]-2-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49p)
Synthesized using the procedure for 49f except using 2-biphenylsulfonyl chloride which was synthesized as previously reported.83 The title compound (57 mg, 58% over two steps) was obtained as a light-orange oil. 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.20 Hz, 1H), 8.00 (d, J = 8.20 Hz, 1H), 7.62 (t, J = 7.54 Hz, 1H), 7.51 (q, J = 7.13, 7.54 Hz, 3H), 7.44–7.37 (m, 2H), 7.37–7.29 (m, 5H), 6.93 (s, 1H), 5.86 (s, 1H), 3.91 (s, 3H), 3.67 (s, 3H), 3.44 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.66, 152.79, 140.89, 138.72, 138.15, 132.77, 132.73, 129.66, 129.43, 128.59, 128.52, 128.39, 128.20, 128.11, 127.94, 127.07, 126.69, 123.16, 122.20, 122.04, 120.40, 61.44, 52.62, 34.96. ESI MS: m/z 494.0 (M + H)+.
Methyl 2-((4-(4′-Chloro-[1,1′-biphenyl]-4-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49q)
Synthesized using the procedure for 49f except using 4′-chlorobiphenyl-4-sulfonyl chloride, which afforded the title compound (284 mg, 80% over two steps) as a pink oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 7.14 Hz, 1H), 7.80 (d, J = 7.14 Hz, 1H), 7.78 (d, J = 7.54 Hz, 2H), 7.54 (d, J = 7.54 Hz, 2H), 7.49–7.44 (m, 2H), 7.44–7.40 (m, 3H), 7.40–7.35 (m, 1H), 7.33 (s, 1H), 6.97 (s, 1H), 3.96 (s, 3H), 3.65 (s, 3H), 3.61 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.73, 154.05, 144.56, 138.08, 137.53, 134.79, 129.97, 129.19, 128.67, 128.48, 128.01, 127.80, 127.40, 126.98, 126.93, 125.64, 123.40, 122.35, 122.33, 61.50, 52.60, 35.03. ESI MS: m/z 528.1 (M + H)+, 550.1 (M + Na)+.
Methyl 2-((4-(2′,4′-Difluoro-[1,1′-biphenyl]-4-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49r)
Synthesized using the procedure for 49f except using 2′,4′-difluoro-biphenyl-4-sulfonyl chloride, which afforded the title compound (75 mg, 58% over two steps) as a purple oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.41 Hz, 1H), 7.83–7.76 (m, 3H), 7.50 (d, J = 8.16 Hz, 2H), 7.46 (d, J = 7.94 Hz, 1H), 7.40 (d, J = 7.94 Hz, 1H), 7.37–7.29 (m, 2H), 6.98 (s, 1H), 6.96–6.86 (m, 2H), 3.96 (s, 3H), 3.66 (s, 3H), 3.61 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.84, 154.01, 139.74, 138.18, 131.40 (dd, J = 4.57, 9.75 Hz), 130.03, 129.43 (d, J = 3.03 Hz), 128.65, 127.76, 127.61, 127.02, 126.96, 125.70, 123.44, 122.37, 122.32, 111.98 (dd, J = 3.71, 21.44 Hz), 105.88–103.01 (m), 61.51, 52.63, 34.95. ESI MS: m/z 530.1 (M + H)+.
Methyl 2-((1-Methoxy-4-(4-phenoxyphenylsulfonamido)naphthalen-2-yl)thio)acetate (49s)
Synthesized using the procedure for 49f except using 4-phenoxybenzenesulfonyl chloride, which afforded the title compound (244 mg, 72% over two steps) as a light-brown oil. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.43 Hz, 1H), 7.80 (d, J = 8.43 Hz, 1H), 7.66 (d, J = 8.14 Hz, 2H), 7.52–7.46 (m, 1H), 7.42–7.32 (m, 4H), 7.23 (s, 1H), 7.17 (t, J = 7.38 Hz, 1H), 6.94 (d, J = 8.52 Hz, 2H), 6.87 (d, J = 8.52 Hz, 2H), 3.95 (s, 3H), 3.69 (s, 3H), 3.66 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.85, 161.65, 155.11, 153.85, 132.59, 130.09, 129.85, 129.59, 128.60, 128.03, 126.90, 126.85, 125.50, 124.81, 123.42, 122.38, 122.31, 120.02, 117.64, 61.50, 52.67, 35.05. ESI MS: m/z 509.8 (M + H)+.
Methyl 2-((4-(4-(4-Fluorophenoxy)phenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49t)
Synthesized using the procedure for 49f except using 4-(4-fluoro-phenoxy)-benzenesulfonyl chloride, which afforded the title compound (93 mg, 72% over two steps) as a yellow oil which solidified upon standing. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.42 Hz, 1H), 7.80 (d, J = 8.42 Hz, 1H), 7.66 (d, J = 8.72 Hz, 2H), 7.51–7.45 (m, 1H), 7.42–7.36 (m, 1H), 7.34 (s, 1H), 7.07–7.00 (m, 2H), 6.94–6.88 (m, 2H), 6.84 (d, J = 8.72 Hz, 2H), 3.95 (s, 3H), 3.69 (s, 3H), 3.66 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 169.87, 161.82, 160.79, 158.36, 153.87, 150.83 (d, J = 2.72 Hz), 132.65, 129.83, 129.66, 128.61, 127.99, 126.88 (d, J = 4.95 Hz), 125.47, 123.39, 122.35 (d, J = 6.41 Hz), 121.67 (d, J = 8.41 Hz), 117.23, 116.86, 116.63, 61.50, 52.69, 35.06. ESI MS: m/z 528.1 (M + H)+, 550.1 (M + Na)+.
Methyl 2-((1-Methoxy-4-(4-phenylpiperazine-1-sulfonamido)naphthalen-2-yl)thio)acetate (49u)
As reported previously,68 to a solution of 1,1′-sulfonyldiimidazole (1.98 g, 10 mmol) in CH2Cl2 (40 mL) was added methyl trifluoromethanesulfonate (1.64 g, 10 mmol) at 0 °C. The solvent was removed after 3 h stirring. To the resulting residue in CH3CN (40 mL) was added 1-phenylpiperazine (1.08 g, 6.67 mmol). After being stirred at room temperature overnight, the reaction mixture was concentrated under reduced pressure, and the crude was purified using flash column chromatography on silica to give 1-((1H-imidazol-1-yl)sulfonyl)-4-phenylpiperazine (1.17 g, 60% yield over 2 steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.30–7.23 (m, 3H), 7.16 (s, 1H), 6.93 (t, J = 7.33 Hz, 1H), 6.89–6.84 (m, 2H), 3.37–3.30 (m, 4H), 3.26–3.20 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 150.21, 136.62, 130.80, 129.34, 121.43, 117.64, 117.20, 48.89, 46.42. ESI MS: m/z 293.0 (M + H)+.
To a solution of 1-((1H-imidazol-1-yl)sulfonyl)-4-phenylpiperazine (85 mg, 0.29 mmol) in CH2Cl2 (3 mL) cooled at 0 °C was added methyl trifluoromethanesulfonate (0.035 mL, 0.32 mmol). After being stirred for 2 h at 0 °C, the reaction mixture was concentrated under reduced pressure to give 3-methyl-1-((4-phenylpiperazin-1-yl)sulfonyl)-1H-imidazol-3-ium as a beige solid which was used in the next step without further purification.
A solution of 3-methyl-1-((4-phenylpiperazin-1-yl)sulfonyl)-1H-imidazol-3-ium (0.29 mmol) and aniline 49a (0.29 mmol) in CH3CN (3 mL) was stirred at 80 °C for 15 h. The reaction mixture was diluted with EtOAc (10 mL), washed with 1 N HCl, 1 N NaOH, H2O, and brine, dried (MgSO4), and filtered. The solvent was removed under reduced pressure, and the crude was purified using flash column chromatography on silica to give 49u (36 mg, 25% over three steps) as a purple solid. 1H NMR (400 MHz, CDCl3) δ 8.12–8.06 (m, 1H), 8.06–8.00 (m, 1H), 7.71 (s, 1H), 7.59–7.52 (m, 2H), 7.22 (t, J = 7.94 Hz, 2H), 6.88–6.81 (m, 3H), 6.75 (s, 1H), 3.98 (s, 3H), 3.74 (s, 2H), 3.68 (s, 3H), 3.43–3.37 (m, 4H), 3.15–3.08 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 169.86, 153.45, 150.61, 129.16, 128.93, 128.69, 128.57, 127.15, 127.07, 123.77, 123.51, 122.71, 121.71, 120.67, 116.73, 61.50, 52.59, 49.15, 46.48, 35.20. ESI MS: m/z 502.1 (M + H)+, 524.1 (M + Na)+.
Methyl 2-((4-(4-Bromobenzamido)-1-methoxynaphthalen-2-yl)thio)acetate (49v)
Synthesized using the procedure for 49f except using 4-bromobenzoyl chloride and Et3N as the base, which afforded the title compound (245 mg, 66%) as a pink solid. 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 8.09 (d, J = 8.38 Hz, 1H), 7.88 (s, 1H), 7.82–7.73 (m, 3H), 7.63–7.56 (m, 2H), 7.56–7.43 (m, 2H), 3.98 (s, 3H), 3.74 (s, 2H), 3.68 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.08, 165.42, 153.22, 133.17, 132.01, 131.74, 131.53, 128.82, 128.59, 128.43, 126.80, 126.77, 123.92, 123.50, 122.67, 121.57, 61.45, 52.57, 35.18. ESI MS: m/z 460.0, 462.0 (M + H)+, 482.0, 484 (M + Na)+.
Methyl 2-((4-((4-Chlorophenyl)methylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetate (49w)
Synthesized using the procedure for 49f except using 4-chlorobenzylsulfonyl chloride, which afforded the title compound (110 mg, 41% over two steps) as a light-pink solid. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.31 Hz, 1H), 7.75 (d, J = 8.31 Hz, 1H), 7.65 (s, 1H), 7.57 (t, J = 7.54 Hz, 1H), 7.50 (t, J = 7.54 Hz, 1H), 7.22–7.16 (m, 4H), 6.73 (s, 1H), 4.37 (s, 2H), 4.00 (s, 3H), 3.75 (s, 2H), 3.70 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.00, 153.34, 135.04, 132.09, 128.91, 128.74, 128.31, 127.98, 127.18, 127.08, 126.88, 123.66, 122.67, 122.59, 121.65, 61.55, 57.41, 52.66, 34.96. ESI MS: m/z 465.8 (M + H)+, 487.8 (M + Na)+.
Methyl 2-((4-(2-(4-Chlorophenyl)acetamido)-1-methoxynaphthalen-2-yl)thio)acetate (49x)
Synthesized using the procedure for 49f except using 4-chlorophenylacetyl chloride and Et3N as the base, which afforded the title compound (208 mg, 69% over two steps) as a light-yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.34 Hz, 1H), 7.82 (s, 1H), 7.49 (m, 1H), 7.45–7.38 (m, 4H), 7.38–7.32 (m, 3H), 3.94 (s, 3H), 3.82 (s, 2H), 3.73 (s, 2H), 3.67 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.16, 169.79, 153.07, 133.95, 132.72, 130.93, 129.52, 128.47, 128.24, 127.90, 126.80, 126.72, 123.43, 123.32, 122.67, 120.80, 61.41, 52.59, 43.65, 35.14. ESI MS: m/z 429.9 (M + H)+, 451.8 (M + Na)+.
Methyl 2-((1-Methoxy-4-(2-phenylacetamido)naphthalen-2-yl)thio)acetate (49y)
Synthesized using the procedure for 49f except using phenylacetyl chloride. Crude was triturated with cold methylene chloride to yield the title compound (154 mg, 49% over two steps) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.38 Hz, 1H), 7.94 (s, 1H), 7.53–7.44 (m, 4H), 7.40 (q, J = 7.27 Hz, 2H), 7.33 (s, 1H), 7.29–7.24 (m, 2H), 3.96 (s, 3H), 3.89 (s, 2H), 3.78 (s, 2H), 3.71 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.10, 169.71, 152.55, 134.54, 129.69, 129.46, 128.70, 128.40, 127.92, 127.64, 126.59, 123.48, 122.57, 122.48, 120.78, 61.40, 52.61, 44.64, 35.16. ESI MS: m/z 396.1 (M + H)+, 418.1 (M + Na)+.
4-Bromo-N-(3-(butylthio)-4-methoxynaphthalen-1-yl)benzenesulfonamide (49z)
Synthesized using the procedure for 49f except using 49c and 4-bromobenzenesulfonyl chloride, which afforded the title compound (55 mg, 57% over two steps) as a light-brown oil which solidified. 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.44 Hz, 1H), 7.77 (d, J = 8.44 Hz, 1H), 7.59 (d, J = 8.36 Hz, 2H), 7.49 (d, J = 8.36 Hz, 2H), 7.46 (d, J = 7.85 Hz, 1H), 7.35 (t, J = 7.85 Hz, 1H), 7.21 (s, 1H), 7.16 (s, 1H), 3.94 (s, 3H), 2.81 (t, J = 7.24 Hz, 2H), 1.58 (p, J = 7.24 Hz, 2H), 1.45 (h, J = 7.24 Hz, 2H), 0.92 (t, J = 7.24 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 152.71, 138.26, 132.23, 128.92, 128.88, 128.65, 127.99, 127.25, 126.93, 126.37, 125.79, 124.49, 122.19, 122.04, 61.01, 31.92, 31.20, 21.95, 13.67. ESI MS: m/z 479.8, 481.8 (M + H)+.
2-((4-(4-Bromophenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetamide (42)
Synthesized using a reported procedure.71 Compound 41 (82 mg, 0.16 mmol) was added to aqueous NH4OH (29%, 3 mL). The mixture was stirred at room temperature for 1 h. Workup included diluting the mixture with EtOAc (10 mL) and washing with H2O (10 mL × 2) and brine (10 mL). Organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure to give 42 (69 mg, 90%) as a pink solid. Crude was used in the next step without further purification. HPLC (method A, tR = 6.50 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 7.95 (d, J = 8.50 Hz, 1H), 7.85 (d, J = 8.50 Hz, 1H), 7.72 (d, J = 8.20 Hz, 2H), 7.60 (d, J = 8.20 Hz, 2H), 7.56–7.50 (m, 2H), 7.43–7.37 (m, 1H), 7.21 (s, 1H), 7.15 (s, 1H), 3.88 (s, 3H), 3.53 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.76, 151.88, 139.67, 132.70, 129.69, 129.18, 129.05, 128.23, 127.46, 127.03, 126.41, 125.24, 124.89, 123.95, 121.82, 61.27, 35.91. ESI MS: m/z 480.7, 482.7 (M + H)+, 502.7, 504.7 (M + Na)+. ESI HRMS: m/z 478.9742 (M – H)−.
2-((1-Methoxy-4-((4-phenoxyphenyl)sulfonamido)naphthalen-2-yl)thio)acetamide (49aa)
Synthesized using the procedure for 42 except using 49s as the starting material, which afforded the title compound (179 mg, quantitative) as a light-purple solid. 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.56–7.46 (m, 2H), 7.41 (t, J = 7.0 Hz, 2H), 7.39–7.33 (m, 1H), 7.23–7.14 (m, 2H), 7.11 (s, 1H), 7.04–6.94 (m, 4H), 3.82 (s, 3H), 3.50 (s, 2H). 13C NMR (125 MHz, DMSO-d6) δ 169.84, 155.74, 130.72, 129.77, 129.55, 128.23, 127.07, 125.57, 125.09, 124.94, 124.55, 121.45, 120.02, 118.17, 109.99, 61.17, 36.13. ESI MS: m/z 495.0 (M + H)+, 517.0 (M + Na)+.
4-Bromo-N-(3-(3-hydroxypropyl)-4-methoxynaphthalen-1-yl)benzenesulfonamide (49bb)
Synthesized using the procedure for 49f except using 49d and 4-bromobenzenesulfonyl chloride, which afforded the title compound (163 mg, 33% over two steps) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.94 (t, J = 7.53 Hz, 2H), 7.70 (d, J = 8.53 Hz, 2H), 7.53 (d, J = 8.53 Hz, 2H), 7.51–7.45 (m, 1H), 7.42–7.35 (m, 1H), 6.86 (s, 1H), 4.47 (s, 1H), 3.79 (s, 3H), 3.37 (t, J = 5.87 Hz, 2H), 2.69–2.60 (m, 2H), 1.59–1.49 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 152.33, 139.44, 132.62, 130.38, 130.34, 129.28, 128.55, 128.23, 127.00, 126.88, 126.78, 126.01, 124.09, 122.21, 62.33, 60.67, 33.79, 25.69. ESI MS: m/z 449.9, 451.9 (M + H)+, 471.9, 473.9 (M + Na)+.
4-Bromo-N-(3-(4-hydroxybutyl)-4-methoxynaphthalen-1-yl)benzenesulfonamide (45)
Synthesized using the procedure for 49f except using 49e and 4-bromobenzenesulfonyl chloride, which afforded the title compound (226 mg, 49% over two steps) as a light-pink solid. HPLC (method A, tR = 6.94 min), purity 96%. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 7.99–7.91 (m, 2H), 7.70 (d, J = 8.43 Hz, 2H), 7.54 (d, J = 8.43 Hz, 2H), 7.49 (t, J = 7.34 Hz, 1H), 7.40 (t, J = 7.34 Hz, 1H), 6.81 (s, 1H), 4.33 (t, J = 5.42 Hz, 1H), 3.78 (s, 3H), 3.37 (q, J = 5.42 Hz, 2H), 2.59 (t, J = 7.01 Hz, 2H), 1.49–1.28 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 152.35, 139.45, 132.62, 130.46, 130.42, 129.33, 128.54, 128.24, 126.97, 126.82, 126.78, 126.02, 124.16, 122.21, 62.36, 60.92, 32.66, 28.85, 26.90. ESI MS: m/z 464.1, 466.1 (M + H)+, 486.1, 488.1 (M + Na)+. ESI HRMS: m/z 462.0378 (M – H)−.
N-(3-(((2H-Tetrazol-5-yl)methyl)thio)-4-methoxynaphthalen-1-yl)-4-bromobenzenesulfonamide (49cc)
Synthesized using a reported procdure.72 To a suspension of 42 (127 mg, 0.26 mmol) and sodium azide (255 mg, 3.9 mmol) in dry CH3CN (5 mL) in a glass tube was added SiCl4 (0.15 mL, 1.3 mmol) via syringe. The tube was sealed, and the stirring reaction mixture was heated to 80 °C for 15 h. The reaction was quenched with 2 M Na2CO3. The solution was then washed with EtOAc (10 mL × 2). The aqueous phase was acidified with 1 N HCl, and the mixture was extracted with EtOAc (15 mL × 5). The combined organic extracts were washed with brine (20 mL), dried (MgSO4), filtered, and concentrated under reduced pressure to give 49cc (66 mg, 50%) as a tan solid. The crude was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 7.93 (d, J = 8.40 Hz, 1H), 7.83 (d, J = 8.40 Hz, 1H), 7.65 (d, J = 8.04 Hz, 2H), 7.55–7.48 (m, 3H), 7.44–7.38 (m, 1H), 7.14 (s, 1H), 6.53 (s, 1H), 4.42 (s, 2H), 3.78 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 153.77, 139.46, 132.64, 130.46, 129.17, 129.07, 128.36, 127.58, 127.06, 126.23, 123.97, 122.53, 122.22, 61.68, 25.09. ESI MS: m/z 505.8, 507.8 (M + H)+, 527.8, 529.8 (M + Na)+.
N-(3-(((2H-Tetrazol-5-yl)methyl)thio)-4-methoxynaphthalen-1-yl)-4-phenoxybenzenesulfonamide (49dd)
Synthesized using the procedure for 49cc except using 49aa as the starting material which afforded the title compound (128 mg, 95%) as a tan solid. 1H NMR (500 MHz, DMSO-d6) δ 10.14 (s, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.56 (t, J = 7.5 Hz, 1H), 7.48–7.43 (m, 1H), 7.40 (t, J = 7.4 Hz, 2H), 7.20 (t, J = 7.3 Hz, 1H), 7.17 (s, 1H), 7.01–6.94 (m, 4H), 4.44 (s, 2H), 3.79 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 160.90, 155.45, 153.57, 134.18, 130.75, 130.55, 129.76, 129.50, 128.35, 127.49, 126.94, 126.14, 125.17, 124.15, 122.62, 122.16, 120.13, 118.27, 61.66, 25.13. ESI MS: m/z 517.9 (M – H)−.
Methyl 3-((1-Methoxy-4-(naphthalene-2-sulfonamido)naphthalen-2-yl)thio)propanoate (49ee)
Synthesized using the procedure for 49f except using 49b and 2-naphthalenesulfonyl chloride, which afforded the title compound (180 mg, 59% over two steps) as a light-pink solid. 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.98 (d, J = 8.39 Hz, 1H), 7.90–7.85 (m, 2H), 7.85–7.80 (m, 2H), 7.80–7.76 (m, 1H), 7.59 (t, J = 7.36 Hz, 1H), 7.53 (t, J = 7.36 Hz, 1H), 7.43 (t, J = 7.39 Hz, 1H), 7.34 (t, J = 7.39 Hz, 1H), 7.19 (s, 1H), 6.98 (s, 1H), 3.89 (s, 3H), 3.64 (s, 3H), 2.94 (t, J = 7.23 Hz, 2H), 2.40 (t, J = 7.23 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 171.84, 153.85, 135.99, 134.83, 131.94, 129.66, 129.43, 129.20, 129.02, 128.91, 128.75, 127.82, 127.78, 127.54, 126.95, 126.72, 125.21, 123.73, 122.39, 122.19, 109.99, 61.20, 51.79, 34.05, 27.65. ESI MS: m/z 481.9 (M + H)+, 503.9 (M + Na)+.
2-((4-(4-Bromophenylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetic Acid (40)
To a solution of 41 (425 mg, 0.85 mmol) in dry THF (2 mL) was added 1N aqueous LiOH (4 mL). The mixture was stirred at room temperature under nitrogen for 1 h. Reaction mixture was diluted with water (10 mL) and washed with EtOAc (10 mL × 2). Aqueous phase was acidified with 1 N HCl and extracted with EtOAc (10 mL × 3). Combined organic extracts were washed with brine, dried (MgSO4), and filtered. The solvent was removed under reduced pressure. Crude was triturated with cold CH2Cl2 to give 40 (305 mg, 74%) as a white/tan solid. HPLC (method A, tR = 6.85 min), purity >99%. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 7.92 (d, J = 8.38 Hz, 1H), 7.87 (d, J = 8.38 Hz, 1H), 7.68 (d, J = 8.08 Hz, 2H), 7.57 (d, J = 8.08 Hz, 2H), 7.51 (t, J = 7.56 Hz, 1H), 7.40 (t, J = 7.56 Hz, 1H), 7.09 (s, 1H), 3.84 (s, 3H), 3.65 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 170.59, 152.07, 139.60, 132.69, 129.83, 129.18, 129.13, 128.28, 127.54, 127.09, 126.57, 124.60, 124.56, 124.01, 121.86, 61.30, 34.63. ESI HRMS: m/z 479.9578 (M – H)−.
2-((4-([1,1′-Biphenyl]-4-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetic Acid (43)
Synthesized using the procedure for 40 except 49n was used as the starting material. Crude was triturated with cold CH2Cl2 to give 43 (88 mg, 70%) as an white solid. HPLC (method A, tR = 7.22 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 12.74 (s, 1H), 10.21 (s, 1H), 7.95 (d, J = 8.44 Hz, 1H), 7.91 (d, J = 8.44 Hz, 1H), 7.79–7.70 (m, 4H), 7.68–7.61 (m, 2H), 7.53–7.48 (m, 1H), 7.45 (t, J = 7.28 Hz, 2H), 7.39 (t, J = 7.28 Hz, 2H), 7.08 (s, 1H), 3.83 (s, 3H), 3.61 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 170.52, 151.84, 144.73, 139.09, 138.87, 129.90, 129.50, 128.93, 128.26, 127.89, 127.78, 127.49, 126.47, 124.56, 124.21, 124.18, 121.77, 61.26, 34.56. ESI HRMS: m/z 478.0787 (M – H)−.
2-((4-([1,1‴-Biphenyl]-3-ylsulfonamido)-1-methoxynaphthalen-2-yl)thio)acetic Acid (44)
Synthesized using the procedure for 40 except 49o was used as the starting material. Crude was triturated with cold CH2Cl2 to give 44 (152 mg, 59%) as a white solid. HPLC (method A, tR = 7.20 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 12.69 (s, 1H), 10.20 (s, 1H), 7.90 (d, J = 8.46 Hz, 1H), 7.86 (d, J = 8.46 Hz, 1H), 7.82 (d, J = 7.81 Hz, 1H), 7.72 (s, 1H), 7.63 (d, J = 7.81 Hz, 1H), 7.55 (t, J = 7.74 Hz, 1H), 7.50–7.44 (m, 1H), 7.43–7.38 (m, 4H), 7.38–7.30 (m, 2H), 7.11 (s, 1H), 3.82 (s, 3H), 3.61 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 170.46, 151.97, 141.49, 140.71, 138.92, 131.42, 130.33, 129.80, 129.49, 129.43, 128.66, 128.24, 127.47, 127.14, 126.43, 125.88, 125.21, 124.64, 124.59, 124.04, 121.77, 61.23, 34.56. ESI HRMS: m/z 478.0788 (M – H)−.
A Representative Procedure for a Single-Pot Ester Hydrolysis and Demethylation with BBr3
2-((1-Hydroxy-4-(thiophene-2-sulfonamido)naphthalen-2-yl)thio)acetic Acid (1)
To a stirred solution of 49f (48 mg, 0.12 mmol) suspended in dry CH2Cl2 (1.5 mL) was added BBr3 (1 M in CH2Cl2, 0.5 mL, 0.5 mmol) dropwise at 0 °C under nitrogen. The mixture was allowed to warm up to room temperature. After 1 h of stirring, the starting material was entirely consumed and the product formed as determined by TLC and MS (ESI–). The mixture was slowly added to a stirred solution of saturated aqueous NH4Cl (20 mL) at 0 °C. The solution was extracted with EtOAc (15 mL × 2). The combined organic extracts were washed with brine (15 mL), dried (MgSO4), and filtered, and the solvent was removed under reduced pressure. The crude was purified using a C18 reverse phase semipreparative HPLC column with solvent A (0.1% of TFA in water) and solvent B (0.1% of TFA in CH3CN) as eluents to give 1 (59) (28 mg, 59%) as a white/tan solid. HPLC (method A, tR = 6.05 min), purity 92%. 1H NMR (400 MHz, DMSO-d6) δ 10.10 (s, 1H), 8.17–8.14 (m, 1H), 7.89–7.84 (m, 2H), 7.52–7.40 (m, 2H), 7.36 (dd, J = 1.32, 3.74 Hz, 1H), 7.09 (s, 1H), 7.07 (dd, J = 3.74, 4.97 Hz, 1H), 3.56 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.54, 152.87, 140.58, 133.57, 132.80, 131.71, 129.80, 128.03, 127.20, 126.22, 125.46, 124.33, 123.61, 122.83, 113.36, 37.25. ESI HRMS: m/z 393.9870 (M – H)−.
2-((1-Hydroxy-4-(methylsulfonamido)naphthalen-2-yl)thio)acetic Acid (2)
Synthesized using the procedure for 1 except 49t was used as the starting material. The title compound (27 mg, 48%) as a white solid after HPLC purification. HPLC (method A, tR = 5.14 min), purity 97%. 1H NMR (400 MHz, DMSO-d6) δ 9.40 (s, 1H), 8.20 (d, J = 8.09 Hz, 1H), 8.14 (d, J = 8.09 Hz, 1H), 7.56 (p, J = 6.86 Hz, 2H), 7.47 (s, 1H), 3.69 (s, 2H), 2.99 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.73, 152.58, 131.89, 129.38, 127.31, 126.30, 125.65, 125.18, 124.32, 122.82, 113.55, 39.79, 37.03. ESI HRMS: m/z 326.0164 (M – H)−.
2-((1-Hydroxy-4-(phenylsulfonamido)naphthalen-2-yl)thio)acetic Acid (3)
Synthesized using the procedure for 1 except 49h was used as the starting material. The title compound (21 mg, 22%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 6.15 min), purity 97%. 1H NMR (400 MHz, DMSO-d6) δ 12.76 (s, 1H), 9.91 (s, 1H), 9.79 (s, 1H), 8.14 (d, J = 8.22 Hz, 1H), 7.88 (d, J = 8.22 Hz, 1H), 7.67–7.61 (m, 2H), 7.61–7.56 (m, 1H), 7.54–7.47 (m, 3H), 7.46–7.39 (m, 1H), 7.01 (s, 1H), 3.51 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.46, 152.56, 140.16, 133.03, 131.56, 129.53, 129.44, 127.25, 127.05, 126.17, 125.46, 124.56, 123.76, 122.75, 113.34, 37.16. ESI HRMS: m/z 388.0319 (M – H)−.
2-((4-(4-Chlorophenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (9)
Synthesized using the procedure for 1 except 49i was used as the starting material. The title compound (48 mg, 20%) was obtained as a white solid after trituration with a mixture of CH3CN:H2O 1:1 and cold CH2Cl2 and without a need for HPLC purification. HPLC (method B, tR = 6.72 min), purity 95%. 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 1H), 10.03 (s, 1H), 9.84 (s, 1H), 8.15 (d, J = 8.12 Hz, 1H), 7.85 (d, J = 8.12 Hz, 1H), 7.66–7.59 (m, 2H), 7.59–7.52 (m, 2H), 7.46 (dt, J = 7.28, 14.85 Hz, 2H), 7.05 (s, 1H), 3.54 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.48, 152.72, 139.06, 137.95, 131.47, 129.66, 129.60, 129.19, 127.15, 126.25, 125.48, 124.24, 123.63, 122.84, 113.38, 37.11. ESI HRMS: m/z 421.9930 (M – H)−.
2-((4-(4-Bromophenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (10)
Synthesized using the procedure for 1 except 41 was used as the starting material. The title compound (134 mg, 45%) was obtained as a white solid after trituration with a mixture of CH3CN:H2O 1:1 and cold CH2Cl2 and without a need for HPLC purification. HPLC (method A, tR = 6.53 min), purity 99%. 1H NMR (400 MHz, DMSO-d6) δ 12.80 (s, 1H), 10.05 (s, 1H), 9.87 (s, 1H), 8.14 (d, J = 8.17 Hz, 1H), 7.84 (d, J = 8.17 Hz, 1H), 7.71 (d, J = 8.48 Hz, 2H), 7.54 (d, J = 8.48 Hz, 2H), 7.48 (t, J = 7.03 Hz, 1H), 7.43 (t, J = 7.03 Hz, 1H), 7.04 (s, 1H), 3.53 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.52, 152.74, 139.45, 132.56, 131.45, 129.66, 129.28, 127.18, 126.92, 126.27, 125.48, 124.21, 123.63, 122.86, 113.38, 37.10. ESI HRMS: m/z 465.9418 (M – H)−.
2-((1-Hydroxy-4-(4-methoxyphenylsulfonamido)naphthalen-2-yl)thio)acetic Acid (11)
Synthesized using the procedure for 1 except 49j was used as the starting material. The title compound (25 mg, 29%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 6.20 min), purity 95%. 1H NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.10 (d, J = 8.07 Hz, 1H), 7.89 (d, J = 7.78 Hz, 1H), 7.52 (d, J = 8.63 Hz, 2H), 7.49–7.36 (m, 2H), 7.00–6.93 (m, 3H), 3.75 (s, 3H), 3.47 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.47, 162.75, 152.44, 131.81, 131.63, 129.49, 129.37, 127.05, 126.18, 125.48, 124.92, 123.93, 122.75, 114.57, 113.35, 56.02, 37.24. ESI HRMS: m/z 418.0424 (M – H)−.
2-((4-(3-Bromophenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (12)
Synthesized using the procedure for 1 except 49k was used as the starting material. The title compound (21 mg, 35%) was obtained as a white/tan solid after trituration with a mixture of CH3CN:H2O 1:1 and cold CH2Cl2 and without a need for HPLC purification. HPLC (method A, tR = 6.69 min), purity 94%. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.15 (d, J = 7.56 Hz, 1H), 7.87–7.78 (m, 2H), 7.75 (t, J = 1.78 Hz, 1H), 7.62–7.56 (m, 1H), 7.52–7.39 (m, 3H), 7.01 (s, 1H), 3.52 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.52, 152.92, 142.07, 135.93, 131.78, 131.49, 129.77, 129.59, 127.18, 126.34, 126.28, 125.53, 124.02, 123.58, 122.89, 122.35, 113.43, 37.24. ESI HRMS: m/z 465.9414 (M – H)−.
2-((4-(2-Bromophenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (13)
Synthesized using the procedure for 1 except 49l was used as the starting material. The title compound (34 mg, 40%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 6.41 min), purity 85%. 1H NMR (400 MHz, DMSO-d6) δ 12.80 (s, 1H), 10.23 (s, 1H), 9.88 (s, 1H), 8.16–8.10 (m, 1H), 8.07–8.02 (m, 1H), 7.86 (dd, J = 1.52, 7.83 Hz, 1H), 7.78 (dd, J = 1.52, 7.83 Hz, 1H), 7.52–7.46 (m, 3H), 7.42 (td, J = 1.33, 7.60 Hz, 1H), 7.05 (s, 1H), 3.46 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.42, 152.60, 139.23, 135.82, 134.64, 131.92, 131.77, 129.49, 128.52, 127.18, 126.32, 125.44, 123.98, 123.78, 122.78, 119.92, 113.54, 37.03. ESI HRMS: m/z 465.9408 (M – H)−.
2-((4-(Benzo[b]thiophene-2-sulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (14)
Synthesized using the procedure for 1 except 49m was used as the starting material. The title compound (13 mg, 22%) was obtained as an orange solid after HPLC purification. HPLC (method B, tR = 7.52 min), purity 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.11 (d, J = 8.14 Hz, 1H), 8.02 (d, J = 8.14 Hz, 1H), 7.89 (t, J = 8.09 Hz, 2H), 7.72 (s, 1H), 7.47 (t, J = 7.56 Hz, 1H), 7.41 (t, J = 7.35 Hz, 2H), 7.38–7.32 (m, 1H), 7.14 (s, 1H), 3.40 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.57, 153.29, 141.31, 141.04, 137.80, 131.73, 129.99, 129.94, 127.65, 127.22, 126.21, 125.86, 125.54, 123.96, 123.54, 123.41, 122.92, 113.33, 37.41. ESI HRMS: m/z 444.0041 (M – H)−.
2-((4-([1,1′-Biphenyl]-4-ylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (16)
Synthesized using the procedure for 1 except 49n was used as the starting material. The title compound (29 mg, 45%) was obtained as a white/tan solid after HPLC purification. HPLC (method B, tR = 8.81 min), purity 99%. 1H NMR (400 MHz, DMSO-d6) δ 9.92 (s, 1H), 8.11 (d, J = 7.93 Hz, 1H), 7.90 (d, J = 7.93 Hz, 1H), 7.76 (d, J = 7.47 Hz, 2H), 7.72–7.62 (m, 4H), 7.50–7.42 (m, 3H), 7.43–7.36 (m, 2H), 7.02 (s, 1H), 3.46 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.52, 152.74, 144.57, 139.00, 138.90, 131.65, 129.57, 129.52, 128.92, 128.00, 127.63, 127.46, 127.08, 126.18, 125.55, 124.53, 123.85, 122.81, 113.42, 37.27. ESI HRMS: m/z 464.0630 (M – H)−.
2-((4-([1,1′-Biphenyl]-3-ylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (17)
Synthesized using the procedure for 1 except 49o was used as the starting material. The title compound (48 mg, 34%) was obtained as a white/tan solid after HPLC purification. HPLC (method A, tR = 6.99 min), purity 95%. 1H NMR (400 MHz, DMSO-d6) δ 12.73 (s, 1H), 9.93 (s, 1H), 9.79 (s, 1H), 8.10 (d, J = 8.25 Hz, 1H), 7.88–7.81 (m, 2H), 7.79 (s, 1H), 7.60–7.50 (m, 2H), 7.50–7.41 (m, 4H), 7.41–7.32 (m, 3H), 7.04 (s, 1H), 3.45 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.44, 152.66, 141.46, 140.82, 139.01, 131.50, 131.30, 130.18, 129.60, 129.55, 128.66, 127.17, 127.07, 126.23, 126.07, 125.51, 125.26, 124.59, 123.73, 122.81, 113.44, 37.16. ESI HRMS: m/z 464.0632 (M – H)−.
2-((4-([1,1′-Biphenyl]-2-ylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (18)
Synthesized using the procedure for 1 except 49p was used as the starting material. The title compound (20 mg, 43%) was obtained as a white/tan solid after HPLC purification. HPLC (method B, tR = 8.95 min), purity 84%. 1H NMR (400 MHz, DMSO-d6) δ 12.77 (s, 1H), 9.76 (s, 1H), 9.70 (s, 1H), 8.12 (d, J = 8.03 Hz, 1H), 7.95 (d, J = 8.03 Hz, 1H), 7.74 (d, J = 8.30 Hz, 1H), 7.58 (t, J = 7.46 Hz, 1H), 7.52 (t, J = 7.46 Hz, 1H), 7.48–7.41 (m, 1H), 7.41–7.34 (m, 1H), 7.23 (q, J = 4.82, 5.81 Hz, 1H), 7.17 (t, J = 7.46 Hz, 3H), 7.01–6.95 (m, 1H), 6.93 (s, 1H), 6.91 (s, 1H), 3.43 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.57, 152.24, 141.36, 139.92, 138.75, 133.21, 132.68, 131.42, 129.62, 129.37, 128.85, 128.36, 127.55, 127.48, 126.97, 126.21, 125.49, 124.57, 123.79, 122.69, 113.65, 37.07. ESI HRMS: m/z 464.0636 (M – H)−.
2-((4-(4′-Chloro-[1,1′-biphenyl]-4-ylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (19)
Synthesized using the procedure for 1 except 49q was used as the starting material. The title compound (36 mg, 25%) was obtained as a white solid after HPLC purification. HPLC (method B, tR = 10.23 min), purity >99%. 1H NMR (400 MHz, DMSO-d6) δ 12.75 (s, 1H), 9.96 (s, 1H), 9.81 (s, 1H), 8.10 (d, J = 8.09 Hz, 1H), 7.88 (d, J = 8.09 Hz, 1H), 7.77 (d, J = 8.24 Hz, 2H), 7.72–7.64 (m, 4H), 7.52 (d, J = 8.24 Hz, 2H), 7.47–7.36 (m, 2H), 6.99 (s, 1H), 3.45 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.45, 152.51, 143.18, 139.25, 137.67, 133.88, 131.56, 129.50, 129.39, 129.27, 128.05, 127.65, 127.09, 126.24, 125.50, 124.54, 123.84, 122.78, 113.44, 37.04. ESI HRMS: m/z 498.0247 (M – H)−.
2-((4-(2′,4′-Difluoro-[1,1′-biphenyl]-4-ylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (20)
Synthesized using the procedure for 1 except 49r was used as the starting material. The title compound (31 mg, 48%) as a white solid after HPLC purification. HPLC (method A, tR = 7.12 min), purity 97%. 1H NMR (400 MHz, DMSO-d6) δ 12.71 (s, 1H), 9.97 (s, 1H), 9.80 (s, 1H), 8.10 (d, J = 8.16 Hz, 1H), 7.86 (d, J = 8.16 Hz, 1H), 7.69 (d, J = 7.72 Hz, 2H), 7.62 (d, J = 7.72 Hz, 2H), 7.59–7.53 (m, 1H), 7.47–7.34 (m, 3H), 7.19 (t, J = 8.31 Hz, 1H), 7.00 (s, 1H), 3.46 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.46, 152.55, 139.44, 138.77, 132.51 (dd, J = 4.88, 9.80 Hz), 131.58, 129.82 (d, J = 2.86 Hz), 129.50, 127.66, 127.06, 126.23, 125.50, 124.50, 123.81, 122.78, 113.47, 112.73 (dd, J = 3.78, 21.26 Hz), 105.15 (t, J = 26.72 Hz), 37.04. ESI HRMS: m/z 500.0441 (M – H)−.
2-((1-Hydroxy-4-(4-phenoxyphenylsulfonamido)naphthalen-2-yl)thio)acetic Acid (21)
Synthesized using the procedure for 1 except 49s was used as the starting material. The title compound (18 mg, 13%) was obtained as a white solid after HPLC purification. HPLC (method B, tR = 10.24 min), purity 99%. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H), 9.87 (s, 1H), 9.83 (s, 1H), 8.14 (d, J = 8.31 Hz, 1H), 7.86 (d, J = 8.31 Hz, 1H), 7.59 (d, J = 8.75 Hz, 2H), 7.50 (t, J = 7.09 Hz, 1H), 7.44 (t, J = 7.81 Hz, 3H), 7.23 (t, J = 7.09 Hz, 1H), 7.05–6.98 (m, 5H), 3.55 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.53, 160.76, 155.60, 152.58, 134.17, 131.57, 130.75, 129.84, 129.76, 127.06, 126.16, 125.48, 125.07, 124.60, 123.78, 122.80, 120.03, 118.33, 113.38, 37.15. ESI HRMS: m/z 480.0579 (M – H)−.
2-((4-(4-(4-Fluorophenoxy)phenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (22)
Synthesized using the procedure for 1 except 49t was used as the starting material. The title compound (36 mg, 44%) as a white solid after HPLC purification. HPLC (method A, tR = 7.17 min), purity 95%. 1H NMR (400 MHz, DMSO-d6) δ 12.78 (s, 1H), 9.83 (s, 1H), 9.79 (s, 1H), 8.11 (d, J = 8.21 Hz, 1H), 7.83 (d, J = 8.21 Hz, 1H), 7.55 (d, J = 8.46 Hz, 2H), 7.44 (dt, J = 6.92, 21.86 Hz, 2H), 7.25 (t, J = 8.52 Hz, 2H), 7.11–7.02 (m, 2H), 6.99–6.96 (m, 2H), 6.95 (s, 1H), 3.51 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.53, 161.09, 152.57, 151.54 (d, J = 2.57 Hz), 134.14, 131.59, 129.86, 129.68, 127.08, 126.19, 125.48, 124.61, 123.80, 122.80, 122.17 (d, J = 8.70 Hz), 117.90, 117.45, 117.22, 113.38, 37.17. ESI HRMS: m/z 498.0485 (M – H)−.
2-((1-Hydroxy-4-(4-phenylpiperazine-1-sulfonamido)naphthalen-2-yl)thio)acetic Acid (23)
Synthesized using the procedure for 1 except 49u was used as the starting material. The title compound (14.5 mg, 48%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 6.55 min), purity 92%. 1H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 9.59 (s, 1H), 8.15 (t, J = 7.91 Hz, 2H), 7.59–7.53 (m, 1H), 7.53–7.48 (m, 2H), 7.20–7.14 (m, 2H), 6.89 (d, J = 7.91 Hz, 2H), 6.77 (t, J = 7.26 Hz, 1H), 3.64 (s, 2H), 3.24–3.16 (m, 4H), 3.14–3.04 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 171.57, 152.02, 150.84, 131.08, 129.40, 128.56, 127.32, 126.28, 125.52, 125.43, 123.76, 122.95, 120.04, 116.48, 113.63, 48.54, 46.36, 37.06. ESI HRMS: m/z 472.1002 (M – H)−.
2-((4-(4-Bromobenzamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (25)
Synthesized using the procedure for 1 except 49v was used as the starting material. The title compound was obtained (8.5 mg, 30%) as a white solid after HPLC purification. HPLC (method A, tR = 6.72 min), purity 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.23 (d, J = 5.36 Hz, 1H), 8.02 (d, J = 8.05 Hz, 2H), 7.83 (d, J = 5.36 Hz, 1H), 7.78 (d, J = 8.05 Hz, 2H), 7.57–7.50 (m, 3H), 3.72 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.74, 165.71, 151.94, 133.90, 131.89, 130.66, 130.30, 128.64, 127.11, 126.17, 126.13, 125.80, 125.47, 123.80, 122.97, 113.65, 37.15. ESI HRMS: m/z 429.9747 (M – H)−.
2-((4-((4-Chlorophenyl)methylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (26)
Synthesized using the procedure for 1 except 49w was used as the starting material. The title compound (15.5 mg, 20%) was obtained as a white solid after HPLC purification. HPLC (method B, tR = 6.22 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 12.86 (s, 1H), 9.84 (s, 1H), 9.55 (s, 1H), 8.20 (d, J = 8.36 Hz, 1H), 8.07 (d, J = 8.36 Hz, 1H), 7.60–7.51 (m, 2H), 7.44 (s, 1H), 7.43–7.38 (m, 4H), 4.49 (s, 2H), 3.71 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.81, 152.35, 133.45, 133.18, 131.79, 129.37, 128.93, 128.72, 127.23, 126.30, 125.63, 124.99, 124.28, 122.79, 113.67, 57.16, 36.96. ESI HRMS: m/z 436.0086 (M – H)−.
2-((4-(2-(4-Chlorophenyl)acetamido)-1-hydroxynaphthalen-2-yl)thio)acetic Acid (28)
Synthesized using the procedure for 1 except 49x was used as the starting material. The title compound (19 mg, 10%) was obtained as a white solid after trituration with a mixture of CH3CN:H2O 1:1 and cold CH2Cl2 and without a need for HPLC purification. HPLC (method B, tR = 5.77 min), purity 93%. 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.23–8.17 (m, 1H), 7.89–7.83 (m, 1H), 7.57–7.50 (m, 3H), 7.47–7.39 (m, 4H), 3.78 (s, 2H), 3.66 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.72, 169.89, 151.36, 135.73, 131.66, 131.49, 129.70, 128.69, 127.15, 127.00, 126.11, 126.07, 125.40, 123.21, 123.02, 113.51, 42.24, 37.21. ESI HRMS: m/z 400.0417 (M – H)−.
2-((1-Hydroxy-4-(2-phenylacetamido)naphthalen-2-yl)thio)acetic Acid (29)
Synthesized using the procedure for 1 except 49y was used as the starting material. The title compound (50 mg, 61%) was obtained as a white solid after trituration with a mixture of CH3CN:H2O 1:1 and cold CH2Cl2 and without a need for HPLC purification. HPLC (method A, tR = 6.07 min), purity 94%. 1H NMR (400 MHz, DMSO-d6) δ 12.78 (s, 1H), 9.97 (s, 1H), 9.66 (s, 1H), 8.26–8.12 (m, 1H), 7.92–7.77 (m, 1H), 7.59–7.47 (m, 3H), 7.42 (d, J = 7.64 Hz, 2H), 7.37 (t, J = 7.40 Hz, 2H), 7.31–7.24 (m, 1H), 3.77 (s, 2H), 3.67 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 171.73, 170.25, 151.26, 136.74, 129.70, 129.58, 128.76, 127.10, 126.97, 126.92, 126.19, 126.06, 125.38, 123.22, 122.99, 113.49, 43.09, 37.13. ESI HRMS: m/z 366.0802 (M – H)−.
4-Bromo-N-(3-(butylthio)-4-hydroxynaphthalen-1-yl)benzenesulfonamide (31)
Synthesized using the procedure for 1 except 49z was used as the starting material. The title compound (10 mg, 8%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 8.44 min), purity 97%. 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 7.40 Hz, 1H), 7.79 (d, J = 7.40 Hz, 1H), 7.59–7.50 (m, 4H), 7.50–7.45 (m, 1H), 7.37 (s, 1H), 7.26 (s, 1H), 6.60 (s, 1H), 3.49 (s, 1H), 2.65 (t, J = 7.24 Hz, 2H), 1.52–1.45 (m, 2H), 1.45–1.35 (m, 2H), 0.90 (t, J = 7.24 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 154.04, 138.33, 132.18, 131.86, 131.23, 128.98, 128.09, 127.90, 126.29, 123.80, 123.47, 122.92, 122.16, 111.25, 36.61, 31.70, 21.74, 13.60. ESI HRMS: m/z 463.9998 (M – H)−.
Methyl 2-((4-(4-Bromophenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetate (32)
To a suspension of 41 (75 mg, 0.15 mmol) in dry CH2Cl2 (1.5 mL) was added BBr3 (1 M in CH2Cl2, 0.4 mL, 0.4 mmol) dropwise at 0 °C. The mixture was allowed to warm up to room temperature and stirred under nitrogen. The starting material was entirely consumed as determined by TLC and analytical HPLC after 30 min. The mixture was again cooled down to 0 °C and MeOH (2 mL) was added. After addition, the mixture was allowed to warm up to room temperature and stirred for 1 h when a new spot formed as monitored by TLC. The mixture was slowly added to a stirring solution of saturated aqueous NH4Cl (15 mL) at 0 °C. The solution was extracted with EtOAc (15 mL × 2). The combined organic extracts were washed with brine (15 mL), dried (MgSO4), and filtered. The solvent was removed under reduced pressure. The crude was purified using a C18 reverse phase semipreparative HPLC column with solvent A (0.1% of TFA in water) and solvent B (0.1% of TFA in CH3CN) as eluents to give 32 (31 mg, 43%) as a white solid. HPLC (method A, tR = 7.45 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.90 (s, 1H), 8.15 (d, J = 8.22 Hz, 1H), 7.85 (d, J = 8.22 Hz, 1H), 7.73 (d, J = 8.58 Hz, 2H), 7.55 (d, J = 8.58 Hz, 2H), 7.52–7.41 (m, 2H), 6.99 (s, 1H), 3.60 (s, 5H). 13C NMR (100 MHz, DMSO-d6) δ 170.21, 152.67, 139.44, 132.58, 131.45, 129.39, 129.23, 127.20, 126.93, 126.30, 125.51, 124.25, 123.67, 122.85, 113.07, 52.64, 36.18. ESI HRMS: m/z 479.9586 (M – H)−.
2-((4-(4-Bromophenylsulfonamido)-1-hydroxynaphthalen-2-yl)thio)acetamide (33)
Synthesized using the procedure for 1 except 41 was used as the starting material. The title compound (24 mg, 42%) as a yellow solid after trituration with a mixture of CH3CN:H2O 1:1 and cold CH2Cl2 and without a need for HPLC purification. HPLC (method B, tR = 9.37 min), purity 97%. 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 9.98 (s, 1H), 8.14 (d, J = 8.15 Hz, 1H), 7.87–7.78 (m, 2H), 7.69 (d, J = 8.16 Hz, 2H), 7.56–7.48 (m, 3H), 7.44 (p, J = 6.86 Hz, 2H), 6.98 (s, 1H), 3.47 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 173.00, 154.82, 139.41, 132.55, 132.15, 131.51, 129.34, 127.54, 126.92, 126.20, 125.68, 123.58, 123.23, 112.74. ESI HRMS: m/z 464.9591 (M – H)−.
4-Bromo-N-(4-hydroxy-3-(3-hydroxypropyl)naphthalen-1-yl)benzenesulfonamide (34)
Synthesized using the procedure for 1 except 49aa was used as the starting material. The title compound (16 mg, 32%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 6.48 min), purity 95%. 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 9.17 (s, 1H), 8.11 (d, J = 8.34 Hz, 1H), 7.82 (d, J = 8.34 Hz, 1H), 7.68 (d, J = 8.50 Hz, 2H), 7.49 (d, J = 8.50 Hz, 2H), 7.38 (t, J = 7.23 Hz, 1H), 7.31 (t, J = 7.23 Hz, 1H), 6.73 (s, 1H), 3.34 (t, J = 6.35 Hz, 2H), 2.61 (t, J = 7.48 Hz, 2H), 1.54 (p, J = 6.68 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 149.29, 139.59, 132.49, 130.41, 129.31, 128.21, 126.80, 126.16, 125.66, 125.40, 123.69, 123.54, 122.46, 122.39, 60.51, 33.15, 26.18. ESI HRMS: m/z 434.0063 (M – H)−.
4-Bromo-N-(4-hydroxy-3-(4-hydroxybutyl)naphthalen-1-yl)benzenesulfonamide (35)
Synthesized using the procedure for 1 except 45 was used as the starting material. The title compound (49 mg, 34%) was obtained as a white solid after HPLC purification. HPLC (method B, tR = 9.60 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 9.13 (s, 1H), 8.12 (d, J = 8.17 Hz, 1H), 7.87 (d, J = 8.17 Hz, 1H), 7.69 (d, J = 8.49 Hz, 2H), 7.51 (d, J = 8.49 Hz, 2H), 7.39 (t, J = 7.31 Hz, 1H), 7.33 (t, J = 7.31 Hz, 1H), 6.67 (s, 1H), 4.36 (s, 1H), 3.42–3.33 (m, 2H), 2.58 (t, J = 5.70 Hz, 2H), 1.45–1.26 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 149.19, 139.58, 132.50, 130.54, 129.39, 128.16, 126.78, 126.20, 125.66, 125.39, 123.64, 123.61, 122.58, 122.46, 61.16, 32.47, 29.39, 26.67. ESI HRMS: m/z 448.0223 (M – H)−.
N-(3-(((2H-Tetrazol-5-yl)methyl)thio)-4-hydroxynaphthalen-1-yl)-4-bromobenzenesulfonamide (36)
Synthesized using the procedure for 1 except 49cc was used as the starting material. The title compound (20 mg, 42%) was obtained as a white solid after HPLC purification. HPLC (method A, tR = 6.45 min), purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.13 (d, J = 8.04 Hz, 1H), 7.75 (d, J = 8.04 Hz, 1H), 7.64 (d, J = 8.62 Hz, 2H), 7.49–7.43 (m, 3H), 7.42–7.36 (m, 1H), 6.91 (s, 1H), 4.25 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 153.78, 139.47, 132.51, 131.79, 130.43, 129.21, 127.45, 126.87, 126.27, 125.59, 124.10, 123.57, 123.08, 26.54. ESI HRMS: m/z 489.9647 (M – H)−.
N-(3-(((2H-Tetrazol-5-yl)methyl)thio)-4-hydroxynaphthalen-1-yl)-4-phenoxybenzenesulfonamide (37)
Synthesized using the procedure for 1 except 49dd was used as the starting material. The title compound (32 mg, 53%) as a white solid after HPLC purification. HPLC (method A, tR = 6.88 min), purity 96%. 1H NMR (500 MHz, DMSO-d6) δ 9.84 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 8.7 Hz, 2H), 7.51–7.46 (m, 1H), 7.44–7.38 (m, 3H), 7.20 (t, J = 7.4 Hz, 1H), 6.99–6.94 (m, 5H), 4.28 (s, 2H). 13C NMR (125 MHz, DMSO-d6) δ 160.70, 155.63, 153.61, 134.27, 131.88, 130.73, 130.54, 129.77, 127.33, 126.16, 125.60, 125.06, 124.51, 123.71, 123.04, 119.98, 118.35, 111.64, 26.59. ESI MS: m/z 503.9 (M – H)−.
3-((1-Hydroxy-4-(naphthalene-2-sulfonamido)naphthalen-2-yl)thio)propanoic Acid (38)
Synthesized using the procedure for 1 except 49ee was used as the starting material. The title compound (23 mg, 15%) was obtained as a white solid after HPLC purification. HPLC (method B, tR = 7.08 min), purity >99%. 1H NMR (400 MHz, DMSO-d6) δ 12.31 (s, 1H), 10.01 (s, 1H), 9.60 (s, 1H), 8.23 (s, 1H), 8.14–8.06 (m, 2H), 8.06–7.97 (m, 3H), 7.80 (d, J = 8.52 Hz, 1H), 7.68 (t, J = 7.36 Hz, 1H), 7.60 (t, J = 7.36 Hz, 1H), 7.43 (p, J = 6.84 Hz, 2H), 6.87 (s, 1H), 2.62 (t, J = 6.83 Hz, 2H), 2.17 (t, J = 6.83 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 172.97, 152.44, 137.07, 134.56, 131.90, 131.48, 129.68, 129.51, 129.23, 129.20, 128.33, 128.20, 128.00, 126.94, 126.22, 125.51, 124.61, 123.96, 122.93, 122.70, 113.33, 34.12, 29.59. ESI HRMS: m/z 452.0630 (M – H)−.
2-((1-Acetoxy-4-(4-bromophenylsulfonamido)naphthalen-2-yl)thio)acetic Acid (46)
Synthesized using a reported procedure.73 A stirred solution of 10 (50 mg, 0.11 mmol) in dry THF (1.5 mL) was cooled to 0 °C. Et3N (31 μL, 0.22 mmol) was added, and the mixture was stirred for 5 min before acetyl chloride (10 μL, 0.14 mmol) was added at 0 °C. The mixture was stirred under nitrogen for an additional 20 min then diluted with EtOAc (10 mL) and washed with H2O (10 mL × 3). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. The crude was subjected to flash column chromatography (hexane/EtOAc 4:1) on silica gel to afford 46 (34 mg, 61%) as a white solid. HPLC (method A, tR = 7.87 min), purity 99%. 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 8.10 Hz, 1H), 7.96 (d, J = 8.48 Hz, 2H), 7.76 (d, J = 8.10 Hz, 1H), 7.71 (d, J = 8.48 Hz, 2H), 7.69–7.59 (m, 2H), 7.37 (s, 1H), 3.63 (s, 2H), 1.79 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.13, 161.35, 147.04, 137.50, 132.14, 131.33, 131.18, 129.73, 129.46, 128.87, 128.46, 126.94, 125.55, 122.76, 121.64, 114.58, 28.48, 24.29. ESI HRMS: m/z 507.9533 (M – H)−.
Fluorescence Polarization-Based Binding Assays
Sensitive and quantitative FP-based binding assays were developed and optimized to determine the binding affinities of small-molecule inhibitors to the recombinant Mcl-1, A1/Bfl-1, Bcl-w, Bcl-2, and Bcl-xL proteins. Protocols for expression and purification of used antiapoptotic proteins and determination of Kd values of fluorescent probes to proteins are provided in the Supporting Information. On the basis of the Kd values, the concentrations of the proteins used in the competitive binding experiments were 90 nM for Mcl-1, 40 nM for Bcl-w, 50 nM for Bcl-xL, 60 nM for Bcl-2, and 4 nM for A1/Bfl-1. The fluorescent probes, Flu-BID and FAM-BID, were fixed at 2 nM for all assays except for A1/Bfl-1, where FAM-BID was used at 1 nM. Then 5 μL of the tested compound in DMSO and 120 μL of protein/probe complex in the assay buffer (100 mM potassium phosphate, pH 7.5; 100 μg/mL bovine gamma globulin; 0.02% sodium azide, purchased from Invitrogen, Life Technologies, supplemented with 0.01% Triton X-100) were added to assay plates (Microfluor 2Black, Thermo Scientific), incubated at room temperature for 3 h, and the polarization values (mP) were measured at an excitation wavelength at 485 nm and an emission wavelength at 530 nm using the plate reader Synergy H1 Hybrid, BioTek. IC50 values were determined by nonlinear regression fitting of the competition curves (GraphPad Prism 6.0 Software). The Ki values were calculated as described previously.77
Solution Competitive Surface Plasmon Resonance Based Assay Using Immobilized Biotin-Labeled Bim BH3 Peptide
The solution competitive SPR-based assay was performed on Biacore 2000. N terminal biotin-labeled Bim BH3 peptide (141–166 amino acids) was immobilized on streptavidin (SA) chip giving density of 1400 RU (Response Units). The preincubated Mcl-1 protein (20 nM) with tested small-molecule inhibitors for at least 30 min was injected over the surfaces of the chip. Response units were measured at 15 s in the dissociation phase, and the specific binding was calculated by subtracting the control surface (Fc1) signal from the surfaces with immobilized biotin-labeled Bim BH3. IC50 values were determined by nonlinear least-squares analysis using GraphPad Prism 6.0 software.
Molecular Modeling
Crystal structure of Mcl-1 in complex with mNoxa BH3 peptide (PDB entry 2NLA) and in silico Schrödinger’s IFD were used to model the binding poses of our designed compounds with Mcl-1. IFD is allowing incorporation of the protein and ligand flexibility in the docking protocol, which consists of the following steps: (i) constrained minimization of the protein with an RMSD cutoff of 0.18 Å, (ii) initial Glide docking of the ligand using a softened potential (van der Waals radii scaling), (iii) one round of Prime side chain prediction for each protein/ligand complex, on residues within defined distance of any ligand pose, (iv) prime minimization of the same set of residues and the ligand for each protein/ligand complex pose, (v) Glide redocking of each protein/ligand complex structure within a specified energy of the lowest energy structure, (vi) estimation of the binding energy (IFDScore) for each output pose. All docking calculations were run in the extra precision (XP) mode of Glide. The center of the grid box of the Mcl-1 was defined by the Val 249 (in h1), Phe 270 (in h2), Val 220 (in h3/h4), and Val 216 (in h4). The size of the grid box was set to 15 Å. Default values were used for all other parameters. Schrödinger’s MC/SD dynamic simulation performs constant temperature calculations that take advantage of the strengths of Monte Carlo methods for quickly introducing large changes in a few degree of freedom and stochastic dynamics for its effective local sampling of collective motions. The MC/SD dynamic simulation time in our study was set to 100 ps by allowing movement of the docked ligand and the residues, which is less than 6 Å to the ligand. The force field used was set to OPLS_2001. Default values were used for all other parameters.
NMR Studies
15N-labeled or 15N, 13C-labeled Mcl-1 proteins for NMR studies were prepared and purified using the same protocol as for unlabeled protein with the exception that the bacteria were grown on M9 minimal media supported with 3 g/L of 13C-glucose and/or 1 g/L of (15NH4)2SO4. Protein samples were prepared in a 20 mM sodium phosphate, 150 mM NaCl, and 1 mM DTT solution at pH 7 in 7% D2O. The binding mode of the compounds has been characterized by recording 1H,15N -HSQC experiments with a 138 μL solution of uniformly 15N-labeled Mcl-1 (75 μM) in the absence and presence of added compounds with the indicated molar ratio concentrations. All spectra were acquired at 30 °C on a Bruker 600 MHz NMR spectrometer equipped with a cryogenic probe, processed using Bruker TopSpin and rNMR,84 and were analyzed with Sparky.85 Plots of chemical shift changes were calculated as ((Δ1H ppm)2 + (0.2(Δ15N ppm))2)0.5 of Mcl-1 amide upon addition of compound. The absence of a bar in a chemical shift plot indicates no chemical shift difference or the presence of a proline or residue that is overlapped or not assigned.
Biotin–Streptavidin Pull-Down Experiment
Human breast cancer 2LMP cells, a subclone of the MDA-MB-231 cell line, were lysed in CHAPS buffer (10 mM HEPES (pH 7.4), 2.5 mM EDTA, 150 mM NaCl, 1.0% CHAP). Precleared cell lysates were incubated with different concentrations of compounds followed by incubation with biotinylated Noxa BH3 peptide (18–43) and streptavidin–agarose beads to pull-down Mcl-1 protein bound to Noxa peptide. Beads were washed with CHAPS buffer, and Mcl-1 protein was eluted by boiling in SDS-PAGE sample buffer and analyzed by Western blotting using Mcl-1 antibody (Santa Cruz).
Cell Culture and Cell Viability Assays
MEFs cells, wild-type and Bax/Bak double knockout, were gifts from Shaomeng Wang at the University of Michigan and were cultured in DMEM (Life Technologies), supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific HyClone). The retroviral transduced lymphoma cells isolated from Eμ-myc transgenic mice were gifts from Ricky W. Johnstone at University of Melbourne, Melbourne, Australia, and cultured as previously described.81 Human leukemia cell lines HL-60, K-562, and MV-4–11 were obtained from American Type Culture Collection (ATCC). The cells were cultured in RPMI 1640 medium (Life Technologies), all supplemented with 10% FBS. MEFs cells were seeded in 12-well plates at 0.5 × 106 cells/well, left to adhere, and then treated for 15 h with increasing concentrations of the compounds. The cells were harvested, washed with phosphate-buffered saline (PBS), and stained with 0.025 mg/mL propidium iodide (MP Biomedicals). The percentage of propodium iodide positive population was determined by flow cytometry and calculated using WinList 3.0. The Mcl-1 and Bcl-2 retroviral transduced lymphoma Eμ-myc cells were seeded in 24-well plates at 0.5 × 106 cells/well. They were treated with different concentrations of tested compounds for 15−18 h. The cells were harvested and stained with violet LIVE/DEAD fixable dead cell stain kit (Invitrogen) according to manufacturer’s protocol. The percentage of fluorescent positive cells was determined by flow cytometry and calculated using WinList 3.0. The effect of the compounds on tested leukemia cells viability was evaluated by CellTiter Glo luminescent cell viability assay (Promega). Cells were plated in 12-well plates at 0.5 × 106 cells/well and treated with various concentrations of the compounds and incubated for 3 days. Cell viability was determined by measuring intracellular ATP levels with the CellTiter Glo reagent and reading the luminescence with the Synergy H1 Hybrid BioTek plate reader. Percent cell growth was calculated relative to DMSO treated cells, and IC50 values were calculated by nonlinear regression analysis using GraphPad Prism 6.0 Software.
Determination of Caspase Activity
HL-60 cells were seeded in 12-well plates at 0.5 × 106 cells/well. After 20 h treatment with different concentrations of the compounds, caspase 3 activity was determined using the fluorometric substrates DEVD-AFC following the protocol of the Caspase-3 fluorometric assay kit (BioVision). Caspase 3 activity is reported as the fold change relative to DMSO treated cells.
Analysis of Apoptosis
To determine the induction of apoptosis, HL-60 cells were plated in 12-well plates at 0.5 × 106 cells/well and treated with various concentrations of tested compounds. After 20 h, cells were harvested, washed with PBS, and treated with annexin-V FITC and propidium iodide using BD annexin V FITC assay kit (BD Biosciences Pharmingen). The percentage of cells undergoing apoptosis was assessed by flow cytometry within 1 h and analyzed using WinList 3.0. Apoptosis was also determined in the presence of Z-VAD-FMK (Bachem), pan caspase-inhibitor. For this purpose sells were pretreated with 100 μM Z-VAD-FMK for about 1 h before adding tested compounds.
Acknowledgments
This research was supported by grants from the U.S. National Cancer Institute, National Institutes of Health R01CA149442, R21CA0158976, and R21NS056915 to Zaneta Nikolovska-Coleska. We are grateful to Kalle Gehring from McGill University, Montreal, Canada, for providing us with the NMR assignment of human Mcl-1 protein. We thank Lei Miao for synthesizing the intermediate for analogue 23, Julie DiBernardo for her help with growing Eμ-myc cells, Ronald Craig for his input in flow cytometric analysis, and George Lund for his help with NMR experimental setup.
Glossary
Abbreviations Used
- Bcl-2
B-cell lymphoma-2
- BH
Bcl-2 homology
- DKO
double knockout
- FP
fluorescence polarization
- IFD
induced fit docking
- MEF
murine embryonic fibroblast
- Mcl-1
myeloid cell leukemia sequence 1
- PI
propidium iodide
- SPR
surface plasmon resonance. Solvent and reagent abbreviations used:
- CH3CN
acetonitrile
- NH4Cl
ammonium chloride
- NH4OH
ammonium hydroxide
- (NH4)2SO4
ammonium sulfate
- Dppf
1,1′-bis(diphenylphosphino)ferrocene
- Pd(PPh3)2Cl2
bis(triphenylphosphine)palladium(II) dichloride
- BBr3
boron tribromide
- Cs2CO3
cesium carbonate
- CuI
copper(I) iodide
- CH2Cl2
dichloromethane
- DTT
dithiothreitol
- EtOAc
ethyl acetate
- EtOH
ethanol
- HCl
hydrogen chloride
- LiOH
lithium hydroxide
- MgSO4
magnesium sulfate
- MeOH
methanol
- NMP
N-methyl-2-pyrrolidone
- Pd(OAc)2
palladium acetate
- NaHCO3
sodium bicarbonate
- Na2CO3
sodium carbonate
- NaCl
sodium chloride
- NaOH
sodium hydroxide
- Na2S2O3
sodium thiosulfate
- Pd(PPh3)4
tetrakis(triphenylphosphine)palladium(0)
- THF
tetrahydrofuran
- Et3N
triethylamine
- TFA
trifluoroacetic acid
- Pd2(dba)3
tris(dibenzylideneacetone)dipalladium(0)
- ZnCl2
zinc chloride
- Z-VAD-FMK
Z-Val-Ala-dl-Asp-fluoromethylketone
Supporting Information Available
Additional experimental details for synthesis of analogues 24, 27, and 30 and intermediates, protein expression and purification, fluorescence polarization-based binding assay, and additional HSQC chemical shift perturbation plots and apoptosis studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ For C.L.: School of Medical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China.
Author Contributions
# These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. The hallmarks of cancer. Cell 2000, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Fulda S.; Debatin K. M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [DOI] [PubMed] [Google Scholar]
- Youle R. J.; Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature Rev. Mol. Cell Biol. 2008, 9, 47–59. [DOI] [PubMed] [Google Scholar]
- Bajwa N.; Liao C.; Nikolovska-Coleska Z. Inhibitors of the anti-apoptotic Bcl-2 proteins: a patent review. Expert Opin. Ther. Pat. 2012, 22, 37–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessene G.; Czabotar P. E.; Colman P. M. BCL-2 family antagonists for cancer therapy. Nature Rev. Drug Discovery 2008, 7, 989–1000. [DOI] [PubMed] [Google Scholar]
- Vogler M.; Dinsdale D.; Dyer M. J.; Cohen G. M. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009, 16, 360–367. [DOI] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C. M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Med. 2013, 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Lessene G.; Czabotar P. E.; Sleebs B. E.; Zobel K.; Lowes K. N.; Adams J. M.; Baell J. B.; Colman P. M.; Deshayes K.; Fairbrother W. J.; Flygare J. A.; Gibbons P.; Kersten W. J.; Kulasegaram S.; Moss R. M.; Parisot J. P.; Smith B. J.; Street I. P.; Yang H.; Huang D. C.; Watson K. G. Structure-guided design of a selective BCL-X(L) inhibitor. Nature Chem. Biol. 2013, 9, 390–397. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Aguilar A.; Chen J.; Bai L.; Liu L.; Meagher J. L.; Yang C. Y.; McEachern D.; Cong X.; Stuckey J. A.; Wang S. Structure-based design of potent Bcl-2/Bcl-xL inhibitors with strong in vivo antitumor activity. J. Med. Chem. 2012, 55, 6149–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Chen J.; Meagher J. L.; Yang C. Y.; Aguilar A.; Liu L.; Bai L.; Cong X.; Cai Q.; Fang X.; Stuckey J. A.; Wang S. Design of Bcl-2 and Bcl-xL inhibitors with subnanomolar binding affinities based upon a new scaffold. J. Med. Chem. 2012, 55, 4664–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C.; Shoemaker A. R.; Adickes J.; Anderson M. G.; Chen J.; Jin S.; Johnson E. F.; Marsh K. C.; Mitten M. J.; Nimmer P.; Roberts L.; Tahir S. K.; Xiao Y.; Yang X.; Zhang H.; Fesik S.; Rosenberg S. H.; Elmore S. W. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Roberts A. W.; Seymour J. F.; Brown J. R.; Wierda W. G.; Kipps T. J.; Khaw S. L.; Carney D. A.; He S. Z.; Huang D. C.; Xiong H.; Cui Y.; Busman T. A.; McKeegan E. M.; Krivoshik A. P.; Enschede S. H.; Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft M. F.; Wei A. H.; Mason K. D.; Vandenberg C. J.; Chen L.; Czabotar P. E.; Willis S. N.; Scott C. L.; Day C. L.; Cory S.; Adams J. M.; Roberts A. W.; Huang D. C. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M.; Contractor R.; Tsao T.; Samudio I.; Ruvolo P. P.; Kitada S.; Deng X.; Zhai D.; Shi Y. X.; Sneed T.; Verhaegen M.; Soengas M.; Ruvolo V. R.; McQueen T.; Schober W. D.; Watt J. C.; Jiffar T.; Ling X.; Marini F. C.; Harris D.; Dietrich M.; Estrov Z.; McCubrey J.; May W. S.; Reed J. C.; Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [DOI] [PubMed] [Google Scholar]
- Chauhan D.; Velankar M.; Brahmandam M.; Hideshima T.; Podar K.; Richardson P.; Schlossman R.; Ghobrial I.; Raje N.; Munshi N.; Anderson K. C. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene 2007, 26, 2374–2380. [DOI] [PubMed] [Google Scholar]
- Chen S.; Dai Y.; Harada H.; Dent P.; Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007, 67, 782–791. [DOI] [PubMed] [Google Scholar]
- Lin X.; Morgan-Lappe S.; Huang X.; Li L.; Zakula D. M.; Vernetti L. A.; Fesik S. W.; Shen Y. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene 2007, 26, 3972–3979. [DOI] [PubMed] [Google Scholar]
- Moulding D. A.; Giles R. V.; Spiller D. G.; White M. R.; Tidd D. M.; Edwards S. W. Apoptosis is rapidly triggered by antisense depletion of MCL-1 in differentiating U937 cells. Blood 2000, 96, 1756–1763. [PubMed] [Google Scholar]
- Marsden V. S.; Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 2003, 21, 71–105. [DOI] [PubMed] [Google Scholar]
- MacCallum D. E.; Melville J.; Frame S.; Watt K.; Anderson S.; Gianella-Borradori A.; Lane D. P.; Green S. R. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005, 65, 5399–5407. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Gojo I.; Fenton R. G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002, 99, 1885–1893. [DOI] [PubMed] [Google Scholar]
- Michels J.; O’Neill J. W.; Dallman C. L.; Mouzakiti A.; Habens F.; Brimmell M.; Zhang K. Y.; Craig R. W.; Marcusson E. G.; Johnson P. W.; Packham G. Mcl-1 is required for Akata6 B-lymphoma cell survival and is converted to a cell death molecule by efficient caspase-mediated cleavage. Oncogene 2004, 23, 4818–4827. [DOI] [PubMed] [Google Scholar]
- Song L.; Coppola D.; Livingston S.; Cress D.; Haura E. B. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol. Ther 2005, 4, 267–276. [DOI] [PubMed] [Google Scholar]
- Qin J. Z.; Xin H.; Sitailo L. A.; Denning M. F.; Nickoloff B. J. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res. 2006, 66, 9636–9645. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y.; Hosotani R.; Wada M.; Lee J. U.; Koshiba T.; Fujimoto K.; Tsuji S.; Nakajima S.; Doi R.; Kato M.; Shimada Y.; Imamura M. Immunohistochemical analysis of Bcl-2, Bax, Bcl-X, and Mcl-1 expression in pancreatic cancers. Oncology 1999, 56, 73–82. [DOI] [PubMed] [Google Scholar]
- Ren L. N.; Li Q. F.; Xiao F. J.; Yan J.; Yang Y. F.; Wang L. S.; Guo X. Z.; Wang H. Endocrine glands-derived vascular endothelial growth factor protects pancreatic cancer cells from apoptosis via upregulation of the myeloid cell leukemia-1 protein. Biochem. Biophys. Res. Commun. 2009, 386, 35–39. [DOI] [PubMed] [Google Scholar]
- Cavarretta I. T.; Neuwirt H.; Untergasser G.; Moser P. L.; Zaki M. H.; Steiner H.; Rumpold H.; Fuchs D.; Hobisch A.; Nemeth J. A.; Culig Z. The antiapoptotic effect of IL-6 autocrine loop in a cellular model of advanced prostate cancer is mediated by Mcl-1. Oncogene 2007, 26, 2822–2832. [DOI] [PubMed] [Google Scholar]
- Chung T. K.; Cheung T. H.; Lo W. K.; Yim S. F.; Yu M. Y.; Krajewski S.; Reed J. C.; Wong Y. F. Expression of apoptotic regulators and their significance in cervical cancer. Cancer Lett. 2002, 180, 63–68. [DOI] [PubMed] [Google Scholar]
- Sieghart W.; Losert D.; Strommer S.; Cejka D.; Schmid K.; Rasoul-Rockenschaub S.; Bodingbauer M.; Crevenna R.; Monia B. P.; Peck-Radosavljevic M.; Wacheck V. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J. Hepatol 2006, 44, 151–157. [DOI] [PubMed] [Google Scholar]
- Cho-Vega J. H.; Rassidakis G. Z.; Admirand J. H.; Oyarzo M.; Ramalingam P.; Paraguya A.; McDonnell T. J.; Amin H. M.; Medeiros L. J. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum. Pathol. 2004, 35, 1095–1100. [DOI] [PubMed] [Google Scholar]
- Khoury J. D.; Medeiros L. J.; Rassidakis G. Z.; McDonnell T. J.; Abruzzo L. V.; Lai R. Expression of Mcl-1 in mantle cell lymphoma is associated with high-grade morphology, a high proliferative state, and p53 overexpression. J. Pathol 2003, 199, 90–97. [DOI] [PubMed] [Google Scholar]
- Backus H. H.; van Riel J. M.; van Groeningen C. J.; Vos W.; Dukers D. F.; Bloemena E.; Wouters D.; Pinedo H. M.; Peters G. J. Rb, mcl-1 and p53 expression correlate with clinical outcome in patients with liver metastases from colorectal cancer. Ann. Oncol. 2001, 12, 779–785. [DOI] [PubMed] [Google Scholar]
- Wuilleme-Toumi S.; Robillard N.; Gomez P.; Moreau P.; Le Gouill S.; Avet-Loiseau H.; Harousseau J. L.; Amiot M.; Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [DOI] [PubMed] [Google Scholar]
- Kaufmann S. H.; Karp J. E.; Svingen P. A.; Krajewski S.; Burke P. J.; Gore S. D.; Reed J. C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [PubMed] [Google Scholar]
- Kitada S.; Andersen J.; Akar S.; Zapata J. M.; Takayama S.; Krajewski S.; Wang H. G.; Zhang X.; Bullrich F.; Croce C. M.; Rai K.; Hines J.; Reed J. C. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with in vitro and in vivo chemoresponses. Blood 1998, 91, 3379–3389. [PubMed] [Google Scholar]
- Gomez-Bougie P.; Bataille R.; Amiot M. The imbalance between Bim and Mcl-1 expression controls the survival of human myeloma cells. Eur. J. Immunol. 2004, 34, 3156–3164. [DOI] [PubMed] [Google Scholar]
- Gomez-Bougie P.; Wuilleme-Toumi S.; Menoret E.; Trichet V.; Robillard N.; Philippe M.; Bataille R.; Amiot M. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res. 2007, 67, 5418–5424. [DOI] [PubMed] [Google Scholar]
- Hussain S. R.; Cheney C. M.; Johnson A. J.; Lin T. S.; Grever M. R.; Caligiuri M. A.; Lucas D. M.; Byrd J. C. Mcl-1 is a relevant therapeutic target in acute and chronic lymphoid malignancies: down-regulation enhances rituximab-mediated apoptosis and complement-dependent cytotoxicity. Clin. Cancer Res. 2007, 13, 2144–2150. [DOI] [PubMed] [Google Scholar]
- Thallinger C.; Wolschek M. F.; Wacheck V.; Maierhofer H.; Gunsberg P.; Polterauer P.; Pehamberger H.; Monia B. P.; Selzer E.; Wolff K.; Jansen B. Mcl-1 antisense therapy chemosensitizes human melanoma in a SCID mouse xenotransplantation model. J. Invest. Dermatol. 2003, 120, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wei S. H.; Dong K.; Lin F.; Wang X.; Li B.; Shen J. J.; Zhang Q.; Wang R.; Zhang H. Z. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother. Pharmacol. 2008, 62, 1055–1064. [DOI] [PubMed] [Google Scholar]
- Guoan X.; Hanning W.; Kaiyun C.; Hao L. Adenovirus-mediated siRNA targeting Mcl-1 gene increases radiosensitivity of pancreatic carcinoma cells in vitro and in vivo. Surgery 2010, 147, 553–561. [DOI] [PubMed] [Google Scholar]
- Abulwerdi F.; Liao C.; Liu M.; Azmi A. S.; Aboukameel A.; Mady A. S.; Gulappa T.; Cierpicki T.; Owens S.; Zhang T.; Sun D.; Stuckey J. A.; Mohammad R. M.; Nikolovska-Coleska Z. A Novel Small-Molecule Inhibitor of Mcl-1 Blocks Pancreatic Cancer Growth In Vitro and In Vivo. Mol. Cancer Ther 2014, 13, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Aikawa K.; Nishida G.; Homma M.; Sogabe S.; Igaki S.; Hayano Y.; Sameshima T.; Miyahisa I.; Kawamoto T.; Tawada M.; Imai Y.; Inazuka M.; Cho N.; Imaeda Y.; Ishikawa T. Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins. J. Med. Chem. 2013, 56, 9635–9645. [DOI] [PubMed] [Google Scholar]
- Friberg A.; Vigil D.; Zhao B.; Daniels R. N.; Burke J. P.; Garcia-Barrantes P. M.; Camper D.; Chauder B. A.; Lee T.; Olejniczak E. T.; Fesik S. W. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013, 56, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T.; Li X.; Chang X.; Liang X.; Zhao Y.; Wu G.; Xie S.; Su P.; Wu Z.; Feng Y.; Zhang Z. 3-Thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (S1) derivatives as pan-Bcl-2-inhibitors of Bcl-2, Bcl-xL and Mcl-1. Bioorg. Med. Chem. 2013, 21, 11–20. [DOI] [PubMed] [Google Scholar]
- Cohen N. A.; Stewart M. L.; Gavathiotis E.; Tepper J. L.; Bruekner S. R.; Koss B.; Opferman J. T.; Walensky L. D. A Competitive Stapled Peptide Screen Identifies a Selective Small Molecule that Overcomes MCL-1-Dependent Leukemia Cell Survival. Chem. Biol. 2012, 19, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi K.; Li R.; Sung S. S.; Wu H.; Liu Y.; Manieri W.; Krishnegowda G.; Awwad A.; Dewey A.; Liu X.; Amin S.; Cheng C.; Qin Y.; Schonbrunn E.; Daughdrill G.; Loughran T. P. Jr.; Sebti S.; Wang H. G. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rega M. F.; Wu B.; Wei J.; Zhang Z.; Cellitti J. F.; Pellecchia M. SAR by interligand nuclear overhauser effects (ILOEs) based discovery of acylsulfonamide compounds active against Bcl-x(L) and Mcl-1. J. Med. Chem. 2011, 54, 6000–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo P. H.; Sivaraman T.; Wan K. F.; Xu J.; Krishnamoorthy J.; Song C. M.; Tian L.; Chin J. S.; Lim D. S.; Mok H. Y.; Yu V. C.; Tong J. C.; Chai C. L. Structural insights into the design of small molecule inhibitors that selectively antagonize Mcl-1. J. Med. Chem. 2010, 53, 2314–2318. [DOI] [PubMed] [Google Scholar]
- Stewart M. L.; Fire E.; Keating A. E.; Walensky L. D. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nature Chem. Biol. 2010, 6, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppidi A.; Doi K.; Edwardraja S.; Drake E. J.; Gulick A. M.; Wang H. G.; Lin Q. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J. Am. Chem. Soc. 2012, 134, 14734–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y.; Kazi A.; Marsilio F.; Luo Y.; Jain S.; Brooks W.; Daniel K. G.; Guida W. C.; Sebti S. M.; Lawrence H. R. Discovery and synthesis of hydronaphthoquinones as novel proteasome inhibitors. J. Med. Chem. 2012, 55, 1978–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad-Zapatero C.; Metz J. T. Ligand efficiency indices as guideposts for drug discovery. Drug Discovery Today 2005, 10, 464–469. [DOI] [PubMed] [Google Scholar]
- Schrödinger Suite 2011 Induced Fit Docking protocol:Glide Version 5.7; Schrödinger, LLC: New York, NY, 2009; ; Prime Version 3.0; Schrödinger, LLC: New York, NY., 2011.
- Czabotar P. E.; Lee E. F.; van Delft M. F.; Day C. L.; Smith B. J.; Huang D. C.; Fairlie W. D.; Hinds M. G.; Colman P. M. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6217–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. F.; Czabotar P. E.; van Delft M. F.; Michalak E. M.; Boyle M. J.; Willis S. N.; Puthalakath H.; Bouillet P.; Colman P. M.; Huang D. C.; Fairlie W. D. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J. Cell Biol. 2008, 180, 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. Y.; Wang S. M. Analysis of Flexibility and Hotspots in Bcl-xL and Mcl-1 Proteins for the Design of Selective Small-Molecule Inhibitors. ACS Med. Chem. Lett. 2012, 3, 308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Moldoveanu T.; Sprules T.; Matta-Camacho E.; Mansur-Azzam N.; Gehring K. Apoptotic regulation by MCL-1 through heterodimerization. J. Biol. Chem. 2010, 285, 19615–19624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanet A. S.; Colobert F.; Broutin P. E. Mild and regioselective iodination of electron-rich aromatics with N-iodosuccinimide and catalytic trifluoroacetic acid. Tetrahedron Lett. 2002, 43, 5047–5048. [Google Scholar]
- Ciattini P. G.; Morera E.; Ortar G. A New, Palladium-Catalyzed Synthesis of Aromatic Mercapturic Acid Derivatives. Tetrahedron Lett. 1995, 36, 4133–4136. [Google Scholar]
- Dai W.; Petersen J. L.; Wang K. K. Synthesis of the parent and substituted tetracyclic ABCD ring cores of camptothecins via 1-(3-aryl-2-propynyl)-1,6-dihydro-6-oxo-2-pyridinecarbonitriles. Org. Lett. 2006, 8, 4665–4667. [DOI] [PubMed] [Google Scholar]
- Eichman C. C.; Stambuli J. P. Zinc-mediated palladium-catalyzed formation of carbon-sulfur bonds. J. Org. Chem. 2009, 74, 4005–4008. [DOI] [PubMed] [Google Scholar]
- Itoh T.; Mase T. A general palladium-catalyzed coupling of aryl bromides/triflates and thiols. Org. Lett. 2004, 6, 4587–4590. [DOI] [PubMed] [Google Scholar]
- Mispelaere-Canivet C.; Spindler J. F.; Perrio S.; Beslin P. Pd-2(dba)(3)/Xantphos-catalyzed cross-coupling of thiols and aryl bromides/triflates. Tetrahedron 2005, 61, 5253–5259. [Google Scholar]
- Riesgo E. C.; Jin X.; Thummel R. P. Introduction of Benzo[h]quinoline and 1,10-Phenanthroline Subunits by Friedlander Methodology. J. Org. Chem. 1996, 61, 3017–3022. [DOI] [PubMed] [Google Scholar]
- Ortar G.; Cascio M. G.; De Petrocellis L.; Morera E.; Rossi F.; Schiano-Moriello A.; Nalli M.; de Novellis V.; Woodward D. F.; Maione S.; Di Marzo V. New N-arachidonoylserotonin analogues with potential “dual” mechanism of action against pain. J. Med. Chem. 2007, 50, 6554–6569. [DOI] [PubMed] [Google Scholar]
- Beaudoin S.; Kinsey K. E.; Burns J. F. Preparation of unsymmetrical sulfonylureas from N,N′-sulfuryldiimidazoles. J. Org. Chem. 2003, 68, 115–119. [DOI] [PubMed] [Google Scholar]
- Greig I. R.; Idris A. I.; Ralston S. H.; van’t Hof R. J. Development and characterization of biphenylsulfonamides as novel inhibitors of bone resorption. J. Med. Chem. 2006, 49, 7487–7492. [DOI] [PubMed] [Google Scholar]
- Li X.; Chu S.; Feher V. A.; Khalili M.; Nie Z.; Margosiak S.; Nikulin V.; Levin J.; Sprankle K. G.; Tedder M. E.; Almassy R.; Appelt K.; Yager K. M. Structure-based design, synthesis, and antimicrobial activity of indazole-derived SAH/MTA nucleosidase inhibitors. J. Med. Chem. 2003, 46, 5663–5673. [DOI] [PubMed] [Google Scholar]
- Wagner J.; von Matt P.; Faller B.; Cooke N. G.; Albert R.; Sedrani R.; Wiegand H.; Jean C.; Beerli C.; Weckbecker G.; Evenou J. P.; Zenke G.; Cottens S. Structure–activity relationship and pharmacokinetic studies of sotrastaurin (AEB071), a promising novel medicine for prevention of graft rejection and treatment of psoriasis. J. Med. Chem. 2011, 54, 6028–6039. [DOI] [PubMed] [Google Scholar]
- Ishizuka N.; Matsumura K.; Sakai K.; Fujimoto M.; Mihara S.; Yamamori T. Structure–activity relationships of a novel class of endothelin-A receptor antagonists and discovery of potent and selective receptor antagonist, 2-(benzo[1,3]dioxol-5-yl)-6-isopropyloxy-4-(4-methoxyphenyl)-2H-chromene-3-carbox ylic acid (S-1255). 1. Study on structure–activity relationships and basic structure crucial for ET(A) antagonism. J. Med. Chem. 2002, 45, 2041–2055. [DOI] [PubMed] [Google Scholar]
- England D. B.; Kerr M. A. Synthesis and cross-coupling reactions of substituted 5-triflyloxyindoles. J. Org. Chem. 2005, 70, 6519–6522. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Bures M.; Praestgaard J.; Fesik S. W. Privileged molecules for protein binding identified from NMR-based screening. J. Med. Chem. 2000, 43, 3443–3447. [DOI] [PubMed] [Google Scholar]
- Medek A.; Hajduk P. J.; Mack J.; Fesik S. W. The use of differential chemical shifts for determining the binding site location and orientation of protein-bound ligands. J. Am. Chem. Soc. 2000, 122, 1241–1242. [Google Scholar]
- Williamson M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
- Nikolovska-Coleska Z.; Wang R.; Fang X.; Pan H.; Tomita Y.; Li P.; Roller P. P.; Krajewski K.; Saito N. G.; Stuckey J. A.; Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal. Biochem. 2004, 332, 261–273. [DOI] [PubMed] [Google Scholar]
- Smits C.; Czabotar P. E.; Hinds M. G.; Day C. L. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure 2008, 16, 818–829. [DOI] [PubMed] [Google Scholar]
- Wei M. C.; Zong W. X.; Cheng E. H.; Lindsten T.; Panoutsakopoulou V.; Ross A. J.; Roth K. A.; MacGregor G. R.; Thompson C. B.; Korsmeyer S. J. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T.; Thompson C. B. Cell death in the absence of Bax and Bak. Cell Death Differ. 2006, 13, 1272–1276. [DOI] [PubMed] [Google Scholar]
- Whitecross K. F.; Alsop A. E.; Cluse L. A.; Wiegmans A.; Banks K. M.; Coomans C.; Peart M. J.; Newbold A.; Lindemann R. K.; Johnstone R. W. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood 2009, 113, 1982–1991. [DOI] [PubMed] [Google Scholar]
- Glaser S. P.; Lee E. F.; Trounson E.; Bouillet P.; Wei A.; Fairlie W. D.; Izon D. J.; Zuber J.; Rappaport A. R.; Herold M. J.; Alexander W. S.; Lowe S. W.; Robb L.; Strasser A. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesan N.; Hunt J. T.; Stein P. D.. Preparation of phenylsulfonamidooxazole and -isoxazole endothelin antagonists. US5514696A, 1996.
- Lewis I. A.; Schommer S. C.; Markley J. L. rNMR: open source software for identifying and quantifying metabolites in NMR spectra. Magn. Reson. Chem. 2009, 47Suppl 1S123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard T. D.; Kneller D. G.. SPARKY 3; University of California: San Francisco, 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.