

Abstract
Bipolar disorder and schizophrenia are two often severe disorders with high heritabilities. Recent studies have demonstrated a large overlap of genetic risk loci between these disorders but diagnostic and molecular distinctions still remain. Here, we perform a combined GWAS of 19,779 BP and SCZ cases versus 19,423 controls, in addition to a direct comparison GWAS of 7,129 SCZ cases versus 9,252 BP cases. In our case-control analysis, we identify five previously identified regions reaching genome-wide significance (CACNA1C, IFI44L, MHC, TRANK1, MAD1L1) and a novel locus near PIK3C2A. We create a polygenic risk score that is significantly different between BP and SCZ and show a significant correlation between a BP polygenic risk score and the clinical dimension of mania in SCZ patients. Our results indicate that first, combining diseases with similar genetic risk profiles improves power to detect shared risk loci and second, that future direct comparisons of BP and SCZ are likely to identify loci with significant differential effects. Identifying these loci should aid in the fundamental understanding of how these diseases differ biologically. These findings also indicate that combining clinical symptom dimensions and polygenic signatures could provide additional information that may someday be used clinically.
Introduction
Bipolar disorder (BP) and schizophrenia (SCZ) are both highly heritable (h2 ~ 0.8), often debilitating psychiatric illnesses that together affect ~2-3% of the adult population worldwide (1, 2). The distinction between BP and SCZ on the basis of clinical features, etiology, family history, and treatment response has been one of the most fundamental and controversial issues in modern psychiatric nosology. While contemporary diagnostic systems distinguish them on the basis of clinical symptomatology, duration, and associated disability, the distinction between Manic-Depressive Illness and Dementia Praecox was originally made largely on the basis of the course of illness in the late 19th century by Emil Kraepelin, who recognized that mood and psychotic symptoms could occur in both disorders (3). But clinical symptoms may not map directly onto underlying molecular mechanisms of disease. For many years, the etiological independence of these two disorders was widely accepted although transitional forms – first labeled schizoaffective disorder by Kasanin in 1933 (4) – were widely recognized. Important support for the Kraepelinian dichotomy was provided from family studies over the last 40 years that suggested at most modest familial co-aggregation of the two disorders (5-7).
However, more recent studies have suggested that the genetic relationship between BP and SCZ might be greater than previously realized. Researchers have found increased risks of affective disorder in the families of schizophrenia patients (8) as well as the reverse (9). The largest of these studies, including data on more than 75,000 affected Swedish families, found that the risk of SCZ was substantially increased in the relatives of BP and vice-versa (10). Because of the availability of information on twin and half-sibling relationships and adopted-away relatives the authors were able to show a substantial genetic correlation between SCZ and BP. However, this study assigned diagnoses on the basis of chart diagnoses, which may be less reliable than direct interviews using standardized instruments. The only twin studies that have examined the genetic correlation between SCZ and BP diagnosed using direct patient interviews have been conducted in the Maudsley twin series (11). However, these studies used non-hierarchical diagnoses, which confound manic syndromes in the course of SCZ with BPD, which might have different genetic influences.
Molecular genetic studies have the potential to more clearly and more powerfully distinguish genetic from environmental factors. Recent molecular genetic studies have identified a substantial polygenic component to SCZ risk involving hundreds to thousands of common alleles of small effect, and this component was shown to also contribute to risk of BP (12). This analysis pointed to risk shared across many genetic markers, but results from individual genome-wide association studies (GWAS) have also implicated specific common shared loci (13). Taking this further, in a meta-analysis of most of the world’s available GWAS data, the Psychiatric Genomic Consortium Bipolar and Schizophrenia Working Groups identified SNPs for both BP and SCZ in CACNA1C, ANK3 and ITIH3-ITIH4 as genome wide significant but not in MHC, ODZ4, TCF4 and other loci that were genome-wide significant for either disorder separately. Additionally, the Cross-Disorder Group of the Psychiatric Genomics Consortium explicitly tested SNPs across five disorders for the best fitting disease model and identified CACNA1C as more significantly associated to a model combining only BP and SCZ than one including other disorders (14).
In addition, diagnoses intermediate between SCZ and BP, such as schizoaffective disorder and BP with psychotic features, comprise individuals who present with admixtures of clinical features common to both disorders. It is not clear whether these disorders are caused by the presence of genetic risk factors for both SCZ and BP, or have separate underlying etiologies (15).
It remains an open question whether the most recent molecular results are capable of dissecting the different symptom dimensions within and across these disorders. One study looked to assess the discriminating ability of SCZ polygenic risk on psychotic subtypes of BP. They identified a SCZ polygenic signature that successfully differentiated between BP and schizoaffective BP type but were unable to identify a significant difference in risk score between BP with and without psychotic features (16).
Our goals here were twofold, to elucidate the shared and differentiating genetic components between BP and SCZ and to assess the relationship between this genetic component and the symptomatic dimensions of these disorders.
Methods
Sample description
This study combines individual genotype data published in 2011 by the PGC Bipolar Disorder and the Schizophrenia Working Groups. Description of the sample ascertainment can be found in the respective publications (17, 18). In addition, four bipolar datasets not included in the primary meta-analysis (although used for the replication phase) are now included: three previously not published bipolar datasets including additional samples from Thematically Organized Psychoses (401 cases, 171 controls), French (451 cases, 1,631 controls), FaST STEP2/TGEN (1,860 cases) and one published dataset Sweden (824 cases, 2,084 controls) (19). The unpublished samples are further described as supplementary information in the original PGC BP study (14). FaST STEP2/TGEN BP cases were combined with GAIN/BIGS BP cases and controls from MIGen (20) to form a single sample (Supplementary Table 2). In the PGC analyses, genotype data from control samples were used in both SCZ and BP GWAS studies.
Independent BP and SCZ datasets with no overlapping genotype data from controls were created by calculating relatedness across all pairs of individuals using an LD pruned set of SNPs directly genotyped in all studies. Controls found in more than one dataset were randomly allocated to balance the number of cases and controls accounting for population and genotyping platform effects. We grouped case-control samples by ancestry and genotyping array into 14 BP samples and 17 SCZ samples (Supplementary Table 1). We further grouped individuals by ancestry to perform a direct comparison of BP and SCZ (Supplementary Table 2).
Genotype data quality control
Raw individual genotype data from all samples were uploaded to the Genetic Cluster Computer hosted by the Dutch National Computing and Networking Services. Quality control was performed on each of the 31 sample collections separately. SNPs shared between platforms and pruned for LD were used to identify relatedness. SNPs were removed if they had: 1) minor allele frequency < 1%, 2) call rate < 98%, 3) Hardy-Weinberg equilibrium (p < 1 × 10−6), 4) differential levels of missing data between cases and controls (> 2%), and 5) differential frequency when compared to Hapmap CEU (> 15%). Individuals were removed who had genotyping rates < 98%, high relatedness to any other individual ( > 0.9), or low relatedness to many other individuals ( > 0.2), or substantially increased or decreased autosomal heterozygosity (|F| > 0.15). We tested 20 MDS components against phenotype status using logistic regression with sample as a covariate. We selected the first four components and any others with a nominally significant correlation (p-value < 0.05) between the component and phenotype. We included these components in our GWAS. This process was done independently for all phenotype comparisons. Imputation was performed using the HapMap Phase3 CEU + TSI data and BEAGLE (21, 22) by sample on random subsets of 300 subjects. All analyses were performed using Plink (23).
Association analysis
The primary association analysis was logistic regression on the imputed dosages from BEAGLE on case-control status with 13 MDS components and sample grouping as covariates. We performed four association tests: 1) a combined meta-analysis of BP and SCZ (19,779 BP and SCZ cases, 19,423 controls) to identify variants shared across both disorders, 2) SCZ only (SCZ n=9,369, vs controls n=8,723), and 3) BP only (BP n=10,410, controls n=10,700) for comparison to dimensional phenotypes and 4) case only BP vs SCZ (SCZ n=7,129, BP n=9,252) to identify loci with differential effects between these two disorders (Table 1). We retained SNPs after imputation with INFO > 0.6. We calculated genomic inflation factors both without normalization for these analyses (λ): 1.26 (BP+SCZ vs controls), 1.19 (SCZ vs controls), 1.15 (BP vs controls) and 1.11 (BP vs SCZ) and normalized to 1,000 cases and 1,000 controls for direct comparison (λ1000 SCZ). Additionally, for the BP+SCZ meta-analysis, we tested heterogeneity between BP vs controls and SCZ vs controls odds ratios using the Cochrane’s Q test. Association regions were defined by an LD clumping procedure for all independent index SNPs with p-value < 5×10−8. We defined the region to include any SNP within 500kb of the index SNP, in LD with the index SNP (r2 > 0.2) and having a p-value < 0.005.
Table 1.
Description of the 4 primary association tests run and sample counts.
| Analysis | “Cases” n |
Samples | “Controls” N |
Samples |
|---|---|---|---|---|
| BP + SCZ | 19,779 | BP + SCZ | 19,423 | SCZ controls + BP controls |
| BP | 10,410 | BP | 10,700 | BP controls |
| SCZ | 9,369 | SCZ | 8,723 | SCZ controls |
| BP vs SCZ | 7,129 | SCZ | 9,252 | BP |
Polygenic analysis
We employed a method used by the International Schizophrenia Consortium (12) and developed by Visscher, Wray and Purcell to calculate both BP polygenic scores in SCZ cases and SCZ polygenic scores in BP cases. Briefly, we defined the SCZ case-control GWAS as our discovery sample and the BP case-control GWAS as our target sample. Based on the discovery sample association statistics, large sets of nominally-associated alleles were selected as “score alleles”, for different significance thresholds. In the target sample, we calculated the total score for each individual as the number of score alleles weighted by the log of the odds ratio from the discovery sample. We repeated this exercise with the BP case-control GWAS as discovery and the SCZ case-control GWAS as the target. We created scores using ten different p-value thresholds (P < 0.0001, P < 0.001, P < 0.01, P < 0.05, P < 0.1, P < 0.2, P < 0.3, P < 0.4, P < 0.5, P < 1). For each threshold, we performed a logistic regression of disease status on the polygenic score covarying for MDS components and sample.
Factor analysis of clinical dimensions of SCZ across multiple datasets
SCZ is clinically heterogeneous, with variation in levels of positive, negative, and affective symptoms, as well as age of onset, course, and outcome (24). Samples included in this study used a variety of structured interviews, symptom checklists, or rating scales to determine the presence of individual clinical features. These instruments are listed in Supplementary Table 6.
Because the individual symptoms assessed differed substantially across sites, we sought to achieve across-site commonality at the level of symptom factors. We therefore constructed quantitative traits common to all instruments and sites, and which could be used as phenotypes of interest in genetic studies. Our approach was stepwise, involving initial exploratory factor analysis of the each instrument in each individual site, followed by harmonization of the different sites by selecting prominent items and factors, and finally, calculation of factor scores using confirmatory factor analysis in each site separately, as follows.
This was done in several steps. First, exploratory factor analysis (EFA) was performed separately in all of the sites that utilized the OPCRIT (Operational Criteria for Psychotic Illness) (25), PANSS (Positive and Negative Syndrome Scale)(26) as follows. For each dataset (i.e. one instrument from each individual site) individual items were excluded if they had > 50% missing data. Remaining missing data was inferred using the method of multiple imputation, as operationalized in Proc Mi in SAS, resulting in five separately imputed datasets for each input dataset. For each item, we used the mean of the corresponding imputed items from these five datasets. These final input variables were entered into EFA using principal component analysis, implemented in SAS using Proc Factor, using VARIMAX rotation (SAS Institute, Cary, N.C.).
The remaining sites had already been factor analyzed, and for these, we used the published factor structures. Prior factor analysis of Lifetime Dimensions of Psychosis Scale (LDPS)(27) in the MGS sample, resulted in three factors: positive, negative, and mood (28). The UCLA sample utilized the Comprehensive Assessment of Symptoms and History (CASH)(29), and had been previously factor-analyzed using a larger sample than that included in the present GWAS (30), which resulted in a clinically meaningful five-factor model of positive, negative, disorganization, manic, and depressive symptoms.
Second, we examined the overall pattern of results across all samples and instruments since the best-fitting models across samples differed in number and composition of factors. This allowed us to identify the most commonly extracted as well as most theoretically justifiable factors. Four such factors were selected in this way – Positive, Negative, Manic, and Depressive.
Third, we attempted to harmonize results across those sites that utilized the same instrument, i.e. the three sites using PANSS and six sites using OPCRIT. For the four selected factors, we compared the EFA loadings in all of these sites on an item-by-item basis. Following convention, we considered an item to load on a given factor if its highest loading was on that factor and was at least 0.4, and if it’s next highest loading was at least 0.2 less than its highest loading. Items which were outliers, i.e. which loaded on a given factor in only one sample without clinical-theoretical justification, and which clearly did not load on that factor in the others, were dropped in all other samples, were included in that factor in all samples. This procedure was followed for the OPCRIT and PANSS sites separately. Factor compositions are described in further detail (28).
Fourth, we attempted to harmonize the disparate instruments. The MGS sample had a single mood factor while the PANSS, OPCRIT, and CASH sites had separate depressive and manic factors. It was therefore split into these two factors. The PANSS sites had separate negative and disorganization factors, which were combined, as these symptoms loaded on a single negative factor in all the other instruments.
When all of the individual items for the final four factors were selected in all sites, we used Confirmatory Factor Analysis in MPLUS to calculate four factor scores in each site separately. The different instruments used have differences in content, objectives, and granularity. For example, the OPCRIT is used to make diagnoses of both psychotic and mood disorders. It contains, therefore, a number of classic manic and depressive symptoms. The PANSS on the other hand, was designed for assessing schizophrenic symptoms and is frequently used in treatment efficacy studies. Its content is therefore geared more towards psychotic agitation and excitement rather than classic manic symptoms. Furthermore, the LDPS comprises 14 more global items, while the OPCRIT has dozens of more fine-grained items and the other instruments are intermediate between these two in granularity.
In order to test the comparability of symptom factors across PANSS and OPCRIT, we performed EFA in the Dublin sample, which used both instruments. Pearson product-moment correlations were calculated for each of the resulting factors across these two instruments. These were the only two instruments that were used in the same sample. All factors except Positive were significantly correlated (r=.48 for depressive, .70 for manic, and .85 for negative symptoms, indicating that the two instruments index the same broad underlying constructs for these dimensions.
Finally, we observed that the distributions of a number of traits in some of the sites were highly skewed and differed across samples (results available on request). This is not surprising, as there was likely to be considerable heterogeneity across sites in item definition, rater training, patterns of help-seeking (affecting age of onset), treatment setting (e.g. ambulatory, institutionalized, etc.) as well as other unobserved patient- or rater-dependent factors. Because this could considerably inflate genetic analyses, we standardized all traits within site, to have mean=0 and SD=1 (treating the individual MGS sites separately).
Deriving clinical dimensions of BP
The BP cases were interviewed using established diagnostic instruments and diagnosed with bipolar disorder according to the RDC (Research Diagnostic Criteria) (31), DSMIII-R (Diagnostic and Statistical Manual of Mental Disorders) DSM-IV or ICD-10. Diagnostic instruments used were the SCID (Structured Clinical Interview for DSM disorders) (32), the SADS-L (Schedule of Affective Disorders and Schizophrenia-Lifetime version) (33), the DIGS (Diagnostic Instrument for Genetic Studies) (34), the MINI (Miniinternational Neuropsychiatric Interview) (35), the ADE (Affective Disorders Evaluation) (36) and the SCAN (Schedules for Clinical Assessment in Neuropsychiatry) (37). Data from these interviews was combined with information from case notes and in some cases supplemented with data from the OPCRIT (operational criteria checklist for psychotic illness) (25). A full description of the instruments used in each of the collaborating centers is given in the supplementary data that accompanied the PGC bipolar disorder meta-analysis (38). We considered subjects experiencing hallucinations or delusions during a manic or depressive episode to have Bipolar Disorder with Psychotic Features.
Results
Analyzing BP and SCZ cases as a single phenotype in GWAS
We analyzed genome-wide data in 19,779 cases (9,369 SCZ plus 10,410 BP) and 19,423 controls consisting of 1.1 million SNP dosages imputed using HapMap Phase 3 (21). Logistic regressions were performed controlling for sample and 13 quantitative indices of ancestry. We identified 219 SNPs in six genomic regions with p-values below the genome-wide significance threshold of 5 × 10−8 (Figure 1a, Table 2, Supplementary Table 3). The most significant SNP (rs1006737, p=5.5 × 10−13, OR=1.12) falls within the gene CACNA1C that was first found to be significant in BP (18, 39), subsequently in SCZ (17, 40) and recently for a best fit model in a 5 disease cross-disorder analysis (14). In our independent disease samples, we find similar odds ratios for both disorders (BP OR=1.127; SCZ OR=1.120). Of the other five genome-wide significant regions, the second most significant SNP is in the major histocompatibility complex (MHC) while the others are in or near the following genes TRANK1, MAD1L1, PIK3C2A, IFI44L. Four have been previously implicated in either SCZ (MHC, MAD1L1) (12, 19) or BP (TRANK1, IFI44L) (41) but the association near PIK3C2A (chr11:17023194-17381287) is novel (Supplementary Figures 1a-f). The region of LD around this SNP includes RPS13, PIK3C2A, NUCB2, KCNJ11, and ABCC8. This locus has not been previously identified through GWAS but has recently been implicated using an alternative approach (42). None of the six genome-wide significant SNPs identified here demonstrated significant heterogeneity in odds ratios between BP and SCZ although MHC had the largest difference in effect size and approached significance (BP OR=0.88, SCZ OR=0.80, p=0.059). However, this test is probably underpowered in meta-analyses of only a few studies (43).
Figure1.
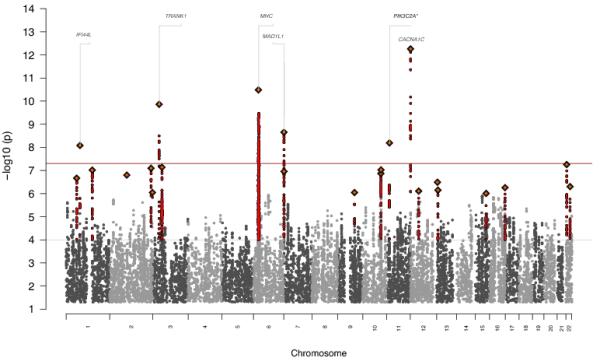
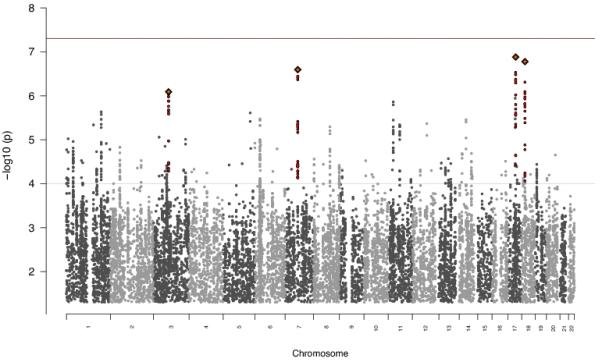
a) Manhattan plot for combined BP + SCZ GWAS identifying 6 genome-wide significant hits including novel associations at PIK3C2A b) Manhattan plot of comparison GWAS
Table 2.
Most significant SNP in 6 genome-wide significant regions from BP+SCZ analysis (* denotes novel locus).
| Closest gene | SNP | Position (hg18) | BP+SCZ P | BP P | SCZ P | Het P |
|---|---|---|---|---|---|---|
| CACNA1C | rs1006737 | chr12:2162951..2290787 | 5.53E-13 | 7.43E-08 | 1.65E-06 | 0.85 |
| MHC | rs17693963 | chr6:27337244..33069339 | 3.28E-11 | 4.28E-04 | 3.27E-09 | 0.06 |
| TRANK1 | rs9834970 | chr3:36817627..36935664 | 1.38E-10 | 3.90E-07 | 7.18E-05 | 0.55 |
| MAD1L1 | rs10275045 | chr7:1834618..2305931 | 2.22E-09 | 2.08E-04 | 1.84E-06 | 0.35 |
| PIK3C2A* | rs4356203 | chr11:17023194..17381287 | 6.46E-09 | 7.36E-05 | 2.14E-05 | 0.70 |
| IFI44L | rs4650608 | chr1:78942596..79066403 | 8.30E-09 | 1.22E-05 | 1.77E-04 | 0.76 |
Examination of variants distinguishing BP and SCZ
To identify loci with differential effects on BP and SCZ, we compared 9,252 BP cases against 7,129 SCZ cases. No SNPs reached genome-wide significance with the smallest p-value at rs7219021 at chr17:44195540 (p=1.31 × 10−7) (Figure 1b). A lack of genome-wide significant findings does not preclude the existence of many small effect loci that in aggregate can significantly discriminate BP from SCZ. We applied a previously used risk profiling approach (12, 44) to our BP vs SCZ data. For each of the nine samples defined by ancestry and array technology (Supplementary Table 2), we computed a risk score using the association data from eight samples as our discovery set and then assessed the ability of those risk scores to predict BP vs SCZ status in the remaining sample. This allowed us to maximize our sample size and ensure no particular sample was disproportionately contributing to the result. Risk scores were calculated for p-value thresholds from 0.001 to 1 as defined in (12). All samples had at least one threshold reaching nominal significance with all but one sample explaining at least 2% of the variance (Figure 2, Supplementary Table 4a-b). These results suggest that we have successfully identified a polygenic signal capable of detecting risk differences between BP and SCZ.
Figure 2.
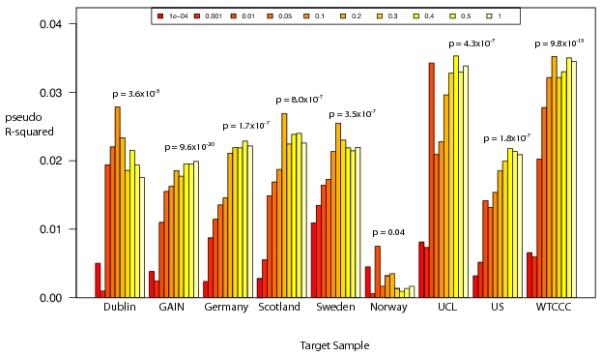
Average pseudo R2 values for polygenic prediction of BP vs SCZ phenotype into the target sample where all other samples were used for discovery.
To investigate further, we took all 22 genome-wide significant loci from a recently submitted SCZ analysis (Ripke et al.) and compared the odds ratios between our independent BP and SCZ datasets. The data are ordered left to right by significance for BP (Figure 3). There is a spectrum of BP effects for statistically significant SCZ loci with loci on the left displaying odds ratios similar in magnitude between the two diseases, while loci on the right, having divergent odds ratios.
Figure 3.
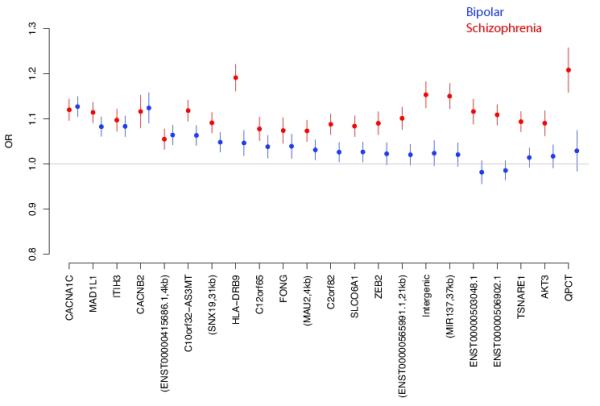
Comparison of odds ratios from independent samples of BP (blue) and SCZ (red) for genome-wide significant loci previously identified in SCZ.
Polygenic scores from BP applied to clinical dimensions in SCZ
Having identified a polygenic signature capable of differentiating BP and SCZ, we sought to identify whether disease specific polygenic scores of one disorder were correlated with symptom dimensions in the other disorder. We had manic, depressive, positive and negative symptom factors from the factor analyses of our SCZ samples described previously (see methods).
We calculated BP polygenic risk scores in our SCZ sample using our full, independent BP dataset. All factor scores were split at the median into two equally sized sets. For all factor scores, now dichotomized, we asked whether polygenic score of BP risk predicted whether SCZ subjects were above or below the median on the symptom factor using logistic regression with sample and MDS as covariates. Risk scores were calculated for 10 p-value thresholds from 0.001 to 1 for each symptom dimension. Polygenic score of BP was associated only with the manic factor in SCZ subjects, with a p-value threshold of 0.3 having significance p=0.003 and pseudo variance explained of ~2% (Figure 4, Supplementary Table 5). Applying the same test to the quantitative mania score yields, a more significant result (p=2.51×10−5). We tested each individual schizophrenia sample independently to ensure no single sample was solely driving the finding. Mania score distributions differed by sample; however, significance was seen across multiple samples and removal of any single sample did not appreciably change the overall result (Supplementary Table 6). The correlation between BP polygenic risk score and mania score was strongest at the high end of the mania distribution, implicating a possible subset of individuals with both high mania scores and high BP polygenic risk scores. To investigate further, we identified a subset of the SCZ cases that included 183 individuals with, and 886 without a schizoaffective disorder diagnosis. Within this subset, schizoaffective individuals had significantly higher mania scores than non-schizoaffective individuals but did not carry significantly higher BP polygenic risk scores. However, this subsample had no significant correlation between BP polygenic risk score and mania score overall leaving us underpowered to assess the affect schizoaffective status has on the overall correlation between BP polygenic risk score and mania score. We additionally sought to understand the effect that individuals with both high BP polygenic score and high manic factor score had on our results. We removed 186 individuals with BP polygenic score and manic factor score one standard deviation from the mean and repeated our analyses. We identify the same six genome-wide significant hits, nearly identical SCZ odds ratios and equal if not slightly more significant discrimination ability in our between disorder BP vs SCZ polygenic analysis (Supplementary Table 7, Supplementary Figures 3-4)
Figure 4.
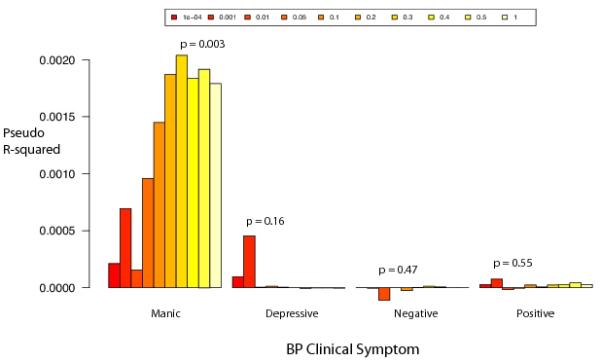
Average pseudo R2 values for predicting SCZ vs controls utilizing odds ratios estimated from BP vs controls GWAS. There are 10 R2 values for each factor score representing 10 different p-value cutoffs for SNPs included in making the risk score (P < 0.0001, P < 0.001, P < 0.01, P < 0.05, P < 0.1, P < 0.2, P < 0.3, P < 0.4, P < 0.5, P < 1).
Polygenic scores from SCZ applied to clinical dimensions in BP
In the BP samples, we were limited to only a dichotomous rating about the presence or absence of psychosis. We calculated SCZ polygenic risk scores using our full independent SCZ dataset in all BP cases and tested for a correlation between presence of psychosis and SCZ polygenic score as described above. No such correlation was observed. (data not shown)
Discussion
Our results present the most detailed comparison to date of the genetic risk underlying BP and SCZ. We identify six genome-wide significant loci associated with a combined BP+SCZ phenotype compared to controls, including a novel locus near PIK3C2A. At the same time, we demonstrate the ability to create a polygenic risk score from a GWAS of BP vs SCZ that significantly discriminates between the two disorders at the level of molecular genetic variants. Additionally, we found a strong correlation between BP polygenic score and the manic symptom dimension in SCZ cases.
Of the six genome-wide significant loci in our BP+SCZ vs controls analysis only two (CACNA1C, MHC) are present at that level in the individual disease analyses (Supplementary Figure 2a-b), highlighting the benefits of combining genetically related disease samples. In all but one region, these loci have near equivalent effect sizes and frequencies between BP and SCZ. The exception is the most significantly associated region found in SCZ (MHC) that has a considerably weaker association in BP. This distinction could point to a biologically relevant difference in disease etiology possibly related to immune function. The most significant result in this study implicates calcium channels as particularly important to risk of both of these disorders. In fact, CACNA1C appears to be more strongly associated to BP and SCZ than other psychiatric disorders including autism spectrum disorder, attention deficit-hyperactivity disorder and major depressive disorder (14). In a joint analysis of these disorders, it was the only genome-wide significant finding where the inclusion of all five disorders was not the most significant model.
We identify no loci that show genome-wide significance for allele frequency differences between BP and SCZ. However, this analysis remains underpowered from smaller sample size and fewer available well matched BP cases and SCZ cases as it is still uncommon for a single site to collect matching disease samples for this type of analysis. We anticipate that larger studies of this type will discover significant loci. We present a comparison of odds ratios in our independent BP and SCZ samples for a set of 22 previously identified genome-wide significant loci (Figure 3). This result implies that there are additional SCZ loci that will also be independently associated BP loci as sample sizes increase, but that there are also loci that are likely to remain SCZ specific and perhaps also BP specific. CACNA1C(39) has already been independently associated in BP and ITIH3-ITIH4 and MAD1L1 have been near the top of the list in the largest BP GWAS performed to date (18). We note two caveats to interpreting these results: 1) there is significant overlap of both the SCZ samples and the control samples with those used to identify these loci in Ripke et al. which will create a small inflation of effect for SCZ and 2) there will be a general deflation of effect in our BP sample and to a smaller extent in our SCZ sample due to winner’s curse.
While BP and SCZ share much of their genetic risk loci, we now report a polygenic component that significantly distinguishes these disorders. As in previous analyses of this type (12) this polygenic component implicates a true underlying genetic architecture difference between BP and SCZ and with larger samples identification of specific loci or biologically relevant gene sets could be uncovered. In addition to the difference in contribution of disease risk from large, rare CNVs we are starting to build a knowledge base to begin to identify disease specific genetic architecture and these analyses should be expanded to include more related diseases. This kind of work could eventually provide clues in the development of molecular diagnostic tools to improve on current methods, which are purely clinical. This is especially important in the case of these SCZ and BP, which are often difficult to distinguish, especially early in the course of illness (45, 46), and in which early diagnosis and treatment could improve outcome (47).
Finally, we present an overlap of a molecular genetic signature and a clinical symptom between BP and SCZ. For the first time, we correlate a BP polygenic signal with a manic symptom dimension in SCZ individuals. This suggests that clinical dimensions of SCZ might be modified by risk variants for other disorders (i.e., “modifier” genes), which might thereby provide treatment targets for these dimensions. It provides further evidence that clinical heterogeneity in schizophrenia is in part due to genetic factors (24). More specifically, it suggests the existence of a mood spectrum that has distinct genetic substrates, and exists to a variable degree in multiple disorders. Evidence such as this could one day help identify individuals who might benefit from a specific course of treatment.
Supplementary Material
References
- 1.Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS medicine. 2005;2(5):e141. doi: 10.1371/journal.pmed.0020141. Epub 2005/05/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Craddock N, Sklar P. Genetics of bipolar disorder: successful start to a long journey. Trends in genetics: TIG. 2009;25(2):99–105. doi: 10.1016/j.tig.2008.12.002. Epub 2009/01/16. [DOI] [PubMed] [Google Scholar]
- 3.Kraepelin E, Diefendorf AR. Clinical psychiatry. The Macmillan company; New York, London: 1907. p. 562. [Google Scholar]
- 4.Kasanin J. The acute schizoaffective psychoses. 1933. The American journal of psychiatry. 1994;151(6 Suppl):144–54. doi: 10.1176/ajp.151.6.144. Epub 1994/06/01. [DOI] [PubMed] [Google Scholar]
- 5.Kendler KS, McGuire M, Gruenberg AM, O’Hare A, Spellman M, Walsh D. The Roscommon Family Study. I. Methods, diagnosis of probands, and risk of schizophrenia in relatives. Arch Gen Psychiatry. 1993;50(7):527–40. doi: 10.1001/archpsyc.1993.01820190029004. Epub 1993/07/01. [DOI] [PubMed] [Google Scholar]
- 6.Maier W, Lichtermann D, Minges J, Hallmayer J, Heun R, Benkert O, et al. Continuity and discontinuity of affective disorders and schizophrenia. Results of a controlled family study. Arch Gen Psychiatry. 1993;50(11):871–83. doi: 10.1001/archpsyc.1993.01820230041004. Epub 1993/11/01. [DOI] [PubMed] [Google Scholar]
- 7.Tsuang MT, Winokur G, Crowe RR. Morbidity risks of schizophrenia and affective disorders among first degree relatives of patients with schizophrenia, mania, depression and surgical conditions. The British journal of psychiatry : the journal of mental science. 1980;137:497–504. doi: 10.1192/bjp.137.6.497. Epub 1980/12/01. [DOI] [PubMed] [Google Scholar]
- 8.Mortensen PB, Pedersen CB, Melbye M, Mors O, Ewald H. Individual and familial risk factors for bipolar affective disorders in Denmark. Arch Gen Psychiatry. 2003;60(12):1209–15. doi: 10.1001/archpsyc.60.12.1209. Epub 2003/12/10. [DOI] [PubMed] [Google Scholar]
- 9.Maier W, Lichtermann D, Franke P, Heun R, Falkai P, Rietschel M. The dichotomy of schizophrenia and affective disorders in extended pedigrees. Schizophr Res. 2002;57(2-3):259–66. doi: 10.1016/s0920-9964(01)00288-2. Epub 2002/09/12. [DOI] [PubMed] [Google Scholar]
- 10.Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373(9659):234–9. doi: 10.1016/S0140-6736(09)60072-6. Epub 2009/01/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardno AG, Rijsdijk FV, Sham PC, Murray RM, McGuffin P. A twin study of genetic relationships between psychotic symptoms. The American journal of psychiatry. 2002;159(4):539–45. doi: 10.1176/appi.ajp.159.4.539. Epub 2002/04/02. [DOI] [PubMed] [Google Scholar]
- 12.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–52. doi: 10.1038/nature08185. Epub 2009/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams HJ, Norton N, Dwyer S, Moskvina V, Nikolov I, Carroll L, et al. Fine mapping of ZNF804A and genome-wide significant evidence for its involvement in schizophrenia and bipolar disorder. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.36. Epub 2010/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013 doi: 10.1016/S0140-6736(12)62129-1. Epub 2013/03/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kendler KS, McGuire M, Gruenberg AM, Walsh D. Examining the validity of DSM-III-R schizoaffective disorder and its putative subtypes in the Roscommon Family Study. The American journal of psychiatry. 1995;152(5):755–64. doi: 10.1176/ajp.152.5.755. Epub 1995/05/01. [DOI] [PubMed] [Google Scholar]
- 16.Hamshere ML, O’Donovan MC, Jones IR, Jones L, Kirov G, Green EK, et al. Polygenic dissection of the bipolar phenotype. The British journal of psychiatry: the journal of mental science. 2011;198(4):284–8. doi: 10.1192/bjp.bp.110.087866. Epub 2011/10/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. Genome-wide association study identifies five new schizophrenia loci. Nature genetics. 2011;43(10):969–76. doi: 10.1038/ng.940. Epub 2011/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature genetics. 2011;43(10):977–83. doi: 10.1038/ng.943. Epub 2011/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergen SE, O’Dushlaine CT, Ripke S, Lee PH, Ruderfer DM, Akterin S, et al. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Molecular psychiatry. 2012;17(9):880–6. doi: 10.1038/mp.2012.73. Epub 2012/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nature genetics. 2009;41(3):334–41. doi: 10.1038/ng.327. Epub 2009/02/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–23. doi: 10.1016/j.ajhg.2009.01.005. Epub 2009/02/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The International HapMap Project Nature. 2003;426(6968):789–96. doi: 10.1038/nature02168. Epub 2003/12/20. [DOI] [PubMed] [Google Scholar]
- 23.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81(3):559–75. doi: 10.1086/519795. Epub 2007/08/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fanous AH, Kendler KS. Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: searching for a framework. Molecular psychiatry. 2005;10(1):6–13. doi: 10.1038/sj.mp.4001571. Epub 2004/12/25. [DOI] [PubMed] [Google Scholar]
- 25.McGuffin P, Farmer A, Harvey I. A Polydiagnostic Application Of Operational Criteria In Studies Of Psychotic Illness - Development and Reliability Of the Opcrit System. Archives of general psychiatry. 1991;48(8):764–70. doi: 10.1001/archpsyc.1991.01810320088015. [DOI] [PubMed] [Google Scholar]
- 26.Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–76. doi: 10.1093/schbul/13.2.261. Epub 1987/01/01. [DOI] [PubMed] [Google Scholar]
- 27.Levinson DF, Mowry BJ, Escamilla MA, Faraone SV. The Lifetime Dimensions of Psychosis Scale (LDPS): description and interrater reliability. Schizophrenia bulletin. 2002;28(4):683–95. doi: 10.1093/oxfordjournals.schbul.a006972. Epub 2003/06/11. [DOI] [PubMed] [Google Scholar]
- 28.Fanous AH, Zhou B, Aggen SH, Bergen SE, Amdur RL, Duan J, et al. Genome-wide association study of clinical dimensions of schizophrenia: polygenic effect on disorganized symptoms. The American journal of psychiatry. 2012;169(12):1309–17. doi: 10.1176/appi.ajp.2012.12020218. Epub 2012/12/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreasen NC, Flaum M, Arndt S. The Comprehensive Assessment of Symptoms and History (CASH). An instrument for assessing diagnosis and psychopathology. Archives of general psychiatry. 1992;49(8):615–23. doi: 10.1001/archpsyc.1992.01820080023004. Epub 1992/08/01. [DOI] [PubMed] [Google Scholar]
- 30.Boks MP, Leask S, Vermunt JK, Kahn RS. The structure of psychosis revisited: the role of mood symptoms. Schizophrenia research. 2007;93(1-3):178–85. doi: 10.1016/j.schres.2007.02.017. Epub 2007/03/27. [DOI] [PubMed] [Google Scholar]
- 31.Spitzer R, Endicott J, Robins E. Research Diagnostic Criteria for a selected group of functional disorders. 3rd edition New York State Psychiatric Institute.; New York: 1978. [Google Scholar]
- 32.Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Archives of general psychiatry. 1992;49(8):624, 9. doi: 10.1001/archpsyc.1992.01820080032005. Epub 1992/08/01. [DOI] [PubMed] [Google Scholar]
- 33.Spitzer R, Endicott J. The Schedule for Affective Disorders and Schizophrenia, Lifetime Version. 3rd edition New York State Psychiatric Institute.; New York: 1977. [Google Scholar]
- 34.Nurnberger JI, Jr., Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, et al. Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Archives of general psychiatry. 1994;51(11):849–59. doi: 10.1001/archpsyc.1994.03950110009002. discussion 63-4. Epub 1994/11/01. [DOI] [PubMed] [Google Scholar]
- 35.Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. The Journal of clinical psychiatry. 1998;59(Suppl 20):22–33. quiz 4-57. Epub 1999/01/09. [PubMed] [Google Scholar]
- 36.Sachs GS. Use of clonazepam for bipolar affective disorder. The Journal of clinical psychiatry. 1990;51(Suppl):31–4. discussion 50-3. Epub 1990/05/01. [PubMed] [Google Scholar]
- 37.Wing JK, Babor T, Brugha T, Burke J, Cooper JE, Giel R, et al. SCAN. Schedules for Clinical Assessment in Neuropsychiatry. Archives of general psychiatry. 1990;47(6):589–93. doi: 10.1001/archpsyc.1990.01810180089012. Epub 1990/06/01. [DOI] [PubMed] [Google Scholar]
- 38.Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nature genetics. 2011;43(10):977–83. doi: 10.1038/ng.943. Epub 2011/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nature genetics. 2008;40(9):1056–8. doi: 10.1038/ng.209. Epub 2008/08/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamshere ML, Walters JT, Smith R, Richards AL, Green E, Grozeva D, et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Molecular psychiatry. 2012 doi: 10.1038/mp.2012.67. Epub 2012/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen DT, Jiang X, Akula N, Shugart YY, Wendland JR, Steele CJ, et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Molecular psychiatry. 2013;18(2):195–205. doi: 10.1038/mp.2011.157. Epub 2011/12/21. [DOI] [PubMed] [Google Scholar]
- 42.Andreassen Ole A., Thompson Wesley K., Schork Andrew J., Ripke Stephan, Mattingsdal Morten, Kelsoe John R., et al. Improved Detection of Common Variants Associated with Schizophrenia and Bipolar Disorder Using Pleiotropy-Informed Conditional False Discovery Rate. PLoS genetics. 2013;9(4) doi: 10.1371/journal.pgen.1003455. Epub 2013/4/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gavaghan DJ, Moore RA, McQuay HJ. An evaluation of homogeneity tests in meta-analyses in pain using simulations of individual patient data. Pain. 2000;85(3):415–24. doi: 10.1016/S0304-3959(99)00302-4. Epub 2000/04/27. [DOI] [PubMed] [Google Scholar]
- 44.Ruderfer DM, Kirov G, Chambert K, Moran JL, Owen MJ, O’Donovan MC, et al. A family-based study of common polygenic variation and risk of schizophrenia. Molecular psychiatry. 2011;16(9):887–8. doi: 10.1038/mp.2011.34. Epub 2011/04/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pope HG, Jr., Lipinski JF., Jr. Diagnosis in schizophrenia and manic-depressive illness: a reassessment of the specificity of ‘schizophrenic’ symptoms in the light of current research. Archives of general psychiatry. 1978;35(7):811–28. doi: 10.1001/archpsyc.1978.01770310017001. Epub 1978/07/01. [DOI] [PubMed] [Google Scholar]
- 46.Ballenger JC, Reus VI, Post RM. The “atypical” clinical picture of adolescent mania. The American journal of psychiatry. 1982;139(5):602–6. doi: 10.1176/ajp.139.5.602. Epub 1982/05/01. [DOI] [PubMed] [Google Scholar]
- 47.Berk M, Hallam KT, McGorry PD. The potential utility of a staging model as a course specifier: a bipolar disorder perspective. Journal of affective disorders. 2007;100(1-3):279–81. doi: 10.1016/j.jad.2007.03.007. Epub 2007/04/17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.