

The integrated activities of Aurora B and CENP-I generate a molecular switch that maintains a robust spindle checkpoint signal at prometaphase kinetochores until they attain mature attachments to microtubules.
Abstract
The RZZ (Rod, ZW10, and Zwilch) complex and Mad1 proteins tightly associate with kinetochores to generate the spindle checkpoint signal, but they are released when a kinetochore forms mature microtubule attachments. Here we demonstrate that the centromere protein CENP-I is required to generate a stable association of RZZ and Mad1 with kinetochores. CENP-I also inhibits their removal by dynein stripping. This regulation of Mad1 and RZZ dissociation functions independently of Aurora B, which regulates their association. We show that the microtubule status of each kinetochore independently dictates the recruitment of Aurora B kinase, kinase activity on a kinetochore substrate, and loading of spindle checkpoint proteins. This dynamic regulation of Mad1 association by Aurora B is only uncovered when CENP-I is depleted, consistent with our finding that CENP-I inhibits the dissociation of Mad1. We conclude that the dual activities of Aurora B and CENP-I generate a molecular switch that maintains a robust spindle checkpoint signal at prometaphase kinetochores until they attain mature attachments to microtubules.
Introduction
The faithful segregation of genetic material during mitosis is critical to safeguard genomic integrity. Defects in this process lead to aneuploidy and cell death and are hypothesized to contribute to cancer development (Rieder and Maiato, 2004; Bharadwaj and Yu, 2004; Kops et al., 2005b). Chromosome segregation depends on kinetochores, large mitosis-specific structures that form on centromeres and make stable attachments to spindle microtubules (Santaguida and Musacchio, 2009; Kops et al., 2010). The spindle checkpoint signal is generated by kinetochores and inhibits mitotic progression until all kinetochores have attachments to the spindle (Musacchio and Salmon, 2007; Burke and Stukenberg, 2008). A single unattached kinetochore is sufficient to generate a mitotic arrest, but the mechanisms that initiate the signal at unattached kinetochores and ensure that it is strong enough to arrest cell cycle progression are incompletely understood.
Spindle checkpoint signaling involves the recruitment of mitotic arrest-deficient (Mad) and budding uninhibited by benzimidazoles (Bub) protein family members to kinetochores (Hoyt et al., 1991; Li and Murray, 1991; Gorbsky et al., 1998; Howell et al., 2004). The key effector of the spindle checkpoint is a complex of Mad1 and Mad2. Elegant structural and biophysical studies have demonstrated that Mad2 can exist in an active closed form (Mad2-c) and an inactive open form (Mad2-o; De Antoni et al., 2005). A dimer of Mad1 is recruited to kinetochores bound to Mad2-c. Once at kinetochores the Mad1–Mad2-c can catalyze the formation of soluble Mad2-o to Mad2-c, which generates a signal that inhibits the anaphase promoting complex, stabilizing important cell cycle substrates including cyclin B and securin (Murray and Kirschner, 1989; Li and Murray, 1991; Yamamoto et al., 1996a,b; Zou et al., 1999).
How kinetochores recruit the Mad1–Mad2-c proteins remains an area of active research. The direct binding site of the Mad1 protein is not known but a complex series of dependencies have been identified. Mad1 recruitment requires Bub1, Bub3, and BubR1 (Chen, 2002). These Bub proteins directly bind the kinetochore protein Knl1 on MELT repeats after they are phosphorylated by Mps1 (Krenn et al., 2012; Shepperd et al., 2012; Yamagishi et al., 2012). Knl1 also recruits the Zwint protein, which is required to recruit the RZZ complex (Kiyomitsu et al., 2007). The RZZ complex is composed of Rod, ZW10, and Zwilch and these three proteins have a second role in recruiting the minus end–directed motor cytoplasmic dynein (Basto et al., 2000; Chan et al., 2000). The Ndc80 complex is also required for Mad1 recruitment (Martin-Lluesma et al., 2002; McCleland et al., 2004). Once chromosomes begin to align to the metaphase plate, Mad1 is stripped from kinetochores by dynein (Howell et al., 2001).
The mitotic serine/threonine kinase Aurora B has been proposed to be at the top of a signaling cascade that regulates Mad1 recruitment. Aurora B acts as part of the chromosome passenger complex and directly phosphorylates proteins within the kinetochore (Vader et al., 2006; Santaguida et al., 2011). Aurora B is required for a spindle checkpoint arrest generated by Taxol, and Aurora B inhibitors prevent Mad1 recruitment to kinetochores of prometaphase cells (Ditchfield et al., 2003; Hauf et al., 2003). Aurora B activity is required to localize Bub1 and BubR1 to kinetochores, and this is at least in part through recruitment of the Mps1 kinase (van der Waal et al., 2012). Aurora B also phosphorylates the protein Zwint to generate a binding site for RZZ to recruit Mad1 and dynein (Wang et al., 2004; Kasuboski et al., 2011).
Surprisingly, cells in microtubule-depolymerizing drugs generate a functional spindle checkpoint in the presence of Aurora kinase inhibitors, though not after injection of function-blocking antibodies (Kallio et al., 2002; Ditchfield et al., 2003; Hauf et al., 2003). Because the inhibitors reduce but do not eliminate kinase activity the current model is that a small amount of Aurora kinase activity generates the spindle checkpoint signal in nocodazole (Santaguida et al., 2011). We previously performed a genetic screen to understand how cells arrest in mitosis with compromised Aurora B activity (Matson et al., 2012). We demonstrated that a complex containing the centromere protein CENP-I is required to signal the spindle checkpoint if Aurora B activity is compromised and that this role is conserved from yeast to humans. CENP-I can also regulate the dynamics of microtubules in the kinetochore after they generate mature attachments requiring Ndc80 (Amaro et al., 2010). However, it is unclear how CENP-I generates the spindle checkpoint signal after inhibition of Aurora B activity.
Unaligned kinetochores nucleate and bundle microtubules into a distinct class of short spindle microtubules known as preformed kinetochore fibers (PreK-fibers; Khodjakov et al., 2003; Tulu et al., 2006; Mishra et al., 2010). These bundles remain closely associated around kinetochores and are distinct from the ordered kinetochore fiber (K-fiber) microtubules that form mature attachments with the Ndc80 complex and facilitate chromosome movements. PreK-fibers exist before K-fibers and have important roles in ensuring the rapid attachment of kinetochores to spindle poles. Recent work out of our laboratory showed they can also recruit additional Aurora B to inner centromeres (Banerjee et al., 2014). However, PreK-fibers have not been implicated in generating spindle checkpoint signals.
Here we demonstrate that CENP-I regulates the dissociation of RZZ and Mad1 from kinetochores, whereas Aurora B dynamically regulates their association rates. CENP-I stabilizes Mad1 at kinetochores by extending its half-life and by inhibiting dynein-mediated stripping of Mad1. This stabilizing activity is required to maintain RZZ and Mad1 at kinetochores with low Aurora B activity and it ensures that a signaling kinetochore recruits a saturating amount of Mad1.
We recently demonstrated that microtubules can regulate Aurora B localization and activity in prometaphase (Banerjee et al., 2014). Here we extend this observation by showing that microtubule stimulation of Aurora B dynamically regulates the association of Mad1 at kinetochores. In addition, we show that the signal generated by microtubule stimulation of Aurora B is contained to a single kinetochore and adjacent kinetochores in the same cell can have distinct signaling events (chromosome autonomy). Our results lead to a model in which Aurora B activity is responsible for the recruitment of RZZ and Mad1 to kinetochores (Fig. 1 A). CENP-I then stabilizes these proteins at kinetochores by greatly enhancing their half-lives and by inhibiting dynein-mediated stripping until mature kinetochore microtubule attachments are formed. Together the local activities of CENP-I and the microtubule stimulation of Aurora B generate a molecular switch that underlies the chromosome autonomous nature of spindle checkpoint signaling.
Figure 1.

Aurora B activity or CENP-I are required to localize Mad1 and ZW10 to unattached kinetochores. (A) Simplified model depicting how Aurora B and CENP-H/I/K function to localize Mad1 at kinetochores. (B and C) Thymidine release assays demonstrating that either CENP-I or Aurora B activity are able to localize Mad1 and ZW10 to unattached kinetochores at the onset of mitosis. After Aurora B inhibition and CENP-I depletion both Mad1 and ZW10 are not at kinetochores. (D) Quantification of Mad1 and ZW10 kinetochore localization from B and C. All cells were treated with 3.3 µM nocodazole. Selected examples of kinetochores without Mad1 or ZW10 are indicated by yellow arrows. Error bars indicate standard deviation. *, P < 0.00005. A.U., arbitrary units. Bars: (white) 5 µm; (yellow) 1 µm.
Results
Kinetochore structure is not dramatically altered 48 h after CENP-I depletion
We examined the levels of a large set of proteins after depleting CENP-I from HeLa cells for 48 h to estimate the overall effect on kinetochore structure. CENP-I was depleted to <5% of control levels by siRNA (Fig. S1, A and B). We did not note any effect on gross chromatin morphology or chromosome structure after CENP-I depletion, although there was an increased number of prometaphase cells after CENP-I depletion as has been shown previously (Fig. S1 C; Liu et al., 2003; Amaro et al., 2010). CENP-I–depleted kinetochores retained CENP-A, Mis12, Knl1, CENP-C, Zwint, Rod, Mps1, Aurora B, P150, dynein, and CENP-F (Fig. S1 D; Matson et al., 2012). However, CENP-I depletion reduced CENP-H, -K, -O, and -P and ∼50% of Hec1 from kinetochores (Matson et al., 2012). Similar results were reported previously after depletion of CENP-H or CENP-K, which are binding partners of CENP-I (Cheeseman and Desai, 2008; Amaro et al., 2010). It is important to not deplete CENP-I for longer than 48 h to avoid the additional depletion of CENP-A, so all experiments are performed at a 48-h time point (Liu et al., 2003, 2006; Okada et al., 2006).
Aurora B activity and CENP-I cooperate to recruit and maintain RZZ and Mad1 at unattached kinetochores
CENP-I is required to send a checkpoint signal in the presence of low Aurora B activity (Matson et al., 2012). We tested whether HeLa cells could recruit the spindle checkpoint proteins RZZ, Mad1, Mad2, and BubR1 to kinetochores depleted of microtubules after Aurora B inhibition, CENP-I depletion, or both. Control or CENP-I–depleted cells were synchronized in S-phase by double thymidine block (Fig. 1 B). The cells were then released and, while still in G2, they were treated with nocodazole to depolymerize microtubules and the proteasome inhibitor MG132 to inhibit precocious mitotic exit. In addition the cells were treated with either the Aurora B inhibitor ZM447439 (ZM) or DMSO as a control. After the cells entered mitosis they were fixed and processed for immunofluorescence to visualize the localization of checkpoint proteins. Control, ZM-treated, and CENP-I–depleted cells all recruited nearly identical levels of Mad1 to unattached kinetochores (Fig. 1, B and D). However, cells depleted of both Aurora B activity and CENP-I had greatly reduced levels of kinetochore-bound Mad1. The RZZ complex protein ZW10 also required either Aurora B activity or CENP-I to localize to kinetochores in nocodazole (Fig. 1, C and D).
We note that the retention of Mad1 at kinetochores treated with Aurora inhibitors in nocodazole is in apparent contradiction to a previous study (Santaguida et al., 2011). However, the author’s overall conclusion that Aurora B is at the top of a signaling cascade that recruits the checkpoint proteins is supported by our findings.
To determine if CENP-I and/or Aurora B activity maintain Mad1 and ZW10 at kinetochores after they are loaded, CENP-I–depleted cells were prearrested in nocodazole for 2 h and then treated with Aurora B inhibitors and MG132 (Fig. S2 A). Cells depleted of either Aurora B activity or CENP-I recruited similar levels of Mad1 and ZW10 to kinetochores as controls (Fig. S2, B and C). However, CENP-I–depleted cells lost virtually all of their Mad1 and ZW10 from kinetochores after 1 h of ZM treatment. Interestingly, Mad1 and ZW10 staining was not dispersed in these cells. Instead they were found on large structures that had completely departed from the kinetochore but seemed to remain stable and in the vicinity of the chromatin for the duration of the experiment (Fig. S2, B and C). These structures also contained Mad2, but not BubR1, whose localization was dependent on Aurora B activity regardless of CENP-I status, as previously reported (Fig. S2, D and E; Ditchfield et al., 2003; Hauf et al., 2003). The structures could also be identified when CENP-I–depleted cells were treated with the structurally distinct Aurora B inhibitor Hesperadin (Fig. S3, A and B; Hauf et al., 2003). We also verified that Mad1 was in these structures using an alternative antibody against Mad1 and cells expressing GFP-Mad1, and we identified the structures using both U20S and 293T cells (Fig. S3, C–G). We conclude that when Aurora B activity is inhibited CENP-I is required to establish and maintain RZZ, Mad1, and Mad2 at kinetochores.
CENP-I is required for the slow dissociation rate of Mad1 at unattached kinetochores
There are two pools of Mad1 at unattached kinetochores in PTK2 cells. There is a highly dynamic pool (half-life of ∼12 s) and a stable pool that has a half-life >15 min (Howell et al., 2004; Shah et al., 2004). We used FRAP to measure the half-life of Mad1 at kinetochores of HeLa cells after Aurora B inhibition or after depletion of CENP-I. Control cells transiently expressing GFP-Mad1 in nocodazole had a highly stable pool of Mad1 that did not recover over the course of the experiment and a dynamic pool, although the dynamic pool of Mad1 in HeLa cells appears approximately twice as large as in PTK2 cells (Fig. 2, A–C; Shah et al., 2004). Inhibiting Aurora B activity did not significantly affect Mad1 recovery, suggesting that high Aurora B activity is not required to retain the stable pool of Mad1 at unattached kinetochores. CENP-I–depleted cells had a larger pool of dynamic Mad1 at kinetochores compared with controls, although the half-life of the dynamic pool did not significantly change (Fig. 2 D). Moreover, the stable population of Mad1 displayed a steady rate of recovery in CENP-I–depleted cells. Consistent with previous analyses we found that Mad1 recovery followed biphasic kinetics that were best fit using the sum of two exponentials (Fig. 2 D; Howell et al., 2004). In control cells the second phase of recovery was extremely slow with a half-life of 145 min, reflecting the remarkably stable nature of this population. However, the slow phase of recovery had a half-life of only 4 min in CENP-I–depleted cells. Thus, the population of stable Mad1 at kinetochores turns over ∼36 times faster in CENP-I–depleted cells than it does in controls. We conclude that CENP-I is required to generate a stable population of Mad1 at unattached kinetochores.
Figure 2.

CENP-I increases the half-life of Mad1 at unattached kinetochores. (A) Images of Mad1-GFP FRAP in control, CENP-I–depleted, and ZM-treated cells arrested in nocodazole. (B) Recovery dynamics of Mad1-GFP after photobleaching demonstrating that CENP-I–depleted cells have a larger initial recovery of Mad1 and a faster turnover of stable Mad1. (C) Total recovery of Mad1-GFP at 120 s after photobleaching. (D) Scatter plot displaying the natural log of the normalized unrecovered fluorescence over time. The biphasic nature of Mad1 recovery is illustrated by overlaid lines. CENP-I–depleted cells have a fast phase of initial Mad1 recovery similar to controls but the pool of stable Mad1 in CENP-I–depleted cells has a greatly decreased half-life relative to control. Red arrows in A indicate FRAP targets. FRAP data are from n = 30 experiments. Error bars indicate standard deviation. *, P < 5 × 10−7; **, P < 2 × 10−11. Bars: (white) 5 µm; (yellow) 1 µm.
CENP-I–depleted cells rapidly lose Mad1 from kinetochores in the presence of microtubules
Up to this point our experiments had been performed in nocodazole, but Mad1 is reported to be absent in CENP-I–depleted cells when microtubules are present, including during early mitosis (Liu et al., 2003; Matson et al., 2012). We measured the rate that Mad1 is lost from CENP-I–depleted kinetochores after exposure to microtubules. Cells were washed out of nocodazole to allow microtubule polymerization and fixed for immunofluorescence, and the amount of Mad1 at kinetochores was quantified at 4-min time points after washout (Fig. 3, A–C). Control cells retained Mad1 at most kinetochores 16 min after nocodazole washout even though bipolar spindles had formed. Loss of Mad1 from kinetochores of control cells was only obvious after 20 min, when strong microtubule bundles consistent with K-fibers appeared. In contrast, Mad1 was lost from most kinetochores of CENP-I–depleted cells 8 min after nocodazole washout and was virtually unobservable after 12 min. Interestingly, at 12 and 16 min many CENP-I–depleted cells had lost Mad1 from kinetochores and this correlated with the accumulation of Mad1 at microtubule foci that are most likely forming spindle poles. When cells were washed out of nocodazole in the presence of ZM Mad1 was retained at kinetochores for up to 20 min (Fig. 3 C). However, we found that ZM treatment also resulted in a slowed rate of microtubule polymerization after nocodazole washout compared with controls and CENP-I–depleted cells (Fig. S3 H). We conclude that the dissociation rate of Mad1 exceeds the association rate in CENP-I–depleted cells when microtubules are present, resulting in the premature dissociation of Mad1 from kinetochores.
Figure 3.

CENP-I–depleted kinetochores lose Mad1 from kinetochores faster than control kinetochores in the presence of microtubules. (A) Control cells retain Mad1 at kinetochores up to 20 min after nocodazole washout. (B) CENP-I–depleted cells lose all Mad1 from kinetochores between 8–12 min after nocodazole washout, even before a bipolar spindle can form. At 16 min, Mad1 can inbriefly be seen at the vertices of microtubule bundles. (C) Quantification of mean Mad1 kinetochore levels across all kinetochores from A and B. Error bars indicate standard deviation. *, P < 10−3; **, P < 10−4; ***, P < 0.05. Indicated statistical significance is between control and CENP-I–depleted groups. A.U., arbitrary units. Bars, 5 µm.
Dynein prematurely strips Mad1 from kinetochores with microtubules in CENP-I–depleted cells
The disappearance of Mad1 from kinetochores of CENP-I–depleted cells and its appearance at spindle poles is consistent with dynein-dependent movements. Dynein does not normally strip RZZ and Mad1 from kinetochores to silence the spindle checkpoint until kinetochores form mature attachments to spindle microtubules (Howell et al., 2001). However, the rapid kinetics of Mad1 loss from unaligned kinetochores in CENP-I–depleted cells suggested that dynein stripping was occurring before proper kinetochore attachments had formed.
To test if CENP-I prevents the premature stripping of Mad1 by dynein we washed cells out of nocodazole to synchronize the stripping of Mad1 from kinetochores and inhibited dynein activity by expressing CC1-GFP. This CC1 fragment of P150/Dynactin inhibits dynein walking activity without affecting dynein localization (Quintyne et al., 1999). Mad1 remained at kinetochores of control cells 10 min after nocodazole washout (Fig. 4 A). In contrast, Mad1 staining was lost from kinetochores and could be visualized at spindle poles in CENP-I–depleted cells. After dynein inhibition both control and CENP-I–depleted cells retained Mad1 at kinetochores after nocodazole washout (Fig. 4, A–C). We conclude that a function of CENP-I is to prevent dynein-mediated stripping of Mad1 at kinetochores that have lateral attachments with microtubules but have not formed mature attachments to the spindle. Furthermore, dynein stripping does not normally carry sufficient Mad1 to poles for it to be localized as discrete foci. Our ability to localize Mad1 to poles in CENP-I–depleted cells is consistent with an increased flow of Mad1 from kinetochores caused by increased dynein loading rates and poor retention of Mad1 at kinetochores.
Figure 4.

CENP-I–depleted kinetochores fail to inhibit dynein-mediated stripping of Mad1. (A) Immunofluorescence images of Mad1 in control and CENP-I–depleted cells 10 min after nocodazole washout, with or without expression of the dynein inhibitor CC1. Control cells retain Mad1 at kinetochores after nocodazole washout, but CENP-I–depleted cells rapidly lose Mad1 from kinetochores and accumulate it at spindle poles in a dynein-dependent manner. (B) Quantification of the total number of Mad1-positive kinetochores in cells from conditions depicted in A. (C) Immunofluorescence images of CENP-I–depleted cells demonstrating that inhibition of dynein does not prevent recruitment of Mad1 to unattached kinetochores, but does prevent loss of Mad1 from kinetochores after nocodazole washout. Centromeres are labeled to demonstrate that Mad1 is at kinetochores. Blue arrows indicate position of spindle poles. Cy5-labeled anti-Mad1 antibody is displayed here in green for ease of viewing. Error bars indicate standard deviation. *, P < 10−7; **, P < 10−3. Bars, 5 µm.
Development of an assay to visualize the association dynamics of spindle checkpoint proteins
Our data demonstrate that CENP-I regulates the dissociation of Mad1 from kinetochores by increasing its half-life and inhibiting premature stripping by dynein. However, we did not detect a role for Aurora B activity in stabilizing existing Mad1 at unattached kinetochores (Fig. 2). This is somewhat surprising because Aurora B activity and CENP-I can each localize RZZ and Mad1 to unattached kinetochores independently of each other (Fig. 1). One explanation is that there are two pathways that can independently recruit RZZ and Mad1: one pathway that requires Aurora B activity and another pathway that requires CENP-I. An alternative and simpler model is that Aurora B activity regulates the association kinetics of RZZ and Mad1 to kinetochores whereas CENP-I regulates their dissociation rates. Because the dissociation rate of stable Mad1 from kinetochores is essentially zero (Fig. 2), even weak Aurora B activity would eventually saturate the kinetochore binding sites. We strongly favor this model where Aurora B regulates the association of Mad1 and CENP-I inhibits its dissociation for three reasons. First, when cells are injected with function-blocking antibodies against Aurora B they lose checkpoint activity even though CENP-I is present, arguing for a single loading pathway (Kallio et al., 2002). Consistent with the idea that low levels of Aurora activity are sufficient to generate a spindle checkpoint signal there is residual Aurora activity in cells treated with Aurora inhibitors and Ipl1 mutants in budding yeast can be rescued through inhibition of PP1 phosphatase (Francisco and Chan, 1994; Santaguida et al., 2010). Second, Aurora B phosphorylates Zwint to drive loading of RZZ, Mad1, and other outer kinetochore proteins, which provides for a direct role for Aurora B in Mad1 recruitment (Kasuboski et al., 2011). Third, our data show that CENP-I regulates two different dissociation reactions: the half-life of Mad1 is reduced as measured by FRAP and dynein can prematurely strip spindle checkpoint proteins in the absence of CENP-I.
We designed a test to determine if Aurora B activity controls the recruitment of RZZ and Mad1 to kinetochores and if the CENP-I pathway regulates their dissociation. Our assay is based on three recent findings. First, recent work in our laboratory showed that Aurora B localization and activity is stimulated by PreK-fibers (Banerjee et al., 2014). Second, in our nocodazole washout assays some kinetochores associate with spindle microtubules or contact/nucleate PreK-fibers that are independent of the spindle, whereas others do not have any microtubule structures associated with them (Figs. 3 and S4 A; Tulu et al., 2006; Mishra et al., 2010). This provides an opportunity to directly visualize chromosome-autonomous regulation of the spindle checkpoint. A critical prediction is that the kinetochores with microtubules should have greater Aurora B protein and activity than the kinetochores of adjacent chromosomes without microtubule contacts. The direct comparison of kinetochores in the same cell eliminates many concerns associated with immunofluorescence artifacts. Third, our identification of methods to increase the dissociation rates of Mad1 (CENP-I depletion) presented an opportunity to identify factors that regulate the kinetochore association rates of RZZ and Mad1, because depletion of CENP-I generates conditions that greatly increase Mad1 turnover dynamics (Fig. 2). We will demonstrate that kinetochores in contact with microtubules recruit Aurora B and stimulate Aurora B activity, which leads to the recruitment of RZZ and Mad1. In contrast, unattached kinetochores in the same cell have low Aurora B activity and require CENP-I to retain checkpoint proteins.
Microtubules at kinetochores recruit Aurora B to centromeres to phosphorylate kinetochores
We first asked whether Aurora B was specifically enriched at kinetochores with associated microtubules during spindle formation after nocodazole washout in unperturbed cells. We found that kinetochores with microtubules (either as small foci or connected to forming poles) had levels of centromere Aurora B that were almost three times higher than kinetochores without detectable microtubules (Fig. 5, A and B; and Fig. S4 B). In addition, the mean Aurora B levels across all centromeres were significantly enhanced (Fig. 5, A and C). Aurora B activity was similarly enriched at kinetochores associated with either PreK-fibers or spindle microtubules, as measured by phosphorylation of the inner centromere Aurora B substrate CENP-A serine 7 (Fig. 5, D and E; and Fig. S4 C). The fact that a threefold enrichment of kinase at kinetochores generated a fourfold increase in kinetochore activity suggests that enrichment of kinase, and not further kinase activation, is the major form of chromosome passenger complex regulation. CENP-I–depleted cells displayed the same pattern of Aurora B localization and activity as controls, arguing that CENP-I has no role in Aurora B localization or activation (Fig. S4, D and E). We conclude that the presence of microtubules at kinetochores correlates with chromosome-autonomous recruitment of Aurora B.
Figure 5.

Centromere Aurora B localization and activity is enhanced by microtubules at kinetochores. (A) Aurora B localizes to all centromeres during prometaphase and in nocodazole. After nocodazole washout, Aurora B specifically localizes to centromeres where kinetochores overlap with microtubules and is reduced or lost at kinetochores without microtubules. (B) After nocodazole washout Aurora B is specifically enhanced at kinetochores with microtubules and is reduced at kinetochores without microtubules. (C) Quantification of Aurora B centromere intensities in nocodazole and after nocodazole washout demonstrating an increase in overall Aurora B staining across all centromeres after nocodazole washout. (D) Aurora B activity as visualized by p(S7)CENP-A phosphorylation in prometaphase, in nocodazole, and after nocodazole washout. Aurora B activity correlates with the presence of microtubules at kinetochores. (E) p(S7)CENP-A phosphorylation levels are high at kinetochores with microtubules and low at kinetochores without microtubules after nocodazole washout. Yellow arrows indicate select examples of kinetochores without detectable microtubules. Blue arrows indicate select examples of kinetochores with associated microtubules. Each image represents multiple Z-slices. Error bars indicate standard deviation. *, P < 0.05; **, P < 5 × 10−7. Noc, nocodazole; A.U., arbitrary units. Bars: (white) 5 µm; (yellow) 1 µm.
Chromosome-autonomous recruitment of RZZ and Mad1to kinetochores is revealed in CENP-I–depleted cells
Having demonstrated that we could generate and visualize chromosome-autonomous localization of Aurora B in a nocodazole washout assay, we used the system to monitor the role of local Aurora B activity in spindle checkpoint signaling. We performed nocodazole washout experiments in both control and CENP-I–depleted cells and stained for Mad1, Mad2, or ZW10. In control cells, all kinetochores retained Mad1, Mad2, and ZW10 (Fig. 6, A and B; and Fig. S4, G–P). This is expected if CENP-I prevents the dissociation of spindle checkpoint proteins and keeps Mad1 levels at kinetochores saturated when there is low Aurora B activity. We reasoned that we could uncover the chromosome-autonomous nature of checkpoint protein recruitment by depleting CENP-I. In fact, kinetochores associated with PreK-fibers or spindle microtubules recruited Mad1, Mad2, and ZW10 in CENP-I–depleted cells, whereas the kinetochores that were not associated with microtubules had fivefold lower amounts of Mad1, Mad2, and ZW10 (Figs. 6 and S4, F–P). Finally we fixed control and CENP-I–depleted cells in early prometaphase and stained for Tubulin, Aurora B, and Mad1 (Fig. S4 Q). We found that although Aurora B localized to kinetochores associated with microtubules in both groups Mad1 could only be localized to kinetochores near spindle microtubules in CENP-I–depleted cells. These data strongly support our argument that CENP-I inhibits the dissociation of spindle checkpoint proteins from signaling kinetochores. They also demonstrate that the enrichment of Aurora B by PreK-fibers and spindle microtubules can dynamically recruit spindle checkpoint proteins.
Figure 6.

Microtubules trigger Mad1 recruitment to individual kinetochores in CENP-I–depleted cells. (A) Control-depleted cells localize Mad1 to kinetochores in prometaphase, during nocodazole treatment, and 10 min after nocodazole washout. CENP-I–depleted cells have no Mad1 at kinetochores in prometaphase but can fully recruit Mad1 to kinetochores in nocodazole. After nocodazole washout, Mad1 is specifically recruited to kinetochores that overlap with microtubules and is absent from kinetochores without microtubules. (B) Quantification of Mad1 kinetochore intensities from A showing that CENP-I–depleted cells fully recruit Mad1 to unattached kinetochores in nocodazole but lose most Mad1 from kinetochores after nocodazole washout. (C) Quantification of Mad1 intensities at kinetochores with or without microtubules after nocodazole washout. Control cells have equal amounts of Mad1 at kinetochores with or without microtubules, whereas CENP-I–depleted cells have fivefold more Mad1 at kinetochores with microtubules. Yellow arrows indicate select examples of kinetochores without microtubules. Blue arrows indicate select examples of kinetochores with associated microtubules. Insets contain multiple Z-sections for clarity. Error bars indicate standard deviation. *, P < 0.00005; **, P < 0.005. Noc, nocodazole; A.U., arbitrary units. Bars: (white) 5 µm; (yellow) 1 µm.
CENP-I–depleted kinetochores fail to retain Mad1 at anti-poleward kinetochores in Monastrol
We sought a nocodazole-independent method to test the hypothesis that CENP-I inhibits the dissociation of Mad1 from kinetochores with premature kinetochore attachments. Cells treated with the Eg5 inhibitor Monastrol generate an ideal situation to test our hypothesis. In these cells, poleward-facing kinetochores are attached to the central pole and lack Mad1, whereas anti-poleward kinetochores have immature attachments to PreK-fibers and recruit Mad1 (shown schematically in Fig. 7 A; Kapoor et al., 2000; Maliga et al., 2002; Cochran et al., 2005). If CENP-I is required for kinetochores with immature microtubule attachments to retain Mad1 then we predict that CENP-I–depleted cells would lack Mad1 at anti-poleward kinetochores in Monastrol.
Figure 7.

Monastrol-treated cells do not retain Mad1 and Mad2 at anti-poleward kinetochores after CENP-I depletion. (A) Cartoon representation of kinetochore–microtubule attachments in Monastrol-treated cells. (B) Immunofluorescence images of cells after siRNA depletion and Monastrol treatment. Control cells retain Mad1 and Mad2 at anti-poleward kinetochores and recruit BubR1 to all kinetochores. CENP-I–depleted cells can still recruit BubR1 to kinetochores but fail to retain Mad1 and Mad2 at anti-poleward kinetochores. (C) Quantification of B. White arrows indicate select examples of anti-poleward facing kinetochores. Error bars indicate standard deviation. *, P < 0.005. Bars: (white) 5 µm; (yellow) 1 µm.
As expected, Mad1 and Mad2 were only observed at anti-poleward kinetochores, whereas BubR1 was found at all kinetochores in control cells treated with Monastrol (Fig. 7, B and C). In contrast, Mad1 and Mad2 were depleted from both poleward and anti-poleward kinetochores in CENP-I–depleted cells treated with Monastrol and BubR1 remained at all kinetochores in CENP-I–depleted cells. Together our data demonstrate that the CENP-I pathway prevents the loss of Mad1 from kinetochores that have not generated mature microtubule attachments.
Mad1 mislocalization in CENP-I–depleted cells is not a result of depletion of Hec1
The kinetochore binding protein Hec1 (also known as Ndc80) is another protein hypothesized to prevent the premature stripping of Mad1 (DeLuca et al., 2003). CENP-I depletion for >72 h prevents localization of Hec1 (Liu et al., 2006), suggesting that CENP-I depletion may be affecting dynein stripping indirectly by reducing kinetochore levels of Hec1. However, we and others have found that the 48-h CENP-I depletion used throughout this study depletes less than half of the kinetochore Hec1 even though CENP-I is >95% depleted from kinetochores (Fig. S5, A–C; Amaro et al., 2010; Matson et al., 2012). However, we assayed whether CENP-I simply recruits Hec1 to stabilize Mad1 (Martin-Lluesma et al., 2002; DeLuca et al., 2003; McCleland et al., 2003). We depleted Hec1 levels to <5% of controls, which is significantly greater than the ∼50% depletion of Hec1 at kinetochores we observed after treatment with CENP-I siRNA (Fig. S5, D and E). CENP-I levels were not affected by Hec1 depletion at kinetochores in prometaphase, in nocodazole, or after nocodazole washout (Fig. S5, F and G). Mad1 was depleted from prometaphase kinetochores after Hec1 depletion, but Mad1 was present at kinetochores in cells treated with nocodazole as previously reported and similar to CENP-I–depleted cells (Fig. S5, H and I; Martin-Lluesma et al., 2002). After nocodazole washout, Hec1-depleted cells lost Mad1 from kinetochores faster than control cells, but not as quickly as CENP-I–depleted cells (Fig. S5, J and K). However, under these conditions we found that kinetochores with microtubules had slightly lower levels of Mad1 than unattached kinetochores (Fig. S5 L). This is the opposite of what we observe after CENP-I depletion. Thus, even when we directly deplete Hec1 to levels significantly lower than those seen after CENP-I depletion, we fail to recapitulate the effects on Mad1 localization observed in CENP-I–depleted cells. We conclude that CENP-I and Hec1 have distinct roles in checkpoint signaling.
Discussion
A single unattached kinetochore is sufficient to generate a spindle checkpoint signal robust enough to arrest the metaphase to anaphase transition. Our data provides a mechanistic framework to understand the on/off nature of this signal. The key event is the localization of RZZ and Mad1 to the kinetochore, which is both necessary and sufficient to generate a spindle checkpoint signal (Maldonado and Kapoor, 2011). The proper localization of proteins to a subcellular structure is a function of the number of available binding sites, the association kinetics, and the dissociation kinetics. We have demonstrated that a pathway requiring CENP-I regulates the dissociation of RZZ and Mad1 from individual kinetochores. We also demonstrated that Aurora B activity regulates the association of RZZ and Mad1 onto each kinetochore.
We suggest that the independent regulation of both association and dissociation reactions is an essential feature of building this tightly controlled molecular switch (Fig. 8 A). Individual kinetochores can exist in three states during prometaphase and metaphase (Fig. 8 B): unattached without PreK-fibers, with lateral (immature) attachments to spindle microtubules or PreK-fibers mediated by dynein, or with properly attached “end-on” to spindle microtubules through the Ndc80 complex. In the first case weak Aurora B activity is sufficient to generate a robust spindle checkpoint signal because CENP-I ensures that the dissociation rate of RZZ and Mad1 is essentially zero. The event that is regulated by CENP-I is not known and could either be the recruitment of a protein receptor or a posttranslational modification to a protein that recruits RZZ. A reasonable candidate is the phosphorylation of Zwint by Aurora B, which is required for kinetochore localization of RZZ (Kasuboski et al., 2011). In this case, CENP-I could inhibit the opposing phosphatase. The spindle checkpoint signal remains robust at kinetochores with lateral attachments because CENP-I continues to inhibit the dissociation of Mad1 from kinetochores. There are two nonexclusive models. First CENP-I may make the attachment of RZZ to kinetochores so tight that it cannot be displaced by dynein. Second, CENP-I could alter the cargo loading of dynein so that it generates PreK-fibers but cannot strip Mad1. In addition, Aurora B signaling is stronger in the presence of immature microtubule attachments, which leads to robust loading of Mad1 (Salimian et al., 2011). After attachments mature, RZZ and Mad1 are quickly removed because these attachments down-regulate Aurora B activity and inhibit CENP-I, which activates dynein stripping (Fig. 8, A and B).
Figure 8.
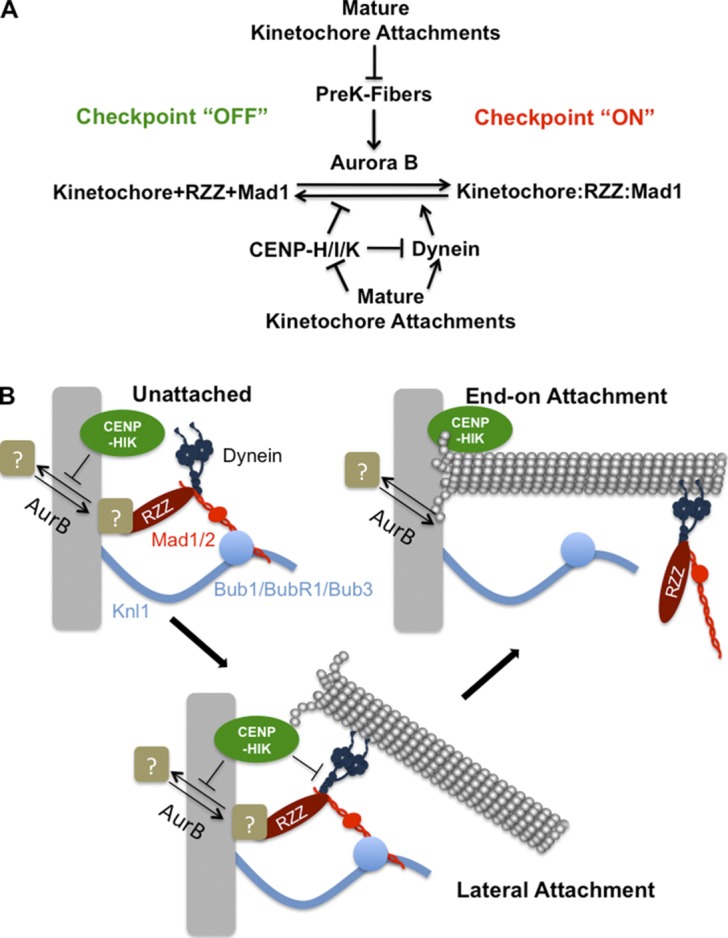
Model for generation and maintenance of the spindle assembly checkpoint. (A) Aurora B activity normally recruits RZZ and Mad1 to unattached kinetochores. PreK-fibers can enhance this process through stimulation of Aurora B activity. CENP-I functions to inhibit their dissociation from kinetochores by stabilizing this interaction and through inhibition of dynein stripping. The formation of mature kinetochore–microtubule attachments suppresses PreK-fibers and extinguishes the stabilizing activity of CENP-I, which allows dynein to strip RZZ and Mad1 from kinetochores. (B) Aurora B and the CENP-H/I/K proteins regulate the recruitment of RZZ by an unknown mechanism, which is shown by recruitment of a receptor that could either be a protein or a posttranslational modification. Kinetochores form early lateral attachments to microtubules through dynein, but the pathway involving CENP-I (CENP-H/I/K) inhibits the dynein-dependent stripping of checkpoint proteins. In this case CENP-I could either inhibit dynein function or make the attachment of RZZ/Mad1 to kinetochores so tight that it can’t be removed by dynein. Upon transition to a mature end-on attachment, CENP-I is turned off and dynein carries checkpoint proteins from kinetochores.
The CENP-H/I/K complex has an established role at the kinetochore in regulating the microtubule dynamics of “end-on” attached microtubules (Amaro et al., 2010). We have previously shown that CENP-I plays no role in the spindle checkpoint signal generated by Taxol, suggesting that CENP-I’s spindle checkpoint function is turned off by the presence of end-on attached microtubules (Matson et al., 2012). Thus a reasonable hypothesis is that the CENP-H/I/K complex locks Mad1 in a stable kinetochore complex until this activity is turned off through the engagement of CENP-H/I/K with end-on attached microtubules (Fig. 8 B).
It is well-established that spindle checkpoint proteins dissociate from kinetochores that form mature microtubule attachments and align to the metaphase plate (Gorbsky et al., 1998; Waters et al., 1998). These experiments have entrenched the concept of chromosome-autonomous dissociation of checkpoint proteins from kinetochores. Our data suggest that the pathway involving CENP-I is the key regulator of RZZ and Mad1 release after microtubule attachment. Moreover, whether the association of RZZ and Mad1 was regulated or constitutive could not previously be measured because the dissociation reaction is so tightly regulated by the CENP-I pathway. However, by depleting CENP-I we enhanced RZZ and Mad1 dissociation and could visualize their association dynamics. Thus our data also demonstrate that the enrichment of Aurora B activity to kinetochores by immature microtubule attachments can dynamically drive recruitment of RZZ and Mad1 to kinetochores.
Our model can explain a confusing observation. Aurora B activity must be high to maintain a spindle checkpoint arrest in Taxol but not nocodazole (Hauf et al., 2003; Ditchfield et al., 2003; Matson et al., 2012). Cells in nocodazole can tolerate the reduced loading of Mad1 by Aurora B inhibition because Mad1 dissociation is inhibited by CENP-I. However, the CENP-I pathway is turned off by the stable kinetochore–microtubule attachments in Taxol-arrested cells and are therefore dependent on continuous Mad1 loading by Aurora B (Matson et al., 2012). Our model cannot fully explain why Aurora B is required during prometaphase to localize RZZ, because it predicts that CENP-I should prevent the removal of RZZ until end-on attachments are generated, like it does in nocodazole (Kasuboski et al., 2011; Kops et al., 2005a). It is possible that there are transient end-on attachments that inhibit CENP-I, because of high phosphorylation of the Ndc80 complex. The recent identification of a direct binding event between Bub1 and Mad1 in Caenorhabditis elegans is an exciting finding (Moyle et al., 2014). However, it is also a potential source of confusion because the levels of Bub1/BubR1 are reduced after inhibition of Aurora B in both prometaphase and nocodazole (Ditchfield et al., 2003; Hauf et al., 2003; Fig. S2). We suggest that there are more molecules of Knl1/Bub1 in the kinetochore than there are RZZ and that RZZ is the limiting component for Mad1 binding. Consistent with this idea, the levels of RZZ, not Bub1, are more closely correlated with Mad1 binding and Xenopus laevis kinetochores have approximately three times more KMN components than RZZ components in nocodazole (Emanuele et al., 2005). There remains much to be learned about the dynamic conversion from lateral to end-on kinetochore–microtubule attachments and how this is coordinated with spindle checkpoint signaling during prometaphase.
An important future direction is to identify how CENP-I controls the dissociation of RZZ and Mad1 from kinetochores. The simplest model is that CENP-I produces a tight binding event between RZZ and kinetochores, which generates an extended half-life and prevents dynein from stripping it off. Alternatively, it is possible that CENP-I has two independent functions: one that increases the stability of the checkpoint complexes at kinetochores and a second that inhibits dynein stripping. CENP-H depletion was shown to increase the stability of K-fiber microtubules in metaphase (Amaro et al., 2010). Thus it is also possible that CENP-I functions to prevent the untimely maturation of lateral attachments. In any case, some event must occur after proper microtubule attachment to allow for stripping of RZZ and Mad1.
Finally, it is established that PreK-fibers can increase the rate of spindle–kinetochore attachment by extending the spindle capture surface (Khodjakov et al., 2003). However, the existence of PreK-fibers has not been considered in terms of spindle checkpoint signaling. Our data suggest that spindle checkpoint mechanisms are exquisitely tuned to work with this class of microtubules. We demonstrate that PreK-fibers can enrich Aurora B kinase at inner centromeres, increase the phosphorylation of adjacent kinetochores, and recruit spindle checkpoint proteins. We also show that CENP-I is required to prevent dynein stripping along PreK-fibers. Yet it has been standard practice for 30 years to trigger mitotic arrest with microtubule-destabilizing drugs. Clearly, when CENP-I is active the basal amount of Aurora B activity in nocodazole is sufficient to generate a checkpoint signal and microtubule-dependent stimulation is not essential. However, in the future the concentration of spindle poisons will need to be carefully noted. There are significant amounts of kinetochore-associated microtubules at 0.33 µM nocodazole and in a recent paper we demonstrate that these can recruit additional Aurora B (Jordan et al., 1992; Banerjee et al., 2014). However, at 3.3 µM nocodazole there are no microtubules around kinetochores or additional Aurora B at centromeres. Thus, depending on the concentration of drug used one can induce or repress additional inner centromere Aurora B recruitment and activity.
Materials and methods
Cell culture, transfections, and immunoblotting
HeLa T-Rex (Invitrogen), U20S, and 293T cells were maintained in DMEM supplemented with 10% FBS. Cells were plated at 30% confluency onto lysine-coated coverslips in 12-well dishes (Corning) overnight. siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) and plasmid transfections were performed using Lipofectamine2000 (Invitrogen) according to the manufacturer’s protocol. A smart pool of siRNA oligos against CENP-I were purchased from Thermo Fisher Scientific (M-029617-01; 5′-GGUACAAGGUGAAUAAUUA-3′, 5′-CAGCAAGACUUAUCAAGAA-3′, 5′-GCUGGUAUUUGGACUAUUU-3′, and 5′-GUGAAGCAUUCCUGUAUAA-3′) and cells were treated with a final concentration of 20 nM siRNA. Experiments were performed 48 h later. Hec1 knockdowns were performed using a custom oligo (5′-GAGUAGAACUAGAAUGUGAUU-3′; QIAGEN). Cells were thymidine arrested for 24 h before treatment with 75 nM siRNA and released into fresh media. 12 h later the cells were treated with an additional 75 nM siRNA and thymidine arrested. After 12 h, the cells were released from thymidine and assayed 8 h later when the majority of the population was in mitosis. For dynein inhibition experiments, the pEGFP-CC1 vector expressing CC1-GFP was provided by K. Pfister (University of Virginia, Charlottesville, VA) and contains the CC1 gene fragment initially reported by T. Schroer (Johns Hopkins University, Baltimore, MD). This vector contains amino acids 217–548 (a region known as CC1) of P150Glued with a C-terminal eGFP fusion expressed under the control of a CMV promoter. Cells in a 12-well dish were transfected with 200 ng pEGFP-CC1 or control plasmid and assayed after 24 h. Monastrol (Tocris Bioscience) was used at 100 µM for 2 h. ZM447439 (Enzo Life Sciences) was used at 2 µM final concentration and Hesperadin (Tocris Bioscience) was used at 100 nM unless otherwise noted.
Cell lysates for Western blotting were generated by scraping cells from the culture plates and pelleting them at 1,000 rpm in a tabletop centrifuge. Pellets were washed once in PBS, resuspended in 2× SDS sample buffer, sonicated, and loaded onto gels.
Nocodazole treatments and nocodazole washout assays
Nocodazole (Sigma-Aldrich) was used at 3.3 µM throughout the study, a concentration sufficient to depolymerize all microtubules, and cells were treated for 2 h unless otherwise indicated. For washout assays the cells were arrested in nocodazole for 2 h and the media were aspirated. The cells were then washed once with PBS and incubated in fresh media for 10 min unless noted otherwise.
Immunofluorescence and quantitative immunofluorescence
Unless otherwise stated, cells on poly-lysine–coated coverslips were fixed in 4% paraformaldehyde and 0.5% Triton X-100 for 20 min and stained in PBST plus 5% BSA. For staining with anti-dynein and anti–Centrin-2 antibodies, cells were fixed in −20°C methanol for 10 min and then washed in PBST and stained in PBST plus 5% BSA. For staining with the anti-ZW10 antibody, cells were first fixed in PBS plus 3.5% paraformaldehyde for 7 min and then extracted in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% BSA, and 0.2% Triton X-100 for 2 min. Staining was then performed in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% BSA. Staining was performed using the following primary antibodies: anti–CENP-I (rabbit polyclonal antibody against full-length human CENP-I), anti-Mad1 (rabbit polyclonal antibody against full-length human Mad1), and anti-BubR1 (rabbit polyclonal antibody against X. laevis BubR1; all obtained from P.T. Stukenberg, University of Virginia, Charlottesville, VA); anti-ZW10 (rabbit), anti-Rod (rabbit), anti–CENP-F (rabbit), anti–CENP-C (rabbit), and anti-Mis12 (rabbit; all gifts from T.J. Yen, Fox Chase Cancer Center, Philadelphia, PA); anti-Zwint (rabbit) and an alternate anti-ZW10 (rabbit) antibody (a gift from G.K. Chan, University of Alberta, Edmonton, Alberta, Canada); anti-Mad1 (rabbit; a gift from P. Meraldi, Swiss Federal Institute of Technology Zurich, Zurich, Switzerland); anti-Hec1 (mouse; GeneTex); anti-centromere antibodies (ACA; human; Antibodies Inc.); anti-Mad2 (rabbit; a gift from G. Gorbsky, Oklahoma Medical Research Foundation, Oklahoma City, OK); anti-Tubulin DM1α (mouse; Sigma-Aldrich); anti–Aurora B (mouse; BD); and anti-pS7CENP-A (rabbit; Cell Signaling Technology).
Quantitative immunofluorescence was performed using ImageJ software (National Institutes of Health) and calculations were performed in Microsoft Excel. In brief, a circular region encompassing one kinetochore was measured and mean gray level intensity was measured for both the experimental antibody signal and ACA, as well as background in both channels. Final intensity was calculated by taking the intensity of the experimental antibody minus background and dividing it by the intensity of the corresponding ACA signal minus background. Approximately 10 kinetochores were measured in 10 cells corresponding to 100 kinetochores per reported intensity value. To measure whether kinetochores had microtubules or not, ACA signals were identified within a Z-series composite image with the tubulin and Mad1 channels turned off. When an ACA dot was identified, the tubulin channel was turned on and kinetochores with signal that could be distinguished from background were considered to have microtubules.
FRAP experiments
HeLa cells were cultured in 2-well chamber slides (Thermo Fisher Scientific) and treated with control or CENP-I siRNA as described in Cell culture, transfections, and immunoblotting. GFP-Mad1 plasmid was a gift from E.D. Salmon (University of North Carolina, Chapel Hill, NC) and consists of full-length human Mad1 N-terminally fused to eGFP and expressed under control of the CMV promoter (Shah et al., 2004). The plasmid was transfected using Lipofectamine2000 24 h before completion of the siRNA knockdown protocol. Nocodazole was added to the media 2 h before FRAP experiments. For the ZM treatment group, ZM was added immediately before FRAP experiments. Mitotic cells were identified by eye based on which GFP-Mad1 dots were minimally visible. Laser power and digital gain were then increased for imaging and FRAP. FRAP analysis was performed using ImageJ and Microsoft Excel. The gray level intensity of the Mad1-GFP target was measured before bleaching, immediately after, and then every 5 s for up to 2 min. Background measurements of gray level intensity taken from an adjacent region of the cytoplasm were subtracted at each time point. FRAP data for ∼30 kinetochores were then averaged for each condition. FRAP analysis was performed as previously described (Howell et al., 2004). FRAP kinetics were determined by calculating a normalized unrecovered fluorescence at each time point: (mean maximal fluorescence recovery − fluorescence at time t)/(mean maximal fluorescence recovery − fluorescence immediately after photobleaching). The natural log of the normalized unrecovered fluorescence was found to fit a double exponential and the fit was calculated using R software (National Institutes of Health). At the conclusion of all FRAP experiments, cells were fixed and stained for CENP-I and immunofluorescence was performed to verify that CENP-I had been depleted.
Microscopy
Microscopy of fixed cells was primarily performed on a DeltaVision deconvolution microscope (GE Healthcare) with a 100× oil immersion objective (NA 1.40; Olympus), using a CoolSNAP HQ2 camera (Photometrics). SoftWoRX was used for image acquisition and deconvolution. Additional fixed cell microscopy (Fig. 6 and Fig. S4, I, J, M, and N) was performed on an Axiovert 200 microscope (Carl Zeiss) with PerkinElmer-RS spinning disk confocal system illuminated by a krypton/argon laser, using a 100× oil immersion objective (NA 1.4; Carl Zeiss), with images acquired by an electron multiplying charge coupled device camera (Hamamatsu) using Velocity software. Photobleaching experiments (Fig. 2) were performed on an LSM700 (Carl Zeiss) with a 63× oil immersion objective (NA 1.40; Carl Zeiss) with heated stage at 37°C and CO2 insert set to 5% CO2. Images were acquired using ZEN software. All imaging of fixed cells was performed at room temperature through ProLong Gold antifade mounting media (Invitrogen). Fluorochromes used in this study include FITC, Cy3, Cy5, and DAPI. Images throughout this study were analyzed using ImageJ.
Antibody production
Full-length human Mad1 was cloned into pET41a upstream of the 6His sequence using the Cold Fusion kit (System Biosciences), transformed into BL21 Escherichia coli, and expressed in 2XYT media with 1 mM IPTG for 5 h at 37°C. Bacteria were pelleted, resuspended in lysis buffer (20 mM Tris, 500 mM NaCl, and 5 mM Imidizole, pH 7.9), lysed via sonication, and centrifuged for 1 h at 16,000 rpm at 4°C. The insoluble pellet was then suspended in room temperature lysis buffer containing 8 M guanidine and centrifuged for 1 h at 16,000 rpm at room temperature. The supernatant was transferred to nickel beads (QIAGEN) and turned end-over-end for 3 h at room temperature. The beads were transferred to a disposable column and washed with 60 bed volumes of wash buffer (20 mM Tris, 500 mM NaCl, and 30 mM Imidizole, pH 7.9) containing 8 M guanidine and then washed with 60 bed volumes of wash buffer containing 6 M urea. Protein was eluted from beads with elution buffer (20 mM Tris, 200 mM NaCl, and 300 mM Imidizole, pH 7.9) containing 6 M urea. Elutions were monitored by Bradford assay (Bio-Rad Laboratories) and purity was determined by running samples of the elutions on a gel and performing Coomassie staining. Elutions that appeared predominantly as a single band on Coomassie staining were immediately flash frozen in liquid nitrogen and sent for antibody production (Cocalico Biologicals) or conjugated to CnBr beads (Roche) to produce an affinity column. Returned rabbit serum was diluted 1:10 with TBS, passed over the affinity column, and then washed with 100 bed volumes of TBS. Bound antibodies were eluted with glycine, pH 2.5, and dialyzed overnight into PBS.
Statistical analyses
All error bars indicate standard deviation. P-values were calculated using Student’s t test or analysis of variance (Microsoft Excel). Log transformation and related analyses were performed using R (R Project).
Online supplemental material
Fig. S1 contains immunofluorescence images demonstrating the status of kinetochores and chromosomes after CENP-I depletion. Fig. S2 contains immunofluorescence images and quantification showing the localization of checkpoint proteins in control and CENP-I–depleted cells after nocodazole treatment and Aurora B inhibition. Fig. S3 displays additional data demonstrating intact Mad1-containing structures away from kinetochores in CENP-I–depleted cells treated with Aurora B inhibitors. Fig. S4 contains immunofluorescence data demonstrating the microtubule-dependent localization of Aurora B and spindle checkpoint proteins. Fig. S5 comprises immunofluorescence data on the effects of Hec1 depletion on the localization of spindle checkpoint proteins. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201307137/DC1.
Supplementary Material
Acknowledgments
We thank P. Meraldi for the Mad1 antibody, G.K. Chan for the Zwint and ZW10 antibodies, and T.J. Yen for the ZW10, CENP-F, CENP-C, Rod, and Mis12 antibodies. We additionally would like to thank E.D. Salmon for the GFP-Mad1 expression construct, K.K. Pfister and T.A. Schroer for the CC1-GFP expression construct, D.J. Burke for extensive help with statistical methods, and D.J. Burke, J.V. Shah, and G.J. Gorbsky for critical review of the manuscript. We thank the University of Virginia Advanced Microscopy Facility for assistance with FRAP experiments.
This work was funded by grants GM063045 to P.T. Stukenberg. D.R. Matson is supported by National Institutes of Health training grants T32GM007267 and T32GM008136. D.R. Matson and P.T. Stukenberg designed and executed the experiments and wrote the manuscript.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- ACA
- anti-centromere antibodies
- Bub
- budding uninhibited by benzimidazoles
- K-fiber
- kinetochore fiber
- Mad
- mitotic arrest deficient
- PreK-fiber
- preformed kinetochore fiber
- ZM
- ZM447439
References
- Amaro A.C., Samora C.P., Holtackers R., Wang E., Kingston I.J., Alonso M., Lampson M., McAinsh A.D., Meraldi P. 2010. Molecular control of kinetochore–microtubule dynamics and chromosome oscillations. Nat. Cell Biol. 12:319–329 10.1038/ncb2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee B., Kestner C.A., Stukenberg P.T. 2014. EB1 enables spindle microtubules to regulate centromeric recruitment of Aurora B. J. Cell Biol. 204:947–963 10.1083/jcb.201307119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R., Gomes R., Karess R.E. 2000. Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nat. Cell Biol. 2:939–943 10.1038/35046592 [DOI] [PubMed] [Google Scholar]
- Bharadwaj R., Yu H. 2004. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 23:2016–2027 10.1038/sj.onc.1207374 [DOI] [PubMed] [Google Scholar]
- Burke D.J., Stukenberg P.T. 2008. Linking kinetochore-microtubule binding to the spindle checkpoint. Dev. Cell. 14:474–479 10.1016/j.devcel.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G.K., Jablonski S.A., Starr D.A., Goldberg M.L., Yen T.J. 2000. Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat. Cell Biol. 2:944–947 10.1038/35046598 [DOI] [PubMed] [Google Scholar]
- Cheeseman I.M., Desai A. 2008. Molecular architecture of the kinetochore–microtubule interface. Nat. Rev. Mol. Cell Biol. 9:33–46 10.1038/nrm2310 [DOI] [PubMed] [Google Scholar]
- Chen R.H. 2002. BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J. Cell Biol. 158:487–496 10.1083/jcb.200204048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran J.C., Gatial J.E., III, Kapoor T.M., Gilbert S.P. 2005. Monastrol inhibition of the mitotic kinesin Eg5. J. Biol. Chem. 280:12658–12667 10.1074/jbc.M413140200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Antoni A., Pearson C.G., Cimini D., Canman J.C., Sala V., Nezi L., Mapelli M., Sironi L., Faretta M., Salmon E.D., Musacchio A. 2005. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol. 15:214–225 10.1016/j.cub.2005.01.038 [DOI] [PubMed] [Google Scholar]
- DeLuca J.G., Howell B.J., Canman J.C., Hickey J.M., Fang G., Salmon E.D. 2003. Nuf2 and Hec1 are required for retention of the checkpoint proteins Mad1 and Mad2 to kinetochores. Curr. Biol. 13:2103–2109 10.1016/j.cub.2003.10.056 [DOI] [PubMed] [Google Scholar]
- Ditchfield C., Johnson V.L., Tighe A., Ellston R., Haworth C., Johnson T., Mortlock A., Keen N., Taylor S.S. 2003. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 161:267–280 10.1083/jcb.200208091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele M.J., McCleland M.L., Satinover D.L., Stukenberg P.T. 2005. Measuring the stoichiometry and physical interactions between components elucidates the architecture of the vertebrate kinetochore. Mol. Biol. Cell. 16:4882–4892 10.1091/mbc.E05-03-0239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco L., Chan C.S. 1994. Regulation of yeast chromosome segregation by Ipl1 protein kinase and type 1 protein phosphatase. Cell. Mol. Biol. Res. 40:207–213 [PubMed] [Google Scholar]
- Gorbsky G.J., Chen R.H., Murray A.W. 1998. Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J. Cell Biol. 141:1193–1205 10.1083/jcb.141.5.1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S., Cole R.W., LaTerra S., Zimmer C., Schnapp G., Walter R., Heckel A., van Meel J., Rieder C.L., Peters J.M. 2003. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 161:281–294 10.1083/jcb.200208092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell B.J., McEwen B.F., Canman J.C., Hoffman D.B., Farrar E.M., Rieder C.L., Salmon E.D. 2001. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 155:1159–1172 10.1083/jcb.200105093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell B.J., Moree B., Farrar E.M., Stewart S., Fang G., Salmon E.D. 2004. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14:953–964 10.1016/j.cub.2004.05.053 [DOI] [PubMed] [Google Scholar]
- Hoyt M.A., Totis L., Roberts B.T. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66:507–517 10.1016/0092-8674(81)90014-3 [DOI] [PubMed] [Google Scholar]
- Jordan M.A., Thrower D., Wilson L. 1992. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J. Cell Sci. 102:401–416 [DOI] [PubMed] [Google Scholar]
- Kallio M.J., McCleland M.L., Stukenberg P.T., Gorbsky G.J. 2002. Inhibition of aurora B kinase blocks chromosome segregation, overrides the spindle checkpoint, and perturbs microtubule dynamics in mitosis. Curr. Biol. 12:900–905 10.1016/S0960-9822(02)00887-4 [DOI] [PubMed] [Google Scholar]
- Kapoor T.M., Mayer T.U., Coughlin M.L., Mitchison T.J. 2000. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 150:975–988 10.1083/jcb.150.5.975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuboski J.M., Bader J.R., Vaughan P.S., Tauhata S.B., Winding M., Morrissey M.A., Joyce M.V., Boggess W., Vos L., Chan G.K., et al. 2011. Zwint-1 is a novel Aurora B substrate required for the assembly of a dynein-binding platform on kinetochores. Mol. Biol. Cell. 22:3318–3330 10.1091/mbc.E11-03-0213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A., Copenagle L., Gordon M.B., Compton D.A., Kapoor T.M. 2003. Minus-end capture of preformed kinetochore fibers contributes to spindle morphogenesis. J. Cell Biol. 160:671–683 10.1083/jcb.200208143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyomitsu T., Obuse C., Yanagida M. 2007. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev. Cell. 13:663–676 10.1016/j.devcel.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Kops G.J., Kim Y., Weaver B.A., Mao Y., McLeod I., Yates J.R., III, Tagaya M., Cleveland D.W. 2005a. ZW10 links mitotic checkpoint signaling to the structural kinetochore. J. Cell Biol. 169:49–60 10.1083/jcb.200411118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G.J., Weaver B.A., Cleveland D.W. 2005b. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 5:773–785 10.1038/nrc1714 [DOI] [PubMed] [Google Scholar]
- Kops G.J., Saurin A.T., Meraldi P. 2010. Finding the middle ground: how kinetochores power chromosome congression. Cell. Mol. Life Sci. 67:2145–2161 10.1007/s00018-010-0321-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V., Wehenkel A., Li X., Santaguida S., Musacchio A. 2012. Structural analysis reveals features of the spindle checkpoint kinase Bub1–kinetochore subunit Knl1 interaction. J. Cell Biol. 196:451–467 10.1083/jcb.201110013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Murray A.W. 1991. Feedback control of mitosis in budding yeast. Cell. 66:519–531 10.1016/0092-8674(81)90015-5 [DOI] [PubMed] [Google Scholar]
- Liu S.T., Hittle J.C., Jablonski S.A., Campbell M.S., Yoda K., Yen T.J. 2003. Human CENP-I specifies localization of CENP-F, MAD1 and MAD2 to kinetochores and is essential for mitosis. Nat. Cell Biol. 5:341–345 10.1038/ncb953 [DOI] [PubMed] [Google Scholar]
- Liu S.T., Rattner J.B., Jablonski S.A., Yen T.J. 2006. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J. Cell Biol. 175:41–53 10.1083/jcb.200606020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M., Kapoor T.M. 2011. Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat. Cell Biol. 13:475–482 10.1038/ncb2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliga Z., Kapoor T.M., Mitchison T.J. 2002. Evidence that monastrol is an allosteric inhibitor of the mitotic kinesin Eg5. Chem. Biol. 9:989–996 10.1016/S1074-5521(02)00212-0 [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma S., Stucke V.M., Nigg E.A. 2002. Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science. 297:2267–2270 10.1126/science.1075596 [DOI] [PubMed] [Google Scholar]
- Matson D.R., Demirel P.B., Stukenberg P.T., Burke D.J. 2012. A conserved role for COMA/CENP-H/I/N kinetochore proteins in the spindle checkpoint. Genes Dev. 26:542–547 10.1101/gad.184184.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland M.L., Gardner R.D., Kallio M.J., Daum J.R., Gorbsky G.J., Burke D.J., Stukenberg P.T. 2003. The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev. 17:101–114 10.1101/gad.1040903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland M.L., Kallio M.J., Barrett-Wilt G.A., Kestner C.A., Shabanowitz J., Hunt D.F., Gorbsky G.J., Stukenberg P.T. 2004. The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochore-microtubule attachment. Curr. Biol. 14:131–137 10.1016/j.cub.2003.12.058 [DOI] [PubMed] [Google Scholar]
- Mishra R.K., Chakraborty P., Arnaoutov A., Fontoura B.M., Dasso M. 2010. The Nup107-160 complex and γ-TuRC regulate microtubule polymerization at kinetochores. Nat. Cell Biol. 12:164–169 10.1038/ncb2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle M.W., Kim T., Hattersley N., Espeut J., Cheerambathur D.K., Oegema K., Desai A. 2014. A Bub1–Mad1 interaction targets the Mad1–Mad2 complex to unattached kinetochores to initiate the spindle checkpoint. J. Cell Biol. 204:647–657 10.1083/jcb.201311015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A.W., Kirschner M.W. 1989. Dominoes and clocks: the union of two views of the cell cycle. Science. 246:614–621 10.1126/science.2683077 [DOI] [PubMed] [Google Scholar]
- Musacchio A., Salmon E.D. 2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8:379–393 10.1038/nrm2163 [DOI] [PubMed] [Google Scholar]
- Okada M., Cheeseman I.M., Hori T., Okawa K., McLeod I.X., Yates J.R., III, Desai A., Fukagawa T. 2006. The CENP-H–I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat. Cell Biol. 8:446–457 10.1038/ncb1396 [DOI] [PubMed] [Google Scholar]
- Quintyne N.J., Gill S.R., Eckley D.M., Crego C.L., Compton D.A., Schroer T.A. 1999. Dynactin is required for microtubule anchoring at centrosomes. J. Cell Biol. 147:321–334 10.1083/jcb.147.2.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder C.L., Maiato H. 2004. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 7:637–651 10.1016/j.devcel.2004.09.002 [DOI] [PubMed] [Google Scholar]
- Salimian K.J., Ballister E.R., Smoak E.M., Wood S., Panchenko T., Lampson M.A., Black B.E. 2011. Feedback control in sensing chromosome biorientation by the Aurora B kinase. Curr. Biol. 21:1158–1165 10.1016/j.cub.2011.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S., Musacchio A. 2009. The life and miracles of kinetochores. EMBO J. 28:2511–2531 10.1038/emboj.2009.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S., Tighe A., D’Alise A.M., Taylor S.S., Musacchio A. 2010. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 190:73–87 10.1083/jcb.201001036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S., Vernieri C., Villa F., Ciliberto A., Musacchio A. 2011. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J. 30:1508–1519 10.1038/emboj.2011.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah J.V., Botvinick E., Bonday Z., Furnari F., Berns M., Cleveland D.W. 2004. Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr. Biol. 14:942–952 [DOI] [PubMed] [Google Scholar]
- Shepperd L.A., Meadows J.C., Sochaj A.M., Lancaster T.C., Zou J., Buttrick G.J., Rappsilber J., Hardwick K.G., Millar J.B. 2012. Phosphodependent recruitment of Bub1 and Bub3 to Spc7/KNL1 by Mph1 kinase maintains the spindle checkpoint. Curr. Biol. 22:891–899 10.1016/j.cub.2012.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulu U.S., Fagerstrom C., Ferenz N.P., Wadsworth P. 2006. Molecular requirements for kinetochore-associated microtubule formation in mammalian cells. Curr. Biol. 16:536–541 10.1016/j.cub.2006.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G., Medema R.H., Lens S.M. 2006. The chromosomal passenger complex: guiding Aurora-B through mitosis. J. Cell Biol. 173:833–837 10.1083/jcb.200604032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Waal M.S., Saurin A.T., Vromans M.J., Vleugel M., Wurzenberger C., Gerlich D.W., Medema R.H., Kops G.J., Lens S.M. 2012. Mps1 promotes rapid centromere accumulation of Aurora B. EMBO Rep. 13:847–854 10.1038/embor.2012.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Hu X., Ding X., Dou Z., Yang Z., Shaw A.W., Teng M., Cleveland D.W., Goldberg M.L., Niu L., Yao X. 2004. Human Zwint-1 specifies localization of Zeste White 10 to kinetochores and is essential for mitotic checkpoint signaling. J. Biol. Chem. 279:54590–54598 10.1074/jbc.M407588200 [DOI] [PubMed] [Google Scholar]
- Waters J.C., Chen R.H., Murray A.W., Salmon E.D. 1998. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141:1181–1191 10.1083/jcb.141.5.1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y., Yang C.H., Tanno Y., Watanabe Y. 2012. MPS1/Mph1 phosphorylates the kinetochore protein KNL1/Spc7 to recruit SAC components. Nat. Cell Biol. 14:746–752 10.1038/ncb2515 [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Guacci V., Koshland D. 1996a. Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. J. Cell Biol. 133:85–97 10.1083/jcb.133.1.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A., Guacci V., Koshland D. 1996b. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J. Cell Biol. 133:99–110 10.1083/jcb.133.1.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H., McGarry T.J., Bernal T., Kirschner M.W. 1999. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science. 285:418–422 10.1126/science.285.5426.418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.