

Abstract
Effective prevention of new HIV infections will require understanding the mechanisms involved in HIV acquisition. HIV transmission across the female genital tract is the major mode of new HIV infections in Sub-Saharan Africa and involves complex processes including cell activation, inflammation, and recruitment of HIV target cells. Activated CD4+ T cells, dendritic cells, and macrophages have been described as targets for HIV at the genital mucosa. Activation of these cells may occur in the presence of sexually-transmitted infections, disturbances of commensal flora, and other inflammatory processes. In this review, we discuss causes and consequences of inflammation in the female genital tract, with a focus on dendritic cells. We describe the central role these cells may play in facilitating or preventing HIV transmission across the genital mucosa, and in the initial recognition of HIV and other pathogens, allowing activation of an adaptive immune response to infection. We discuss studies that investigate interventions to limit dendritic cell activation, inflammation and HIV transmission. This knowledge is essential in the development of novel strategies for effective HIV control including microbicides and pre-exposure prophylaxis.
Keywords: Activation, Cytokines, Dendritic cells, HIV, Inflammation
Introduction
The Joint United Nations Programme for HIV/AIDS (UNAIDS) report for 2012 - “Together we will end AIDS”-estimated that 34 million people worldwide were living with HIV in 2011, and 23.5 million in sub-Saharan Africa. In the same year, 2.5 million people acquired new infections,1 highlighting the urgent need for interventions to prevent transmissions.
Most HIV transmissions occur via the mucosal surfaces of the genital and gastrointestinal tracts.2 Women are at higher risk of HIV acquisition compared with their male counterparts. In heterosexual discordant relationships, male-to-female HIV transmission is approximately 8-fold greater than female-to-male transmission during vaginal intercourse.3–6 Several behavioural and biological factors have been proposed to explain differences in HIV acquisition between men and women, including differences in sex hormones, stage of HIV infection, age of index patient, condom use, male circumcision, presence of genital ulcers, mucosal area exposed during sexual intercourse, areas of vulnerability at the transformation zone of the cervix, dose of HIV transmitted in female versus male genital secretions, and geographical location.4 It is estimated that 30 – 40% of annual worldwide infections occur through HIV invasion of the female genital tract via exposure to virus-containing semen.7 In the female genital tract, HIV penetration and infection occurs through vaginal, ectocervical, and endocervical mucosa, but the relative contribution of each site to successful transmission remains unknown. While the single-layer epithelial lining of the endocervix offers the best opportunity for HIV to cross the barrier, the relatively large surface area of the ectocervix and vagina may offer HIV a greater chance of penetrating the epithelium.7, 8
The mechanisms by which HIV crosses the genital epithelium are complex. It has been suggested that during sexual transmission, cell-free or cell-associated HIV traverses the female genital mucosa through transcytosis and/or trans-epithelial emigration of infected dendritic cells (DC), among other routes.9, 10 Transcytosis of cell-free HIV-1 across epithelial surfaces of the genital tract occurs through an epithelial transcellular vesicular pathway, where HIV-1 virions are captured on the surface of the epithelium by epithelial cells into vesicles and are liberated on the stromal side of the epithelium.11 Many viral and host components have been implicated in this process. These include viral envelope glycoprotein (gp) 41 and gp120, glycosphingolipids, the co-receptor CCR5, and the heparin sulfate proteoglycan attachment receptors, among others. The transcytosed virus can then bind to underlying CD4 cells leading to an effective infection. Only a small percentage of viruses cross the epithelial barrier by transcytosis.12, 13
Transepithelial migration of HIV occurs through cell-associated viruses (i.e. HIV infected host cells).9 Seminal cells containing HIV particles may become trapped in the mucus covering the epithelial surface.14 Upon contact with the epithelium, these cells can release free viruses (which are able to penetrate between epithelial cells), be captured by tissue-resident DCs, including Langerhans cells (LCs), and ultimately become internalised in the endocytic compartment.14, 15 These cells may become activated and migrate into the basal compartments where they can transfer the virus to CD4+ T cells by trans-infection.16 During coitus, mechanical abrasions of mucosal surfaces induced by intercourse could allow free or cell-associated virus in semen to penetrate the epithelium and have direct access to DCs, macrophages or CD4+ T cells that lie underneath the stroma.7
Of the potential target cells present in the genital tract, DCs, macrophages and CD4+ T cells are the most likely cells infected with HIV infection.17 Although all these cell types can become infected with HIV to varying degrees, CD4+ T cells are the most susceptible to HIV infection. DCs become infected with HIV both in vivo and in vitro, albeit at low levels, and can migrate to draining lymph nodes where antigen presentation occurs leading to activation of CD8+ and CD4+ T cells which can in some instances control viral replication during established infection.18 DCs, macrophages, and epithelial cells of the genital mucosa can recognise pathogens and are able to produce inflammatory cytokines or chemokines, which contribute to an immune response. The recognition of pathogens and the production of cytokines and chemokines can lead to activation and recruitment of target cells to the genital mucosa, which although is an integral process in normal host immunity, in the case of HIV, can in turn increase the odds of HIV capture and transmission due to HIV targeting activated cell.
In this review, we discuss causes and consequences of inflammation and their role in HIV acquisition in the female genital tract, with a specific focus on the role of DCs in driving genital tract inflammation. While this review focuses only on the role of DCs in driving inflammation, it is important to note that other innate cells such as macrophages and neutrophils may also play a critical role in inflammation and HIV transmission in the female genital tract. We will discuss possible ways of regulating genital tract inflammation through this focus on DCs, which may have an impact on HIV transmission.
DC subsets and pathogen binding receptors
DCs were discovered in the 1970s, and today represent a diverse and rare population of leukocytes with several subsets that differ in their origin, maturation state and anatomic location.19, 20 DCs are derived from hematopoietic bone marrow progenitor cells and are usually found in an immature state and the first to respond to invading pathogens. DCs become activated upon pathogen recognition leading to upregulation of activation markers and cytokine production.21 DCs can be infected by a wide variety of pathogens including bacteria and viruses. Recognition of pathogens by DCs and subsequent infection occur through various receptors expressed on these cells. The type of innate response, mostly mediated by DCs, may dictate the type of adaptive immune response to an infection with a pathogen. Activation of an adaptive response by DCs is achieved through antigen presentation of pathogen-derived peptides to cells of the adaptive immune system. The ability of DCs to produce cytokines and to upregulate expression of major histocompatibility complex (MHC) class I and II molecules and co-stimulatory markers, makes them the primary and most efficient activators of adaptive immunity.22
DC subsets differentially express pathogen-binding receptors depending on their function and location.23 The type of pathogen recognition receptor (PRR) triggered may determine the outcome of an innate immune response, as binding to different receptors may result in distinct immune outcomes.24 PRRs are expressed mainly on innate cells and are centrally involved in recognition of invading pathogens and induction of immune responses.25, 26 They bind to conserved structures on microbial species known as pathogen-associated molecular patterns (PAMPs) or damage associated molecular patterns (DAMPs), which are danger signals resulting from necrotic, stressed or apoptotic cells.27 These receptors include Toll-like receptors (TLR), nucleotide-binding oligomerisation domain (Nod)-like receptors (NLR), C-type lectin receptors (CLR) such as DC-specific intracellular adhesion molecule 3 (ICAM-3)-grabbing non-integrin (DC-SIGN), mincle, dectin-1, mannose receptor (MR); retinoic-acid-inducible protein RIG-1-like receptor (RLR), complement receptors (CR), and scavenger receptors.28, 29 Of these PRRs, TLRs have been studied most extensively. There are 10 characterised TLRs (1 to 10) in humans, which are located either on the plasma membrane or within endosomes recognising a wide variety of PAMPs. TLRs 1, 2, 4, 5, 6 and 10 are mainly expressed on the plasma membrane and recognise microbial membrane components such as lipids, lipoproteins and proteins. TLRs 3, 7, 8 and 9 are expressed in endosomal compartments where they recognise nucleic acid components of (typically) viral origin.30, 31 Twenty-three human NLR genes have been identified, but the physiological function of most of these receptors remains poorly understood.32, 33 The well-characterised members of the NLR family are NOD-containing protein 1 (NOD1) and NOD2, which recognise distinct structural motifs derived from peptidoglycans (PGN).34 Other receptors expressed by DCs that are relevant to pathogen binding and signalling include chemokine receptors.35 Chemokine receptors bind chemokines, which are necessary for recruitment of cells during inflammation and homeostasis. These receptors are expressed by leukocytes including DCs, macrophages and CD4+ T cells. Several chemokine receptors have been described with varying functions in the immune response to pathogens.35 Here, we focus mainly on the chemokine receptors CCR5 and CXCR4 because they are co-receptors for HIV entry into target cells.36
Three major subsets of DCs have been identified according to their origin, function and location: myeloid DCs (mDC), plasmacytoid DCs (pDC) and Langerhans cells (LCs) (Table 1).
Table 1.
DC subtypes, identification markers and major HIV related receptors expressed.
| Cell type | Identification markers | Major PRRs | Major HIV receptors | binding |
|---|---|---|---|---|
| Myeloid DCs | CD11c | TLRs 2, 3, 4, 5, 7,8 NOD1, NOD2 DC-SIGN, Mannose receptor |
CCR5, CXCR4, DC-SIGN | |
| Plasmacytoid DCs | CD123 | TLRs 2, 3, 6, 7, 8, 9 | CCR5, CXCR4 | |
| Langerhans Cells | CD1a or langerin | TLRs 1, 3, 6, 7, 10 | CCR5, langerin |
Myeloid DCs
These cells are also referred to as conventional or classical DCs and are characterized by the surface expression of CD11c (CD11chigh) and the absence of CD123 expression (CD123low) (Table 1).37 mDC originate from common myeloid progenitors and are found in blood in an immature state. These cells migrate into tissues in response to chemotactic cytokines and contribute to the local inflammation.38 mDC express a wide range of PRRs for the recognition and phagocytosis of pathogens, leading to the production of cytokines and chemokines, and the upregulation of surface co-stimulatory molecules. Commonly studied PRRs include TLRs 2, 4, 5, 6, 7, 8, NOD1 and NOD2. DC-SIGN, CCR5 and CXCR4 are also highly expressed on these cells and are of relevance to HIV infection.39 mDCs can be further subdivided into mDC1 [expressing CD11c and CD1c (BDCA-1)] and mDC2 [expressing CD11c and CD141 (BDCA-3)].40 These subsets are functionally distinct: CD11c+CD141+ mDC2 are more efficient than mDC1 at capturing exogenous antigens for cross presentation on MHC class I molecules to CD8+ T cells. mDC1 and mDC2 have been reported to induce T helper (Th) type 1 and Th2 T cell differentiation, respectively.41–43 mDC1 and mDC2 both express a wide range of TLRs, albeit at different levels. mDC1 express high levels of TLR4, 5 and 7, while mDC2 express high levels of TLRs 3 and 8. These differences in PRR expression may explain their different roles in the immune response to infection.
Plasmacytoid DCs
In contrast to mDC, pDC express high levels of CD123 (CD123high) and low levels of CD11c (CD11clow) (Table 1). They can also be identified by the expression of CD303 or BDCA-2.37, 40 pDC originate from a common lymphoid progenitor as pre-pDC expressing CD123 and require granulocyte-macrophage colony stimulation factor (GM-CSF) to differentiate into immature cells.44 pDCs are principally found in blood and migrate to tissues in response to chemotactic cytokines and contribute to the local inflammation process.38 pDC play a major role in the immune response to viral infections, as the major producers of anti-viral type I interferons. pDC exhibit several additional characteristics that make them effective during viral infections.20, 45 They contain MHC class I molecules in their early endosomal compartments which facilitate antigen presentation from the exogenous processing pathway to CD8+ T cells. In addition, the late endosomal compartment of pDCs contain MHC class II molecules for viral antigen presentation to CD4+ T cells. An extensive endoplasmic reticulum (ER) compartment facilitates high-capacity secretion of anti-viral factors, including type I interferons. The secretion of type I interferons, IL-6, and the expression of CD70 upon stimulation with CpG (a TLR9 ligand) make pDC efficient activators of plasma B cells and antibody responses.46, 47 These cells are central to anti-viral immunity due to the high density expression of receptors that recognise or bind viral components and production of type 1 interferons. These receptors include chemokine receptors CCR5 and CXCR4, TLRs 3, 7 and 9.39, 48 They also express other TLRs, including TLRs 1, 2, 4, 6 and 10, which can recognise bacterial components.49, 50
Langerhans cells
These are DCs found in the skin and the mucosal epithelium of the vagina and ectocervix. Langerhans cells (LCs) are often defined by the expression of CD1a and/or langerin, a C-type lectin receptor (Table 1).51 LCs are thought to originate from bone marrow hematopoietic precursors but are maintained by local precursors present in the skin.52, 53 LCs are usually the first antigen presenting cells to encounter pathogens in the genital tract. Their high motility allows them to emigrate from tissues to draining lymph nodes upon contact with the antigens.54, 55 LCs express PRRs including TLRs and chemokine receptors CCR5. TLRs expressed by LCs include TLRs 1, 3, 6, 7 and 10, but little or no TLRs 2, 4 and 5.56, 57 LCs are also characterised by the presence of Birbeck granules that are involved in the cytolytic breakdown of pathogens.58 Birbeck granules are rod-shaped subdomain cytosolic organelles of the endocytic recycling compartment in LCs.59
Innate receptor signaling and generation of an immune response
DCs play an important role in antigen capture and presentation to T cells, critical to immune surveillance. Upon recognition of the ligand expressed on pathogens by PRR on DCs, a signaling cascade is initiated, which involves either phosphorylation or de-phosphorylation of different adapter molecules and transcription factors.60 This signaling cascade is necessary for the generation of an immune response, including cytokine/chemokine production and maturation of innate cells. Adapter molecules associated with TLR signaling include MyD88, TIR-domain containing adapter–inducing IFN-β translocating chain associated membrane protein (TRIF), and TIR domain-containing adapter proteins (TIRAP).61, 62 All TLRs (except TLR3) signal through MyD88. TLR3 signals through TRIF, while TLR4 can signal through either MyD88 or TRIF. All of this signaling via these adaptor molecules culminates in the activation of the transcription factor nuclear factor kappa B (NF-κB), which translocates into the nucleus for the induction of pro-inflammatory genes.60 The resulting inflammation is a critical early step in controlling the invading microbe while an adaptive immune response is initiated. Although TLRs are essential for protective immunity against infection, inappropriate TLR responses may contribute to acute and chronic inflammation, as well as to systemic autoimmune diseases.25
In contrast to TLRs, NLRs and CLRs utilize different signaling pathways, typically via the caspase recruitment domain family, member 9 (CARD9).63 NLRs can also signal via interacting protein 2 and TANK-binding kinase 1. CLRs signal via Raf1, and FcR-gamma converging to CARD9 for signaling. Other transcription factors involved in PRR signaling pathways include activator protein-1 (AP-1), mitogen-activated protein kinase (MAPK) and IFN regulatory factors (IRF).64–66 Activated transcription factors translocate into the nucleus and initiate transcription of immune response genes, such as those for cytokines and chemokines. The initial production of cytokines, chemokines, and other molecules that result from activating these signaling pathways then serve to shape and determine the type of subsequent adaptive immune response generated.67
Cytokines
Several cytokines mediate inflammation and innate immune responses. These cytokines include IL-12, IL-10, IL-6, IL-1β, TNF-α, and IFN-α,67, 68 and play important roles in the development of inflammation and the polarization of naïve T cells into Th1, Th2, Th17 or regulatory T cells.69–71 The type of PRR triggered ultimately determines the cytokines that are produced by innate cells, and therefore the outcome of the innate response. For example, the triggering of TLRs 7 and 9 on pDC by viral components leads to elevated production of IFN-α, while triggering of cytosolic receptors such as NLRs may lead to the formation of an inflammasome complex resulting in the production of IL-1β through a caspase-1-dependent pathway.72, 73 Together this provides an innovative way for the immune system to link the type of pathogen to an appropriate innate and eventually adaptive immune response.
Chemokines
Chemokines are chemotactic cytokines that are important in innate immune responses to pathogens and facilitate leukocyte migration and positioning, to ensure that the correct immune cells arrive at the correct locations, and are involved in additional processes such as leukocyte degranulation.74 There are many chemokines with diverse and overlapping functions in immunity.75–77 These chemokines include MIP-1α, MCP-1, MIP-1β, CXCL12/SDF-1 and RANTES among others. These are secreted in response to infection and their expression can be enhanced by other inflammatory cytokines such as IL-1β. The primary role of chemokines is to create a microenvironment that acts as a chemotactic gradient, attracting immune cells from the blood to the site of the infection, which is often in tissue.78, 79 Inflammatory chemokines include CCL3 (MIP-1α) and CCL2 (MCP-1) that bind to CCR1 and CCR2, respectively.
DC activation and maturation
Most DCs are immature in phenotype and undergo maturation only upon pathogen encounter. Maturation comprises morphological and functional changes associated with activation of innate cells by microbial stimuli such as TLR agonists.20 Immature DCs are efficient phagocytes and once activated, these cells mature by increasing expression of maturation markers, while at the same time, their phagocytic capacity is reduced. Some of the better-studied maturation markers on DCs include CD86, CD80, CD83, and CD40. CD80 and CD86 belong to the B7 family of co-stimulatory molecules and provide co-stimulation during antigen presentation by binding to two molecules on T cells: CD28 for activation, and CTLA-4 for inhibition of T cell responses.80 CD83 is a maturation marker that is expressed on DCs and upregulated upon activation.81 CD40 belongs to the TNF receptor superfamily and is expressed by innate cells such as DCs and monocytes, and binds to CD40 ligand (CD154) expressed on T cells, thereby activating the T cells.82, 83 While CD86 and CD80 are required for T cell activation during antigen presentation, CD40 is required for long-term DC activation, cytokine production and T cell polarization.80, 82, 84
Inflammation in the genital mucosa
Genital immune responses are similar in some ways to responses in blood or other mucosal sites, but are also distinct, due to differences in cellular composition, local cytokine milieu, microbiome complexity and microbial burden. Inflammation generally results from increased cytokine and/or chemokine production by cells that have recognised the presence of pathogens, signalling infection. The main goal of inflammation is the increased recruitment of cells to the site of infection to perform effector functions that lead to pathogen clearance. Cytokines orchestrate an inflammatory response by inducing cell death of inflammatory tissues and modifying vascular endothelial permeability.85 These also facilitate the recruitment of more immune cells to the site of infection, thus amplifying the inflammatory response. Since DCs are among the first immune cells to recognise and respond to infection with genital tract pathogens, they are likely to contribute substantially to genital tract inflammation.
Sexually transmitted infections (STIs) are recognized as major causes of inflammation in the genital mucosa. Neisseria gonorrhoeae, Chlamydia trachomatis and Mycoplasma genitalium have all been well described to cause inflammation, as well as clinically important sequelae such as pelvic inflammatory disease in women.86 Trichomonas vaginalis, and to a lesser extent bacterial vaginosis (not an STI but a perturbance of local microbial flora) have also been implicated in causing inflammation through several mechanisms including the disruption of the epithelial barrier.87 Ulcerative STIs comprising Herpes simplex virus (HSV), Haemophilus ducreyi (chancroid) and Treponema pallidum (syphilis) have all been characterized as pro-inflammatory sexually-acquired conditions that manifest themselves both systemically and in the genital tract.88–90 STI-causing pathogens are either extracellular or intracellular and activate DCs through binding to surface and intracellular PRRs such as TLRs, NLRs and CLRs that recognise PAMPs on these pathogens. DCs and other PRR-expressing cells in the genital tract, such as macrophages, neutrophils and epithelial cells,91 can become activated and produce inflammatory cytokines/ chemokines upon direct binding to these genital pathogens. Neisseria gonorrhoeae and Chlamydia trachomatis have both been shown to stimulate DCs and monocytes in vitro leading to secretion of pro-inflammatory cytokines.92, 93 Ulcerative STIs, by definition, are those that lead to breaches in the epithelial barrier. Trauma caused on genital epithelium during sexual activity, as well as vaginal practices such as douching, may also lead to inflammation.94, 95
HIV acquisition in the genital mucosa
The vaginal and ectocervical compartments are comprised of multi-layered, stratified epithelial cells lacking tight junctions, while the endocervix is protected by a polarised single layer of columnar epithelial cells separated by tight junctions.17 HIV enters the lower female genital tract mostly through the vagina and ectocervix, which represent an extensive surface area when compared with the endocervix.17
Inflammation causes the migration of HIV target cells, including CD4+ DCs, macrophages and T cells, as well as other immune cells such as neutrophils, CD8+ T cells and natural killer (NK) cells to the epithelium as a natural process to mediate host defence.7 In the rabbit vaginal irritation model, vaginal inflammation was shown to enhance trafficking of immune cells to the mucosa and enhance activation of these cells, both of which have also been associated with increased risk for HIV acquisition and transcription in infected cells.96 The presence of microbes such as STI-causing pathogens in the genital mucosa has been associated with an increased risk of HIV-1 transmission in several epidemiologic studies.97,98
DCs play a dual role in determining the outcome of HIV infection
DCs can facilitate HIV transmission by getting infected by HIV directly, and then transferring the virus to CD4+ T cells. Alternatively, DCs could indirectly facilitate infection of other target cells by producing cytokines that enhance the risk of infection, by either recruiting more potential target cells (increasing target cell density) or activating target cells so they are easier to infect. The fate of HIV captured by DCs may depend on the activation state of these cells or on binding receptors. The expression of CCR5 and langerin on LCs enables these cells to capture, become infected by, or disseminate HIV.99 In vitro experiments using skin explants have shown that LCs are mostly susceptible to R5-tropic viruses.100 CXCR4 expression on LCs is still controversial. This may be due to differences in sites where these cells were isolated. While expression of CXCR4 on genital tract LCs has been reported by some,101 others have shown that CXCR4 is not expressed on these cells in the skin.102 Others were able to induce an increase in CXCR4 expression on epithelial LCs after culture with granulocyte-macrophage colony-stimulating factor (GM-CSF).103 Activation of LCs in vitro downregulates langerin expression which makes these cells more susceptible to infection.58 In a human skin explant model, TNF-α and PAM3, a ligand for TLR2/1, have been reported to increase HIV capture by LCs, possibly in a langerin-independent manner, and increase transmission.104 HSV-2 has been shown to decrease langerin expression on LCs or competed with HIV for binding to langerin thereby increasing the risk of HIV transmission.105 LCs can also play a role in inhibiting HIV transmission. HIV binding by langerin rapidly internalises the virus to Birbeck granules leading to virus degradation. When LCs capture or internalise HIV, they rapidly migrate from the epithelium to the sub-mucosal layer where they can transfer the virus to CD4+ T cells. They can also migrate to lymph nodes where they can present HIV-derived antigens to CD4+ or CD8+ T cells generating an adaptive immune response which leads to the destruction of HIV-1 infected cells.106 HIV-infected CD4+ T cells in the mucosa can also traffic to the lymph nodes where more HIV replication occurs resulting in more infected target cells. Thus, LCs can either inhibit or promote HIV transmission, depending on their activation status and expression of langerin.
Whereas LCs reside in the genital mucosa where they mediate initial contact with HIV, mDC and pDC are predominantly found in blood and can be recruited to the tissues in response to inflammatory signals. Several studies in animal models have reported that pDC are rapidly recruited to inflamed tissues such as genital mucosa via high endothelial venules (HEV), where they recognize HIV, secrete IFN-α or take up antigen and migrate to draining lymph nodes for antigen presentation.107, 108 HIV-1 can infect pDC directly via receptors such as CCR5, CXCR4 and CD4, resulting in IFN-α production. 109, 110, 111, 112 Alternatively, HIV may bind and enter mDC by endocytosis mediated by DC-SIGN, CCR5 and CXCR4, with CD4 as co-receptor.113, 114 While blood mDC and pDC have been reported to be more frequently infected with HIV compared with monocytes and resting CD4+ T cells in vitro ,115 other studies reported that these cells are only rarely infected with HIV-1 and are not activated by HIV to produce cytokines or mature for antigen presentation.110, 116 The minimal infection of mDC in these studies was attributed to high expression of a restriction factor SAMHD1, which is a deoxynucleoside triphosphate triphosphohydrolase that prevents reverse transcription of HIV RNA.117 The lack of mDC maturation following binding to HIV may also be due to concurrent binding to DC-SIGN, which is known to inhibit TLR stimulation.118, 119 However, mDC are known to transfer HIV to CD4+ T cells in the absence of productive infection or become dysfunctional during HIV infection.39, 120
Immature DCs in tissues can more efficiently capture and internalise the virus when compared to mature DCs.121 Virus captured by mature DCs or retained for longer periods by maturing DCs can efficiently be transferred to CD4+ T cells through trans-infection. DCs bind and internalise HIV through CCR5, CXCR4, langerin or DC-SIGN (Fig. 1). The binding of HIV to one or more of these receptors may result in different outcomes. Binding of HIV-1 gp120 to DC-SIGN leads to the internalisation of the virus and transfer to the early endosomal compartment.122 In this compartment, if not degraded, the virus can be transported to the sub-epithelial mucosa or lymph nodes where T cells may get trans-infected through interaction with DCs. Transfer of virus from DCs to T cells can also occur independently of the DC-SIGN.123 Once HIV is internalised by DCs (Fig. 1), PRRs such as TLR7 and TLR9, expressed intracellularly, recognise the presence of HIV and initiate an antiviral immune response including the secretion of IFN-α and other inflammatory cytokines by pDCs, and DC maturation.
Figure 1.
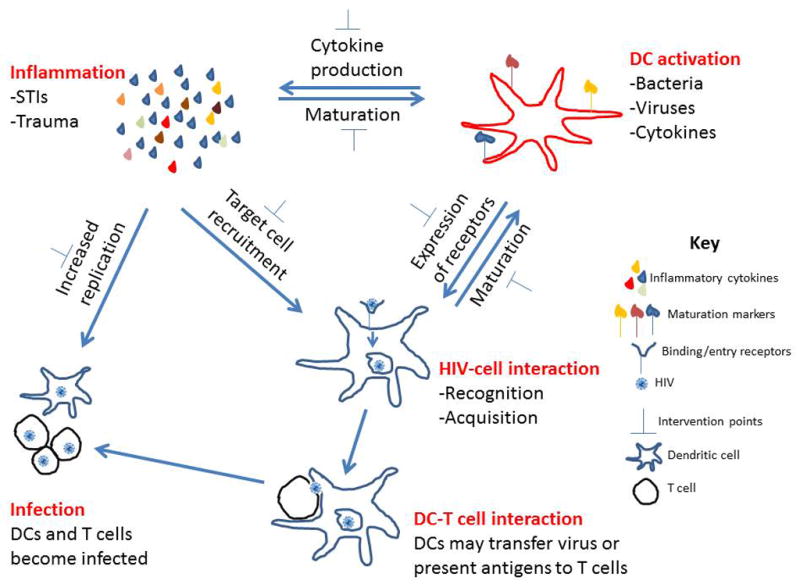
Relationship between inflammation, DC activation and HIV infection. Inflammation and DC activation can increase the risk of HIV transmission, while HIV acquisition by DC can also lead to DC activation and inflammation. DCs acquire HIV and can either become infected or transfer the virus to T cells, which become infected. Inflammation can enhance HIV replication in infected cells. Anti-inflammatory mediators can be used to reduce inflammation and DC activation, which lead to a decrease in risk of HIV transmission.
Depending on the cytokines produced by DCs, and the type of cells these cytokines recruit to the genital mucosa in the presence of HIV and/or STIs, these cytokines can either be beneficial (i.e. prevent) or detrimental to (i.e. promote) HIV transmission. The production of IFN-α and IL-12 pDC and/or mDC may result in the activation of T cells and killing of virus-infected cells as has been shown in vitro,124 while other cytokines such as TNF-α and IL-8 can lead to an increased replication of HIV in infected cells, and increased recruitment of target cells to the site of infection, thereby increasing the pool of susceptible target cells for newly budding HIV virions in human skin or explant cells.104, 125 STIs are known to form a lethal synergy with HIV, where HIV can exploit the inflammation caused by STIs to propagate itself and establish infection. Thus, inflammation in the genital tract resulting from recognition of pathogens is a double-edged sword that needs to be tightly regulated. While genital inflammation may increase risk for HIV infection, the process of inflammation is necessary in the control of infection with other mucosal pathogens such as tuberculosis and HPV.126, 127 The relative contribution of each leukocyte to the general inflammation in the genital tract is unclear and is an important area for future studies. Current evidence suggests that genital tract inflammation may be more of a “foe” than a “friend” in the context of HIV transmission. A fine balance between the level of inflammation that is beneficial or detrimental to host immunity against pathogens may need to be established.
The role of sex hormones in DC activation and HIV acquisition in women
Gender differences in risk for HIV infection and pathogenesis following infection may be attributed to several factors, including sex hormones. Differences in HIV shedding in genital secretions and transcription of HIV genes have been associated with the menstrual cycle in women and levels of progesterone.128, 129 Hormones may have both protective and enhancing roles in HIV infection and this may be related to changes in immune cell function as well as thickness of the epithelial lining. In macaques, administration of estriol prior to SIV challenge was shown to thicken the vaginal epithelial resulting in protection against SIV infection.130 Estrogen has also been shown to directly reduce the susceptibility of CD4+ T cells and macrophages to HIV infection in vitro through binding to estrogen receptors and altering HIV entry into these cells.131 Progesterone containing Depot medroxyprogesterone acetate (DMPA) is a commonly used injectable contraceptive in high HIV prevalence regions, particularly in Sub-Saharan Africa. This hormone was shown to significantly reduce the production of inflammatory cytokines by peripheral blood DCs in response to TLR stimulation, as well as prevented down-regulation of CCR5 and CXCR4 on activated T cells and increased HIV transcription in in vitro cell cultures.132 Women using this hormone also expressed lower cytokine levels in blood and genital secretions compared with non-users.132 Hormonal contraceptive use was associated with increased risk of HIV transmission in younger women compared with older women, possibly due to increased cervical ectopy in younger women.133, 134 Hormonal contraceptive use and fluctuations in hormonal levels during menstrual cycle lead to changes in female genital tract immunology and epithelial thickness, which may have serious consequences on risk of HIV transmission.
Reducing inflammation to prevent HIV transmission
If genital inflammation is associated with increased risk of HIV transmission, strategies to safely regulate inflammation may be able to curb the spread of HIV. These strategies could involve preventing, identifying and treating the causes of genital tract inflammation, inducing more natural anti-inflammatory mediators, or using exogenous anti-inflammatory drugs. Since one of the major causes of genital tract inflammation are STIs and most of them are treatable, active diagnosis and treatment of individuals with STIs could have a profound impact on HIV transmission. Despite this, population-wide approaches to STI prevention and treatment have demonstrated mixed efficacy in achieving this goal and have proven to be operationally difficult to implement effectively.135–141 HSV is possibly the exception, as it infects a wide range of cells including leukocytes, epithelial cells and nerve cells. The virus can remain latent in nerve cells until getting reactivated when the immune system is suppressed.142 It has been reported that even after treatment of HSV lesions and healing, HSV-induced inflammation still persists in the genital tract for up to 20 weeks.143 A vaccine to prevent HSV acquisition in adolescents could have a huge impact on preventing subsequent rates of HIV infection.
Natural anti-inflammatory mediators that balance the potential harm caused by inflammation include soluble tumour necrosis factor receptors (sTNF-Rs), IL-1 receptor antagonist (IL-1Ra), IL-10, and regulatory T cells (Tregs).144 Among the anti-inflammatory regulatory molecules, the role of the IL-10 family of cytokines in human immunology and in the response to viral and bacterial pathogens has been well described.145 IL-10 is secreted by leukocytes including macrophages, monocytes, DCs and T cells.144 Innate cells secrete IL-10 in response to TLR stimulation and during clearance of apoptotic cells, while T cells secrete IL-10 in response to T cell receptor triggering.146, 147 IL-10 functions by binding to the IL-10 receptors (IL-10R1 and IL-10R2) and uses the Janus Kinase family members and Signal Transducers and Activators of Transcription (STAT) transcription factors to mediate its effects.148 The main inhibitory effects of IL-10 are exerted on monocytes/macrophages, by inhibiting the production of pro-inflammatory cytokines and downregulating the expression of MHC II and CD86 in response to LPS or IFN-γ stimulation.149–151 We have also observed that IL-10 inhibits IL-6 and CD40 expression by monocytes and DCs in response to TLR7/8 and TLR4 ligands. 152 IL-10 also inhibits T cell proliferation and cytokine production by both Th1 and Th2 cells.153, 154 The inhibitory effect of IL-10 on chlamydia-induced cytokine production by human epithelial cells has also been described.155 In the context of antiviral responses, IL-10 decreases the expression of MHC I on DCs, and increases deletion of mature DCs by NK cells.156 Culturing mature DCs with HIV resulted in a significant increase in IL-10 production leading to a decrease in expression of CD83, HLA-DR and HLA class I, while the opposite effect was observed when immature DCs were co-cultured with HIV.156 On the other hand, IL-10 enhances the expression of other regulatory molecules such as IL-1Ra and sTNF-R.157, 158
IL1Ra and sTNFR both directly inhibit the stimulatory effects of IL-1 and TNF-α via receptor competition. IL-1Ra is a member of the IL-1 family and is a naturally occurring anti-inflammatory protein, which competitively blocks the binding of IL-1 to type I and II IL-1 receptors without inducing any signalling activity or intracellular response.159 The action of TNF-α is mediated by two receptors (TNF-R1 and TNF-R2).160 sTNF-Rs serve as antagonists of cell surface receptors for TNF-α and have a dual role in the control of inflammation in this environment. Binding of sTNF-Rs to TNF-α can either reduce the “toxic” effects of TNF-α or serve as a carrier and stabiliser of TNF-α. Thus, natural immune regulatory molecules are necessary and present to control immune activation and inflammation.
The female genital tract is in constant contact with both commensal and introduced microbes, most of which do not elicit an active immune response. Because of the commensals and its primary role in fertility, this mucosal environment is immune privileged (tolerogenic), particularly with respect to T cell populations. Tregs expressing FoxP3 and CD25 are a subset of T cells that form part of the natural anti-inflammatory mechanisms and can actively suppress both cellular activation and inflammation via direct and indirect mechanisms. Tregs actively suppress the activation or persistence of an immune response, and prevent pathological self-reactivity that leads to autoimmune disease, by mechanisms that include IL-10 and TGF-β secretion.161–165 While this subset has not been well-characterized in the female genital tract of humans, this population is common at other mucosal surfaces such as the gut .166 Treg development is opposed to that of another inflammatory Th subset called Th17 cells. Both Treg and Th17 subsets are required for effective host immunity, and their relative balance is likely to influence levels of inflammation. Both of these T cell subsets are also targets of HIV.167 In addition to Tregs, pathogen-specific T cells can upregulate molecules with regulatory functions, such as IL-10 and CTLA-4, limiting their inflammatory potential. Therefore, the relevance of the Th subset balance in HIV exposed mucosa includes their influence on the inflammatory status of the mucosal environment, and their presence as HIV targets. HIV infection is known to impair the capacity of DCs to induce Tregs by limiting CD25 and FOXP3 expression in a caspase-dependent manner, possibly due to preferential killing of Tregs by HIV-infected DC.168 Tregs can also down-modulate the capacity of DCs to activate effector T cells through IL-10, and inhibition of the expression of co-stimulatory molecules on DCs.169–171 In combination with vaccines and microbicides to prevent infection, immune modulatory strategies that induce Tregs that dampen inflammation may have beneficial effects by limiting HIV transmission.
Natural anti-inflammatory mediators are ultimately unable to sufficiently control high genital tract inflammation in the presence of STIs or other inflammatory conditions to reduce HIV transmission. Addition of exogenous anti-inflammatory mediators is an intervention that may further augment the body’s natural defences. Anti-inflammatory medication, including corticosteroids and non-steroidal anti-inflammatory drugs (NSAIDs), have been licenced for use in humans to treat various inflammatory conditions,172, 173 and may possibly have a role to play in reducing genital tract inflammation, and HIV transmission. Monoclonal antibodies have also been used to limit inflammation especially in autoimmune conditions, for example, the use of Infliximab and other anti-TNF monoclonal antibodies have clinical benefit in Crohn’s disease.174–176 Although these strategies to dampen inflammation by TNF are useful to reduce symptoms of autoimmunity, it should be noted that important side effects have been described for anti-TNF treatment, which include reactivation of latent tuberculosis, pneumonia, meningitis and sepsis among others.177
Anti-inflammatory mediators can exert the following effects: (i) a decrease in pro-inflammatory cytokine production, (ii) a decrease in the activation of DCs and T cells, and (iii) a reduction in the expression of some binding/entry receptors (Fig. 1). Decreases in inflammation, receptor expression and activation of DCs and other target cells can have profound effects on HIV transmission in the female genital tract.98 Animal models have shown that Glycerol monolaurate (GML), a compound with anti-inflammatory properties, can inhibit simian immunodeficiency virus (SIV) transmission in the genital tract.178 GML exhibits its anti-inflammatory effect by inhibiting immune activation and cytokine/chemokine production such as IL-8 and MIP-3α, thereby reducing DC recruitment to the genital tract.178–180 Because of these anti-inflammatory properties, GML was evaluated for its ability to prevent SIV acquisition in the genital tract of rhesus macaques. All GML-exposed animals were completely protected from repeated high dose SIV challenges, compared to no protection in the untreated control animals.178 These experiments support a model in which the initial inflammatory cascade forms a central feature of subsequent risk of HIV acquisition. The use of anti-inflammatory agents in HIV control in humans should therefore be investigated further.
Conclusion and future perspectives
DC activation results in cytokine production and inflammation, and inflammation can reciprocally fuel DC activation. Both immune activation and inflammation are risk factors for HIV transmission. PRRs and chemokine receptors are centrally involved in the process of HIV recognition and acquisition. Inflammation that results from the presence of HIV and other pathogens in the genital mucosa can be regulated by various anti-inflammatory mediators – whether they are induced naturally or exogenous. Until recently, the use of microbicides to prevent HIV transmission has been largely ineffective. Some of the earlier microbicides, such as nonoxynol-9 (N-9) and cellulose sulphate, increased risk of HIV acquisition by causing genital inflammation or micro-abrasions.181, 182 The new generation microbicides, which include potent anti-retroviral drugs such as 1% tenofovir topical gel showed moderate but encouraging success in reducing risk of HIV acquisition, although efficacy of this approach has been shown to be influenced by pre-existing genital inflammation.183 Women in this 1% tenofovir gel trial with pre-existing genital inflammation were at increased risk of HIV acquisition, irrespective of whether they were using the tenofovir gel or placebo.184 Strategies to prevent HIV infection at the genital mucosa (such as vaccines or microbicides) may be substantially improved on safely limiting inflammation at the genital mucosa, thereby reducing the risk of HIV transmission.
In summary, DC activation, inflammation, and HIV transmission are interrelated. Whereas cell activation and inflammation may be beneficial in controlling other pathogens, these host responses inadvertently increase the risk of HIV transmission by increasing the availability of HIV target cells in the female genital tract. Identifying and treating causes of genital tract inflammation by adding anti-inflammatory agents or mediators to the female genital tract could make an important contribution to controlling HIV transmission. The counter side to this approach is that inflammation in the genital mucosa serves an important function in protection against other pathogens. More effective strategies to control HIV transmission in the female genital tract will entail a comprehensive understanding of factors – cell activation and inflammation in particular – that lead to sub-optimal efficacy of anti-HIV microbicides or vaccines.
Acknowledgments
MSS is supported by the Columbia University-Southern African Fogarty International Training and Research Program (AITRP) funded by the Fogarty International Center, National Institute of Health (grant # D43TW00231).
Footnotes
Conflict of interest: The authors declare that there is no conflict of interest.
References
- 1.UNAIDS. UNAIDS report on the global AIDS epidemic: "Together we will end AIDS". WHO Library; 2012. [Google Scholar]
- 2.Shattock RJ, Moore JP. Inhibiting sexual transmission of HIV-1 infection. Nature reviews Microbiology. 2003;1:25–34. doi: 10.1038/nrmicro729. [DOI] [PubMed] [Google Scholar]
- 3.Comparison of female to male and male to female transmission of HIV in 563 stable couples. European Study Group on Heterosexual Transmission of HIV. . Bmj. 1992;304:809–13. doi: 10.1136/bmj.304.6830.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boily MC, Baggaley RF, Wang L, et al. Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies. The Lancet infectious diseases. 2009;9:118–29. doi: 10.1016/S1473-3099(09)70021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Padian NS, Shiboski SC, Glass SO, Vittinghoff E. Heterosexual transmission of human immunodeficiency virus (HIV) in northern California: results from a ten-year study. American journal of epidemiology. 1997;146:350–7. doi: 10.1093/oxfordjournals.aje.a009276. [DOI] [PubMed] [Google Scholar]
- 6.Royce RA, Sena A, Cates W, Jr, Cohen MS. Sexual transmission of HIV. The New England journal of medicine. 1997;336:1072–8. doi: 10.1056/NEJM199704103361507. [DOI] [PubMed] [Google Scholar]
- 7.Hladik F, Hope TJ. HIV infection of the genital mucosa in women. Current HIV/AIDS reports. 2009;6:20–8. doi: 10.1007/s11904-009-0004-1. [DOI] [PubMed] [Google Scholar]
- 8.Pendergrass PB, Belovicz MW, Reeves CA. Surface area of the human vagina as measured from vinyl polysiloxane casts. Gynecologic and obstetric investigation. 2003;55:110–3. doi: 10.1159/000070184. [DOI] [PubMed] [Google Scholar]
- 9.Carreno MP, Chomont N, Kazatchkine MD, et al. Binding of LFA-1 (CD11a) to intercellular adhesion molecule 3 (ICAM-3; CD50) and ICAM-2 (CD102) triggers transmigration of human immunodeficiency virus type 1-infected monocytes through mucosal epithelial cells. Journal of virology. 2002;76:32–40. doi: 10.1128/JVI.76.1.32-40.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoddard E, Ni H, Cannon G, et al. gp340 promotes transcytosis of human immunodeficiency virus type 1 in genital tract-derived cell lines and primary endocervical tissue. Journal of virology. 2009;83:8596–603. [Google Scholar]
- 11.Shen R, Drelichman ER, Bimczok D, et al. GP41-specific antibody blocks cell-free HIV type 1 transcytosis through human rectal mucosa and model colonic epithelium. Journal of immunology. 2010;184:3648–55. doi: 10.4049/jimmunol.0903346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bobardt MD, Chatterji U, Selvarajah S, et al. Cell-free human immunodeficiency virus type 1 transcytosis through primary genital epithelial cells. Journal of virology. 2007;81:395–405. doi: 10.1128/JVI.01303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bomsel M. Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nature medicine. 1997;3:42–7. doi: 10.1038/nm0197-42. [DOI] [PubMed] [Google Scholar]
- 14.Maher D, Wu X, Schacker T, Horbul J, Southern P. HIV binding, penetration, and primary infection in human cervicovaginal tissue. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11504–9. doi: 10.1073/pnas.0500848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller CJ, Li Q, Abel K, et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. Journal of virology. 2005;79:9217–27. doi: 10.1128/JVI.79.14.9217-9227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavrois M, Neidleman J, Kreisberg JF, Greene WC. In vitro derived dendritic cells trans-infect CD4 T cells primarily with surface-bound HIV-1 virions. PLoS pathogens. 2007;3:e4. doi: 10.1371/journal.ppat.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen R, Richter HE, Smith PD. Early HIV-1 target cells in human vaginal and ectocervical mucosa. American journal of reproductive immunology. 2011;65:261–7. doi: 10.1111/j.1600-0897.2010.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Streeck H, Nixon DF. T cell immunity in acute HIV-1 infection. The Journal of infectious diseases. 2010;202 (Suppl 2):S302–8. doi: 10.1086/655652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. The Journal of experimental medicine. 2012;209:653–60. doi: 10.1084/jem.20111457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palucka K, Banchereau J, Mellman I. Designing vaccines based on biology of human dendritic cell subsets. Immunity. 2010;33:464–78. doi: 10.1016/j.immuni.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 22.Steinman RM. Dendritic cells and the control of immunity: enhancing the efficiency of antigen presentation. The Mount Sinai journal of medicine, New York. 2001;68:160–6. [PubMed] [Google Scholar]
- 23.Kavanagh DG, Bhardwaj N. A division of labor: DC subsets and HIV receptor diversity. Nature immunology. 2002;3:891–3. doi: 10.1038/ni1002-891. [DOI] [PubMed] [Google Scholar]
- 24.Kollmann TR, Crabtree J, Rein-Weston A, et al. Neonatal innate TLR-mediated responses are distinct from those of adults. Journal of immunology. 2009;183:7150–60. doi: 10.4049/jimmunol.0901481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. International immunology. 2009;21:317–37. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo EK. Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Current opinion in infectious diseases. 2008;21:279–86. doi: 10.1097/QCO.0b013e3282f88b5d. [DOI] [PubMed] [Google Scholar]
- 27.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of leukocyte biology. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 28.Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochemical and biophysical research communications. 2009;388:621–5. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 29.Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. The Biochemical journal. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 30.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 31.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–50. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Ting JP, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–7. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye Z, Ting JP. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Current opinion in immunology. 2008;20:3–9. doi: 10.1016/j.coi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Moreira LO, Zamboni DS. NOD1 and NOD2 Signaling in Infection and Inflammation. Frontiers in immunology. 2012;3:328. doi: 10.3389/fimmu.2012.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horuk R. Chemokine receptors. Cytokine & growth factor reviews. 2001;12:313–35. doi: 10.1016/s1359-6101(01)00014-4. [DOI] [PubMed] [Google Scholar]
- 36.D'Souza MP, Harden VA. Chemokines and HIV-1 second receptors. Confluence of two fields generates optimism in AIDS research. Nature medicine. 1996;2:1293–300. doi: 10.1038/nm1296-1293. [DOI] [PubMed] [Google Scholar]
- 37.MacDonald KP, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DN. Characterization of human blood dendritic cell subsets. Blood. 2002;100:4512–20. doi: 10.1182/blood-2001-11-0097. [DOI] [PubMed] [Google Scholar]
- 38.Randolph GJ, Ochando J, Partida-Sanchez S. Migration of dendritic cell subsets and their precursors. Annual review of immunology. 2008;26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254. [DOI] [PubMed] [Google Scholar]
- 39.Lore K, Smed-Sorensen A, Vasudevan J, Mascola JR, Koup RA. Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. The Journal of experimental medicine. 2005;201:2023–33. doi: 10.1084/jem.20042413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dzionek A, Fuchs A, Schmidt P, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. Journal of immunology. 2000;165:6037–46. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 41.Ebner S, Hofer S, Nguyen VA, et al. A novel role for IL-3: human monocytes cultured in the presence of IL-3 and IL-4 differentiate into dendritic cells that produce less IL-12 and shift Th cell responses toward a Th2 cytokine pattern. Journal of immunology. 2002;168:6199–207. doi: 10.4049/jimmunol.168.12.6199. [DOI] [PubMed] [Google Scholar]
- 42.Hata M, Takahara S, Tsuzaki H, et al. Expression of Th2-skewed pathology mediators in monocyte-derived type 2 of dendritic cells (DC2) Immunology letters. 2009;126:29–36. doi: 10.1016/j.imlet.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi Y, Ishii Y, Hata-Suzuki M, et al. Comparative analysis of circulating dendritic cell subsets in patients with atopic diseases and sarcoidosis. Respiratory research. 2013;14:29. doi: 10.1186/1465-9921-14-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. The Journal of experimental medicine. 1997;185:1101–11. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Pucchio T, Chatterjee B, Smed-Sorensen A, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nature immunology. 2008;9:551–7. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw J, Wang YH, Ito T, Arima K, Liu YJ. Plasmacytoid dendritic cells regulate B-cell growth and differentiation via CD70. Blood. 2010;115:3051–7. doi: 10.1182/blood-2009-08-239145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 48.McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. Journal of virology. 2005;79:17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernandez JC, Arteaga J, Paul S, Kumar A, Latz E, Urcuqui-Inchima S. Up-regulation of TLR2 and TLR4 in dendritic cells in response to HIV type 1 and coinfection with opportunistic pathogens. AIDS research and human retroviruses. 2011;27:1099–109. doi: 10.1089/aid.2010.0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. Journal of immunology. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 51.van de Ven R, van den Hout MF, Lindenberg JJ, et al. Characterization of four conventional dendritic cell subsets in human skin-draining lymph nodes in relation to T-cell activation. Blood. 2011;118:2502–10. doi: 10.1182/blood-2011-03-344838. [DOI] [PubMed] [Google Scholar]
- 52.Katz SI, Tamaki K, Sachs DH. Epidermal Langerhans cells are derived from cells originating in bone marrow. Nature. 1979;282:324–6. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 53.Mende I, Karsunky H, Weissman IL, Engleman EG, Merad M. Flk2+ myeloid progenitors are the main source of Langerhans cells. Blood. 2006;107:1383–90. doi: 10.1182/blood-2005-05-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnston LJ, Halliday GM, King NJ. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. The Journal of investigative dermatology. 2000;114:560–8. doi: 10.1046/j.1523-1747.2000.00904.x. [DOI] [PubMed] [Google Scholar]
- 55.Kissenpfennig A, Henri S, Dubois B, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–54. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 56.van der Aar AM, Sylva-Steenland RM, Bos JD, Kapsenberg ML, de Jong EC, Teunissen MB. Loss of TLR2, TLR4, and TLR5 on Langerhans cells abolishes bacterial recognition. Journal of immunology. 2007;178:1986–90. doi: 10.4049/jimmunol.178.4.1986. [DOI] [PubMed] [Google Scholar]
- 57.Flacher V, Bouschbacher M, Verronese E, et al. Human Langerhans cells express a specific TLR profile and differentially respond to viruses and Gram-positive bacteria. Journal of immunology. 2006;177:7959–67. doi: 10.4049/jimmunol.177.11.7959. [DOI] [PubMed] [Google Scholar]
- 58.Valladeau J, Ravel O, Dezutter-Dambuyant C, et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12:71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- 59.Mc Dermott R, Ziylan U, Spehner D, et al. Birbeck granules are subdomains of endosomal recycling compartment in human epidermal Langerhans cells, which form where Langerin accumulates. Molecular biology of the cell. 2002;13:317–35. doi: 10.1091/mbc.01-06-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 61.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nature immunology. 2001;2:835–41. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 62.Weighardt H, Jusek G, Mages J, et al. Identification of a TLR4- and TRIF-dependent activation program of dendritic cells. European journal of immunology. 2004;34:558–64. doi: 10.1002/eji.200324714. [DOI] [PubMed] [Google Scholar]
- 63.Bertin J, Guo Y, Wang L, et al. CARD9 is a novel caspase recruitment domain-containing protein that interacts with BCL10/CLAP and activates NF-kappa B. The Journal of biological chemistry. 2000;275:41082–6. doi: 10.1074/jbc.C000726200. [DOI] [PubMed] [Google Scholar]
- 64.Takaoka A, Yanai H, Kondo S, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–9. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 65.Liu W, Ouyang X, Yang J, et al. AP-1 activated by toll-like receptors regulates expression of IL-23 p19. The Journal of biological chemistry. 2009;284:24006–16. doi: 10.1074/jbc.M109.025528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peroval MY, Boyd AC, Young JR, Smith AL. A critical role for MAPK signalling pathways in the transcriptional regulation of toll like receptors. PloS one. 2013;8:e51243. doi: 10.1371/journal.pone.0051243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van der Meide PH, Schellekens H. Cytokines and the immune response. Biotherapy. 1996;8:243–9. doi: 10.1007/BF01877210. [DOI] [PubMed] [Google Scholar]
- 68.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine & growth factor reviews. 2002;13:413–21. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 69.Swain SL. T-cell subsets. Who does the polarizing? Current biology : CB. 1995;5:849–51. doi: 10.1016/s0960-9822(95)00170-9. [DOI] [PubMed] [Google Scholar]
- 70.Mikhalkevich N, Becknell B, Caligiuri MA, Bates MD, Harvey R, Zheng WP. Responsiveness of naive CD4 T cells to polarizing cytokine determines the ratio of Th1 and Th2 cell differentiation. Journal of immunology. 2006;176:1553–60. doi: 10.4049/jimmunol.176.3.1553. [DOI] [PubMed] [Google Scholar]
- 71.Dorhoi A, Reece ST, Kaufmann SH. For better or for worse: the immune response against Mycobacterium tuberculosis balances pathology and protection. Immunological reviews. 2011;240:235–51. doi: 10.1111/j.1600-065X.2010.00994.x. [DOI] [PubMed] [Google Scholar]
- 72.Abdul-Sater AA, Said-Sadier N, Ojcius DM, Yilmaz O, Kelly KA. Inflammasomes bridge signaling between pathogen identification and the immune response. Drugs of today. 2009;45 (Suppl B):105–12. [PMC free article] [PubMed] [Google Scholar]
- 73.Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 74.Mackay CR. Chemokines: immunology's high impact factors. Nature immunology. 2001;2:95–101. doi: 10.1038/84298. [DOI] [PubMed] [Google Scholar]
- 75.Lebre MC, Burwell T, Vieira PL, et al. Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: a role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunology and cell biology. 2005;83:525–35. doi: 10.1111/j.1440-1711.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 76.Luther SA, Bidgol A, Hargreaves DC, et al. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. Journal of immunology. 2002;169:424–33. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 77.Mueller SN, Hosiawa-Meagher KA, Konieczny BT, et al. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science. 2007;317:670–4. doi: 10.1126/science.1144830. [DOI] [PubMed] [Google Scholar]
- 78.Moser B, Willimann K. Chemokines: role in inflammation and immune surveillance. Annals of the rheumatic diseases. 2004;63(Suppl 2):ii84–ii9. doi: 10.1136/ard.2004.028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 2000;95:3032–43. [PubMed] [Google Scholar]
- 80.Ellis JH, Burden MN, Vinogradov DV, Linge C, Crowe JS. Interactions of CD80 and CD86 with CD28 and CTLA4. Journal of immunology. 1996;156:2700–9. [PubMed] [Google Scholar]
- 81.Kruse M, Rosorius O, Kratzer F, et al. Inhibition of CD83 cell surface expression during dendritic cell maturation by interference with nuclear export of CD83 mRNA. The Journal of experimental medicine. 2000;191:1581–90. doi: 10.1084/jem.191.9.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miga AJ, Masters SR, Durell BG, et al. Dendritic cell longevity and T cell persistence is controlled by CD154-CD40 interactions. European journal of immunology. 2001;31:959–65. doi: 10.1002/1521-4141(200103)31:3<959::aid-immu959>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 83.Hernandez MG, Shen L, Rock KL. CD40-CD40 ligand interaction between dendritic cells and CD8+ T cells is needed to stimulate maximal T cell responses in the absence of CD4+ T cell help. Journal of immunology. 2007;178:2844–52. doi: 10.4049/jimmunol.178.5.2844. [DOI] [PubMed] [Google Scholar]
- 84.Yasumi T, Katamura K, Yoshioka T, et al. Differential requirement for the CD40-CD154 costimulatory pathway during Th cell priming by CD8 alpha+ and CD8 alpha- murine dendritic cell subsets. Journal of immunology. 2004;172:4826–33. doi: 10.4049/jimmunol.172.8.4826. [DOI] [PubMed] [Google Scholar]
- 85.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 86.McGowin CL, Radtke AL, Abraham K, Martin DH, Herbst-Kralovetz M. Mycoplasma genitalium Infection Activates Cellular Host Defense and Inflammation Pathways in a 3-Dimensional Human Endocervical Epithelial Cell Model. The Journal of infectious diseases. 2013;207:1857–68. doi: 10.1093/infdis/jit101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thurman AR, Doncel GF. Innate immunity and inflammatory response to Trichomonas vaginalis and bacterial vaginosis: relationship to HIV acquisition. American journal of reproductive immunology. 2011;65:89–98. doi: 10.1111/j.1600-0897.2010.00902.x. [DOI] [PubMed] [Google Scholar]
- 88.Koelle DM, Posavad CM, Barnum GR, Johnson ML, Frank JM, Corey L. Clearance of HSV-2 from recurrent genital lesions correlates with infiltration of HSV-specific cytotoxic T lymphocytes. The Journal of clinical investigation. 1998;101:1500–8. doi: 10.1172/JCI1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sellati TJ, Wilkinson DA, Sheffield JS, Koup RA, Radolf JD, Norgard MV. Virulent Treponema pallidum, lipoprotein, and synthetic lipopeptides induce CCR5 on human monocytes and enhance their susceptibility to infection by human immunodeficiency virus type 1. The Journal of infectious diseases. 2000;181:283–93. doi: 10.1086/315209. [DOI] [PubMed] [Google Scholar]
- 90.Hobbs MM, Paul TR, Wyrick PB, Kawula TH. Haemophilus ducreyi infection causes basal keratinocyte cytotoxicity and elicits a unique cytokine induction pattern in an In vitro human skin model. Infection and immunity. 1998;66:2914–21. doi: 10.1128/iai.66.6.2914-2921.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of toll-like receptor 4-mediated signaling. Journal of immunology. 2002;168:2424–32. doi: 10.4049/jimmunol.168.5.2424. [DOI] [PubMed] [Google Scholar]
- 92.Gervassi A, Alderson MR, Suchland R, Maisonneuve JF, Grabstein KH, Probst P. Differential regulation of inflammatory cytokine secretion by human dendritic cells upon Chlamydia trachomatis infection. Infection and immunity. 2004;72:7231–9. doi: 10.1128/IAI.72.12.7231-7239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Vliet SJ, Steeghs L, Bruijns SC, et al. Variation of Neisseria gonorrhoeae lipooligosaccharide directs dendritic cell-induced T helper responses. PLoS pathogens. 2009;5:e1000625. doi: 10.1371/journal.ppat.1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weiler AM, Li Q, Duan L, et al. Genital ulcers facilitate rapid viral entry and dissemination following intravaginal inoculation with cell-associated simian immunodeficiency virus SIVmac239. Journal of virology. 2008;82:4154–8. doi: 10.1128/JVI.01947-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hladik F, McElrath MJ. Setting the stage: host invasion by HIV. Nature reviews Immunology. 2008;8:447–57. doi: 10.1038/nri2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trifonova RT, Bajpai M, Pasicznyk JM, Chandra N, Doncel GF, Fichorova RN. Biomarkers of leukocyte traffic and activation in the vaginal mucosa. Biomarkers : biochemical indicators of exposure, response, and susceptibility to chemicals. 2007;12:608–22. doi: 10.1080/13547500701600670. [DOI] [PubMed] [Google Scholar]
- 97.Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sexually transmitted infections. 1999;75:3–17. doi: 10.1136/sti.75.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Keller MJ, Herold BC. Impact of microbicides and sexually transmitted infections on mucosal immunity in the female genital tract. American journal of reproductive immunology. 2006;56:356–63. doi: 10.1111/j.1600-0897.2006.00436.x. [DOI] [PubMed] [Google Scholar]
- 99.de Witte L, Nabatov A, Pion M, et al. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nature medicine. 2007;13:367–71. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 100.Kawamura T, Kurtz SE, Blauvelt A, Shimada S. The role of Langerhans cells in the sexual transmission of HIV. Journal of dermatological science. 2005;40:147–55. doi: 10.1016/j.jdermsci.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 101.Prakash M, Kapembwa MS, Gotch F, Patterson S. Chemokine receptor expression on mucosal dendritic cells from the endocervix of healthy women. The Journal of infectious diseases. 2004;190:246–50. doi: 10.1086/422034. [DOI] [PubMed] [Google Scholar]
- 102.Zaitseva M, Blauvelt A, Lee S, et al. Expression and function of CCR5 and CXCR4 on human Langerhans cells and macrophages: implications for HIV primary infection. Nature medicine. 1997;3:1369–75. doi: 10.1038/nm1297-1369. [DOI] [PubMed] [Google Scholar]
- 103.Tchou I, Misery L, Sabido O, et al. Functional HIV CXCR4 coreceptor on human epithelial Langerhans cells and infection by HIV strain X4. Journal of leukocyte biology. 2001;70:313–21. [PubMed] [Google Scholar]
- 104.de Jong MA, de Witte L, Oudhoff MJ, Gringhuis SI, Gallay P, Geijtenbeek TB. TNF-alpha and TLR agonists increase susceptibility to HIV-1 transmission by human Langerhans cells ex vivo. The Journal of clinical investigation. 2008;118:3440–52. doi: 10.1172/JCI34721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Jong MA, de Witte L, Taylor ME, Geijtenbeek TB. Herpes simplex virus type 2 enhances HIV-1 susceptibility by affecting Langerhans cell function. Journal of immunology. 2010;185:1633–41. doi: 10.4049/jimmunol.0904137. [DOI] [PubMed] [Google Scholar]
- 106.Ballweber L, Robinson B, Kreger A, et al. Vaginal langerhans cells nonproductively transporting HIV-1 mediate infection of T cells. Journal of virology. 2011;85:13443–7. doi: 10.1128/JVI.05615-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moniz RJ, Chan AM, Kelly KA. Identification of dendritic cell subsets responding to genital infection by Chlamydia muridarum. FEMS immunology and medical microbiology. 2009;55:226–36. doi: 10.1111/j.1574-695X.2008.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kwa S, Kannanganat S, Nigam P, et al. Plasmacytoid dendritic cells are recruited to the colorectum and contribute to immune activation during pathogenic SIV infection in rhesus macaques. Blood. 2011;118:2763–73. doi: 10.1182/blood-2011-02-339515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fong L, Mengozzi M, Abbey NW, Herndier BG, Engleman EG. Productive infection of plasmacytoid dendritic cells with human immunodeficiency virus type 1 is triggered by CD40 ligation. Journal of virology. 2002;76:11033–41. doi: 10.1128/JVI.76.21.11033-11041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fonteneau JF, Larsson M, Beignon AS, et al. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. Journal of virology. 2004;78:5223–32. doi: 10.1128/JVI.78.10.5223-5232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beignon AS, McKenna K, Skoberne M, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. The Journal of clinical investigation. 2005;115:3265–75. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haupt S, Donhauser N, Chaipan C, et al. CD4 binding affinity determines human immunodeficiency virus type 1-induced alpha interferon production in plasmacytoid dendritic cells. Journal of virology. 2008;82:8900–5. doi: 10.1128/JVI.00196-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Geijtenbeek TB, Kwon DS, Torensma R, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–97. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 114.Geijtenbeek TB, van Kooyk Y. DC-SIGN: a novel HIV receptor on DCs that mediates HIV-1 transmission. Current topics in microbiology and immunology. 2003;276:31–54. doi: 10.1007/978-3-662-06508-2_2. [DOI] [PubMed] [Google Scholar]
- 115.Cameron PU, Handley AJ, Baylis DC, et al. Preferential infection of dendritic cells during human immunodeficiency virus type 1 infection of blood leukocytes. Journal of virology. 2007;81:2297–306. doi: 10.1128/JVI.01795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frank I, Kacani L, Stoiber H, et al. Human immunodeficiency virus type 1 derived from cocultures of immature dendritic cells with autologous T cells carries T-cell-specific molecules on its surface and is highly infectious. Journal of virology. 1999;73:3449–54. doi: 10.1128/jvi.73.4.3449-3454.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Laguette N, Sobhian B, Casartelli N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–7. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Geijtenbeek TB, van Kooyk Y. Pathogens target DC-SIGN to influence their fate DC-SIGN functions as a pathogen receptor with broad specificity. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2003;111:698–714. doi: 10.1034/j.1600-0463.2003.11107803.x. [DOI] [PubMed] [Google Scholar]
- 119.Geijtenbeek TB, Van Vliet SJ, Koppel EA, et al. Mycobacteria target DC-SIGN to suppress dendritic cell function. The Journal of experimental medicine. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sabado RL, O'Brien M, Subedi A, et al. Evidence of dysregulation of dendritic cells in primary HIV infection. Blood. 2010;116:3839–52. doi: 10.1182/blood-2010-03-273763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fahrbach KM, Barry SM, Ayehunie S, Lamore S, Klausner M, Hope TJ. Activated CD34-derived Langerhans cells mediate transinfection with human immunodeficiency virus. Journal of virology. 2007;81:6858–68. doi: 10.1128/JVI.02472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Altfeld M, Fadda L, Frleta D, Bhardwaj N. DCs and NK cells: critical effectors in the immune response to HIV-1. Nature reviews Immunology. 2011;11:176–86. doi: 10.1038/nri2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Boggiano C, Manel N, Littman DR. Dendritic cell-mediated trans-enhancement of human immunodeficiency virus type 1 infectivity is independent of DC-SIGN. Journal of virology. 2007;81:2519–23. doi: 10.1128/JVI.01661-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. Journal of immunology. 2004;172:61–9. doi: 10.4049/jimmunol.172.1.61. [DOI] [PubMed] [Google Scholar]
- 125.Narimatsu R, Wolday D, Patterson BK. IL-8 increases transmission of HIV type 1 in cervical explant tissue. AIDS research and human retroviruses. 2005;21:228–33. doi: 10.1089/aid.2005.21.228. [DOI] [PubMed] [Google Scholar]
- 126.Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal immunology. 2011;4:271–8. doi: 10.1038/mi.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stanley MA. Immune responses to human papilloma viruses. The Indian journal of medical research. 2009;130:266–76. [PubMed] [Google Scholar]
- 128.Baeten JM, Lavreys L, Overbaugh J. The influence of hormonal contraceptive use on HIV-1 transmission and disease progression. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2007;45:360–9. doi: 10.1086/519432. [DOI] [PubMed] [Google Scholar]
- 129.Reichelderfer PS, Coombs RW, Wright DJ, et al. Effect of menstrual cycle on HIV-1 levels in the peripheral blood and genital tract. WHS 001 Study Team. Aids. 2000;14:2101–7. doi: 10.1097/00002030-200009290-00005. [DOI] [PubMed] [Google Scholar]
- 130.Smith SM, Mefford M, Sodora D, et al. Topical estrogen protects against SIV vaginal transmission without evidence of systemic effect. Aids. 2004;18:1637–43. doi: 10.1097/01.aids.0000131393.76221.cc. [DOI] [PubMed] [Google Scholar]
- 131.Rodriguez-Garcia M, Biswas N, Patel MV, et al. Estradiol reduces susceptibility of CD4+ T cells and macrophages to HIV-infection. PloS one. 2013;8:e62069. doi: 10.1371/journal.pone.0062069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huijbregts RP, Helton ES, Michel KG, et al. Hormonal contraception and HIV-1 infection: medroxyprogesterone acetate suppresses innate and adaptive immune mechanisms. Endocrinology. 2013;154:1282–95. doi: 10.1210/en.2012-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kleinschmidt I, Rees H, Delany S, et al. Injectable progestin contraceptive use and risk of HIV infection in a South African family planning cohort. Contraception. 2007;75:461–7. doi: 10.1016/j.contraception.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 134.Morrison CS, Chen PL, Kwok C, et al. Hormonal contraception and HIV acquisition: reanalysis using marginal structural modeling. Aids. 2010;24:1778–81. doi: 10.1097/QAD.0b013e32833a2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Grosskurth H, Mosha F, Todd J, et al. Impact of improved treatment of sexually transmitted diseases on HIV infection in rural Tanzania: randomised controlled trial. Lancet. 1995;346:530–6. doi: 10.1016/s0140-6736(95)91380-7. [DOI] [PubMed] [Google Scholar]
- 136.Wawer MJ, Sewankambo NK, Serwadda D, et al. Control of sexually transmitted diseases for AIDS prevention in Uganda: a randomised community trial. Rakai Project Study Group. Lancet. 1999;353:525–35. doi: 10.1016/s0140-6736(98)06439-3. [DOI] [PubMed] [Google Scholar]
- 137.Ghys PD, Diallo MO, Ettiegne-Traore V, et al. Effect of interventions to control sexually transmitted disease on the incidence of HIV infection in female sex workers. Aids. 2001;15:1421–31. doi: 10.1097/00002030-200107270-00012. [DOI] [PubMed] [Google Scholar]
- 138.Kamali A, Quigley M, Nakiyingi J, et al. Syndromic management of sexually-transmitted infections and behaviour change interventions on transmission of HIV-1 in rural Uganda: a community randomised trial. Lancet. 2003;361:645–52. doi: 10.1016/s0140-6736(03)12598-6. [DOI] [PubMed] [Google Scholar]
- 139.Celum C, Wald A, Hughes J, et al. Effect of aciclovir on HIV-1 acquisition in herpes simplex virus 2 seropositive women and men who have sex with men: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:2109–19. doi: 10.1016/S0140-6736(08)60920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Watson-Jones D, Weiss HA, Rusizoka M, et al. Risk factors for herpes simplex virus type 2 and HIV among women at high risk in northwestern Tanzania: preparing for an HSV-2 intervention trial. Journal of acquired immune deficiency syndromes. 2007;46:631–42. doi: 10.1097/QAI.0b013e31815b2d9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gregson S, Adamson S, Papaya S, et al. Impact and process evaluation of integrated community and clinic-based HIV-1 control: a cluster-randomised trial in eastern Zimbabwe. PLoS medicine. 2007;4:e102. doi: 10.1371/journal.pmed.0040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kaushic C, Roth KL, Anipindi V, Xiu F. Increased prevalence of sexually transmitted viral infections in women: the role of female sex hormones in regulating susceptibility and immune responses. Journal of reproductive immunology. 2011;88:204–9. doi: 10.1016/j.jri.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 143.Zhu J, Hladik F, Woodward A, et al. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nature medicine. 2009;15:886–92. doi: 10.1038/nm.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sabat R. IL-10 family of cytokines. Cytokine & growth factor reviews. 2010;21:315–24. doi: 10.1016/j.cytogfr.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 145.Mege JL, Meghari S, Honstettre A, Capo C, Raoult D. The two faces of interleukin 10 in human infectious diseases. The Lancet infectious diseases. 2006;6:557–69. doi: 10.1016/S1473-3099(06)70577-1. [DOI] [PubMed] [Google Scholar]
- 146.Chung EY, Liu J, Homma Y, et al. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity. 2007;27:952–64. doi: 10.1016/j.immuni.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O'Garra A. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity. 2009;31:209–19. doi: 10.1016/j.immuni.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sabat R, Grutz G, Warszawska K, et al. Biology of interleukin-10. Cytokine & growth factor reviews. 2010;21:331–44. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 149.de Waal Malefyt R, Haanen J, Spits H, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. The Journal of experimental medicine. 1991;174:915–24. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. Journal of immunology. 1991;147:3815–22. [PubMed] [Google Scholar]
- 151.Fiorentino DF, Zlotnik A, Vieira P, et al. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. Journal of immunology. 1991;146:3444–51. [PubMed] [Google Scholar]
- 152.Shey MS, Maharaj NR, Liebenberg LJ, et al. Role of inflammatory cytokines and TLR ligands in modulating genital tract-derived dendritic cell activation. IAS 2013: 7th IAS Conference on Pathogenesis, Prevention and Treatment; 30 June - 3 July 2013; Kuala Lumpur, Malaysia. Abstract TUPE228. [Google Scholar]
- 153.Del Prete G, De Carli M, Almerigogna F, Giudizi MG, Biagiotti R, Romagnani S. Human IL-10 is produced by both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and inhibits their antigen-specific proliferation and cytokine production. Journal of immunology. 1993;150:353–60. [PubMed] [Google Scholar]
- 154.Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. The Journal of experimental medicine. 1996;184:19–29. doi: 10.1084/jem.184.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yilma AN, Singh SR, Fairley SJ, Taha MA, Dennis VA. The anti-inflammatory cytokine, interleukin-10, inhibits inflammatory mediators in human epithelial cells and mouse macrophages exposed to live and UV-inactivated Chlamydia trachomatis. Mediators of inflammation. 2012;2012:520174. doi: 10.1155/2012/520174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Alter G, Kavanagh D, Rihn S, et al. IL-10 induces aberrant deletion of dendritic cells by natural killer cells in the context of HIV infection. The Journal of clinical investigation. 2010;120:1905–13. doi: 10.1172/JCI40913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hart PH, Hunt EK, Bonder CS, Watson CJ, Finlay-Jones JJ. Regulation of surface and soluble TNF receptor expression on human monocytes and synovial fluid macrophages by IL-4 and IL-10. Journal of immunology. 1996;157:3672–80. [PubMed] [Google Scholar]
- 158.Jenkins JK, Malyak M, Arend WP. The effects of interleukin-10 on interleukin-1 receptor antagonist and interleukin-1 beta production in human monocytes and neutrophils. Lymphokine and cytokine research. 1994;13:47–54. [PubMed] [Google Scholar]
- 159.Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annual review of immunology. 1998;16:27–55. doi: 10.1146/annurev.immunol.16.1.27. [DOI] [PubMed] [Google Scholar]
- 160.Brockhaus M, Schoenfeld HJ, Schlaeger EJ, Hunziker W, Lesslauer W, Loetscher H. Identification of two types of tumor necrosis factor receptors on human cell lines by monoclonal antibodies. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:3127–31. doi: 10.1073/pnas.87.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. The Journal of experimental medicine. 2003;198:1875–86. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. The Journal of experimental medicine. 1998;188:287–96. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunological reviews. 2006;212:256–71. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 164.Annacker O, Asseman C, Read S, Powrie F. Interleukin-10 in the regulation of T cell-induced colitis. Journal of autoimmunity. 2003;20:277–9. doi: 10.1016/s0896-8411(03)00045-3. [DOI] [PubMed] [Google Scholar]
- 165.Joetham A, Takeda K, Taube C, et al. Naturally occurring lung CD4(+)CD25(+) T cell regulation of airway allergic responses depends on IL-10 induction of TGF-beta. Journal of immunology. 2007;178:1433–42. doi: 10.4049/jimmunol.178.3.1433. [DOI] [PubMed] [Google Scholar]
- 166.Mizrahi M, Ilan Y. The gut mucosa as a site for induction of regulatory T-cells. Current pharmaceutical design. 2009;15:1191–202. doi: 10.2174/138161209787846784. [DOI] [PubMed] [Google Scholar]
- 167.McKinnon LR, Kaul R. Quality and quantity: mucosal CD4+ T cells and HIV susceptibility. Current opinion in HIV and AIDS. 2012;7:195–202. doi: 10.1097/COH.0b013e3283504941. [DOI] [PubMed] [Google Scholar]
- 168.Presicce P, Moreno-Fernandez ME, Rusie LK, Fichtenbaum C, Chougnet CA. In vitro HIV infection impairs the capacity of myeloid dendritic cells to induce regulatory T cells. PloS one. 2012;7:e42802. doi: 10.1371/journal.pone.0042802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. European journal of immunology. 2000;30:1538–43. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 170.Kryczek I, Wei S, Zou L, et al. Cutting edge: induction of B7-H4 on APCs through IL-10: novel suppressive mode for regulatory T cells. Journal of immunology. 2006;177:40–4. doi: 10.4049/jimmunol.177.1.40. [DOI] [PubMed] [Google Scholar]
- 171.Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. Journal of immunology. 2004;172:4676–80. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- 172.Engelhardt G. Pharmacology of meloxicam, a new non-steroidal anti-inflammatory drug with an improved safety profile through preferential inhibition of COX-2. British journal of rheumatology. 1996;35 (Suppl 1):4–12. doi: 10.1093/rheumatology/35.suppl_1.4. [DOI] [PubMed] [Google Scholar]
- 173.Anderson KE, Johnson TW, Lazovich D, Folsom AR. Association between nonsteroidal anti-inflammatory drug use and the incidence of pancreatic cancer. Journal of the National Cancer Institute. 2002;94:1168–71. doi: 10.1093/jnci/94.15.1168. [DOI] [PubMed] [Google Scholar]
- 174.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. The New England journal of medicine. 1997;337:1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 175.D'Haens G, Van Deventer S, Van Hogezand R, et al. Endoscopic and histological healing with infliximab anti-tumor necrosis factor antibodies in Crohn's disease: A European multicenter trial. Gastroenterology. 1999;116:1029–34. doi: 10.1016/s0016-5085(99)70005-3. [DOI] [PubMed] [Google Scholar]
- 176.D'Haens GR. Infliximab (Remicade), a new biological treatment for Crohn's disease. Italian journal of gastroenterology and hepatology. 1999;31:519–20. [PubMed] [Google Scholar]
- 177.Antoni C, Braun J. Side effects of anti-TNF therapy: current knowledge. Clinical and experimental rheumatology. 2002;20:S152–7. [PubMed] [Google Scholar]
- 178.Li Q, Estes JD, Schlievert PM, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–8. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Peterson ML, Schlievert PM. Glycerol monolaurate inhibits the effects of Gram-positive select agents on eukaryotic cells. Biochemistry. 2006;45:2387–97. doi: 10.1021/bi051992u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Schlievert PM, Strandberg KL, Brosnahan AJ, et al. Glycerol monolaurate does not alter rhesus macaque (Macaca mulatta) vaginal lactobacilli and is safe for chronic use. Antimicrobial agents and chemotherapy. 2008;52:4448–54. doi: 10.1128/AAC.00989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fichorova RN, Tucker LD, Anderson DJ. The molecular basis of nonoxynol-9-induced vaginal inflammation and its possible relevance to human immunodeficiency virus type 1 transmission. The Journal of infectious diseases. 2001;184:418–28. doi: 10.1086/322047. [DOI] [PubMed] [Google Scholar]
- 182.Mesquita PM, Cheshenko N, Wilson SS, et al. Disruption of tight junctions by cellulose sulfate facilitates HIV infection: model of microbicide safety. The Journal of infectious diseases. 2009;200:599–608. doi: 10.1086/600867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Abdool Karim Q, Abdool Karim SS, Frohlich JA, et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science. 2010;329:1168–74. doi: 10.1126/science.1193748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Roberts L, Passmore JS, Williamson C, Little F, Naranbhai V, Sibeko S, Walzl G, Abdool Karim Q, Abdool Karim S. Genital Tract Inflammation in Women Participating in the CAPRISA Tenofovir Microbicide Trial Who Became Infected with HIV: A Mechanism for Breakthrough Infection?. Conference on Retroviruses and Opportunistic Infections; Boston, MA. Feb 27 - March 2, 2011; Abtract number Y-137. [Google Scholar]