

Abstract
A predisposition for thoracic aortic aneurysms leading to acute aortic dissections can be inherited in families in an autosomal dominant manner. Genome-wide linkage analysis of two large unrelated families with thoracic aortic disease, followed by whole exome sequencing of affected relatives, identified causative mutations in TGFB2. These mutations, a frameshift mutation in exon 6 and a nonsense mutation in exon 4, segregated with disease with a combined LOD score of 7.7. Sanger sequencing of 276 probands from families with inherited thoracic aortic disease identified two additional TGFB2 mutations. TGFB2 encodes the transforming growth factor beta-2 (TGF-β2) and the mutations are predicted to cause haploinsufficiency for TGFB2, but aortic tissue from cases paradoxically shows increased TGF-β2 expression and immunostaining. Thus, haploinsufficiency of TGFB2 predisposes to thoracic aortic disease, suggesting the initial pathway driving disease is decreased cellular TGF-β2 levels leading to a secondary increase in TGF-β2 production in the diseased aorta.
Thoracic aortic aneurysms can lead to acute aortic dissections or ruptures, and death due to these complications is common1. A number of genetic syndromes predispose to thoracic aortic disease, including Marfan syndrome (MFS, MIM 154700), Loeys-Dietz syndrome (LDS, MIM 609192, 608967, 610168, 610380) and Aneurysms-Osteoarthritis Syndrome (AOS, MIM 613795)2-4. Systemic complications are shared among these syndromes, including skeletal, craniofacial and skin manifestations. LDS is caused by mutations in the genes encoding the TGF-β receptors type I and II (TGFBR1 and TGFBR2, respectively), which bind TGF-β and initiate cellular signaling. AOS results from mutations in SMAD3 (SMAD family members 3), which encodes a protein critical for cellular signaling downstream of the TGF-β receptors after ligand binding. Despite the fact that the majority of mutations in these genes are predicted to disrupt proper function of these signaling proteins, diseased aortic tissue from these patients show increased nuclear phosphorylated SMAD2 (pSMAD2) in aortic smooth muscle cells (SMCs), suggesting increased TGF-β cellular signaling3, 4. Although MFS is caused by mutations in FBN1, encoding an extracellular matrix protein called fibillin-1, excessive TGF-β signaling is also evident in aortic tissue from MFS patients and mouse models5, 6.
We sought to identify the defective gene in two unrelated large American (TAA288) and French (MS239) families with autosomal dominant thoracic aortic aneurysms and aortic dissections with decreased penetrance, along with intracranial aneurysms (ICAs) and subarachnoid hemorrhages (SAHs), after sequencing of the known genes failed to identify causative mutations (Fig. 1a)7. Genome wide linkage analysis followed by exome sequencing was pursued to identify the defective gene in these families. In TAA288, linkage analysis using 50K GeneChips Hind array from Affymetrix was done using samples from 9 family members and multipoint log10 of odds (LOD) scores greater than 2.0 were obtained for a locus on chromosome 1 (Fig. 1b). Similar analysis of the MS239 family using 1056 microsatellites was done using samples from 18 family members and LOD scores greater than 1.4 were obtained on chromosomes 1, 4, 12, and 20 (Fig. 1c). A locus at chromosome 1q41 was shared between the two families. Exome sequencing was pursued in both families using DNA from two distantly related affected individuals (1/16 coefficient of relatedness) from TAA288 and 3 affected and 1 unaffected family members from MS239 (circled individuals in Fig. 1a). The sequencing results were filtered based on novel, heterozygous rare variants shared between affected relatives within a family that were non-synonymous, nonsense, and insertion/deletion variants. This filtering strategy identified 16 rare variants in TAA288 and 5 rare variants in MS239 (Supplementary Table 1). Rare variants in TGFB2 were present in both families and TGFB2 fell within the 1q41 locus identified for the causative gene in both families (Fig. 1b and 1c). The TGFB2 rare variant in TAA288 was a 5 base pair deletion (c.1021_1025delTACAA) in exon 6 of the gene, leading to a frameshift in the translation of the transcript and a premature stop codon (p.Tyr341Cysfs*25; Fig. 1d, Supplementary Fig. 1). This variant had a two point linkage LOD score of 3.3 (theta = 0.0) and was not present in 3795 control exomes based on bioinformatic analysis and visual inspection of the sequencing data. The TGFB2 variant in MS239 was a nonsense variant in exon 4 (p.Cys229*). Complete segregation with thoracic aortic disease was found in the family with a LOD score of 4.4 (theta = 0.0). This nonsense rare variant was not present in over 10,000 chromosomes (http://evs.gs.washington.edu/EVS/). In fact, there are no TGFB2 rare variants predicted to disrupt translation of the protein in the database (Fig. 1d). Thus, these data provide strong evidence that the identified TGFB2 mutations are responsible for the inherited vascular disease in both families.
Figure 1.

Identification of TGFB2 as the causative gene responsible for thoracic aortic disease in families TAA288 and MS239. (a) Pedigrees of family TAA288 and MS239 with the legend indicating the disease and mutation status of the family members. The age at diagnosis of aortic root enlargement and/or dissection (“dx”) is shown in years and “d” indicates age at death. A single asterisk indicates individuals whose DNA was used in genome-wide mapping. DNA from the circled individuals was used for exome sequencing. (b) Parametric two-point and multi-point LOD score profile for thoracic aortic aneurysms and dissections (TAAD) across the human genome in family TAA288 based on the Affymetrix 50K GeneChips Hind array data. The parametric two-point (grey) and multi-point (red) LOD scores are on the y-axis and are correlated to physical location of human chromosome on the x-axis. (c) Parametric two-point LOD score profile for TAAD across the human genome in family MS239. (d) Schematic representation of the TGFB2 gene and the protein domains and preproprotein proteolytic processing sites for mature TGF-β2. Boxes represent exons 1-7 with the untranslated regions (UTRs) and the open reading frame designated. The domains of the protein are designated using orange, blue and pink. The proteolytic sites of TGF-β2 preproprotein are marked with scissors symbol. Proteolytic cleavage sites remove the signal peptides from the amino-terminus and release the mature TGF-β2 from the latent associated peptide. The TGFB2 mutations identified in this study are indicated in pink type. Below the gene diagram are the rare variants found in the NHLBI exome sequencing variant server (http://evs.gs.washington.edu/EVS/); blue type designates variants predicted to be possibly or probably damaging by PolyPhen-2 analysis and black type designates variants predicted to be benign.
To determine the frequency and spectrum of TGFB2 mutations in individuals with thoracic aortic disease, the 7 exons and flanking intronic regions were sequenced using DNA from French probands (62 familial cases and 74 sporadic cases from a Marfan referral clinic) and American probands (214 affected probands in families with two or more members with thoracic aortic disease, including 30 families who also had members with intracranial aneurysms, and 57 sporadic thoracic aortic disease cases)7(Supplementary Table 2). Two further mutations were identified in the French familial cases: a nonsense p.Glu102* in MS1756 and frameshift duplication (c.873_888dup) leading to p.Asn297* in MS211; additional family members were not available for analysis (Supplementary Fig. 2). Therefore, TGFB2 mutations were identified in 2 out of 276 probands with familial thoracic aortic disease. Genome SNP array data on 898 patients with thoracic aortic disease, including 88 patients with inherited thoracic aortic disease, were analyzed for genomic rearrangements using CNVPartition and PennCNV and the B allele frequencies and logR ratio files visually inspected. No deletions or duplications involving TGFB2 were identified8.
The TGFB2 nonsense mutations and the frameshift duplication in exon 5 are predicted to lead to degradation of the message and thus cause haploinsufficiency for TGFB2. The mutation identified in TAA288, p.Tyr341Cysfs*25, is a deletion of 5 base pairs in exon 6, causing a frameshift in protein translation and addition of 25 aberrant amino acids. Since the nonsense codon that terminates protein translation occurs in exon 7, it is not expected to cause nonsense mediated decay9. Analysis of the TGFB2 transcript confirmed that similar levels of both mutant and wildtype transcript were present in RNA harvested from aortic tissue and fibroblasts from affected family members (III:11 and IV:1; Fig. 2a; Supplementary Table 2). The TGF-β family members are synthesized as peptides that form disulfide cross-linked homodimers (proprotein), which are cleaved by furin immediately prior to secretion. TGFB2 expression levels were similar in SMCs and dermal fibroblasts explanted from affected family members in TAA288 and control cells (Fig. 2b), but immunoblot analysis of the TGF-β2 proprotein in cellular lysates from these cells showed reduced TGF-β2 proprotein levels in the mutant cells compared with the wildtype cells and no evidence of the truncated protein (Fig. 2c; Supplementary Fig. 3). These data suggest that although the deleted transcript is expressed, the mutant protein is rapidly degraded by an endoplasmic reticulum-associated degradation pathway, resulting in decreased cellular levels of wildtype TGF-β2.
Figure 2.
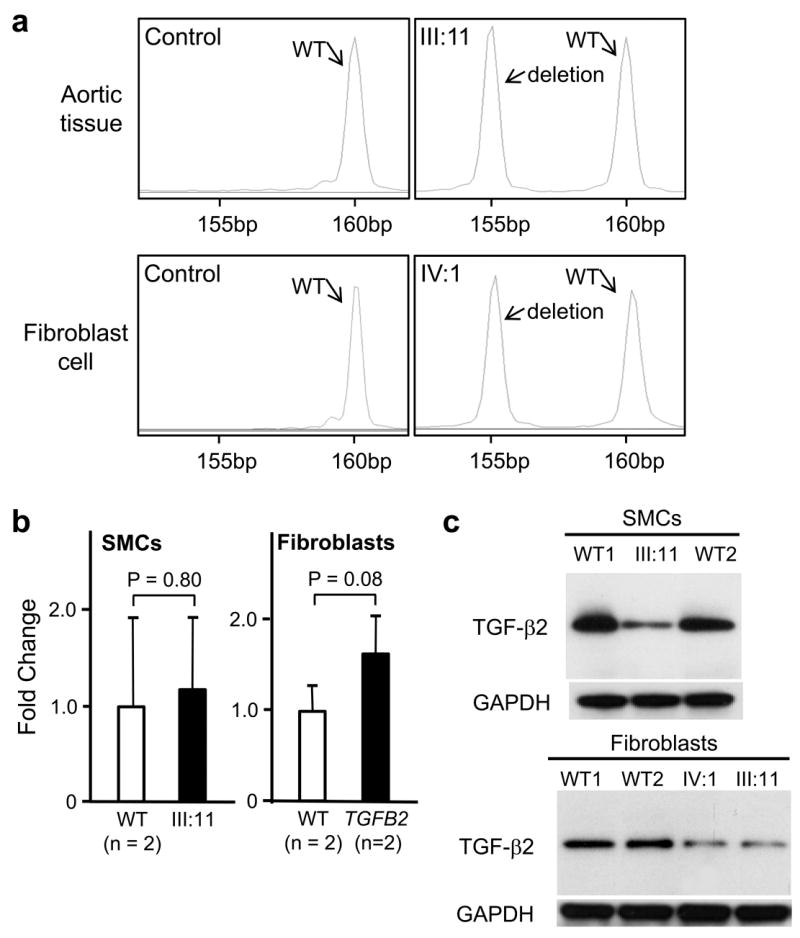
Transcript and protein analysis of the TGFB2 mutation p.Tyr341Cysfs*25 in exon 6. (a) RT-PCR of RNA extracted from normal ascending aortic specimens (n=2) or fibroblast cell cultures (n=2) showed only one PCR product at 160 bp. RT-PCR of RNA extracted from ascending aortic specimen of a patient (III:11) or fibroblast cell culture (IV:1) from family TAA288 demonstrated that the transcript from both normal and 5 bp deleted allele were present. (b) Expression of TGFB2 was quantified in aortic SMCs and dermal fibroblasts explanted from TGFB2 mutation carriers and matched controls using quantitative-PCR. TGFB2 expression levels between patients and controls were similar in both SMCs and fibroblasts. TGFB2 levels are standardized to GAPDH messages. The relative expression values were determined via the ΔΔCt method, and assays were performed in triplicate. Data are expressed as mean ± standard error of the mean for pooled experimental results. (c) Immunoblot analysis of TGF-β2 proprotein in cellular lysates of aortic SMCs and fibroblasts from patients (III:11 and IV:1, TAA288 family) and two normal controls. A protein band at 47kD, the molecular weight of the proprotein of TGF-β2, was identified with immunoblot analysis in both the patient and control lysates using a polyclonal antibody specific for TGF-β2. Decreased intensity of this band was found in patients’ cells, both SMCs and fibroblasts, when compared with control cells.
The aortic pathology associated with TGFB2 mutations was assessed using aortic tissue from patients MS239 III:16 and MS1756 II:8. The aortic pathology typical for thoracic aortic disease was found, which is characterized by fragmentation and loss of elastin fibers and accumulation of proteoglycans in the tunica media (Fig. 3a). Assessment of nuclear pSMAD2 in medial SMCs from the TGFB2 mutant aortas showed increased staining when compared with control aortas (Fig. 3b). TGFB1, TGFB2 and TGFB3 expression levels were assessed by Q-PCR using aortic tissue RNA from III:11 in family TAA288 and two matched controls (Fig. 3c). Surprisingly, only TGFB2 expression was increased in the aortic tissue (Fig. 2b). Immunoblot analysis of protein lysates from these same aortas demonstrated a corresponding increase in TGF-β2 proprotein levels in the mutant aorta (Fig. 3d). Immunostaining of TGF-β2 in aortic tissue from patients MS239 III:16 and MS1756 II:8 also showed increased intensity of the signal in patients’ aortas compared with control aorta (Fig. 3b). Thus, the aortas from patients with TGFB2 mutations show enhanced TGF-β signaling associated with a paradoxical increase in TGFB2 expression and TGF-β2 protein levels.
Figure 3.
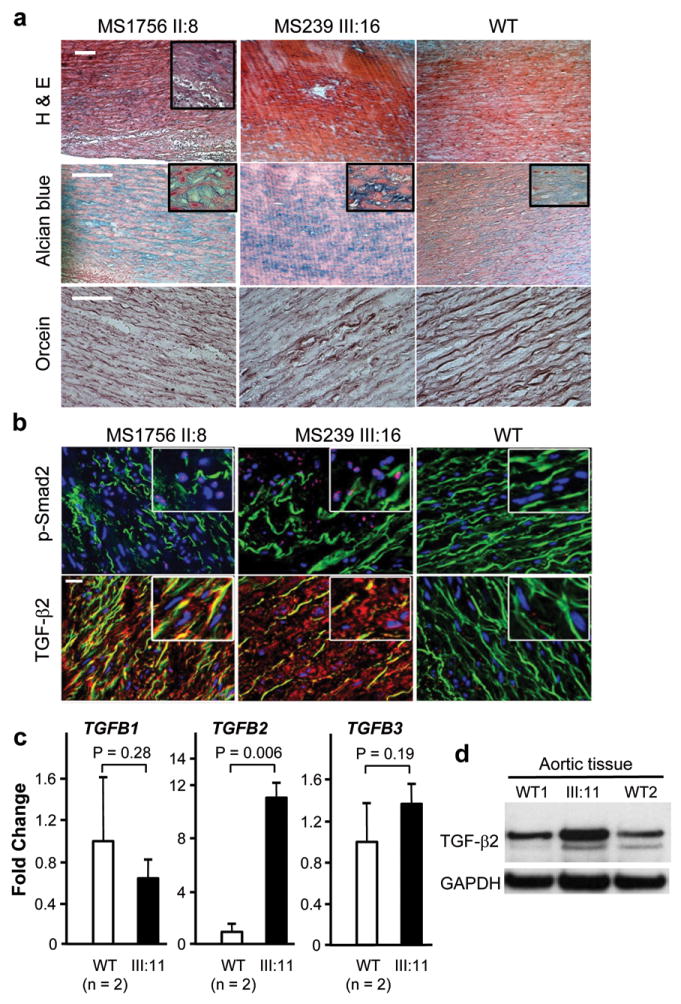
Aortic pathology and assessment of TGF-β signaling in patients with TGFB2 mutations. (a) Histology of aortic media from a dissection case (left column) with a TGFB2 mutation resulting in p.Glu102* (II:8, MS1756), an aneurysm case (middle column) with a TGFB2 mutation resulting in p.Cys229* (III:16, MS239 family), and a control (right column). Hematoxylin-eosin (H&E) staining displays disorganization of the aortic media with loss of SMCs and alcian blue reveals proteoglycan accumulation (blue). Note the loss, disarray and fragmentation of elastin fibers versus control with orcein staining. The white scale bars in the upper left pictures are 100μm. (b) Immunohistofluorescent staining for the same patients and control samples. Elastin fibers appear in green (autofluorescence), while nuclei are counterstained blue. Nuclear pSMAD2 staining (pink, upper panel) is present in the disease tissues and is absent in the control aorta. The lower panel shows the intense immunostaining for TGF-β2 using a polyclonal antibody specific for TGF-β2. Staining for pSMAD2 and TGF-β2 is located in the most disorganized areas of the aortic wall. The white scale bar in the upper left picture is 20μm. (c) The expression of TGFB1, TGFB2 and TGFB3 was assayed by Q-PCR using RNA isolated from the aorta of III:11 from TAA288. The expression of TGFB2 was increased over 10-fold when compared to aortic tissue isolated from controls aortas. Gene expression levels are standardized to GAPDH. The relative expression values were determined via the ΔΔCt method, and assays were performed in triplicate. Data are expressed as mean ± standard error of the mean for pooled experimental results. (d) Immunoblot analysis of the mutant and control aortas showed increased levels of the 47 kD TGF-β2 protein band in the patient’s aorta compared with two control aortas using a TGF-β2 specific antibody. Data are expressed as mean ± standard error of the mean for experimental results.
The clinical features in TGFB2 mutation carriers were assessed and found to be similar to other syndromes causing thoracic aortic disease. The median age at aortic disease presentation was 35 years, with the majority of affected family members presenting with aneurysms at the level of the sinuses of Valsalva (Fig. 4a,b; Table 1; Supplementary Table 3). A 60 year old male underwent surgical repair after his aneurysm enlarged from 47 to 50 mm over six years, and a 35 year old male underwent repair with an aortic root diameter of 54 mm. Aortic dissection was documented in an obligate mutation carrier in MS239 (III:12, female 56 years), and in the probands of MS1756 (II:8, male, 47 years) and MS211 (III:14, male, 31 years). Three women who were obligate carriers presented with cerebrovascular disease; TAA288-II:1 had a ruptured anterior communicating artery aneurysm at age 54 years, and TAAII:3 and MS239-III:2 died of SAH presumably due to ruptured ICAs. Fusiform dilatation and tortuosity of cerebrovascular arteries were present in mutation carriers (Fig. 4c,d). Additional cardiovascular features included interventricular septal defect present in an obligate carrier from MS239 (III:12), mitral regurgitation requiring surgical repair at the age of 43 years in IV:2 from MS239, and dilated cardiomyopathy prior to aortic root aneurysm formation in III:3 from TAA288. Inguinal hernias, typically bilateral and requiring surgical repair, were common in mutation carriers, including women. Skeletal features, joint laxity, and skin striae similar to those observed in MFS patients were found in some individuals with TGFB2 mutations but were insufficient in any individual to meet the diagnostic criteria of MFS (Table 1, Fig. 4e,f)10. Ectopia lentis was absent in these families. The affected family members examined by clinical geneticists (13 out of 19 mutation carriers) did not have a particular facial appearance or the craniofacial or skin findings observed in LDS patients. Prominent degenerative joint disease described in AOS patients was not present, although age- and trauma-related osteoarthritis was present.
Figure 4.

Clinical features associated with TGFB2 mutations. (a) CT scan imaging of the aorta of patient III:16 from family MS239 demonstrates dilatation predominating at the level of the sinuses of Valsalva (50 mm). (b) CT scan of a normal aorta with a diameter of 34 mm at the level of the sinuses of Valsalva. (c) Three dimensional CT scan from patient MS239, III:16 showing mild tortuosity (arrows) of cerebral arteries compared to normal control (d). Minimal arachnodactyly is evident in individual TAA288 III:8 based on positive wrist (Walker) sign (e) but negative thumb (Steinberg) sign (f).
Table 1.
Comparison of the clinical features observed in patients with TGFB2 mutations and patients with TGFBR2, FBN1, or SMAD3 mutations.
| Feature | Frequency, no. individuals/total (%)a | |||
|---|---|---|---|---|
| TGFB2 | TGFBR2 | FBN1 | SMAD3 | |
| Cardiovascular: | ||||
| Aortic root aneurysm | 14/19 (74) | 54/69 (78) | 178/226 (79) | 28/39 (72) |
| Aortic dissection | 3/23 (13) | 10/71 (14) | 23/243 (10) | 13/39 (33) |
| Cerebrovascular disease | 3/10 (30) | 0 b | 0 b | 6/16 (38) |
| Arterial tortuosity | 3/5 (60) | 5/25 (20) | NA | 8/16 (50)c |
| Mitral valve prolapse | 3/19 (16) | 15/66 (21) | 105/232 (45) | 18/36 (50)d |
| Skeletal: | ||||
| Pectus deformity | 7/16 (44) | 31/69 (45) | 144/243 (59) | 12/33 (36) |
| Arachnodoctyly | 8/13 (62) | 28/70 (41) | 137/240 (57) | 13/33 (39) |
| Spondylolisthesis | 1/7 (14) | 5/47 (11) | 15/223 (7) | 10/26 (38) |
| Scoliosis | 4/15 (27) | 21/68 (31) | 122/240 (51) | 22/36 (61) |
| Flat feet | 11/15 (73) | 8/58 (14) | 108/243 (44) | 30/33 (91) |
| Protrusio acetabularis | 1/8 (12) | 3/47 (6) | 79/221 (36) | 7/20 (35) |
| Joint hyperflexibility | 10/15 (67) | 45/59 (76) | 168/243 (69) | 3/31 (10) |
| High arched palate | 9/15 (60) | 32/67 (48) | 151/243 (62) | 15/28 (54)d |
| Cutaneous: | ||||
| Striae atrophicae | 8/15 (53) | 24/66 (36) | 139/241 (58) | 17/32 (53) |
| Operated hernia | 6/17 (35) | 13/67(19) e | 40/239 (17)e | 17/40 (43) |
| Pulmonary: | ||||
| Pneumothorax | 1/17 (6) | 3/69 (4) | 10/240 (4) | NA |
| Dural ectasia | 3/5 (60) | 4/40 (10) | 64/220 (29) | NA |
The frequency of clinical features associated with TGFBR2, FBN1 and SMAD3 are based on previous publications 11, 17, 22. Provided are the number of individuals with the feature/total number of individuals assessed for the particular feature (percentage);
based on medical history (imaging to screen for asymptomatic vascular disease not done);
number of patients with tortuosity of the cerebral arteries;
designated in publication as abnormal palate;
number of individuals with recurrent hernias;
NA- data not available.
We report a new etiology for familial thoracic aortic disease due to loss of function mutations in the TGFB2 gene. Aortic dilatation occurs at the level of the sinuses of Valsalva, similar to what is observed with mutations in FBN1, TGFBR1, TGFBR2, and SMAD3. The occurrence of ICAs and SAHs in three obligate carriers is most likely a manifestation of the TGFB2 mutation since similar cerebrovascular disease occurs in individuals with TGFBR1, TGFBR2, or SMAD3 mutations. Lastly, some systemic features overlapping with Marfan syndrome are also observed, as is the case with other syndromic forms of thoracic aortic disease (Table 1) 11. It should be underscored that no aortic dissections occurred younger than 31 years of age or with minimal aortic enlargement.
Mice with specific genetic disruption of each of the genes for the three isoforms of TGF-β (Tgfb1, Tgfb2 and Tgfb3) have discrete phenotypes, indicating a distinct role for each of the TGF-β isoforms. Tgfb2-/- mice die shortly after birth and have ascending aortas that are comparatively small and thin walled and other development defects involving the heart, lung, craniofacial, limb, spinal column, eye, inner ear, and urogenital systems12. The developmental processes disrupted by loss of TGF-β2 include epithelial to mesenchymal transformation (EMT) and differentiation of neural crest-derived components. These mice also show dysmorphic calvaria, extensive cleft palate, defective mandibles, and retrognathia; only retrognathia was observed in one TGFB2 mutation carrier. Based on our findings, haploinsufficiency of TGF-β2 does not disrupt cardiovascular development, suggesting that reduction of TGF-β2 level does not disrupt proper migration or homing of cells during development. Rather, decreased levels of TGF-β2 predispose to adult-onset vascular diseases involving the sinuses of Valsalva and the cerebrovascular arteries. SMCs in these two vascular beds are derived from two different origins, the secondary heart field and neural crest cells, respectively13. Thus, the clinical features suggest that decreased TGF-β2 levels lead to a defect in differentiation, proliferation or viability of cells in these arteries. The differentiation of neural crest cells into vascular smooth muscle cells is dependent on TGF-β signaling and secondary heart field EMT is specifically TGF-β2 dependent12, 14, and decreased levels of TGF-β2 may disrupt proper differentiation of SMCs in these arteries. Relevant to this speculation, it is notable that SMCs explanted from patients with TGFBR2 mutations are relatively de-differentiated in culture when compared to control SMCs, and fail to differentiate to the same extent as control SMCs when exposed to TGF-β115.
Our results provide evidence that haploinsufficiency of TGFB2 is an initiating step in the pathogenic events leading to thoracic aortic disease. A subset of the SMAD3 mutations are frameshift mutations that are predicted to cause SMAD3 haploinsufficiency16, 17. The missense mutations in TGFBR1 and TGFBR2 have either been shown or are predicted to disrupt kinase function, which is critical for propagating TGF-β signaling4, 15, 16, 18, 19. Despite the fact that these mutations are predicted to decrease TGF-β signaling, these defective genes trigger poorly understood signaling events that lead to increased nuclear pSMAD2 immunostaining in diseased tissue. Immunostaining for pSMAD2 is also increased in aortas of thoracic aortic disease patients without mutations in these genes5. One event contributing to increased pSMAD2 staining may be epigenetic modifications of the promoter of SMAD2, resulting in constitutive activation of this signaling protein20. Another possible contributing event is that the relative resistance of the SMCs to TGF-β signaling due to the gene mutations may paradoxically increase signaling through other TGF-β signaling proteins. It is notable that deletion of Tgfbr2 in mouse cranial neural crest cells leads to elevated expression of TGF-β2 and TGF-β type III receptor and activation of SMAD-independent cellular pathways21. Our study identified a similar increase in TGF-β2 levels in the diseased tissue in response to TGFB2 haploinsufficiency that is not present in explanted SMCs and fibroblasts. Although our data support the conclusion that the initiating event leading to vascular diseases due to mutations in TGFB2 is decreased TGF-β2 levels, further work is warranted to unravel the pathways that are secondarily altered to cause a paradoxical increase in TGFB2 expression and increased pSmad2 signaling in diseased aortic tissue.
Online Methods
Whole Genome Linkage Analysis
For the American TAA288 family, genomic DNA from nine family members was analyzed using a 50K GeneChips Hind array from Affymetrix following manufacturer’s protocol. Multipoint linkage analyses of the Affymetrix 50K SNP array data was performed with the MERLIN program. An autosomal dominant model for TAAD with a disease-gene frequency of 0.001 was assumed. Two-point linkage analysis with candidate variant status was performed in the families with TGFB2 mutations. The minor allele frequency of candidate mutation as 0.0001 and penetrance of TAAD as 0.90 were assumed. Log of odds (LOD) scores were calculated with MLINK program of the computer software FASTLINK, version 3.P.23
For the French MS239 family, genomic DNA from 18 members were analyzed with 1056 microsatellites (deCODE high-density marker set) in multiplex reactions with fluorescently-labeled primers24. All related family members were given an affected, unaffected or unknown status as provided by referring clinicians and in agreement with clinical data. Parametric linkage analyses were performed with the following parameters: (1) autosomal dominant transmission of the disease trait, (2) reduced penetrance of 0.9 for heterozygotes, and (3) frequency of the disease allele of 0.001%. We used Pedcheck to detect Mendelian inheritance errors. SuperLink v1.5 and SimWalk v2.91 programs were used to compute two-point and multipoint LOD scores25-27. All these programs were run with the easyLinkage Plus v5.00 package28.
Exome Sequencing
Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols. Two micrograms of DNA from two affected individuals in family TAA288 were used for construction of the shotgun sequencing library as described previously using adaptors for paired-end sequencing.29, 30 Exome sequences were captured by SeqCap EZ version 2.0 (Roche) and recovered according to manufacturer’s directions. Enriched libraries were then sequenced on a HiSeq using manufacturer protocols. Reads were mapped to the reference human genome (UCSC hg19) with BWA (Burrows-Wheeler Aligner)31, and variant detection and genotyping are performed using the UnifiedGenotyper (UG) tool from GATK.32. Single nucleotide variants (SNVs) and indels were filtered to >8X and quality >30. Annotation of variants was performed using the SeattleSeq server (http://gvs.gs.washington.edu/SeattleSeqAnnotation/). The identified variants were then filtered against exome data from non-affected control individuals for indel and SNV calls to identify novel non-synonymous, splice, nonsense or indel variants in affected individuals. These variants were considered as a candidate mutation.
DNA samples from 3 affected and one unaffected individuals in family MS239 were sequenced by Integragen S.A. (Evry, France). The adapter-ligated library was prepared from 3 μg of DNA with the “Paired-End kit PE Sample Prep” (Illumina). DNAs were subjected to the exome capture procedure using the Agilent “SureSelect Human All Exon Kit” (V3, 50Mb) according to manufacturer’s protocols. Paired-end sequencing was performed on an Illumina HiSEQ2000 and the short reads (75 bp) were aligned to the Human Genome (UCSC hg19) using CASAVA 1.8. Image analysis and base calling were performed using Illumina Real Time Analysis (RTA) Pipeline version 1.14 with default parameters. The alignment algorithm used was ELANDv2. SNVs and indels were filtered to >8X and quality >30. Variation annotation was performed with the in-house pipeline. Identified variants were then filtered against a further in-house 1500 exome data set to identify novel heterozygous variants that were present in the 3 affected subjects and absent in their healthy relative. These variants were considered as candidate mutations.
DNA Sequencing
Bidirectional Sanger DNA sequencing assays were performed using primers designed 60-120 base pairs from the variants or intron-exon boundaries to confirm candidate variants or sequence candidate genes on probands from unrelated TAAD familes (Supplementary Table 2). Polymerase chain reaction (PCR) amplifications were carried out using HotStar Taq™ DNA polymerase (Qiagen Inc.Valencia, CA). PCR products were treated with EXOSAP-IT (Affymetrix, Inc. OH) to digest the primers and followed with sequencing PCR using the BigDye™ sequencing reaction mix (Applied Biosystems, CA). The sequencing PCR products were purified using the BigDye XTerminator kit (Applied Biosystems, CA) and then loaded on an ABI3730xl sequencing instrument using the Rapid36 run module or on an ABI3500xLDx sequencing instrument.. DNA sequencing results were analyzed using the Mutation Surveyor software (SoftGenetics, PA) or SeqScape v2.7 (Applied Biosystem, CA).
Allele-Specific TGFB2 mRNA Quantification
Allele specific TGFB2 mRNA quantification was performed on mRNA extracted from ascending aortic tissues from patients or control using a TRIzol RNA extraction kit (Life Technologies, Grand Island, NY) and cDNAs were synthesized using MMLV-RT kit (Life Technologies) and random hexamer according to the manufacturer’s protocol33. RT-PCR was performed using primers flanking TGFB2 mutation and a fluorescently labeled primers 34. The PCR products were assayed using an ABI3730xl sequencing instrument using the Rapid36 run module and analyzed by GeneMap4.0 software (Life Technologies). The ratio of allele specific expression of TGFB2 mRNA was obtained based on the ratio of peak areas corresponded to wildtype and mutant alleles.
Quantitative PCR Analysis
Total cellular RNA was extracted from SMCs, fibroblast cells, or aortic specimens using TRI reagent according to the manufacturer’s protocol (Sigma-Aldrich, MO, USA). Q-PCR assays were carried out using pre-designed TaqMan reagents from Life Technologies following the manufacturer’s protocols, and were performed on a ABI Prism 7000 Sequence Detection System (Life Technologies, CA, USA).
Histology Analysis
Aneurysmal ascending aortic and dissection specimens were collected during aortic surgery (Bichat University Hospital). Normal thoracic aorta was obtained from a normal organ transplant donor with the authorization of the French Biomedicine Agency. Aneurysmal tissue was sampled in the outer curvature, the most dilated part of the ascending aorta. The aortic specimens were fixed in 4% (v/v) buffered paraformaldehyde for 48 h at room temperature, embedded in paraffin, and serial sections (5 μm thickness) were obtained from each specimen. The samples were histologically examined after Hematoxylin-Eosin, Alcian Blue (for visualization of proteoglycans) and orcein (for visualization of elastic fibres) staining. Immunohistofluorescence studies were performed as previously described20 using the following primary antibodies: mouse monoclonal anti-TGFβ2 that does not recognize TGFβ1 or TGFβ3 (ab36495 from Abcam, Paris, France), rabbit monoclonal anti pSmad2 (Santa Cruz Biotechnology Inc., CA, USA). Secondary antibodies used were Alexa Fluor 555 anti mouse and anti-rabbit (Invitrogen, Life Technologies SAS, Saint Aubin, France). Nuclei were counterstained by Hoechst staining (Invitrogen, Life Technologies SAS, Saint Aubin, France).
Protein Analysis
To isolate proteins from the whole tissue, a piece of cryopreserved tissue (10mg or less) was placed in protein extraction buffer (30mM Hepes, 2.5mM EGTA, 2.5mM EDTA, 20mM KCl, 40mM β-glycerophosphate, 40mM NaF, 4mM NaPPi, 1 mM PMSF, 10 mM NaF, 1 mM Na3VO4, 10 ug/ml aprotinin,10 ug/ml leupeptin and 10 ug/ml pepstatin) and homogenized on the Polytron PT-10-35 homogenizer 3 times for 15 seconds each. Samples were then centrifuged, and the pellet fraction discarded. To isolate proteins from SMCs, cells from one patient (III:11) and two normal controls were washed twice with ice-cold PBS and lysed in RIPA buffer (50 mm of Tris, pH 7.5, 150 mm of NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) supplemented with protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail (Sigma). Protein concentration was determined by the Bio-Rad protein assay.
Total lysates from either cells or tissues (10 μg) were separated by SDS–PAGE with 12.5% Tris–HCl gel (Ready Gel, Bio-Rad, Hercules, CA, USA), followed by transfer to polyvinylidene difluoride membranes (Immobilon-P, Millipore, Bedford, MA, USA). Membranes were incubated in blocking buffer (5% nonfat milk in T-PBS) for 1 h and immunoblotted with primary antibody (polyclonal anti-TGF-β2 from R&D System) diluted in 5% BSA (Sigma-Aldrich, MO, USA). Membranes were probed with horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Western blots were visualized by the enhanced chemiluminescence technique (Amersham ECL Western Blotting Detection Reagents, GE Healthcare, Piscataway, NJ, USA). Densitometry was performed using ImageJ software. Each band was quantified three independent times, and results shown were pooled from at least two separate immunoblots.
Statistical Analysis of RNA and Protein Expression
All values are expressed as means ± standard error of the mean (standard deviation divided by the square root of the number of separate experimental replicates). Statistical differences between TGFB2 and control RNA or protein expression were analyzed by a Student’s t-test. Differences were considered statistically significant at values of P <0.05. Data shown was pooled from at least two experimental replicates.
Supplementary Material
Acknowledgments
The authors are extremely grateful to the families involved in this study and the medical professionals who aided in the collection of clinical data from the families. We would like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung Cohorts Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Heart Cohorts Sequencing Project (HL-103010), the Broad Institute Sequencing Project (HL-102925), the Northwest Genomics Center Sequencing Project (HL-102926, D.A.N, M.J.R, and J.S.) and the Family Studies Project Team. We thank JM. Serfaty for providing vascular imaging and L. Louedec and Z. Ren for technical assistance.
The following sources provided funding for these studies: RO1 HL62594 (D.M.M.), P50HL083794-01 (D.M.M.), UL1 RR024148 (CTSA), Vivian L. Smith Foundation (D.M.M.), TexGen Foundation (D.M.M.), Richard T. Pasani Funds (D.M.M.), GIS-Maladies rares (C.B.), PHRC AOM09093 (G.J.), PHRC AOM 10108 (C.B.), ANR 2010 BLAN 1129 from the French National Research Agency (G.J.) and the EU FP-7-integrated project ‘Fighting Aneurysmal Disease’ (FAD, www.fighting-aneurysm.org/) (J.B.M.)
Footnotes
Author contributions
C.B., D.M.M., and G.J. planned the project, designed the experiments, oversaw all aspects of the research and wrote the manuscript. C.B., S.L.L. R.L.R.-C., M.V., D.G., M.G., and M.A. performed linkage analyses and analyzed exome sequencing data. M.J.R., J.S., and D.A.N. did the exome sequencing in TAA288. D.G., A.H.L. and N.H. analyzed exome sequencing data and performed Sanger sequencing analysis with H.d’I. L.G., D.G. and C.S.K. performed the TGFB2 transcript and protein assays. J.B.M. performed histological and imunostaining analyses with MS.G. S.P. did the genomic deletion analysis. E.S.R., B.G., C.M., A.C.B. and L.G. provided clinical and pedigree data. D.D., D.M.M., and G.J. provided clinical data and performed the sample collection. A.V. revised the manuscript.
Competing financial interests
The authors declare no competing financial interests.
ACCESSION Number
The TGFB2 mRNA reference sequence is available at NCBI (NM_003238.3).
Reference List
- 1.Hiratzka LF, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease: Executive Summary. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 2.Faivre L, et al. Pathogenic FBN1 mutations in 146 adults not meeting clinical diagnostic criteria for Marfan syndrome: further delineation of type 1 fibrillinopathies and focus on patients with an isolated major criterion. Am J Med Genet A. 2009;149A:854–860. doi: 10.1002/ajmg.a.32809. [DOI] [PubMed] [Google Scholar]
- 3.Loeys BL, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 4.van de Laar IM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 5.Gomez D, et al. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol. 2009;218:131–142. doi: 10.1002/path.2516. [DOI] [PubMed] [Google Scholar]
- 6.Habashi JP, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regalado E, et al. Autosomal dominant inheritance of a predisposition to thoracic aortic aneurysms and dissections and intracranial saccular aneurysms. Am J Med Genet A. 2011;155A:2125–2130. doi: 10.1002/ajmg.a.34050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prakash SK, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010;87:743–756. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva AL, Romao L. The mammalian nonsense-mediated mRNA decay pathway: to decay or not to decay! Which players make the decision? FEBS Lett. 2009;583:499–505. doi: 10.1016/j.febslet.2008.12.058. [DOI] [PubMed] [Google Scholar]
- 10.Loeys BL, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 11.Attias D, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120:2541–2549. doi: 10.1161/CIRCULATIONAHA.109.887042. [DOI] [PubMed] [Google Scholar]
- 12.Sanford LP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 14.Chen S, Lechleider RJ. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–1202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- 15.Inamoto S, et al. TGFBR2 Mutations Alter Smooth Muscle Cell Phenotype and Predispose to Thoracic Aortic Aneurysms and Dissections. Cardiovasc Res. 2010;88:520–529. doi: 10.1093/cvr/cvq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regalado ES, et al. Exome Sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van de Laar IM, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49:47–57. doi: 10.1136/jmedgenet-2011-100382. [DOI] [PubMed] [Google Scholar]
- 18.Mizuguchi T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stheneur C, et al. Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders. Hum Mutat. 2008;29:E284–E295. doi: 10.1002/humu.20871. [DOI] [PubMed] [Google Scholar]
- 20.Gomez D, et al. Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc Res. 2011;89:446–456. doi: 10.1093/cvr/cvq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwata J, et al. Modulation of noncanonical TGF-beta signaling prevents cleft palate in Tgfbr2 mutant mice. J Clin Invest. 2012;122:873–885. doi: 10.1172/JCI61498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faivre L, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cottingham RW, Jr, Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 24.Kong A, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 25.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fishelson M, Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18(Suppl 1):S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 27.Weeks DE, Sobel E, O’Connell JR, Lange K. Computer programs for multilocus haplotyping of general pedigrees. Am J Hum Genet. 1995;56:1506–1507. [PMC free article] [PubMed] [Google Scholar]
- 28.Lindner TH, Hoffmann K. easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–407. doi: 10.1093/bioinformatics/bti009. [DOI] [PubMed] [Google Scholar]
- 29.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng SB, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Depristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He R, et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 2006;131:671–678. doi: 10.1016/j.jtcvs.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 34.Guo DC, Milewicz DM. Methodology for using a universal primer to label amplified DNA segments for molecular analysis. Biotechnol Lett. 2003;25:2079–2083. doi: 10.1023/b:bile.0000007075.24434.5e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.