

Abstract
Schistosomiasis is the second most important parasitic disease in the world in terms of public health impact. Globally, it is estimated that the disease affects over 200 million people and is responsible for 200,000 deaths each year. The three major schistosomes infecting humans are Schistosoma mansoni, S. japonicum, and S. haematobium. Much immunological research has focused on schistosomiasis because of the pathological effects of the disease, which include liver fibrosis and bladder dysfunction. This Unit covers a wide range of aspects of maintaining the life cycles of these parasites, including preparation of schistosome egg antigen, maintenance of intermediate snail hosts, infection of the definitive and intermediate hosts, and others. The Unit primariiy focues on S. mansoni, but also includes coverage of S. japonicum and S. haematobium life cycles.
Keywords: Schistosomiasis, snail, mansoni, japonicum, haematobium
INTRODUCTION
The trematode parasites in the family Schistosomatidae (phylum Platyhelminthes) infect a wide range of vertebrates. Three species of the genus Schistosoma are of major medical importance: S. mansoni, S. japonicum, and S. haematobium. This unit is revised (from Lewis, 1999) to cover all three species although more emphasis is placed on the Schistosoma mansoni life cycle since it is more frequently maintained in the laboratory. Among the far-ranging investigations in the immunology of schistosomiasis are studies in vaccine development, immunopathology of granulomatous inflammation and fibrosis, eosinophil function, and in vivo regulation of TH1 and TH2 responses.
Subject Group: Laboratory Organisms and Animal Models • Models of Infectious Disease • Immunology • Animal Models • Host-Pathogen Interactions
2009. This unit describes maintenance and collection procedures for various stages of Schistosoma spp. that have immunologic interest, including infection of mice and hamsters with cercariae (see Basic Protocol 1, Alternate Protocol 1, Alternate Protocol 2, Basic Protocol 2, Basic Protocol 3); collection of cercariae (see Support Protocols 1–3); preparation, culture, and cryopreservation/thawing of in vitro–derived schistosomules (see Basic Protocols 4–6, 4 and Alternate Protocol 3); preparation of in vivo–derived schistosomules (see Alternate Protocol 4); and collection of adult worms (see Basic Protocol 7) and eggs (see Basic Protocols 8 and 9). Included also are techniques for preparing soluble egg antigen (SEA; see Basic Protocol 10), one of the more commonly used schistosome antigenic preparations. Since part of the life cycle of all schistosomes involves a snail host a discussion is given of the basic steps that are important in maintaining the snail intermediate host (see Support Protocol 4), and infecting the snails with schistosome miracidia (see Support Protocol 5).
Often, problems in experiments can be traced back to improper snail and parasite maintenance, or lack of attention to detail during mammalian exposure to the infective stage (cercaria) of the parasite.
For reference, a general life cycle for Schistosoma spp. infecting humans is shown in Figure 19.1.1. The figure also shows the various parasite stages described in the text. The reader is directed to specific sections in the text for more details on specific stages of the life cycle.
Figure 19.1.1.

The life cycle of human schistosomes (image courtesy of the Centers for Disease Control and Prevention, DPDx). The figure depicts several life cycle stages that are mentioned in the text. For information on time to development of particular stages (e.g. lung schistosomules, adult worms), see the text for these specific stages. Key collection points mentioned in the text include: miracidia, cercariae, schistosomules, adult worms, and eggs. Approximate measurements for the various stages are: cercaria length (body plus tail), 500 μm; adult female worm length, 12 mm; adult male worm length, 9 mm; egg (length × width), 140 × 60 μm; miracidium (length × width), 140 × 55 μm.
BIOHAZARD CONSIDERATIONS
The schistosome’s infectious stage for humans is the cercarial stage. Depending on the maintenance temperature, Schistosoma mansoni cercariae can emerge from Biomphalaria glabrata snails 3 to 4 weeks after exposure to miracidia, and continue to emerge throughout the life of the snail. Schistosoma haematobium cercariae emerge from Bulinus spp. snails about5 to 6 weeks after they are exposed to miracidia. Cercariae of S. japonicum take much longer to develop in Oncomelania hupensis ssp. snails, approximately 3 months after snails are exposed to miracidia. Since cercariae can penetrate intact skin within 1 to 2 min of exposure, it is imperative that proper precautions be maintained to prevent contaminated water from coming into contact with skin. Workers should wear protective latex gloves when handling exposed snails and cercariae. As a rule, treating all snails as if they were shedding cercariae is a good laboratory practice to adopt.
Cercariae can be killed on contact with 70% alcohol, so placing alcohol squirt bottles throughout the laboratory is a good precautionary measure. Hand sanitizer that contains alcohol as the active ingredient (e.g. Purell) can be placed in strategic locations in the laboratory for accidental exposures to cercariae. Containers of bleach can also be kept at strategic places in the laboratory to be used for discarding cercariae and contaminated materials. Hot water (≥50°C) also kills cercariae within a few seconds.
The investigator should be aware that symptomatology for exposure to Schistosoma spp. is not always clearly defined, and various phases can be misdiagnosed. Mild cutaneous lesions can occur at the site of cercarial entry a few days after exposure, but these skin rashes are more frequently associated with repeated exposures. On heavy initial infections, a febrile response with coughing and shortness of breath can occur as a result of the transit of the organisms through the lungs (2 to 3 weeks after exposure). A more serious, acute response (Katayama fever) occurs after deposition of eggs by the female worms, ~5 weeks after infection. Fever, accompanied by eosinophilia, and gastrointestinal symptoms (e.g., diarrhea and abdominal pain) can be prominent features. The chronic phase in severe infections (S. mansoni and S. japonicum), which may take years to develop, leads to progressive portal fibrosis, portal hypertension, hepatosplenomegaly, and hepatic failure. The disease results from reactions to the embolized eggs, with subsequent fibrosis. Although rare, central nervous system involvement can also occur. With S. haematobium infections, chronic disease can manifest in the form of hematuria, kidney dysfunction, ureteral/bladder dysfunction, and predispose the infected to bladder cancer. The current drug of choice for treating schistosomiasis infections is praziquantel (Biltricide from Miles Laboratories), a very effective and well tolerated drug, with few side effects.
The definitive diagnosis for Schistosoma spp. infection depends on finding characteristic eggs for each species in the feces or urine. In the U. S. A., the Centers for Disease Control (Atlanta, GA.) has developed a very sensitive ELISA for the detection of schistosome infections. For serological analysis of a suspected infection, contact Patricia Wilkins, Chief of the Reference Diagnostics Laboratory, at (404) 718-4101(. It is a good practice to collect and store sera at −70°C from anyone who plans to work with schistosomes, so a comparison can be made with sera collected after accidental or suspected cercarial exposure.
BASIC PROTOCOL 1: PERCUTANEOUS EXPOSURE OF MICE TO SCHISTOSOMA MANSONI CERCARIAE VIA THE TAIL
The most frequently used mammalian host for experimental purposes is the laboratory mouse. Although there is some mouse strain variation regarding the percentage of S. mansoni cercariae that develop to adulthood, all mouse strains so far tested are susceptible to infection. The natural route of exposure to cercariae is by skin penetration, which is usually the preferred route for experimental purposes. Subcutaneous or intraperitoneal injection of cercariae (see Alternate Protocol 2) can also be used, if necessary, but injection methods are often less reliable than percutaneous exposure in the reproducibility of the level of infections achieved. In addition, injection introduces the cercarial tail, a structure to which the host is not normally exposed. Percutaneous exposure of mice is usually done through tail or abdominal skin. Tail skin exposure offers several advantages. First, with the proper restraining devices the mice do not need to be anesthetized. Second, one can estimate the number of cercariae that actually penetrate, after the mouse’s tail is removed from the exposure tube, whereas it is difficult to estimate the success of cercarial penetration after abdominal exposure.
Materials
Cercariae (see Support Protocol 1)
Conditioned water (see recipe)
Mice
12 × 75–mm glass or plastic test tubes
Exposure racks: e.g., test tube racks of height such that 12 × 75–mm tubes are flush with top of rack (see Figure 19.1.2)
Broome plastic restraining devices for mice (Harvard Apparatus) (see in Figure 19.1.2)
Adhesive tape (Johnson & Johnson, 0.5-in. width, Zonas porous)
Counting dish: 90 × 50–mm glass evaporating dish (Pyrex 3180) scored with a diamond pen
Dissecting microscope
Additional reagents and equipment for mouse handling and restraint (UNIT 1.3) and iodine staining and counting of cercariae (see Support Protocol 3)
Figure 19.1.2.

Materials for mouse tail exposure to schistosome cercariae. Shown are a base and upper level of fluorescent light panels (available in standard hardware stores), separated by spacers and anchored with Velcro strips. The height is adjusted to 75 mm, to accommodate 75 × 12 mm test tubes (not shown). A single mouse will be restrained in the Broome-type restrainer, with its tail extending from the bottom of the test tube that contains cercariae. Variations of these restrainers can be purchased commercially, or they can be engineered from plastics companies. Restrainers with diameter openings of about 25 mm can restrain mice up to 20 grams, whereas larger ones (up to 30 mm dia openings) are needed for larger mice.
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Procedure
-
Within 5 hr of harvesting cercariae (see Support Protocol 1), pipet them into a 12 × 75–mm glass or plastic test tube and add conditioned water to ~10 mm from the top of the tube. Place the tube in a test tube rack of height such that the top of the tube is flush with the top of the rack.
Penetration of cercariae into mouse tail skin drops precipitously after 5 hr post emergence. The number of cercariae used per mouse depends on the experiment. For establishing chronic infections (e.g., >12 weeks), use 25 to 30 cercariae; for testing vaccines, use 100 to 150; for maximum egg and adult worm yields (~7 weeks), use 180 to 200. One can expect that approximately 40% of the inoculum can be harvested as adult worms. -
Place a mouse in a plastic restraining tube (see Figure 19.1.2), with its tail extending from the bottom of the tube (Figure UNIT 1.3). Attach small pieces of adhesive tape to the base of the tail to help anchor it in place.
A piece of absorbent wiper (e.g., Kimwipes) placed in the bottom of the restraining tube can prevent mouse urine, which kills cercariae, from contaminating the cercarial suspension. -
Wipe the mouse tail using a gauze sponge moistened with conditioned water to clean away debris, then insert the tail into the tube Figure by placing the restraining tube on top of the exposure rack (Figure 19.1.2).
Wiping the tail before immersing it in the cercarial suspension will help remove any oils from bedding or other debris that can hinder penetration into skin. -
Expose mouse to cercariae for 1 hr, then remove the mouse from the restraining tube and return it to its cage without wiping the tail.
Most cercariae will penetrate within 30 min of exposure, but 1 hr allows enough time for maximal numbers to penetrate. -
To estimate the number of cercariae that have successfully penetrated the skin after the 1-hr exposure period, empty the contents of the exposure tube (s) into a counting dish, rinse with 2 to 3 ml water, and stain the cercariae with iodine (see Support Protocol 1). With a dissecting microscope, count all intact cercariae, plus the bodies that have separated from the tails.
A sketch of a cercaria is shown in Figure 19.1.3. Detached cercarial tails should not be counted. Under ideal conditions >90% of the cercariae will have penetrated. Allow 5 to 7 days before collecting schistosomules (see Alternate Protocol 4), and 6 to 7 weeks before collecting adult worms (see Basic Protocol 7) or eggs (see Basic Protocol 9).
Figure 19.1.3.

Diagram of a Schistosoma mansoni cercaria. Since both body and tail are contractile, the overall length of this stage varies considerably, usually between 300 and 500 μm.
ALTERNATE PROTOCOL 1: ABDOMINAL PERCUTANEOUS EXPOSURE OF MICE TO SCHISTOSOMA MANSONI CERCARIAE
Unlike exposure of mice to cercariae via the tail (see Basic Protocol 1), abdominal exposure requires that the animals be anesthetized for approximately 1 hr. Determining the proper anesthetic dosage depends on the strain, weight, and age of the mouse. Using drugs containing sodium pentobarbital, a starting dosage is ~60 mg/kg body weight.
Materials
Mice
Scale to weigh mice
Sodium Pentobarbital as anesthetic for cercarial exposure (see recipe)
Animal clippers, fitted with a #40 blade
Conditioned water (see recipe)
Gauze sponges
10-cm watch glass or custom-made slotted boards for abdominal skin exposure (slots ~1-in. width)
Cercariae (see Support Protocol 1)
Dissecting microscope
Sieve (made from PVC tubing measuring 10 mm in diameter × 20 mm high glued to a stainless steel wire mesh of 45-μm size, Newark wire)
Petri dish (60 × 15 mm)
Pasteur pipettes
18-mm high × 10-mm wide stainless steel ring (7 mm i.d.; can be obtained from standard plumbing supply vendors) (optional)
Strong light source (desk lamp)
Additional reagents and equipment for intraperitoneal injection of mice (UNIT 1.6)
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails or infected animals. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Procedure
Weigh the mouse, calculate the dosage of anesthetic needed (e.g. Sodium Pentobarbital at 60–80 mg/kg body weight), and inject appropriate volume intraperitoneally (UNIT 1.6).
-
Once the animal is sufficiently anesthetized, shave the abdomen with animal clippers.
Care should be taken to use sharp blades and to shave the abdomen as cleanly as possible. Residual stubble that is too long can trap air and impede cercarial penetration. Wipe the abdomen with a gauze sponge moistened with conditioned water.
-
Place the mouse on its back in a 10-cm watch glass, or in a slotted restraining device, so that involuntary movements will not disturb the cercarial suspension.
A suitable restraining device can easily be constructed from wood. Using a 0.75-in. plywood base, attach seven 36-in. long, 0.5-in. wide wooden strips, each separated by a 1-in. gap. The resulting board (dimensions 36 × 9.5–in.) is suitable for exposing ~30 mice at the same time. -
Pipet the desired cercarial inoculum in 1–5 drops of water onto the shaved abdomen. Alternatively, one can place a stainless steel ring on the abdomen, then pipet the cercarial suspension into the ring with a Pasteur pipette.
If the cercariae need to be concentrated, pour the suspension first over a sieve containing a stainless steel wire mesh of 20-μm. While the cercariae are still in suspension, gently collect them from the top of the screen with a Pasteur pipette and transfer to a petri dish. Caution should be exercised when concentrating cercariae, however, since at densities >2,000/ml they tend to clump together. This will make it difficult to expose the mouse to a specified number of cercariae. -
Expose the mouse to cercariae for ~1 hr (see Basic Protocol 1 for additional detail). If using a steel ring, remove the cercarial suspension from the ring with a Pasteur pipette. Keep the mouse warm throughout the procedure with a warming lamp or heated pad. Place the mouse back into its cage without washing or wiping the exposure site.
Since one may not remove all the non-penetrating cercariae with the Pasteur pipette, determining the number of non-penetrating cercariae is less reliable for abdominal exposure than for tail exposure. Allow 5 to 7 days before collecting schistosomules (see Alternate Protocol 4) and 6 to 7 weeks before collecting adult worms (see Basic Protocol 7) or eggs (see Basic Protocol 9).
ALTERNATE PROTOCOL 2: INJECTION OF MICE WITH SCHISTOSOMA MANSONI CERCARIAE
As an alternative to percutaneous exposure (see Basic Protocol 1 and Alternate Protocol 1), injecting cercariae is an acceptable practice if the level of the patent infection is not a critical matter.
Materials
Cercariae (see Support Protocol 1)
Mice
1-ml plastic syringes and 21-gauge disposable hypodermic needles
Counting dish: 90 × 50–mm glass evaporating dish (Pyrex 3180) scored with a diamond pen
Dissecting microscope
Additional reagents and equipment for iodine staining and counting of cercariae (see Support Protocol 3) and injection of mice (UNIT 1.6)
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Procedure
-
To estimate the injection dose, pull up a suspension of cercariae into a 1-ml plastic syringe fitted with a 21-gauge. needle. Express the suspension into a counting dish, stain with iodine, and count using a dissecting microscope (see Support Protocol1).
Several aliquots should be counted and averaged to calculate the inoculum. -
Inject mice subcutaneously or intraperitoneally (UNIT 1.3) with the appropriate dose, using a 1-ml plastic syringe with a 21-gauge needle.
The number of cercariae used per mouse depends on the experiment. For establishing chronic infections (e.g., >12 weeks), use 25 to 30 cercariae; for testing vaccines, use 100 to 150; for maximum egg and adult worm yields, use 180 to 200. Allow 5 to 7 days before collecting schistosomules (see Alternate Protocol 4) and 6 to 7 weeks before collecting adult worms (see Basic Protocol 3) or eggs (see Basic Protocol 4).
SUPPORT PROTOCOL 1: COLLECTING SCHISTOSOMA MANSONI CERCARIAE FROM INFECTED SNAILS
Numerous procedures in a schistosomiasis laboratory necessitate counting cercariae. One such procedure is the exposure of small mammals such as mice or hamsters to cercariae for infection, where it is important to obtain an accurate estimate of the number of cercariae to which each animal is exposed. S. mansoni cercariae normally take ~ 4 weeks to develop from miracidia in Biomphalaria glabrata. Before exposing laboratory mammals to schistosomes, Biomphalaria glabrata snails liberating (or “shedding”) cercariae are placed in glass beakers under a strong light. After cercariae mature within the snail, they are then shed into the surrounding water. In field conditions, cercariae typically emerge in greatest numbers in the daytime. In the laboratory and for experimental purposes, investigators can adjust lighting conditions to take advantage of maximal release of cercariae at a time of the investigator’s choosing. Some cercariae will still emerge in the dark. The greatest amount of cercarial shedding typically occurs 1–2 weeks after initial shedding, but absolute numbers of cercariae vary between snails and are dependent upon many factors. It is good practice to keep shedding records of snails and track the average number of cercariae produced per snail. Such records can help inform if parameters such as light and temperature should be adjusted.
CAUTION: Containment procedures are extremely important when handling cercariae, and personnel should wear latex gloves, lab coats and take other precautions to insure that no water contact occurs with skin. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Materials
Infected Biomphalaria glabrata snails (see Support Protocol 5)
100 ml beakers
Featherweight forceps (Ward’s Biological Supply, #14 V 0520) or small fish net
Conditioned water (see recipe)
Iodine solution (see recipe)
Incubator fitted with a strong light source
Filtration screen apparatus (if available) consisting of a 300 ml funnel with glass support (Millipore #XX1004703) and a 47 mm diameter stainless steel support screen (47μm mesh size) (see Figure 19.1.4)
Eppendorf blue 101- to 1000-μl plastic pipet tips (or equivalent universal tip VWR # 83007-376) and 100–1000 μl pipette (e.g. Eppendorf Research)
Counting dish: 90 × 50–mm glass evaporating dish (Pyrex 3180) scored with a diamond pen
Dissecting microscope
Figure 19.1.4.

Components of the unit useful for filtering cercariae. The stainless steel screen has 47μm pore size openings, sufficient to allow passage of cercariae. The screen is placed inside of the lower filter portion and the clamp upper and lower parts together.
Procedure
-
To collect cercariae for experimental purposes, place the snails that are shedding cercariae into 100-ml or larger beakers with conditioned water at a density of 1 snail per 2 ml water.
If maintained at 25° to 26°C, B. glabrata snails exposed to S. mansoni miracidia (see Support Protocol 3) can begin to liberate (shed) cercariae between 3.5 and 5 weeks. Cercarial release can be determined by placing the snails individually in small glass vials and exposing them to light for 1 to 2 hr. Cercariae can easily be seen using a dissecting microscope. High yields of cercariae can be isolated when infected snails are kept in the dark 24 hours before shedding them. This can lead to high mortality in the snail colony over multiple shedding periods, so patent snails must continually be added to the snail colony. -
Place the beaker under a strong, isolated light source (e.g., in a 26°C incubator) for 1–2 hours, taking care not to overheat the snails.
Placing the beaker in an incubator or water bath that is kept ~2°C above the temperature of the aquarium in which the snails are routinely maintained will enhance shedding. With featherweight forceps or a small fish net, remove the snails from the beaker and return them to their aquarium.
-
Gently pour the contents of the beaker through the filtration screen apparatus and into a clean beaker.
This 47-μm-pore-size screen effectively traps snail feces and other debris while allowing cercariae to pass through. -
Gently swirl the cercarial suspension with a tip fastened to a pipette, and withdraw several 200-μl aliquots.
Eppendorf blue pipet tips (101- to 1000-μl capacity) or an equivalent universal tip should be used for this procedure. Most commercially available plastic pipet tips with maximum capacity of <1000 μl have bores too small for easy passage of the cercariae, and physical damage to cercariae can occur as a result, reducing their infectivity. It is important to mix the suspension gently (do not create a vortex) to evenly distribute cercariae before withdrawing aliquots. Place each aliquot in a separate counting dish and add 1 to 2 ml of water and a few drops of iodine solution to kill and stain the cercariae.
-
Count all of the intact cercariae in the dish using a dissecting microscope.
Several aliquots should be counted since it is difficult to maintain a homogeneous suspension of live cercariae. Use the average of the aliquots as the inoculum. Maintain records of the number of cercariae produced per snail, which is an important indicator of the productivity of the life cycle. Note: If the original cercarial suspension is approximately 2,000/ml and above, the cercariae tend to clump together, making an accurate count even more difficult to achieve. The suspension should then be further diluted with conditioned water to prevent clumping and improve counting accuracy. Use cercariae to infect mice (see Basic Protocol 1, Alternate Protocol 1, or Alternate Protocol 2) within 5 hr of harvesting.
BASIC PROTOCOL 2: ABDOMINAL PERCUTANEOUS EXPOSURE OF MICE TO SCHISTOSOMA JAPONICUM CERCARIAE
Since the pathology of S. japonicum infection in a mouse is considerably greater on a worm pair basis than that of a S. mansoni infection, low cercariae numbers (e.g., 20–30 per mouse) are usually used in experimental situations. It is best to have cercariae from at least 10 snails to expose mice, to ensure mixed sex infections. Cercariae of S. japonicum are difficult to manipulate and this is related to the extremely sticky nature of the cercariae. Since they readily adhere to any plastic or glass surface, it is best to handle them in the meniscus of a hairloop or similar tool.
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Materials
Mice
Scale to weigh mice
Sodium Pentobarbital as anesthetic for cercarial exposure (see recipe)
Animal clippers, fitted with a #40 blade
Conditioned water (see recipe)
Gauze sponges
10-cm watch glass or custom-made slotted boards for abdominal skin exposure (slots ~1-in. width)
Cercariae (see Support Protocol 2)
Dissecting microscope
Fine forceps or small hypodermic needle (e.g., 25-gauge) placed on a syringe
Petri dishes (60 × 15 mm)
Pasteur pipettes
Hairloop (approximately 2×4 mm) attached to a 23-gauge needle fitted onto a 1ml syringe
Additional reagents and equipment for intraperitoneal injection of mice (UNIT 1.6)
Strong light source (desk lamp)
Procedure
Weigh the mouse, calculate the dosage of anesthetic needed (e.g. Sodium Pentobarbital at 60 mg/kg body weight), and inject appropriate volume intraperitoneally (UNIT 1.6).
-
Once the animal is sufficiently anesthetized, shave the abdomen with animal clippers.
Care should be taken to use sharp blades and to shave the abdomen as cleanly as possible. Residual stubble that is too long can trap air and impede cercarial penetration. Wipe the abdomen with a gauze sponge moistened with conditioned water.
-
Place the mouse on its back in a 10-cm watch glass, or in a slotted restraining device, so that involuntary movements will not disturb the cercarial suspension.
A suitable restraining device can easily be constructed from wood. Using a 0.75-in. plywood base, attach seven 36-in. long, 0.5-in. wide wooden strips, each separated by a 1-in. gap. The resulting board (dimensions 36 × 9.5–in.) is suitable for exposing ~30 mice at the same time. -
Cercariae shed from multiple snails in the petri dish (Support Protocol 2) should be gently swirled to ensure mixing of both sexes prior to collection. With the aid of a dissecting microscope, pick up the cercariae in the meniscus of the hairloop and count them before placing them on the shaved and moistened abdomen of an anesthetized mouse. For most purposes, a good exposure number is 20–25 cercariae per mouse. Expose the mouse to cercariae for ~1 hr. Keep the mouse warm throughout the procedure with a warming lamp or heated pad. Once the mouse has recovered sufficiently, place it back into its cage without washing or wiping the exposure site.
Since only a drop or two of cercariae is placed on the abdomen, determining the number of non-penetrating cercariae is not possible for abdominal exposure. Allow 3 to 5 days before collecting schistosomules (see Alternate Protocol 4) and 6 to 7 weeks before collecting adult worms (see Basic Protocol 7) or eggs (see Basic Protocol 9).
SUPPORT PROTOCOL 2: COLLECTING SCHISTOSOMA JAPONICUM CERCARIAE FROM INFECTED ONCOMELANIA HUPENSIS SSP. SNAILS
Cercariae of S. japonicum emerge from Oncomelania hupensis ssp. snails, approximately 3 months after snails are exposed to miracidia. To obtain S. japonicum cercariae in large numbers, it is necessary to crush snails in order to dissect the secondary sporocysts that contain cercariae. Therefore, snails cannot be used at a later time for cercarial production, as is the case with Bulinus and Biomphalaria snails. Each positive Oncomelania hupensis ssp. snail, though, can typically yield between 300–500 infective cercariae.
CAUTION: Containment procedures are extremely important when handling cercariae, and personnel should wear latex gloves, lab coats and take other precautions to insure that no water contact occurs with skin. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Materials
Infected Oncomelania hupensis ssp. snails (see Support Protocol 5)
Featherweight forceps (Ward’s Biological Supply, #14 V 0520)
Petri dish (60 × 15 mm)
Conditioned water (see recipe)
Strong light source (desk lamp)
Fine-tip dissecting forceps and a small gauge needle
Dissecting microscope
NOTE: Primary sporocysts are difficult to see in the headfoot tissues of an exposed Oncomelania hupensis ssp. snail; however at about 3 months after exposure one can see developing secondary sporocysts through the shell (apex area), provided that the shell is not too eroded or covered in algae. The algae can be gently scraped off by using fine-tipped forceps while holding the snail with featherweight forceps.
Procedure
-
Crush snails at about 3.5 months after exposure to miracidia. Place a few snails, each spaced out on an inverted plastic petri dish top, and gently crush the shells by placing the bottom of the dish on top of the snails and pressing down.
By spacing the snails prior to crushing, one can easily identify the infected snails from uninfected ones.It is possible to obtain a few cercariae from the snails by shedding alone, but for large numbers of cercariae it is necessary to crush them and dissect out the tissues. -
Add a small drop of water to the crushed snail and observe cercariae. Using fine forceps and a small gauge needle, separate out and discard broken shell fragments from the snail soft tissue. If the snail is infected, the body tissue close to the apex will appear transparent with dark banding. Place the infected snail tissue into a small petri dish and add a few drops of conditioned water to keep the tissue moist. Some cercariae will release themselves from sporocysts while in the petri dish. In order to release more cercariae, tease the secondary sporocysts apart using fine-tipped forceps.
The most infective (mature) cercariae will be those that swim to the top of the water and hang there. Use cercariae to infect mice (see Basic Protocol 2) within 5 hr of harvesting.
BASIC PROTOCOL 3: ABDOMINAL PERCUTANEOUS EXPOSURE OF HAMSTERS TO SCHISTOSOMA HAEMATOBIUM CERCARIAE
One of the best small animal models for S. haematobium is the hamster (Moore and Meleney, 1954). However, hamsters do not exhibit the urogenital form of chronic schistosomiasis typically found in humans, and adult worms will mature in the mesenteric veins draining the large intestine. For the best results using percutaneous exposure with S. haematobium in hamsters, the abdomen is the preferred skin site for exposure.
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Materials
Hamsters
Scale to weigh hamster tissues
Sodium Pentobarbital as anesthetic for cercarial exposure (see recipe)
Conditioned water (see recipe)
Animal clippers, fitted with a #40 blade
Gauze sponges
10-cm watch glass
Petri dishes (100 × 25 mm)
Cercariae (see Support Protocol 3)
Dissecting microscope
Fine-tipped (drawn) glass Pasteur pipettes
Sieve (made from PVC tubing measuring 10 mm in diameter × 20 mm high glued to a stainless steel wire mesh of 20-μm size, Newark wire)
18-mm high × 10-mm wide stainless steel ring (7 mm i.d.; can be obtained from standard plumbing supply vendors) (optional)
Additional reagents and equipment for intraperitoneal injection (UNIT 1.6)
Strong light source (desk lamp), and anaesthesia (UNIT 1.4).
Procedure
Weigh the hamster, calculate the dosage of anesthetic needed (e.g. Nembutal [Sodium Pentobarbital] at 60 mg/kg body weight), and inject appropriate volume intraperitoneally (UNIT 1.6).
-
Once the animal is sufficiently anesthetized, shave the abdomen with animal clippers.
Care should be taken to use sharp blades and to shave the abdomen as cleanly as possible. Residual stubble that is too long can trap air and impede cercarial penetration. Wipe the abdomen with a gauze sponge moistened with conditioned water.
Place the hamster on its back in a 10-cm watch glass, so that involuntary movements will not disturb the cercarial suspension.
-
Using a fine-tipped glass pipette and under a dissecting microscope, draw up cercariae and collect them in a small sieve with a 20-μm mesh screen until a predetermined number of cercariae are counted. Express the suspension onto the hamster’s abdomen. For most purposes, a good exposure number is 350 cercariae per hamster. Alternatively, one can place a stainless steel ring on the abdomen, then pipet the cercarial suspension into the ring with a Pasteur pipette. Expose the hamster to cercariae for ~30 minutes. If using a steel ring, remove the cercarial suspension from the ring with a Pasteur pipette. Keep the hamster warm throughout the procedure with a warming lamp or heated pad. Place the hamster back into its cage without washing or wiping the exposure site.
If the cercariae need to be concentrated before counting out the inoculum, pour the suspension first over a sieve containing a stainless steel wire mesh of 20-μm. While the cercariae are still in suspension, gently collect them from the top of the screen with a Pasteur pipette and transfer to a petri dish. Caution should be exercised when concentrating cercariae, however, since at densities >2,000/ml they tend to clump together. This will make it difficult to expose the hamster to a specified number of cercariae.It is not possible to assess accurately the percentage of cercariae that penetrate the abdominal skin once they are applied; however, when the hamsters are perfused, one can expect that about 30% of the estimated number of cercariae applied to the skin to be recovered as adult worms. Allow 3.5 to 4 months before collecting adult worms (see Basic Protocol 7) or eggs (see Basic Protocol 9).
SUPPORT PROTOCOL 3: COLLECTING SCHISTOSOMA HAEMATOBIUM CERCARIAE FROM INFECTED SNAILS
Schistosoma haematobium cercariae emerge from Bulinus spp. snails around 5–6 weeks after they are exposed to miracidia. Collecting S. haematobium cercariae from Bulinus spp. can be performed as described for the S. mansoni life cycle (see Support Protocol 1). This protocol describes another method where infected snails are placed dry in a petri dish before adding water. The period of dryness and addition of water represent two types of stimuli that seem to aid in the release of cercariae from snails. The researcher should experiment with different methods to determine the optimal method for collecting cercariae.
CAUTION: Containment procedures are extremely important when handling cercariae, and personnel should wear latex gloves, lab coats and take other precautions to ensure that no water contact occurs with skin. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Materials
Infected Bulinus spp. snails (see Support Protocol 5)
Plastic container with wire mesh screen glued on (3 mm × 3 mm square openings) and lid for cleaning snails
Deck sprayer nozzle with tubing (can be purchased at a hardware store)
Sieve (made from PVC tubing measuring 10 mm in diameter × 20 mm high glued to a stainless steel wire mesh of 20-μm size, Newark wire)
100 ml beakers
Conditioned water (see recipe)
Petri dish (100 × 25 mm)
Featherweight forceps (Ward’s Biological Supply, #14 V 0520) or small fish net
Other materials as described in Support Protocol 1
Procedure
-
Collect infected Bulinus spp. snails onto a mesh screen and gently rinse them using a deck sprayer nozzle connected to a faucet (control the water pressure moderately to prevent damaging the snails). Spray water for several minutes from different angles to remove any rotifers from the snails. Transfer snails to a petri dish. Keep snails dry for approximately 1 hour and add a minimum amount of water to cover the bottom of the petri dish. Leave the snails in the petri dish with water for 1–2 hrs and before checking for cercariae in the water.
If maintained at 25° to 26°C, B. truncatus snails exposed to S. haematobium miracidia (see Support Protocol 5) can begin to liberate (shed) cercariae between 5 and 6 weeks. Cercarial release can be determined by placing the snails individually in small glass vials and exposing them to light for 1 to 2 hr. Cercariae can easily be seen using a dissecting microscope. High yields of cercariae can be isolated when infected snails are kept in the dark 24 hours before shedding them. This can lead to high mortality in the snail colony over multiple shedding periods, so patent snails must continually be added to the snail colony.If more cercariae are required, the petri dish with snails can be left under a table lamp. The bright light and some heat generated by lamp may result in higher yield of cercariae. However, it should be noted that some snail mortality will occur due to high cercarial shedding. -
Carefully decant the water containing the cercariae into a new petri dish. Remove any unwanted matter such as snail feces, mud and algal particles using a Pasteur pipet. Collect cercariae from several snail petri dishes into one petri dish and add more water into the petri dishes with snails. Place the snails under the table lamp so that more cercariae can be collected after exposing the first few hamsters. Cercariae can be collected periodically in this manner by decanting water into a new petri dish.
If the cercariae need to be concentrated, pour the suspension first over a sieve containing a stainless steel wire mesh of 20-μm. While the cercariae are still in suspension, gently collect them from the top of the screen with a Pasteur pipette and transfer to a petri dish. Caution should be exercised when concentrating cercariae, however, since at densities >2,000/ml they tend to clump together. This will make it difficult to expose the hamster to a specified number of cercariae Use cercariae to infect hamsters (see Basic Protocol 3) within 5 hr of harvesting.
SUPPORT PROTOCOL 4: SNAIL PROPAGATION AND MAINTENANCE
The snail species most often used for maintaining Schistosoma mansoni is Biomphalaria glabrata. Several detailed reviews have described the maintenance features of this snail for the most efficient production of the parasite (Bruce et al., 1971; Lewis et al., 1986; Liang et al., 1987). Reports in the literature have also described the maintenance of Oncomelania hupensis ssp. (Bruce et al., 1971; Liang et al., 1987; Moloney et al., 1987) and Bulinus spp.(Moore et al., 1953;Liang, 1974; Liang et al., 1987 ; Sodeman et al., 1973; Najarian, 1964), but these snails are not maintained as frequently as Biomphalaria spp.. For the investigator interested in developing life cycles of S. mansoni, S. haematobium, or S. japonicum, the NIH maintains a supply contract whereby infected or uninfected snails, infected mice or hamsters, or molecular reagents derived from life cycle stages can be obtained free of charge. Contact the Schistosomiasis Resource Center at 301-881-3300 (extension 31) or the Parasitology and International Programs Branch Division of Microbiology and Infectious Diseases of the National Institute of Allergy and Infectious Diseases (NIAID) at (240) 627-3314. The following discussion gives a brief description of techniques for rearing these snails in the lab. Much information is given for maintenance of B. glabrata, but many aspects of propagation and maintenance can be applied to Bulinus spp. and Oncomelania hupensis ssp. as well. Particular aspects for the care of the latter species are noted below.
Materials
Biomphalaria glabrata snails
Conditioned water (see recipe)
Romaine lettuce
Cyanobacteria (Nostoc spp.; Ward’s Biological Supply;optional)
Autoclaved mud, as nutrient source for growth of Nostoc (see recipe, optional)
Prepared snail food (see recipe, optional)
10- or 30-gallon (equivalent to 45- or 135-liter) aquaria with under-gravel filters and standard immersible aquarium heaters
Plastic mouse cages (polycarbonate, 11″ L × 9″ W × 6″ H, equivalent to 28 × 23 × 15 cm or 18.5″ L × 10″ W × 6″H, equivalent to 47 × 25 × 15 cm)) or
small plastic snail rearing containers (pans) (12″ L × 8.5″ W x × 2.5″ H in., equivalent to 30.5 × 22 × 6.5 cm)
400-ml beakers
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Aquaria versus other snail-rearing containers
B. glabrata can easily adapt to almost any container, provided that one maintains simple precautions to prevent overfeeding and overcrowding. For very small operations, a few snails kept in 400-ml beakers may suffice if enough infected snails are available to assure an approximately equal ratio of male to female cercariae. Retaining populations of 30 to 40 shedding snails should be adequate for most needs.
For newly hatched snails and those of <3- to 4-mm diameter, small rearing pans and/or petri dishes offer a space-saving alternative to large tanks. For snails of >4-mm diameter, plastic mouse cages of 11 × 9 × 6 in. (length × width × height; equivalent to 28 × 23 × 15 cm), or shallow containers of 12 × 8.5 × 2.5 in. (30.5 × 22 × 6.5 cm), are almost ideal and most practical for various reasons, provided sufficient controlled-temperature space is available and the water is changed twice weekly. Glass lids can be placed over these containers to prevent snails from escaping. Snails >4-mm diameter can also be housed effectively in larger numbers in mouse cages of size 18.5″ L × 10″ W × 6″H (7 × 25 × 15 cm). With aeration and proper water changes, snails can be regularly maintained. Snails can be size-sorted through sieves after a few weeks of growth in these containers to maintain equivalent sizes. Newly hatched snails should be maintained in shallow containers, separate from the adult snails.
Aquaria of 30-gallon (135-liter) capacity offer several advantages for maintaining pre-patent snails. First, their temperature can easily be controlled by aquarium heaters, meaning that controlled-temperature rooms are not necessary. Second, if under-gravel filters are used, the water usually does not need changing for at least 4 weeks. Maintaining shedding snails in large aquaria, however, may present an unacceptable exposure risk to personnel when retrieving the snails for collecting cercariae.
Separate tanks of uninfected and S. mansoni-infected snails should be maintained.
Snail crowding
Crowding of snails substantially affects their growth (Chernin and Michelson, 1957; Wright, 1960). It is obvious that as they grow in size they should be maintained in less crowded conditions. Snails >5 mm in diameter grow best if they are maintained at a density of no greater than 10 per liter, provided that the water is changed twice per week. Overcrowding can result in increased mortality rates, slower growth rates, and reduction in the number of cercariae produced. This can be offset somewhat by maintaining snails in a large (at least 10-gallon) aquarium with an under-gravel filter and continuous aeration.
Several ingenious methods have been developed to limit crowding and make the cleaning of snail tanks easier. One that has been particularly useful in the authors’ laboratory is to maintain snails on a nylon net suspended in a container of conditioned water (Rowan, 1958). This allows easy transfer of the entire snail population to a fresh container when needed, although care should be taken to clean the net periodically to reduce buildup of snail mucus. One can sort snails from populations of mixed size and separate them into groups of similar size. This is useful if slow growth in large aquaria is occurring, most likely due to older snails outcompeting younger snails. Mixed size snails can be sorted by passing them through sieves of decreasing size (a tiered system of mesh opening sizes 4.75 mm, 3.35 mm, and 2.36 mm works well).
Water Temperature
Strict temperature control for growth of B. glabrata is perhaps the most important factor for both snail and parasite propagation. For large-scale production, water temperature ranges between 26° and 28°C promote vigorous growth of both snails and parasites. Newly-hatched B. glabrata can grow to about 5 mm (shell diameter) in 3–4 weeks (or sooner) if the temperature is maintained at 26°C, providing other growth conditions are also optimal. Exposure to temperatures >28°C for any length of time increases snail mortality rates. Although the snail can withstand temperatures of 20°C and below, several laboratories maintain them at ~23° to 24°C. There have been occasions when infected snails have lost their schistosome infections, and this appears to happen more frequently at the lower temperatures. Although mortality rates of infected snails are reduced at 23° to 24°C, for greatest production of cercariae a temperature range of ~26°C is preferred with most S. mansoni/B. glabrata strain combinations (Stirewalt, 1954). If the snails are maintained continually at 26°C, some snails can begin to shed cercariae by 28–30 days after they are exposed to miracidia. Depending on the size of the room where snails are housed, a small to medium sized space heater can be used to help regulate heat and water temperature. It is good practice to assure air temperature is about 2°C higher than the desired water temperature.
Light
For convenience, most laboratories maintain uninfected B. glabrata snails under a regulated light/dark cycle, although there is little experimental evidence that maintenance in constant dark or light conditions appreciably affects growth. During the pre-patent period, there is likewise little evidence that variation in light cycles affects the maturation of the parasite. Light has a definite influence, however, on release of cercariae from snails. For maximal cercarial harvests it is best to maintain patent snails in the dark and to subject them to light for the harvest of cercariae, even though there is some evidence that such “forced” shedding increases snail mortality. All patent snails will shed cercariae regularly in the water, even in the dark, but the level of shedding, as mentioned above, can be partially controlled by the light/dark periodicity used in their maintenance. If a dark room is not available, covering infected snail tanks with foil can serve as an alternative for limiting light exposure.
Water quality
Along with water temperature, the quality of water that ensures growth and reproduction of the snails is a critical component of rearing snails for any trematode life cycle. Numerous water sources have been adopted by laboratories for rearing Biomphalaria spp. Water that is suitable for rearing Biomphalaria spp. is likely suitable for Bulinus and Oncomelania hupensis ssp. as well. Water sources for propagating snails are usually dictated by cost. For small-scale operations, many laboratories rely on commercially available “spring” water, or distilled water containing a combination of salts (Cohen et al., 1980; see recipe for conditioned water below). Larger operations, which may need to use hundreds of gallons of water per week, usually pass tap water through a charcoal filter and subsequently “condition” the water by aerating it (bubbling air through the water column) for 1 to 3 days before use. This reduces chlorine concentration to acceptable levels. Charcoal filtration may also help eliminate toxicity problems for the snails caused by the presence of copper tubing in the water supply system. Whatever the source of water a laboratory uses, the water should be tested first to determine if it has a detrimental effect on uninfected snails. Investigators should monitor the movements of the snails and the mortality rates for indications of suboptimal water quality. Snails that are placed in water with high chlorine levels will not glide smoothly on the surface of the tank, will struggle to retain their natural balance, and eventually withdraw into their shells and die Any negative effect(s) on the snails’ normal movement and feeding behavior will undoubtedly be noticed within a few hours or days.
Snail/parasite strain combinations
Investigators should keep in mind that neither B. glabrata nor S. mansoni are genetically homogeneous, with strain differences occurring in both that can affect parasite production (Richards and Shade, 1987). When rearing snails, it is sometimes seen that inadvertent selection can lower susceptibility to the parasite (Lewis et al., 1986). This results in a need for greater numbers of personnel and more space to offset this lower susceptibility. For this reason it is desirable to maintain a separate population of snails, derived from a known highly susceptible stock, which serve as breeders only. Snail embryos can be collected from plastic or styrofoam strips placed in the aquarium, on which the adult snails will preferentially lay their egg masses. Breeder snails can also be removed from containers after a large number of egg masses are laid. These containers do not need much maintenance and snails will hatch out in 1–2 weeks. Maintaining the life cycle with cercariae from large numbers of snails helps retain the genetic heterogeneity while maintaining a normal sex ratio in the parasite population.
Living contaminants
A wide variety of microorganisms and metazoans are known that can create problems in snail maintenance and parasite production. There are several invertebrates that can interfere with the growth of Biomphalaria glabrata and/or the development and release of cercariae from the infected snails in a laboratory setting. Among the most common are rotifers, ostracods and oligochaetes. Field-collected snails may harbor a wide variety of contaminants, some of which, if left unchecked, may have a deleterious effect on a laboratory-maintained snail colony. A variety of techniques have been suggested to control or eliminate these contaminants, but in many cases one may have to completely restart the snail colony with uncontaminated snails or egg masses.
Problems caused by bacterial overgrowth are largely attributed to either over-feeding the snails or not removing dead snails from the aquarium. Cloudy conditions and rank odors from the tank usually are attributable to bacterial overgrowth. Water-borne fungi occasionally can build up on the shells of snails, limiting their mobility and ability to feed. This is usually seen as a slimy covering on the shell, and can be removed by Q-tip, water spray, or other mechanical means.
Metazoans probably cause more problems in life cycles than any other type of living contaminant. Ostracods and oligochaetes have both been found to cause problems in life cycle maintenance (Lo, 1967; Liang et al., 1973). Ostracods can either attack the bodies of the snails or disturb them enough to cause them to withdraw into their shells and prevent the snails from feeding. Ostracod eggs can be harbored in the snail and passed through the intestines. Continuous removal of snail feces may eventually eliminate the problems caused by these organisms. Little is known about the interaction of oligochaetes and Biomphalaria spp., but they are frequently found as contaminants of snails collected from the field. Michaelson (1964) reported that the oligochaete, Chaetogaster limnaei, had a dramatic effect on infection of the snail, but that they could be eliminated by immersing the snail in 1% urethane for 10–20 minutes.
One of the more common problems, especially in reducing cercarial production and activity, is the presence of rotifers. Rotifers (phylum Rotifera) are free-living organisms, found mostly in fresh water, which possess a crown of cilia on trochal disks resembling revolving wheels (see Figure 19.1.5). Those of the order Bdelloidea are especially common in aquaria and readily attach to solid surfaces such as the snail shells. They are roughly the same size as schistosome cercariae (~500 μm in length). At least one species is known to produce a water-soluble substance that can reversibly paralyze cercariae, thus leading to spurious results when mammals are exposed (Stirewalt and Lewis, 1981). This is probably one of the most underappreciated problems in the schistosome life cycle, and care must be taken to control rotifer populations as much as possible. One effective removal method of rotifers is to direct a forceful spray of water onto the surface of the snail’s shell. The force of the stream is provided by a perfusion pump attached with tubing and a 20–22 gauge needle (or by a commercial dental water pic). A deck sprayer with a high pressure nozzle also works well for removing rotifers from groups of snails. If the snails are shedding cercariae, one must use protective measures to avoid exposure to infectious water. Alternatively, one may use a cotton-tipped swab to wipe the snail shell surface to reduce the rotifers to manageable levels. Additional reduction in rotifer contamination in the overall snail colony can be accomplished by incubating snail egg masses in a 1% solution of Clorox in conditioned water for 10 min at room temperature, then washing the egg masses extensively. Embryos from egg masses treated this way will hatch normally. Periodic examination of snail shell surfaces under a dissecting microscope is highly recommended to prevent rotifer problems. Whatever the means of mechanical removal used, the procedure should be repeated whenever rotifers are observed building up again on the shells.
Figure 19.1.5.

Sketch of a rotifer representative of the family Bdelloidea. The organisms are very contractile, and readily invert their trochal disks. When the disks are everted (as shown here) the movement of the cilia will resemble revolving wheels.
Food
A large number of food sources have been reported for the growth of B. glabrata. Some laboratories use romaine lettuce as the staple, plus a prepared supplement (Standen, 1951). This nutrient-rich prepared food (see recipe) is an ideal supplement to the mud, algae (Nostoc) and lettuce that is normally fed to adult snails, particularly when Nostoc production is low. Snails 4 mm in diameter and larger will migrate to the gel and eat it vigorously. Snails less than 4 mm prefer the algae, so it is not recommended that the gel preparation be used to feed small snails. Also, this food source must be dispensed in small quantities to avoid fouling of the container with bacterial overgrowth.
Fresh lettuce is used for mature snails, but lettuce that has been wilted by heat is easier to consume for newly hatched and immature snails (1–3 mm). Each laboratory should experiment to determine the amount of lettuce that its snail population needs. One rule that many laboratories adopt, especially if snails are maintained around 26°C water temperature, is that the snails should have no more food in their tanks than they can consume in 1 day. Over-feeding snails (and not regularly cleaning the containers) can lead to bacterial overgrowth (as evidenced by cloudy and foul-smelling water) if food is not consumed in a reasonable period of time.
Probably the best food source, especially for newly hatched and juvenile snails, is Nostoc sp. (Cyanobacteria) that is grown over a layer of autoclaved mud. Nostoc produced this way, described best by Bruce and Liang (1992), supports rapid growth of baby snails and is readily consumed by snails of all ages. The amount of time and space needed to grow Nostoc (see below), and ready availability of a reliable source of mud containing the proper nutrients, may be limiting factors for some laboratories. Laboratories that take the effort to grow Nostoc by this method, however, can reap huge rewards in the supply of large numbers of snails to infect. For details about preparing mud for best Nostoc growth, the investigator should review Bruce and Liang (1992). Immature snails will also eat dried fish food flakes readily.
Propagation and maintenance of Bulinus spp. as a host for S. haematobium
Much work in maintaining S. haematobium has used Bulinus truncatus truncatus snails as an intermediate host. This subsection will focus on this species but other Bulinus spp. (ex. B. globosus) can be maintained the same way. B. t. truncatus grow well under the same conditions as those described for Biomphalaria spp. snails. There are a few differences which are important to consider when working with B. t. truncatus.
The egg clutches laid by adult Bulinus spp. snails are smaller than those laid by B. glabrata, and there will be fewer embryos within each egg clutch. Typically, 20 or fewer embryos are contained within the egg clutches, compared to 30 or more embryos for many of the B. glabrata egg clutches. Bulinus spp. snails like to lay eggs on surfaces of material in the growth aquaria (lettuce, squares of plastic garbage bags)
Growth of Bulinus, measured by the time to reach maturity, is usually slower than of B. glabrata.
The optimal size of B. t. truncatus snails exposed to S. haematobium miracidia is around 2–3 mm in diameter, whereas for exposure of B. glabrata to S. mansoni, optimal size is around 5–8 mm in diameter.
The number of S. haematobium cercariae one can obtain at any one time from one B. t. truncatus snail (300–500) is considerably lower than the number of S. mansoni cercariae obtained from one B. glabrata snail during its lifetime (2000–4000), all conditions being equal.
The headfoot surface of B. t. truncatus, in relation to its total body size, is substantially greater than that of B. glabrata. B. t. truncatus snails adhere to solid surfaces more firmly than do B. glabrata. These differences have practical consequences when handling the two snail species with forceps and when cleaning their aquaria. B. glabrata can be dislodged easily from hard surfaces (tank, tray or lettuce, etc.) with small forceps, whereas attempting to dislodge B. t. truncatus in the same way can result in damage to the body of the snail if it is not done carefully.
Suport Protocol 5: Propagation and maintenance of Oncomelania hupensis ssp. as a host for S. japonicum
Very few researchers maintain Oncomelania hupensis ssp. snails in laboratories over long periods (unless they have unlimited access to snails in nature). This is mainly due to the exacting nutritional requirements of these snails compared to Biomphalaria spp. and Bulinus spp. Oncomelania hupensis ssp. cultures are kept under 24-hour lighting conditions. Oncomelania hupensis ssp. snails are not hermaphroditic, so males must be mixed with females to obtain viable eggs. The lifespan of uninfected Oncomelania hupensis ssp. in the laboratory is 1–2 years under optimal conditions. In general, trays (described below) should be changed once a week. Fifty-to-100 snails can be maintained per tray. With all 4 subspecies listed here, snail pairs will produce the greatest numbers of eggs for about the first 6 months after they mature, after which egg production begins to diminish.
During their lifespan, many Oncomelania hupensis spp. crawl onto the lid of the container and hang there indefinitely. This is normal behavior, since they are amphibious in nature. Hanging snails should be removed and placed back into the water when the containers are routinely changed (there is no need to do so more frequently)
Materials
Suitable shallow plastic pans (or aquaria) for maintenance of snails (see above)
Glass lids for snail containers
Conditioned water
Oncomelania hupensis ssp. snails (20–30 pairs [male + female])
Petri dish of Nostoc sp. and mud (see recipe)
Petri dish of Navicula pelliculosa diatoms (see recipe)
Lime (pulverized limestone)
Children’s clay
Procedure
-
In a shallow pan containing the equivalent of one-quarter to one-half of a petri dish of algae and one petri dish of diatoms, add water to about 1″ depth. Also add a small amount of lime to the container.
Care must be taken to place the diatoms into the container. This can be achieved by first decanting the water in the diatom petri dish into one corner of the snail-rearing container and subsequently scooping out small amounts of mud/diatoms and placing them in another corner of the container. -
Add 20–30 pairs (male + female) of adult Oncomelania hupensis ssp. snails (the outer lip of the shell [varix] is usually thickened in sexually mature snails).
Determining male sex is a critical step in breeding Oncomelania hupensis ssp. snails. Male snails can be distinguished from females by the presence of a verge (penis). To find the verge, place an adult snail in a horizontal position, and insert the apex of the shell into children’s clay that has been attached to the inside rim of a petri dish. The snail’s operculum should be facing up. Flood the petri dish with water. Once the head of the snail extends, place the petri dish under a dissecting microscope to identify the verge (see Figure 19.1.6), which presents as a structure situated between the mantle collar and the neck of the snail, but may not extend past the shell opening.Size alone is not a practical way to determine the sex of these snails, since both sexes of O. h. hupensis, O. h. nosophora, and O. h. formosana are about the same size (around 8 mm length) when they are fully grown. O. h. quadrasi full-grown males (around 5 mm length) are only slightly smaller than full-grown females (6 mm).
Figure 19.1.6.

A male adult Oncomelania hupensis hupensis snail. The apex of the snail has been anchored in children’s clay, and the headfoot is extended. Extending from the right edge of the shell opening is a flesh-colored structure (the verge).
Support Protocol 6: Collecting, isolating, and hatching Oncomelania hupensis spp. eggs
Unlike Biomphalaria spp. and Bulinus spp. snails, Oncomelania hupensis ssp. snails do not lay their eggs in a clutch (group), but lay their eggs individually on surfaces. The eggs are most often covered with a fine layer of mud, grains of sand or other debris, making them more difficult to see, isolate and harvest in large numbers.
Oncomelania hupensis ssp. eggs will hatch approximately 16 days after they are laid. Once eggs have hatched, the juvenile snails* should be transferred to another petri dish containing water and a small amount of algae and diatoms. A Pasteur pipette can be used to pick up the freshly hatched snails. Care should be taken with the amount of algae and diatoms used in the petri dish to prevent their overgrowth in the dish. Within 2–3 days, the juvenile snails should be transferred into a fresh dish with algae and fresh diatoms. Over time, the snails can be segregated based on their size to allow better growth rates.
Provided that the conditions for growth are optimal, one can obtain adult Oncomelania hupensis spp. snails by 1–2 months after hatching. Once the snails (male and female) are placed in their containers and maintained at ~ 24°C, the containers should be changed once every 10–14 days. With a fresh container of mature snails and under the above conditions, one should begin to see eggs in about 1 month, although some O. hupensis subspecies (e.g., O. h. hupensis) typically take about two months to lay their eggs.
Materials
Suitable shallow plastic pans (or aquaria) for maintenance of snails (see above)
Glass lids for snail containers
Conditioned water
Oncomelania hupensis ssp. snails (20–30 pairs [male + female])
Featherweight forceps (Ward’s Biological Supply, #14 V 0520) or small fish net
Fine sieve (approx. 0.5 mm pore size)
Medium size sieve (approx. 1 mm pore size)
Petri dishes (60 × 15 mm)
Small spatula
Dissecting microscope
Pasteur pipettes
Petri dish of Nostoc sp. and mud (see recipe)
Petri dish of Navicula pelliculosa diatoms (see recipe)
Procedure
Pour the contents of the container through a fine sieve or household strainer (approximately 0.5 mm pore size), so that the adult snails will be collected in the sieve. All the mud, eggs, and other inclusions will pass through.
-
Gently spray the container and the surface of the mud mounds with a stream of water to dislodge the upper mud surface. Pass the mud/water through the sieve, spray the remaining mud, and disperse it sufficiently so that the fine mud can be passed through the sieve.
Depending on the subspecies, several to many eggs may be attached to the bottom of the container (appearing as small soil-colored specks approximately 1 mm in diameter), and these can be removed gently with a small spatula and placed in a petri dish containing water alone. O. h. hupensis usually do not attach eggs to any hard surfaces, but they are laid within the top layers of the mud mound. O. h. chiui and O. h. quadrasi will lay more eggs on the bottom surface of the container. Place the adult snails into a fresh container with algae and diatoms.
With the dispersed mud in the filtrate, pass the filtrate through a sieve with pore size 0.5 mm, so that the eggs and mud particles of approximately the same size and larger will be trapped. Gently agitate the sieve while partially immersed in a pan of water to get rid of the fine-grained mud particles.
-
Empty the contents of the sieve into a petri dish and add conditioned water.
Many of the Oncomelania hupensis ssp. eggs will be covered with a fine layer of mud and are sometimes difficult to distinguish from other particulates in the preparation (see Figure 19.1.7). However, there are some key ways to distinguish them from the surrounding particles. The eggs covered in mud are less dense than other particulate material and will move more readily on slight agitation of the petri dish. If the eggs of O. h. chiui or O. h. quadrasi were recovered by spatula from the surface of the container, they will have one flat side, instead of being entirely oval, as are those deposited by O. h. hupensis on the mud mound. Figure 19.1.8 shows a typical egg recovered from a mud mound (oval, opaque) and one recovered from the hard surface of a container (flat side). Eggs will also move more readily than grains of sand toward the center of the petri dish upon gentle swirling. The eggs should be removed with a glass Pasteur pipette and placed in a petri dish, with conditioned water only.
Figure 19.1.7.
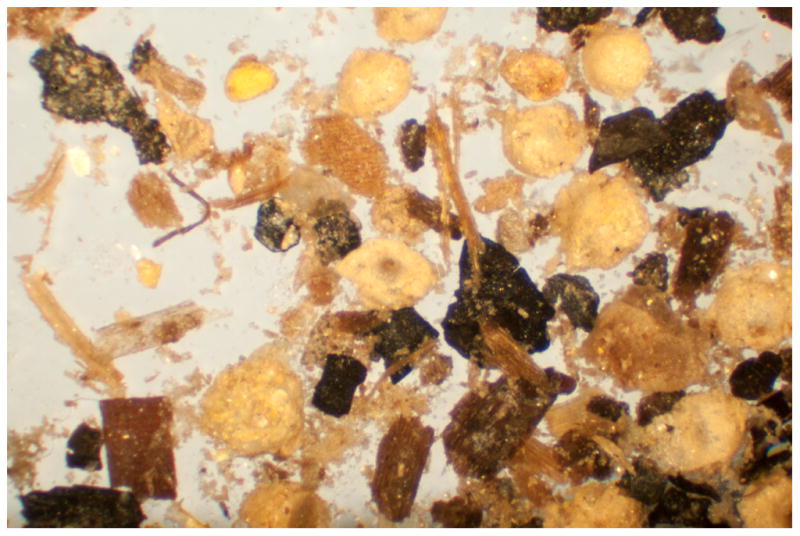
Typical collection of soil in which Oncomelania hupensis ssp. will lay their eggs. Magnified here approximately 150X, it is difficult to distinguish Oncomelania hupensis ssp. eggs from the surrounding granules, without agitating the dish and observing the less dense eggs moving more easily than the granules.
Figure 19.1.8.

Three Oncomelania hupensis ssp. eggs. The eggs are covered in mud. Two of them were removed from the plastic surface of their pan, hence the flat surface of the egg can easily be observed.
Support Protocol 7: Changing containers of snails
Biomphalaria spp., Bulinus spp., and Oncomelania hupensis spp. snails can be maintained in a wide variety of containers as mentioned above. Whatever the container or aquarium used, once the water has been established as conducive to the snails’ growth and reproduction, water should be changed periodically to reduce build-up of snail and food by-products. Of particular importance is keeping water quality at a level in which there is no bacterial or other contaminant overgrowth that can cause noxious conditions for the snails. If the water is cloudy and foul-smelling, it should be changed completely. Keeping water continuously aerated by using an air bubbler with an aquarium pump will reduce the need to change the water so frequently.
Materials
Shallow pan or aquarium
Glass lids for snail containers
Air bubbler
Aquarium pump
Conditioned water
Fine sieve for removing snails
Petri dish of algae/mud (see recipe)
Petri dish of diatoms (see recipe)
Lime (pulverized limestone)
Procedure
Remove uneaten pieces of lettuce, gel snail food, and mud/algae plates from the container/aquarium.
Carefully remove snails by pouring the water over a fine sieve and place them in one or more containers/aquaria (depending on the number of snails) containing conditioned water (see below).
-
Once snails are removed and the container emptied of old lime, mud, and snail feces, scrub the inner surface of the container/aquarium with gauze pads dipped in lime to get rid of scum.
Do not use soap or any other detergents to clean snail pans Rinse well with tap water, and refill with conditioned water.
-
Add a teaspoon or two of lime, and then put snails back into the container and feed with romaine lettuce and a scoop of mud/algae.
Snails usually do well if changed into a completely fresh container of conditioned water. For new laboratories, performing partial changes (leaving some of old water/contents behind) may be preferable until one is assured that completely fresh changes of conditioned water do not increase mortality in the colony. One of the more common problems necessitating frequent water changes is the presence of dead snails in the population. This is especially true in the case of infected snail populations that are actively producing cercariae, where the mortality rate is usually considerably higher than in uninfected snails. The soft tissues of dead snails are ready substrates for overgrowth of bacteria and protozoa. Fouling of the tank can occur rapidly if unchecked and will affect the health of the remaining snails.
SUPPORT PROTOCOL 8: INFECTION OF SNAILS WITH SCHISTOSOMA SPP. MIRACIDIA
Large numbers of miracidia can be obtained from the livers of mice infected for 7 weeks with 180–200 S. mansoni cercariae per mouse, or 20–30 S. japonicumcercariae per mouse. Miracidia can be obtained from eggs from feces of infected mammals, but they usually do not hatch as quickly in water as do those from tissues (liver and intestines). Hamsters infected for 3.5 to 4 months with S. haematobium will have most of the recoverable eggs in the intestinal walls, rather than in the liver. S. japonicum miracidia penetrate readily into the tissues of Oncomelania hupensis spp. snails, as do S. mansoni and S. haematobium miracidia into their respective snail hosts. The percentage of snails developing a patent infection (with its corresponding geographic strain of parasite) with the below procedure is about 50%. The exception to this is O.h.hupensis, 70% of which can develop patent infections.
Snails can be exposed to miracidia either en masse, or individually. For best results, use miracidia < 3 hr after hatching from eggs. When collecting miracidia, avoid collecting within the first 15 minutes, as some may hatch soon after blending infected tissue, due to mechanical damage. Miracidia isolated 15–20 minutes after adding conditioned water will infect snails at a higher rate. Eggs prepared from the minced livers or intestines/feces and isolated with use of the stainless steel screens (see Basic Protocol 9) are sufficient for harvesting miracidia for use in snail exposures. Eggs can be suspended in petri dishes with conditioned water at room temperature for hatching and collecting miracidia. Allowing them to hatch in a petri dish works well, but it may be difficult to clean up the preparation well enough to obtain the miracidia easily. The procedure below makes use of a darkened side-arm flask, with a light directed to the uncoated side arm, for concentrating the phototactic miracidia. Eggs will hatch within a few minutes, and miracidia can be collected using a fine-tipped Pasteur pipette. The snails can then be infected as in steps 8 to 9, below. This procedure allows the preparation of a miracidial suspension relatively free from tissue debris. It is a small scale procedure for isolating eggs mainly used for infecting snails. For large-scale egg isolation, the investigator is referred to Basic Protocol 9.
Materials
S. mansoni–infected mice (see Basic Protocol 1, Alternate Protocol 1, or Alternate Protocol 2), S. japonicum-infected mice (see Basic Protocol 2), or S. haematobium-infected hamsters (see Basic Protocol 3)
Sodium pentobarbital with heparin (see recipe)
1.2% (w/v) NaCl in H2O
Conditioned water (see recipe)
Juvenile Biomphalaria glabrata snails, 5 to 8 mm diameter (see Support Protocol 4)
Juvenile Oncomelania hupensis ssp. snails, 4–6 mm in length (see Support Protocol 4)
Juvenile Bulinus truncatus truncatus snails, 2–3 mm in length (see Support Protocol 4)
Warring blender with 300 ml stainless steel container with blades
50 ml Centrifuge tubes
Tabletop centrifuge
1L Side-arm flask with all but the top few millimeters of the side arm painted black (see Figure 19.1.9)
Light source (stereomicroscope lamp)
Pasteur pipettes (regular and fine-tipped)
Hand-held counter
Petri dishes (100 ×25 mm, 60 × 15 mm)
400 ml beaker
Nostoc algae (see recipe) and diatoms (see recipe)
Shallow pan or aquarium
Glass lids for snail containers
Additional reagents and equipment for intraperitoneal injection of mice (UNIT 1.6)
Figure 19.1.9.

1-liter side arm flask, painted black with the exception of approx 20 mm of the upper section of its side arm. Once eggs are deposited in the flask and it is filled with aged tap water, with a light source directed at the clear side arm, miracidia will concentrate in the clear side arm within 20–30 minutes. This allows easy collection, with a Pasteur pipette, of a relatively clean miracidial preparation
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Euthanize the mouse by intraperitoneal injection (UNIT 1.6) of sodium pentobarbital at 250 mg/kg body weight with heparin (see recipe).
Remove the liver and/or small and large intestines with feces. Rinse tissues in 1.2% NaCl. If using the intestines, cut open and rinse out the intestinal contents with repeated washes using 1.2% NaCl.
-
Place livers and/or intestines/feces, in perfusion fluid or 1.2% saline in a stainless steel container and blend in a Warring blender for 30 sec. Centrifuge the homogenate in 50 ml tubes for 5 min at 1000 rpm, room temperature.
A series of decreasing size sieves can also be used to isolate eggs (see Basic Protocol 9) after blending tissues. Miracidia can be collected from eggs and used to infect snails as in steps 7–9. -
Pour off the supernatant, add 5 ml pre-warmed conditioned water, and shake the tube vigorously for a few seconds.
For best hatching use conditioned water warmed to 25° to 27°C. Pour the suspension into a darkened side-arm flask, making sure that the water fills the unpainted section. Dilute the suspension at least 100-fold in conditioned water.
-
Direct a light source at the exposed, unpainted part of the side arm, taking care not to overheat the side arm.
Since the miracidia are phototropic and possess cilia, they will swim to the lit area of water in the side arm after hatching. Under these conditions miracidia will begin collecting in the water within the unpainted side arm in 10 to 20 min, at which point they can be removed by a Pasteur pipette. -
Using a Pasteur pipette, withdraw the miracidia from the side-arm and place in a petri dish. With a fine-tipped Pasteur pipette and using a dissecting microscope, withdraw the appropriate number of miracidia and place with the snails in a small volume of water. Add conditioned water back into the side-arm flask to keep the volume constant.
Regardless of the procedures employed for collecting miracidia, the level of exposure can vary, depending on the susceptibility of the snails to the strain of parasite. For B. glabrata snails that are highly susceptible, an average of 5 miracidia/snail is sufficient to ensure that a high percentage of the snails will develop a patent infection. For B. t. truncatus and O. hupensis ssp., an average of 10 miracidia/snails is recommended\ -
Add the miracidia to a 400 ml beaker or petri dish containing juvenile snails in conditioned water.
For best results, subspecies of Oncomelania hupensis should be exposed to miracidia once they have reached about ½ the length of their eventual size. O.h.quadrasi should be exposed at no less than 3 mm in length, whereas O. h. nosophora, O.h.hupensis, and O.h. formosana should be exposed at no less than 4 mm in length. In determining the appropriate size of O. hupensis ssp., one can also check for the thickened shell wall along the aperture (aperture wall is not thick and curling outward in small/juvenile snails)If exposing snails individually, place the appropriate number of miracidia in a small vial and add 2 to 3 ml conditioned water and a single snail. Keep snails in the presence of miracidia for at least 2 hr to ensure miracidial penetration. Then return snails to their respective aquaria.
for other applications, concentrated miracidia can be obtained by repeatedly collecting them into 50 ml centrifuge tubes (on ice). Centrifuge tubes for 10 minutes at 100 × g, 4°C. The resultant pellet can be frozen for later use.
BASIC PROTOCOL 4: IN VITRO TRANSFORMATION OF SCHISTOSOMA SPP. CERCARIAE TO SCHISTOSOMULES BY VORTEXING
Cercariae, once in the body of the definitive host, rapidly undergo marked physiological and ultrastructural changes to adapt to the host’s internal environment. These changes (among many others) include loss of the glycocalyx, development of a heptalaminate tegumental membrane, loss of water tolerance, and evacuation of secretory glands, resulting in the schistosomule (or schistosomulum) stage. Methods to prepare schistosomules in vitro usually incorporate an initial step to agitate the organisms sufficiently so that the tails become detached from the body. This is done by vigorous shaking or vortexing of the cercariae, or by repeatedly passing the organisms through a needle and syringe (see Alternate Protocol 3). The below two mechanical procedures, combined with Percoll separation, will yield a clean preparation by separating the cercarial tails from the bodies. Schistosomules prepared by the below procedures, and incubated at 37°C will gradually undergo morphological and physiological changes. By 24 hours in culture, the organisms will resemble (in most respects) cercariae that have penetrated and resided in the skin for about 1 hour.
Schistosomules have been studied extensively, primarily since they have been recognized as major targets for immune attack in the irradiated cercarial model of immunity. It should also be noted that alternative methods have been developed to isolate the migrating form in vivo, usually from the lungs (see Alternate Protocol 4).
Materials
Suspension of Schistosoma spp. cercariae (see Support Protocols 1–3)
50-ml plastic centrifuge tubes
Pasteur pipettes
Dulbecco’s minimum essential medium (DMEM; Life Technologies), or RPMI 1640 (Life Technologies), 4°C
Percoll gradient suspension (see recipe), 4°C
Refrigerated centrifuge
250-ml tissue culture flasks
37°C, 5% CO2 incubator
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, snails, or any material associated with snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Vortex cercariae to produce schistosomules
-
1
Place cercariae in a 50-ml plastic centrifuge tube and leave on ice for 30 min.
-
2
Centrifuge the tube 2 min at 100 × g, 4°C.
-
3
Withdraw and discard all but 2 to 3 ml of water from the top of the pellet.
-
4
Add 3 ml of 4°C DMEM or RPMI 1640 to the tube.
-
5
Cap the tube and vortex 45 sec at high speed.
-
6
Place tube on ice for 3 min, then vortex again for 45 sec.
Purify and culture schistosomules
-
7
Place 40 ml of 4°C Percoll gradient suspension into a 50-ml centrifuge tube. With a Pasteur pipette, gently layer up to 10 ml of the cercarial suspension onto the top of the Percoll gradient suspension.
-
8
Centrifuge 15 min at 500 × g, 4°C.
-
9
Withdraw and discard all but the lowest 10 ml from the tube.
-
10
Resuspend the pellet in the residual solution and add DMEM or RPMI 1640 to 50 ml final volume.
-
11
Centrifuge 5 min at 100 × g, 4°C.
-
12
Wash the final pellet twice, each time by resuspending with 50 ml 4°C DMEM or RPMI 1640, centrifuging 5 min at 100 × g, 4°C, then removing the supernatant.
-
13
Place the resulting organisms into 250-ml tissue culture flasks in 100 ml DMEM or RPMI 1640 at a density of <500 organisms/ml. Incubate at 37°C in a 5% CO2 incubator (for long term culture, see basic protocol 5).
Antibiotics should be added if incubation is longer than 8 to 12 hr.The organisms at this point will steadily undergo changes rendering them incapable of skin penetration. Caution should still be exercised with them however, since (at least for the first 12 to 24 hr of culture) a small percentage might still have this capability.
ALTERNATE PROTOCOL 3: IN VITRO TRANSFORMATION OF SCHISTOSOMA SPP. CERCARIAE TO SCHISTOSOMULES BY NEEDLE AND SYRINGE
Separating the tails from the bodies of cercariae using a needle and syringe is an alternative to vortexing (see Basic Protocol 2), and is an equally valid method for the initial phase of preparing schistosomules. Once this phase is completed, the schistosomules can be placed on the Percoll gradient and purified as above (see Basic Protocol 4).
Additional Materials (also see Basic Protocol 4)
Suspension of Schistosoma spp. cercariae (~1000 cercariae/ml; see Support Protocols 1–3)
50-ml plastic centrifuge tubes
10-ml plastic syringes with 22-gauge disposable hypodermic needles
Pasteur pipettes
Dulbecco’s minimum essential medium (DMEM; Life Technologies), or RPMI 1640 (Life Technologies), 4°C
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Place ~10 ml cercarial suspension, at a concentration of ~1000 cercariae/ml, in a 50-ml plastic centrifuge tube.
-
Using a 10-ml syringe with a 22-gauge needle, fill the syringe with the suspension and repeatedly pass it through the needle.
From 10 to 15 passages should be sufficient. Allow the cercarial suspension to settle for 2 to 3 min, then withdraw and discard all but the lowest 3 ml of the suspension.
Add DMEM or RPMI 1640 to the resulting pellet, resuspend, purify by Percoll gradient separation, and culture (see Basic Protocol 4).
ALTERNATE PROTOCOL4: COLLECTION OF SCHISTOSOMA MANSONI AND SCHISTOSOMA JAPONICUM SCHISTOSOMULES FROM MOUSE LUNGS
Migrating schistosomules are most readily obtained from the lungs of mice a few days after cercarial exposure. Under a dissecting microscope they are roughly morphologically similar in appearance to a cercarial body. Depending on the length of residence in the lungs, however, they can be approximately twice the length of a cercarial body (with an approximate minimum length of 150 μm and a maximum length of 400 μm), and assume a more vermiform appearance. It is now known that the number of lung schistosomules that can be collected at any one time does not necessarily reflect the number that will eventually mature in the mouse. The interesting morphologic and physiological changes that they undergo in the lungs, combined with the evidence that this is probably the stage most vulnerable to immune attack, have made lung schistosomules the object of considerable study. The protocol described here is a simple method to obtain this stage for culture. The day for peak recovery from the lungs will depend on the mouse strain used, whether or not the mouse has been immunized, and the route of infection. Schistosoma mansoni lung schistosomules can be obtained in greatest numbers from mice ~5–7 days after cercarial penetration of the skin (see Basic Protocol 1, Alternate Protocols 1 and 2). Those of S. japonicum can be obtained in greatest numbers from mice ~3–5 days post-exposure to cercariae (see Basic Protocol 2).
Materials
Schistosoma mansoni-infected mice, exposed 5–7 days prior to procedure (see Basic Protocol 1, Alternate Protocol 1, or Alternate Protocol 2) or S. japonicum-infected mice, exposed 3–5 days prior to procedure (see Basic Protocol 2)
Sodium pentobarbital with heparin (see recipe)
Dissecting instruments (iris scissors and forceps)
RPMI 1640 (Life Technologies) containing 10 U heparin/ml
10-ml syringe and 22-gauge needles
60 × 15 mm petri dishes
RPMI 1640 medium (Life Technologies) containing 100 U/ml penicillin and 100 mM streptomycin
Small beaker (30 ml)
37°C, 5% CO2 incubator
50-ml plastic centrifuge tubes
30-mesh stainless steel screen (~500 μm openings) or cheese cloth Refrigerated centrifuge
Dissecting microscope
Fine-tipped Pasteur pipettes
Additional reagents and equipment for intraperitoneal injection of mice (UNIT 1.6)
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, snails, or any material associated with snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Procedure
A 5 to 7 days (3–5 days for S. japonicum) after cercarial exposure (>500 cercariae), euthanize the mouse with an intraperitoneal injection (UNIT 1.6) of 250 mg/kg body weight pentobarbital with heparin.
With dissecting scissors expose the thoracic cavity and perfuse the lungs by injecting 5 to 10 ml RPMI 1640 containing 10 units heparin/ml into the right ventricle of the heart with a 10 ml syringe and 22-gauge needle. Lungs will turn whitish in color when done properly.
Remove the lungs and place them in a petri dish with a minimal volume of RPMI 1640 medium. Remove excess tissue that may have been removed with the lungs (heart fragments, esophagus, etc.).
-
Place the lungs in a small beaker with 1 to 2 ml RPMI 1640 medium with penicillin and streptomycin. Mince the lungs into fine tissue fragments with iris scissors.
The lung tissue must be chopped finely enough to allow the schistosomules to migrate out of the capillary beds and into the tissue culture medium. -
Place the suspension in a 50-ml centrifuge tube, bring to 50 ml with RPMI 1640/penicillin/streptomycin, seal the tube, and incubate 3 hr at 37°C.
This time is sufficient to allow immature worms to exit the lungs. Resuspend the organisms, and pass the suspension through a crude (30 mesh) screen or cheesecloth into another 50 ml centrifuge tube. Centrifuge the filtrate 3 min at 100 × g, 4°C.
Remove most of the supernatant, then wash the pellet once by adding RPMI 1640/penicillin/streptomycin, centrifuging at 100 × g, 4°C, then removing the supernatant. Place the final pellet in 2 to 3 ml RPMI 1640/penicillin/streptomycin in a 60 × 15-mm diameter petri dish.
-
Using a dissecting microscope, count the schistosomules in the entire dish. With a fine-tip Pasteur pipette, collect the schistosomules and place them into culture containers for experimental purposes.
The schistosomules will be about twice the length of the cercarial body, but they are more vermiform in appearance. Recovery of more schistosomules can be achieved by reincubating the minced lung tissues in RPMI 1640 at 37°C for an additional 18 to 24 hr.
BASIC PROTCOL 5: CULTURING SCHISTOSOMULES
Transformed schistosomules can be cultured in vitro in a complex medium (Basch, 1981). These in vitro cultured schistosomules do not grow at the same rate as those in a permissive host, nor will they become patent adults. With good technique, about 50% of them will mature with fully formed guts, and 10% will develop into sexually distinct male and female worms. Hundreds of thousands of worms can be easily maintained, providing a vast amount of parasite material, allowing for genetic manipulation through RNAi and transgenesis. It is possible to increase the percentage of schistosomules forming guts and growing properly (50% versus 20%) by supplementing the media with conditioned media during the first week of culture. With the method below, schistosomules can be cultured for at least two months. The biggest obstacle to success is fungal/bacterial contamination, probably a reflection of the fact that the parasites originate from non-sterile snails. Therefore, it may be necessary to use more than the suggested concentration of antibiotic/antimycotic. This does not appear to affect parasite growth.
Materials
37°C, 5% CO2 incubator
Biosafety cabinet/Tissue culture hood
Schistosomules (see Basic Protocol 4, Alternate Protocols 3 and 4)
6-well tissue culture plates
Schistosomulum Wash (SW) (see recipe)
Schistosomule Wash+Tween (SWAT) (see recipe)
Schistosomulum Medium (SM) (see recipe)
100–1000 μl pipette (e.g. Rainin P-1000) and compatible tips
Packed heparinized mouse red blood cells
15 ml conical centrifuge tube
Table top centrifuge
Procedure
-
Place 10,000–50,000 schistosomules in a single well of a six well plate, with 4 ml of medium (SM). We use tissue-cultured treated plates.
Do not add red blood cells at this stage. Two days after the initial culture is set-up, tilt the 6-well plate at an angle and take out at least 3 ml of the “conditioned medium” being sure to leave the schistosomules behind. Filter the conditioned medium with a syringe filter to avoid fungal contamination problems with the cultures.
Add 3 ml of SWAT and gently pipet the schistosomules in the SWAT using a P-1000 pipette. Overly vigorous pipetting will cause schistosomules to float on the surface of the liquid.
Let the schistosomules settle (1–2 minutes) and remove as much of the added SWAT as possible, being sure to leave the schistosomules behind.
Add 3 ml of fresh SM plus 1 ml of conditioned medium.
-
Supplement the culture with ~50 μl of packed heparinized mouse red blood cells.
Red blood cells should be washed with SW once prior to use. Add approximately 1 ml of heparinized mouse blood plus 10 ml of SW in a 15 ml centrifuge tube and centrifuge at 3000 rpm for 5 minutes. Continue to feed and wash the schistosomules every other day starting on a Monday. Supplement 3 ml of fresh SM with 1 ml of conditioned medium for as long as the conditioned medium lasts (~3–4 media changes). Once the conditioned medium is exhausted, add 4 ml of fresh SM and ~50 μl of packed heparinized mouse red blood cells to each well after removing the SWAT (after washing).
BASIC PROTCOL 6: CRYOPRESERVING AND THAWING SCHISTOSOMULES
Under some circumstances, maintenance of Schistosoma spp. life cycles must be suspended or an unforeseen event disrupts the ability to maintain snails or infect animals. It is possible to begin life cycles of Schistosoma spp. parasites by thawing cryopreserved preparations of schistosomules (James and Farrant, 1977; James, 1981; Stirewalt et al., 1984; Cooper et al., 1989). Cercariae that have been converted to schistosomules are cryopreserved and stored in liquid nitrogen for future use. These preparations can be thawed and injected into the definitive host to continue a suspsended life cycle. A method of cryopreserving schistosomules with ethylene glycol was first reported by James (1981) and later adapted by Stirewalt et al. (1984) and Cooper et al. (1989). The procedure below describes a crude method for cryopreserving strains when starting with less than 50,000 cercariae. This method does not completely remove all cercarial tails but the cercarial body has undergone partial transformation to the schistosomule stage, even with the tail attached. One can start with the tails detached, if need be, by vortex or syringe passage techniques (see Basic Protocol 4, Alternate Protocols 3 and 4) prior to incubation, but it is not a necessary step to ensure recovery of viable organisms.
Materials
Schistosoma spp. cercariae (see Support Protocols 1, 2, and 3)
50 ml centrifuge tubes
Refrigerated centrifuge
Dulbecco’s minimum essential medium (DMEM; Life Technologies), or Roswell Park Memorial Institute medium 1640 (RPMI, Life Technologies), 37°C
15 ml centrifuge tubes or another small tube that has volume graduations (1 ml)
Parafilm
Water bath set at 37°C
Ethylene glycol (20% and 60% in DMEM or RPMI)
Pasteur Pipettes
Aluminum weighing pans with aluminum foil lids
Chromerge cleaning solution (VWR, chromium trioxide diluted in sulfuric acid)
Long forceps
Dissecting microscope
Procedure
Cryopreserving schistosomules
Centrifuge cercariae for 10 min at 100 × g, 4°Cin 50 ml centrifuge tubes.
Pour off all but 2–3 ml of the water and replace with 47 ml pre-warmed (37°C culture medium (RPMI 1640 or DMEM).
-
Gently agitate the pellet so that the organisms are dispersed. A few minutes of gentle agitation is usually enough to disperse the organisms.
One can also disperse the organisms in the water before adding the culture medium but the pellet seems to be less sticky when culture medium is added. Seal the capped tube with parafilm and and place it horizontally in a 37°C water bath for 3 hrs.
During the incubation period, prepare the cryopreservation solutions. Make two concentrations of ethylene glycol (20%, 60%) are in culture medium. Place the 20% ethylene glycol solution at 37°C and the 60% solution on ice.
-
for the last 30 minutes of the incubation period, position the tube vertically so that the organisms settle to the bottom. Once settled, transfer the organisms with a Pasteur pipette to a 15 ml centrifuge tube.
The idea is to have the organisms in a 1ml volume at the end of the 3 hr incubation period. Draw supernatant down to make a volume of 1 ml Schistosomule suspension At the end of 3 hr incubation, add 1ml of the 20% ethylene glycol solution to the 1ml of schistosomules, and mix thoroughly. The concentration of ethylene glycol will now be 10%. Keep the tube upright.
Incubate for 10 min at 37C.
At the end of 10 min, discard 1ml of supernatant, then place the tube on ice for 5 min.
-
At the end of 5 min, add 1ml of the cold 60% solution and mix thoroughly. This will make the final solution 35% ethylene glycol. Place the tube upright on ice for 10 minutes.
The effectiveness of ethylene glycol diminishes somewhat after a few months, so using a fresh bottle every so often is desirable. -
Withdraw and discard 1 ml of supernatant and pour schistosomules into an aluminum weighing pan (pre-cleaned with chromic acid and water). An aluminum foil cover is placed on the suspension and the entire pan is quickly plunged into liquid nitrogen. The pan is transferred into a long-term liquid nitrogen storage vessel, and kept under the surface of the liquid nitrogen during its storage.
Try to cryopreserve in as little volume as possible to concentrate the schistosomules. It is critical to acid-treat the aluminum pans a few minutes before freezing in order to remove any surface film that tends to make the freezing solution “bead up”, rather than spread out into a thin film before freezing. After acid treatment the pan is washed thoroughly with distilled water and dried before use. The almost instantaneous transfer of heat, both on freezing and on thawing, is very important. Therefore, the method is more effective when using aluminum pans rather than conventional plastic tubes.
Thawing procedure
Pre-warm 150ml of culture medium (e.g. DMEM, RPMI) to 41°C. Place this medium in a beaker, dish, or other large-mouth container.
With long forceps remove the pan of organisms from liquid nitrogen, plunge it into the culture medium, and agitate the pan a few seconds. Remove the cover from the pan as quickly as possible after thawing.
Pour the suspension into 50 ml centrifuge tubes and spin the organisms for 3–5 min at about 1000 rpm.
Draw up the pellet of organisms with a Pasteur pipette, examine aliquots under a dissecting scope, count and inject into the animal. We use intramuscular injection. This seems to give us better recovery, for some reason, than subcutaneous injection.
BASIC PROTOCOL 7: COLLECTION OF ADULT SCHISTOSOME WORMS FROM THE PORTAL VENOUS SYSTEMS OF MICE AND HAMSTERS
One of the more common procedures in a schistosome life cycle laboratory is the collection of adult worms from the definitive mammalian host. For many experiments it is important to collect and count the number of mature schistosomes that arise from cercarial exposure. For S. mansoni and S. japonicum infections, this is usually done between 6 and 8 weeks after exposure—shortly after the time mature worms (see Figure 19.1.10) have migrated into the mesenteric venous system of the mammal and have begun to produce eggs Figure In a bisexual worm infection in the mouse, adult S. mansoni and S. japonicum typically reside in the mesenteric veins, from which they can be harvested by perfusion of the portal venous system.
Figure 19.1.10.
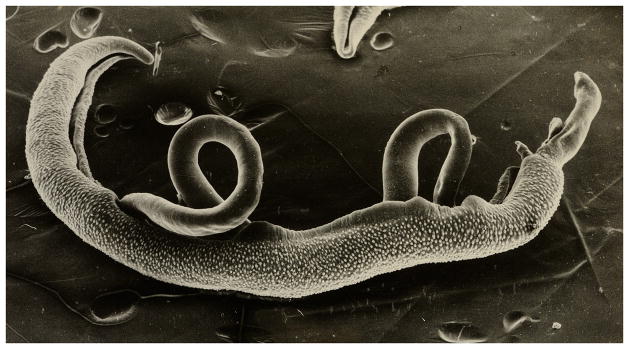
Electron micrograph of adult male and female Schistosoma mansoni in copula. The female is residing in the gynecophoral canal of the male worm. Length of males ranges from 6 to 13 mm; that of females ranges from 10 to 20 mm.
Materials
S. mansoni–infected mice (see Basic Protocol 1, Alternate Protocol 1, or Alternate Protocol 2) or S. japonicum-infected mice (see Basic Protocol 2)
Sodium pentobarbital with heparin (see recipe)
Dissecting instruments
Perfusion fluid (see recipe)
Peristaltic perfusion pump (e.g., Masterflex Console Drive/Cole-Palmer Inst. Co.)
Silicone tubing (e.g., Masterflex 96420-14) fitted with a 20-gauge (1″) needle
Foot-pedal for peristaltic pump (e.g., Treadlite II/Linemaster Switch Corp.)
Small plastic pans (7″ W × 11″ L × 2.5″ H) for collecting perfusate
100 × 25 mm plastic gridded petri dish
Fluorescent light box
Pasteur pipettes
Dissecting microscope
Additional reagents and equipment for intraperitoneal injection of mice (UNIT 1.6)
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
-
Euthanize the mouse with an intraperitoneal injection (UNIT 1.6) of 250 mg/kg body weight sodium pentobarbital with heparin.
Sodium pentobarbital–based drugs affect release of the adult worms from the mesenteries (so-called hepatic shift), making recovery of the worms easier. Clip and remove the skin from the abdominal and thoracic cavity.
With dissecting scissors, open the abdominal cavity, cut the diaphragm, and expose the left half of the thoracic cavity. Sever the ribs near the spinal column.
-
Sever the hepatic portal vein, insert the 20-gauge needle attached to the peristaltic perfusion pump into the descending aorta, and pump perfusion fluid through the needle (Figure 19.1.11).
Using a foot pedal to operate the peristaltic pump greatly facilitates the perfusion process by leaving both hands free to perform the procedure. Perfusing out every adult worm without taking additional steps is difficult, since some worms may still be trapped in the mesenteries, and others may have been swept into the liver. One may see worms trapped in the liver by removing the liver after perfusion and compressing it between two glass plates. If every worm must be accounted for, one must examine the mesenteries using a dissecting microscope. Collect the perfusate (worm suspension), which exits from the hepatic portal vein, in a petri dish, small plastic pan, graduated cylinder or any other large-mouth container.
-
Place the worm suspension over a fluorescent light box and withdraw individual adult worms with a Pasteur pipette. Add perfusion fluid to the worms in a gridded petri dish and count under a dissecting microscope.
Adult worms thus perfused may still be in copula, although some will be unpaired, and some may still be immature at 6 to 8 weeks post infection. Immature female worms, never having paired with a male, will be less than half the length of a mature female and will contain little or no pigment. Occasionally a female worm may be completely hidden within the gynecophoral canal of the adult male, making counting difficult. Dissection of any suspected pair with fine forceps can reveal whether or not a female is present.
Figure 19.1.11.

Sketch of perfusion apparatus for collecting adult Schistosoma mansoni from the mesenteric venous system. A 20-gauge needle, fitted onto the end of Tygon tubing, is placed in the descending aorta. Adult worms are collected in the perfusate from the severed portal vein.
Support Protocol 9: Perfusion of adult Schistosoma haematobium worms from hamsters
Collection of S. haematobium adult worms is much the same principle as that described for collection of adult S. mansoni worms from mice above (e.g., perfusion from the portal venous system). The best time for recovery of adult worms is approximately 3.5–4 months after cercarial exposure. This amount of time allows for recovery of fully mature worms and for maximum egg recovery from the tissues.
Materials
S. haematobium-infected hamsters (see Basic protocol 3)
Sodium pentobarbital with heparin (see recipe)
70% ethanol
Dissecting instruments
Screen with clamps for anchoring hamster that can be placed over collecting tray
Perfusion fluid (see recipe)
Silicone tubing (e.g., Masterflex 96420-14) fitted with a 21-gauge (2″) needle
Peristaltic perfusion pump (e.g., Masterflex Console Drive/Cole-Palmer Inst. Co.)
Foot-pedal for peristaltic pump (e.g., Treadlite II/Linemaster Switch Corp.)
Small plastic pan or tray for collecting perfusate
Vacuum flask with tubing connected to a suction device
Suction device (double-sided PVC tubing and outlet tubing connected to a sieve [PVC tubing measuring 10 mm in diameter × 20 mm high glued to a stainless steel wire mesh of 45-μm size, Newark wire])
Dissecting microscope
Petri dish (100 × 25 mm)
Physiological saline: 0.85% (w/v) NaCl, sterile
Fine-tipped dissecting forceps
Procedure
Euthanize the hamster with an intraperitoneal injection of 300 mg/kg body weight sodium pentobarbital+heparin.
Moisten the fur with warm water or 70% ethanol to reduce the amount of hair that collects in the perfusate. Dissect the skin from the abdominal wall.
Make an incision in the abdominal wall to expose the viscera.
Cut through the diaphragm and the hamster’s left rib cage to expose the descending aorta.
Anchor the carcass on a support, or screen, over the collecting vessel, allowing free use of both hands while perfusing.
-
Insert a 21-gauge needle (fitted to the end of the silicone tubing) into the descending aorta.
To help stabilize the needle in the descending aorta, the needle can be threaded through skin and tissue before placing into the aorta. Pump a small amount of fluid to verify that the needle is inserted adequately, as evidenced by a swelling of the portal vein.
Slit a small hole in the portal vein with forceps, about 5 mm below its entrance into the liver.
-
With the 21 gauge needle still in place in the aorta, pump perfusion fluid through the venous system.
The perfusate, which will include many of the adult worms, can be collected in the underlying collecting vessel or pan. Alternatively, one attach a vacuum line to a suction device with a 45-μM sieve to collect the perfusate as it exits the portal vein. Worms can be collected on the mesh screen of the sieve. -
Once the mesenteries are cleared of blood, one should examine the mesenteric vasculature (using a dissecting microscope) to locate other worms. Dissect worms gently from vasculature and place in a petri dish with 0.85 % saline and a few drops of hamster blood.
In contrast to S. mansoni adult worm collection from mice (or hamsters), roughly 50% of the S. haematobium adult worms still remain in the mesenteric veins, even after prolonged perfusion. Therefore, it is imperative to perform this dissection to collect all worms. Most of the worms will be in the mesenteric veins of the cecum (especially fibrotic Sigmoid colon) and large intestine; they are rarely found in the mesenteries of the small intestine. When adult worms are located in the mesenteries, they can be recovered by opening the mesentery vein wall with fine forceps and gently removing them through the opening. Most worms can be recovered by this procedure without breaking them.
BASIC PROTOCOL 8: ENUMERATION OF SCHISTOSOME EGGS FROM MOUSE TISSUES
Schistosome eggs can be counted after perfusion to provide some index of the level of infection intensity. Eggs are also routinely collected from tissues for injection into other animals, or for preparing antigens. Knowing the number of schistosome eggs in tissues, along with adult worm burdens, gives a measure of the fecundity of the worms. For a reliable estimate of fecundity, however, it is crucial to distinguish and record the numbers of mature versus immature female worms upon perfusion (see Basic Protocol 7). Mature females will be darkly pigmented and immature females will be smaller and have little pigment. Most Schistosoma spp. eggs (see Figure 19.1.12) will either be located in the liver or intestines/feces. Tissues can be frozen indefinitely before digestion. It is difficult however to obtain accurate egg counts from formalin-fixed tissues.
Figure 19.1.12.
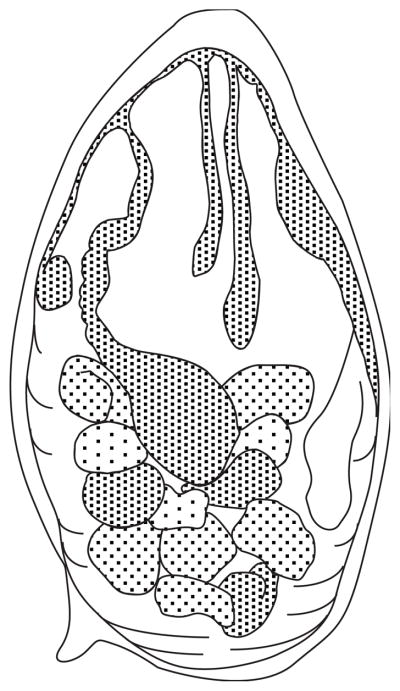
Sketch of a Schistosoma mansoni egg. Approximate dimensions are 140 μm (length) by 60 μm (width).
Materials
S. mansoni-infected mice (see Basic Protocol 1, Alternate Protocol 1, or Alternate Protocol 2), S. japonicum-infected mice (Basic Protocol 2) or S. haematobium-infected hamsters (Basic Protocol 3)
Sodium pentobarbital with heparin (see recipe)
Dissecting instruments
Scale to weigh mouse tissues
4% (w/v) KOH
400-ml glass beakers
37°C warm room or incubator
Dissecting microscope or compound microscope with 3× to 4× objective
2–20 μl pipette (e.g. Rainin P-20) and compatible tips
Additional reagents and equipment for intraperitoneal injection of mice (UNIT 1.6)
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Euthanize the mouse/hamster by intraperitoneal injection (UNIT 1.6) of 250–300 mg/kg body weight sodium pentobarbital with heparin.
-
Dissect out the tissues to be digested from the mice/hamsters. Determine wet weight of tissues.
Before weighing, intestines should be cut open to expose the lumen and the intestinal contents cleaned out by repeated rinsing with 1.2% NaCl (see Basic Protocol 5, annotation to step 2). Place individual tissues in 4% KOH, with tissues occupying no more than 10% of the total volume.
-
Incubate tissues at 37°C for 16 to 24 hr.
A CO2 incubator is inappropriate, since absorbed CO2 will neutralize the KOH. -
Mix the suspension thoroughly and count the eggs in several aliquots using a dissecting microscope or a compound microscope with a 3× to 4× objective.
A simplified method for egg counting is to pipet and spread 20 μl of the thoroughly mixed egg suspension onto a microscope slide and count the eggs using a dissecting microscope. As a reference, one can expect 2000 to 3000 eggs per gram of liver per worm pair from an 8-week-infected mouse. It is convenient to dilute the egg suspension appropriately so that 20 to 100 eggs will be contained in one sample.
BASIC PROTOCOL 9: COLLECTION OF SCHISTOSOME EGGS AND INJECTION INTO MICE TO INDUCE PULMONARY GRANULOMA FORMATION
Eggs are routinely collected from tissues for injection into other animals or for preparing antigens. Schistosome eggs are collected for experimental use primarily from either livers or intestinal tissue/feces of heavily infected animal hosts. For the greatest yield of S. mansoni/S. japonicum eggs, it is best to use heavily infected animals at 7 to 8weeks post infection. These mice will have less fibrosis and a higher percentage of viable eggs in the tissues than those with longer infections. In general, tissues from outbred mice exposed to 180 to 200 cercariae each will yield large numbers of eggs with this infection period. With practice, one can obtain about 20,000 mature eggs/liver at this level of infection. Harvesting eggs from the intestinal walls of mice is possible, but only after the intestines are cleaned of feces and rinsed in copious amounts of 1.2% NaCl. At the level of infection above, one can expect to obtain roughly half the number of eggs from an individual mouse’s intestines as from its liver. The procedure for collection of S. haematobium eggs from the tissues of hamsters is similar to that described for the collection of S. mansoni eggs from mice (above). In practice, however, the majority of recoverable S. haematobium eggs are found in the walls of the cecum and large intestine/feces, rather than in the liver, even though it is obvious on gross examination that the liver contains many granulomatous lesions. In this respect, the situation is very different from that in the S. mansoni-infected mouse. One can usually obtain up to 50,000 mature schistosome eggs from a single hamster using the abdominal exposure method (from hamsters exposed to around 350 S. haematobium cercariae 3.5–4 months earlier [see Basic Protocol 3]). As a crude estimation, 1 ml of the settled egg pellet (from S. mansoni and S. haematobium infections) will contain approximately 800,000 eggs.
The procedure for collection of S. japonicum eggs from mouse tissues is essentially the same as that used for collecting S. mansoni eggs from mouse tissues. Recoverable S. japonicum eggs will be found in the liver and in the intestinal walls/feces of infected mice (see Basic Protocol 2). S. japonicum eggs are smaller than those of S. mansoni. As a consequence, 1 ml of the settled egg pellet will contain approximately 1,000,000 eggs.
Since most of the pathology in schistosomiasis is directly attributable to host reactions to the eggs and egg-associated antigens, immunologists have directed a great deal of attention to this life cycle stage. Schistosome eggs have been used intact to stimulate egg-induced pathology, primarily in a pulmonary model of granuloma formation, and extracts of eggs are routinely used in a variety of experiments to dissect the host response at this clinically important stage. This procedure describes the isolation of eggs from the livers of S. mansoni-infected mice. In order to obtain the cleanest preparation of eggs, the mice should first be perfused of adult worms, or the final egg preparation may contain many unwanted adult worm fragments.
Materials
Schistosoma mansoni–infected mice (see Basic Protocol 1, Alternate Protocol 1, or Alternate Protocol 2), S. japonicum-infected mice (Basic Protocol 2), or S. haematobium-infected hamsters (Basic Protocol 3)
Sodium pentobarbital with heparin (see recipe)
Dissecting instruments
1.2% (w/v) NaCl, 4°C
Warring blender, with variable speed control and 300 ml container for blending
Stainless steel sieves of decreasing pore sizes (Newark Wire Cloth; mesh openings of 420 μm, 180 μm, 105 μm, and 45 μm- see Figure 19.1.13)Spray apparatus (2-gal deck sprayer pump-type, typically found in hardware stores)
Glass petri dishes (100 mm diameter) with flat bottoms
Pasteur pipettes
Falcon #2340 cell strainer, 40 μm nylon (see Figure 19.1.14) or sieve (made from PVC tubing measuring 10 mm in diameter × 20 mm high glued to a stainless steel wire mesh of 45-μm size, Newark wire)
Light box
15 ml conical centrifuge tubes
Physiological saline: 0.85% (w/v) NaCl, sterile
Uninfected mice
Mouse restrainer
Alcohol swabs
1-ml disposable syringe with 23- or 25-gauge needle Gauze pads
Additional reagents and equipment for enumeration of eggs (see Basic Protocol 4), mouse handling and restraint (UNIT 1.3), immunohistochemistry (UNIT 21.4), and intraperitoneal injection of mice (UNIT 1.6)
Figure 19.1.13.

Stainless steel sieves stacked (top to bottom) with pore sizes 420 μm, 180 μm, 105 μm, and 45 μm. Once the tissue homogenate is placed in the top-most sieve, repeated washing of the column with 1.2% NaCl results in eggs trapped on the surface of the lowest sieve, relatively free of contaminating larger particulate matter.
Figure 19.1.14.

Falcon 2340 Cell Strainer. These commercially-available screens work well for concentrating schistosome eggs. Pore size of 40 μm allows retention of the eggs on the bottom of the screen, while extraneous, smaller matter passes through.
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, infected snails, or any material associated with infected snails or infected animals. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
Isolate eggs
-
1
Euthanize mice/hamsters by intraperitoneal injection (UNIT 1.6) of 250–300 mg/kg body weight sodium pentobarbital with heparin.
-
2
Dissect out tissues (liver and/or intestine) and rinse thoroughly in 4°C 1.2% NaCl.
It is important to cut the intestines open to expose the lumen and clean out the intestinal contents with repeated rinses of NaCl. Many investigators place the livers and/or intestines at 4°C overnight in 1.2% NaCl, followed by a 1-hr incubation at 37°C prior to mincing the tissues. Others include a step of protease and/or collagenase digestion to further enhance release of the eggs from the tissues.
Purify eggs
-
3
Mince the tissues with scissors, suspend in 1.2% NaCl, then place in a Warring blender. Blend tissues for 30 sec at low speed. Pass the blended material through a crude sieve (420 μm opening). Collect the tissue remaining on top of the sieve and blend again for 30 sec, this time at medium speed. Pass the resulting material through the sieve as before, collect the remaining tissue from the upper surface of the sieve, and blend for 30 sec at the highest speed setting. Pass the tissue through the crude sieve an additional time.
-
4
Pass the filtrate through the series of stainless steel sieves with decreasing pore sizes, using a spray apparatus filled with 1.2% NaCl to rinse the filtrate through the successive screens.
Agitate the sieves throughout the entire process to ensure that most of the eggs will pass through to the lowest sieve. For best results, re-homogenize the homogenate trapped on the top sieve in the Warring blender and pass the homogenate through the sieves again, using the technique described.Followed repeated washes through the upper sieves, the eggs will be retained on the final sieve (pore size of 45 μm). -
5
Wash the eggs off the final sieve into a glass petri dish and add 1.2% cold NaCl so that it is about half full. To enrich the population for mature eggs, gently swirl the dish over a light box (for better visibility). With gentle swirling, concentrate the eggs in the middle of the dish (mature eggs will concentrate in the center of the vortex). Place a Falcon #2340 40 μm nylon cell strainer (or sieve with equivalent pore size) in a second glass petri dish half full of cold 1.2% NaCl next to the plate being swirled, and continuously pipet mature eggs into the strainer until no more eggs can be seen in the center of the petri dish.
The initial egg suspension will contain eggs of several stages of maturation (see figure 19.1.15). Cold NaCl will keep most of the eggs from hatching into miracidia.For best results eggs should be collected using a petri dish with a perfectly flat bottom. -
6
When no more eggs can be concentrated in the middle of the petri dish upon swirling, discard the contents of the dish (immature eggs, small pieces of liver, etc.). Pour the collected suspension of enriched eggs from the strainer to its petri dish and repeat the swirling/collection procedure, using a fresh petri dish of 1.2% NaCl and the cleaned out strainer for the next cycle. Use the pipette to (gently) rinse remaining eggs from the strainer into its petri dish, and continue to use the same strainer for all cycles. Add enough cold 1.2% NaCl to the petri dish each time eggs are removed to keep the volume constant, and repeat swirling and concentrating the eggs until no more can be concentrated in the middle of the dish.
-
7
After 3 to 4 cycles of these purification steps, most of the tissue debris and immature eggs will be eliminated. The final egg population will be enriched in mature eggs (see Figure 19.1.16). After the last cycle, gently pipette the clean, mature eggs from the strainer (while tipping it slightly onto its side to facilitate pipetting the eggs) into a 15 ml conical centrifuge tube, on ice. Fill to the top with cold 1.2% NaCl and allow eggs to settle to the bottom of the tube. Finally, count eggs (see Basic Protocol 4). Alternatively, one may gently rinse the strainer of eggs with a small volume (>10 ml) of cold 1.2% NaCl into a clean petri dish and then pipette into the 15 ml tube, rinsing the petri dish with another 5 ml of NaCl and adding this to the tube.
Although it is best to inject fresh eggs into the mouse vein, eggs can be kept at 4°C for several days in 0.85% NaCl without appreciable loss in viability.At this point, purified eggs can be transferred to conditioned water for hatching and collection of miracidia (see Support Protocol 5)
Figure 19.1.15.

Sample of S. mansoni eggs collected from the surface of the wire cloth retaining sieve (45-μm pore size). The suspension contains considerable amounts of extraneous tissue at this stage in purification.
Figure 19.1.16.

Sample of S. mansoni eggs after 3–4 cycles of swirling for concentration of mostly mature eggs.
Inject eggs into mouse
-
8
Restrain mouse in a suitable restrainer, with the tail exposed. Clean the tail with a sterile alcohol swab.
-
9
Using a 1-ml disposable syringe with a 23- or 25-gauge needle, inject 2000 to 5000 eggs in 0.25 ml sterile physiological saline into one of the lateral tail veins.
It is important to inject the eggs as rapidly as possible, since they settle in the syringe quickly. -
10
Upon withdrawing the needle, press sterile gauze on the injection site and, once hemostasis is assured, return the mouse to its cage.
-
11
At the appropriate time post injection, euthanize mouse with an intraperitoneal injection of 250 mg/kg body weight of sodium pentobarbital with heparin and remove lungs for histology studies (UNIT 21.4) or other experiments.
In a naive mouse, peak lung granuloma formation occurs ~14 days after egg injection. In mice sensitized previously with egg antigens, or in mice with a patent infection, peak lung granuloma formation occurs around 6 days post injection.
BASIC PROTOCOL 10: PREPARATION OF SCHISTOSOMA SPP. CRUDE SOLUBLE EGG ANTIGEN
One of the more common schistosome antigenic preparations is soluble egg antigen (SEA), a complex, crude homogenate obtained from purified mature eggs isolated from the tissues of the definitive host. The use of SEA has been critical in dissecting immunologically-driven responses to the eggs in an active infection. The use of SEA has been important in experimental immunology studies for its strong Th-2 polarizing activity. Techniques for SEA isolation were first described by Boros and Warren (1970). As with any crude extract of a multicellular organism, it consists of a complex array of components, such as proteins, glycoproteins, polysaccharides and glycolipids.
Materials
Purified Schistosoma spp. eggs (see Basic Protocol 9, steps 1 to 7)
Phosphate-buffered saline (PBS; APPENDIX 2), pH 7.4 (4°C)
Hand-held Potter-Elvehjem glass homogenizer (15-ml capacity) with tight pestle, prechilled
Dissecting microscope
15 or 50 ml centrifuge tubes
Refrigerated centrifuge
Ultracentrifuge
10-ml disposable syringe with 0.2-μm pore-size syringe filter
CAUTION: Schistosomes are a biohazard. Workers should wear latex gloves at all times when handling schistosomal suspensions, snails, or any material associated with snails. Carefully review the discussion of Biohazard Considerations at the beginning of this unit before proceeding.
-
Suspend purified eggs in 5 to 7 ml of 4°C PBS at a concentration of 100,000 eggs/ml.
Other buffers can be chosen, depending on the needs of the investigator to enrich the final solution in components such as glycoproteins or glycolipids. A protease inhibitor (leupeptin, at 10 μg/ml) is sometimes included in the extraction buffer. -
Either in a cold room or on ice, homogenize the eggs with the prechilled glass Potter-Elvejem hand-held homogenizer, using a tight pestle.
The percentage of intact eggs remaining at various stages of the procedure gives an indication of the success of homogenization. To determine this, place a small drop of the suspension on a microscope slide and examine at 40×. Make a crude determination of the percentage of eggs that have been broken apart. Intact eggs can be easily differentiated from the clear, empty shells. It may take 10 to 15 min of homogenization depending on the number of plunging cycles and the force used (~400 to 500 repeated cycles of grinding) to break apart most of the eggs. -
Once at least 95% of the eggs have been broken apart, centrifuge the crude mixture for 20 min at 2000 × g, 4°C. Retain the supernatant and keep at 4°C.
The pellet (crude SEA) at this point will still contain large numbers of intact eggs that can be re-homogenized as above. Withdraw the crude supernatant and ultracentrifuge 90 min at 100,000 × g, 4°C.
-
Sterilize the final supernatant (SEA) by passing through a 0.2-μm filter, then determine protein concentration. Store the final preparation at −70°C.
In the authors’ experience, the protein concentration of the purified supernatant (starting from a crude prep) equates to 1–1.3mg per ml. Starting with 50 livers from S. mansoni-infected mice and ending with about 10 ml final purified prep, therefore a final yield of 10–13 mg SEA can be expected.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2; for suppliers, see APPENDIX 5.
Anesthetic for cercarial exposure
6.5 mg sodium pentobarbital
10 ml 100% ethanol
20 ml propylene glycol
70 ml distilled water
Store for up to 6 months at room temperature
Dilute prepared drug (65 mg/ml) 1:4 in 1X PBS, pH 7.2 (APPENDIX 2). Inject mice intraperitoneally with 0.05 ml/10 g body weight (81 mg/kg body weight).
Alternatively, commercially available Nembutal (50 mg/ml sodium pentobarbital, Henry Schein) can be used. For mice, dilute Nembutal to 10 mg/ml in 1X PBS and then give at a dose of 60 mg/kg body weight. For hamsters, dilute to 30 mg/ml in 1X PBS and give at a dose of 60 mg/kg body weight
Sodium pentobarbital with heparin
For euthanizing mice, use commercially available Euthasol (390 mg/ml Sodium pentobarbital, 50 mg/ml phenytoin sodium, Virbac Animal Health) and dilute to working solution (24.5 mg/ml):
8 ml euthasol
2.5 ml heparin (10,000 U/ml, Fisher Scientific)
75 ml H2O
42 ml PBS
Store at room temperature for ~ 6 months
Inject each mouse with 0.3 ml (250–300 mg/kg body weight)
For euthanizing hamsters, use powdered sodium-pentobarbital:
45 ml 65 mg/ml sodium pentobarbital (above)
2.5 ml heparin (10,000 U/ml)
Store for up to 6 months at room temperature
Use at a dose of 300 mg/kg body weight
Conditioned water
Water suitable for snail maintenance can usually be obtained by passing tap water through any of several readily available activated charcoal filters, followed by bubbling air through the water column (aquarium stones) for 2 to 3 days. In some cases, dictated either by cost or the need for only small volumes, an alternative water source (artificial conditioned water, 10× solution) can be prepared as follows (see also Cohen et al., 1980 for a modified recipe).
Add 2 to 3 liters distilled H2O to a 6-liter flask
Add, in the following sequence (waiting for each to dissolve before adding the next):
3.33 g CaCl2
7.38 g MgSO4.7H2O
0.258 g K2SO4
0.3 ml of 0.15g FeCl3·6H2O per 50 ml
Allow to stand ~1 hr
Add 2.52 g NaHCO3
Stir a few minutes and bring volume to 6 liters with distilled H2O
Check pH, which should be 7.0 ± 0.5
Dilute aliquots to 1× before use
Iodine solution
4 g potassium iodide
2 g iodine
100 ml H2O
Store indefinitely at room temperature
Percoll gradient suspension
24 ml Percoll
4 ml 10× Eagle’s minimum essential medium (EMEM; Life Technologies)
1.5 ml penicillin-streptomycin (10,000 U per ml penicillin/10,000 μg per ml streptomycin; Life Technologies)
1 ml 1 M HEPES in 0.85% (w/v) NaCl
9.5 ml distilled H2O
Store up to 5 days at 4°C
Perfusion fluid (0.85% sodium chloride + 0.75% sodium citrate)
7.5 g trisodium citrate dihydrate
8.5 g sodium chloride
1 liter H2O
Store up to 7 days at 4°C
Prepared snail food
Materials
1L beaker
500 ml distilled water
8 g barley grass powder (available at health food stores)
2.0 g wheat germ (available at grocery stores)
2.0 g fish food (Tetramin large tropical flakes, available at pet stores)
1.0 g powdered milk (available at grocery stores)
2.0 g sodium alginate (Alginic acid-sodium salt from Sigma, catalog # A-2033, medium viscosity sold as a fine powder)
7″× 9″× 6″ plastic pans
Glass plates
1 liter 2% calcium chloride solution (Sigma catalog #C-4901, anhydrous)
Procedure
-
In a 1-liter beaker, heat 500 ml of distilled water to near boiling in a microwave, with continuous stirring.
Do not boil at any time, but continue to heat on the hot plate. -
First, add 8 g barley grass powder to the hot water while stirring. Pulverize wheat germ, fish food and powdered milk to a fine powder in the mortar and pestle (do each separately), then gradually add the powder to the barley grass suspension, continuing to heat and stir. After ingredients are in suspension, add the sodium alginate very gradually.
Most, but not all of the alginate will go into suspension; some alginate will form clumps, but these will not cause problems with the final product. -
Continue to stir for a few minutes. Pour the hot suspension into flat pans to cool to room temperature. For 500 ml of food, two 7″× 9″× 6″ pans will provide adequate area for a 0.5″ depth gel. Allow the suspension to cool at room temperature for 2–3 hours without disturbing the pans. After the suspension is cool and partially solidified, gently flood each pan with the 2% calcium chloride (CaCl2) solution until the gel is well covered. Place the pans in the refrigerator (4°C) overnight.
In order not to disturb the gel, slowly pour the calcium chloride solution over a glass plate onto the surface of the gel. The gel will shrink after 2–3 hours in the cold CaCl2. -
Pour off the CaCl2, and rinse the gel 1–2 times with deionized water. The gel is now firm enough to hold with a gloved hand while rinsing. Store the remaining gel at 4°C in the 7″x 9″x 6″ pan until use. The gel may be fed to snails by pinching off a section approximately 1″x 1″ per 50 snails.
A daily diet of 3 to 4 g of the final preparation should provide sufficient nourishment for 40 snails. Be careful not to overfeed the snails. If the gel is not cleared within a day or two, bacteria may begin to grow and foul the water. The gel snail food will be useable for about 1 week before it deteriorates. Any remaining gel should be disposed of and a new preparation made each week. The gel can also be frozen for later use.
Support Protocol 10: Preparation of mud base medium for growth of Nostoc sp. algae and Navicula pelliculosa diatoms
For growth in the laboratory, especially of the fastidious Oncomelania hupensis ssp. snails and juvenile B. glabrata and Bulinus spp., the cyanobacterium Nostoc sp. serves as a nutritious and readily consumed food source. Romaine lettuce, which is a staple for growth of Biomphalaria spp. and Bulinus spp. snails, is not suitable or adequate for the diet of Oncomelania hupensis ssp. snails because they need a source of food appropriate for their feeding behavior (functional morphology of radula shape and orientation determine food specificity) and that is easily digestible.
When maintaining Oncomelania hupensis ssp. under laboratory conditions, it is important to provide them with an appropriate diet. It has been found that the diatom Navicula pelliculosa is an important food source, in addition to providing them with cyanobacterial cultures such as Nostoc sp. Navicula cultures initially purchased through scientific suppliers can be grown in the laboratory by maintaining them in petri dishes with the same mud mounds that support Nostoc sp. growth. A petri dish of diatoms is sufficient to feed 60–100 Oncomelania hupensis ssp. snails.
The proportions of dried mud, lime and chicken manure needed for good growth of Nostoc sp. and Navicula pelliculosa will likely vary, depending on the richness of the soil obtained. A small amount of clay may be necessary for cohesion of the mud mound that will be placed in the petri dishes. The following describes the optimal proportions of each component for the soil. Trial and error will be the rule, rather than the exception, to accommodate the apparent richness (or lack thereof) of soils in other regions. The soil and site chosen ideally should be one where there is considerable sedimentation (e.g., a stream bed bottom) or topsoil. Soil should be obtained where no known herbicides or pesticides have been used.
Materials
Mud, or soil source
Chicken manure
Calcium carbonate (pulverized limestone)
Clay
0.06% sodium nitrate solution prepared with conditioned water (only needed if the soil is collected from nutrient-poor locations)
Stainless steel baking pan (10″ × 15″ × 3″)
Spatulas
Autoclaved conditioned water
Plastic petri dishes (100 × 25 mm)
Nostoc sp. (stock cultures can be obtained from Ward’s Biological Supply, Rochester, N.Y.)
Diatoms (Navicula pelliculosa purchased from Ward’s Scientific)
Procedure
Strain collected mud or soil through a series of crude screens to remove rocks and other large debris. Once it is of a rather fine consistency, it should be completely dried before use.
Mix 3 kg of dried mud with 90 g lime (pulverized limestone), and 30 g dried chicken manure. To this mixture, add enough conditioned water to make a paste. Place the mud mixture in a large stainless steel baking pan and cover with aluminum foil. The depth of the mud should be no more than about 100 mm. Autoclave for a continuous 2 hours.
-
Once the mud is autoclaved and cooled to room temperature, use a sterile spatula (spatulas should be wiped down periodically with gauze drenched in pure alcohol) to place about 40 g of moist mud in the center of a petri dish. Form a smooth and solid mud mound about 15 mm high and 60 mm in diameter. If the mud has dried too much during autoclaving and needs some additional liquid to make it spreadable, add a few ml of sterile 0.06% nirtrate solution and mix thoroughly.
To expedite the spreading process, one can use two curved sterile spatulas to stir a third to half of the mud in the steel pan (adding the sterile 0.06% nitrate solution as needed) before spreading it into the petri dishes. This ensures consistency of the ingredients in the mud that is placed in each petri dish. -
Add about 5–10 ml of a Nostoc suspension to seed the mud plate for new growth and cover the mud mound with 0.06% nitrate water. Be sure not to flood the petri dish with liquid, so that the lid does not become wet with the growth medium.
The Nostoc inoculum can be a mix of older seed plates and reconstituted dried Nostoc -
Cover and place the plates under fluorescent lighting (40 watt, cool-white fluorescent) at 25–27°C for 1–3 weeks. For best results, the lights should be about 1 foot above the petri dishes.
The preparation is suitable for feeding to the snails once a solid mat of the Nostoc has grown over the surface. A healthy mat should be dark green and may be bubbly across the top (see Figure 19.1.17).
Figure 19.1.17.

Freshly seeded mud base plate with Nostoc sp. (left). On the right is an identical plate placed under a fluorescent light, at room temperature, for 3 weeks. Notice the dark mat of Nostoc sp. on the surface of the plate.
Preparation of Diatoms (Navicula pelliculosa) in Mud for Oncomelania hupensis ssp. snails
Procedure
Agitate the test tube of Navicula sp. diatoms and pour approximately 8–10 ml of suspension into a petri dish of mud based medium prepared in a petri dish (see above). Pour enough autoclaved water to cover the mound of mud.
-
Cover the petri dish with the lid and store under fluorescent lighting (40 watt, cool-white fluorescent) at 22–24°C for 1–2 weeks. The light should be placed a foot above the petri dishes.
During the propagation period of the diatoms, particular attention should be paid to replace any water that is evaporated. The exposure of the mud mound in the petri dish to air over prolonged periods is detrimental to the diatom culture.One can also use Navicula diatoms from older petri dishes to seed new cultures. Decant 1/3 of the old diatom inoculum into each new petri dish containing a mud mound and cover the mound with conditioned water.
Schistosomule Culture Media
Schistosomule Wash Buffer (SW)
500 ml RPMI (Cellgro, 15-040)
5 ml Hepes Buffer (Cellgro, 25-060-CI)
10 ml Antibiotic/Antimycotic (Invitrogen, 15240-062)
Store for one month at 4°C
Schistosomule Wash Buffer plus Tween (SWAT)
SW + 0.5% Tween 20
Schistosomule Medium (SM)
First, make a. 20X solution of Lactalbumin hydrolysate/glucose:
2.5 g lactalbumin hydrolysate (Sigma, L9010)
2.5 g glucose (Sigma, G5400)
Mix in 250 ml Basal Medium Eagle (Gibco, 21010046)
Filter sterilize
Store for one month at 4°C
To make 1 liter of SM:
50 ml 20X Lactalbumin hydrolysate/glucose
0.5 ml Hypoxanthine (1 mM) (−20°C) (Sigma, H9377)
1 ml Serotonin (1 mM) (−20°C) (Sigma, H9523)
1 ml Insulin (8mg/ml) (4°C) (Sigma, I0516)
1 ml Hydrocortisone (1 mM) (−20°C) (Sigma, H0888)
1 ml Triiodothyronine (0.2 mM) (−20°C) (Calbiochem, 64245)
5 ml MEM Vitamins (100X) (−20°C) (Invitrogen, 11120-052)
50 ml Schneiders Medium (Drosophila) (+4°C) (Invitrogen, 11720067)
10 ml Hepes Buffer (+4°C) Triiodothyronine
100 ml Human Serum (thaw at 37°C prior to use) (Gemini, 100-512)
20 ml Antibiotic/Antimycotic (Invitrogen, 15240-062)
QS to 1 liter with Basal Medium Eagle (Gibco, 21010046)
Filter sterilize
Store for one month at 4°C
COMMENTARY
Background Information
It is not surprising that a wide range of immune reactions accompany a schistosome infection (Cheever and Yap, 1997). Schistosomes present a bewildering array of antigens to their definitive hosts, beginning with the penetration enzymes of the cercariae and progressing to tegumental (and subtegumental) antigens of the immature and mature worms, regurgitated products from the worm’s digestive tract, and egg-associated products.
Although immune responses can occur to antigens of exclusively cercarial origin, the schistosomule is the transitional stage of the parasite, which is of most interest from a vaccine perspective. It is easily collected from the lungs of experimental animals a few days after cercarial exposure (Perez et al., 1974; Sher et al., 1974; Lewis and Colley, 1977; Gobert et al., 2007). This stage is often used in assays to measure the activity of serum components or cell types in parasite-killing experiments, and methods have been developed to produce this stage in quantity (Colley and Wikel, 1974; Ramalho-Pinto et al., 1974). In the past, much study centered on which age of the schistosomule was most vulnerable to immune destruction, and the site at which this killing occurred. These may well vary depending on the definitive species studied and the method of immunization (Damian, 1984; Pearce and James, 1986). For the irradiated cercarial model of immunity in the mouse—the model that is best characterized—schistosomules passing through the lungs are the ones most vulnerable to immune elimination (Wilson and Coulson, 1989). It is appropriate for the investigator to decide which age of the schistosomule, if not several, to test in an in vitro killing assay system (Lewis et al., 1990). Schistosomules that are processed by in vitro methods gradually undergo changes during culture to resemble early schistosomules removed from intact animals (Stirewalt, 1974). The timing of the changes in the intact animal, however, is considerably faster than for in vitro–derived schistosomules (Stirewalt et al., 1983), a fact that should be kept in mind when trying to equate in vitro schistosomule killing experiments with immune events in the definitive host.
Since the first publication of the characterization of soluble egg antigens (SEA; Boros and Warren, 1970), work has centered on defining the components involved in stimulating granuloma formation (Lukacs and Boros, 1991). This is a daunting task, since crude SEA is a complex mixture of proteins, glycoproteins, polysaccharides, and glycolipids. Recent studies have implicated discreet egg components (e.g. glycoproteins) and provided great insight into schistosome egg components involved in immune responses (Everts et al., 2009; Steinfelder et al., 2009; deWalick et al., 2011; Meevissen et al., 2011; Pearce, 2005). Methods to homogenize the eggs have changed little since the early 1970s. Hand-held glass homogenizers seem the best tools for breaking apart the very tough egg shell. In the author’s’ experience, electrically powered homogenizers have not worked as well. In the initial stages of preparation, some investigators have enriched for certain components by varying extraction buffers (Lustigman et al., 1985; Weiss et al., 1986). Although a great deal is known about responsiveness to the crude and purified fractions, studies have also investigated recombinant egg antigens in an effort to dissect responsiveness to antigens in the formation of the granuloma (Cai et al., 1996).
Critical Parameters and Troubleshooting
Successful collection of parasite stages from the mammalian host depends upon a basic understanding of the schistosome’s life cycle, and especially upon the capacity to produce active cercariae. Efforts to maximize the production of cercariae, and expose the mammalian host so that maximal percentages mature to the adult worm stage, will reduce many problems and maximize cost efficiency.
Many frustrating problems can appear when working with schistosome life cycles. Many of these problems can be averted by applying the principles of snail rearing described in Support Protocol 4. A troubleshooting guide for many of the more common problems is shown in Table 19.1.1.
Table 19.1.1.
Troubleshooting Guide
| Problem | Possible causes | Solution |
|---|---|---|
| Low cercarial penetration rate | Old cercariae | Expose animals within 1–2 hr of shedding |
| Snails contaminated with rotifers | Mechanically remove rotifers from snail shells | |
| Inappropriate animal bedding/chemical inhibition of cercariae | Eliminate softwood bedding, wipe mouse tails prior to cercarial exposure | |
| Suboptimal water quality | Use charcoal-filtered and conditioned water | |
| Mechanical trauma to cercariae or cercariae difficult to manipulate (S. japonicum) | Use large-bore pipet tip for aliquots or hair loop to transfer cercariae | |
| Excessive hair stubble on abdominal penetration site | Shave abdominal exposure site completely to eliminate excess stubble | |
| Dry abdominal exposure area | Adequately moisten abdomen with conditioned water before cercarial exposure | |
| Low adult worm yields | Poor cercarial penetration | See above |
| Incomplete perfusions | Use greater volume of perfusion fluid, revise technique (insertion of needle, incision of portal vein, dissect worms from mesenteries) | |
| Low cercariae yields | Snail maintenance temperature not optimum | Adjust aquarium temperature |
| Snails contaminated with rotifers | Mechanically remove rotifers from snail shells | |
| Snail/schistosome incompatibility | Select for more susceptible snail line | |
| Poor shedding of snails/collection of cercariae | Leave snails in dark before shedding under a light source; leave snails under light while collecting cercariae | |
| High snail mortality | Foul or suboptimal water | Change water more frequently and/or reduce chlorine levels further |
| Overfeeding | Reduce food levels | |
| Water Temperatures >28°C | Reduce water temperature to ≤26°C | |
| Over infection | Expose snails individually to 3–5 miracidia/snail | |
| Metazoan contamination | Mechanically remove contaminants | |
| Snail crowding | Reduce snail density | |
| Copper tubing in plumbing | Replace with plastic tubing | |
| Low snail fecundity | Poor collection of egg masses or eggs | Provide substratum for egg laying (Biomphalaria spp., Bulinus spp.); Sift through mud and isolate from containers (Oncomelania hupensis ssp.) |
| Immature snails | Establish populations of breeder snails | |
| Oncomelania hupensis ssp. laying low number of eggs | Check for bias of male snails in breeder populations | |
| Slow snail growth rates | Crowding of snails | Prevent over-population of a single growth container; group snails in containers by size |
| Insufficient food source(s) | Supplement primary food source with other food types (algae, fish food flakes, etc.) |
Adopting techniques to ensure reproducible infections in laboratory animals will go far in circumventing problems in experiments. Mammals should be exposed to cercariae soon after shedding from snails, since the rate at which cercariae lose infectivity is influenced by the time elapsed after release from the snail. Penetration into mouse tail skin drops precipitously after 5 hr post-emergence. Whether or not all newly released cercariae are equally infectious is not known, although penetration rates for S. mansoni in experimental animals of up to 100% of cercariae have been reported.
If low cercarial penetration rates are noticed, one of the most frequently overlooked problems may not be parasite-related, but attributable to laboratory animal maintenance procedures. If laboratory animals are housed on softwood bedding, cercarial penetration rates will be low, due to oils toxic to cercariae that are in the softwood shavings (Campbell and Cuckler, 1961). Low penetration rates of cercariae will mean low and extremely variable recovery of adult worms. For this reason, laboratory animals should be maintained on bedding composed either of hardwood chips alone or of one of the paper or corn cob–based bedding materials, or in suspended cages. When exposing mice to S. mansoni cercariae via the tail, it is recommended that mouse tails be wiped with conditioned water before exposure. This measure helps remove any toxic oils that may accumulate on tail skin.
Using an exposure method whereby one can monitor the success of cercarial penetration, such as tail exposure of mice (Basic Protocol 1), will help an investigator track down problems that may be incorrectly attributed to such factors as parasite strain differences.
Of the several parameters used to measure infection intensity, most experiments in schistosomiasis rely on estimating the adult worm burden. Techniques to collect and count the adults are relatively straightforward, although perfusion should be followed with careful dissection of the mesenteries (especially important for S. haematobium-infected hamsters), since some adult worms may be difficult to dislodge upon perfusion and others may adhere to the surface of tissues outside the portal-vein area (Smithers and Terry, 1965; Duvall and DeWitt, 1967). Care should also be taken to record the degree of maturity of the female worms, since this forms the basis for estimating the fecundity of the worms.
Like the harvesting of adult worms, counting eggs in the tissues presents few technical difficulties. Most Schistosoma spp. eggs will either be located in the liver or intestines/feces. Potassium hydroxide digestion of tissues, under controlled conditions, followed by counting several aliquots of eggs, is sufficient for estimating tissue egg burden (Cheever, 1970). Problems in estimating fecundity can arise in infections of ≥10-week duration due to shunting of worm pairs to the lungs.
Studies based on the pathogenesis of schistosomiasis center their attention on reactions to the intact egg or egg components. Injection of intact eggs for use in the “pulmonary model” of granulomatous inflammation offers an insight into the window of granulomatous reactions, primarily because the reactions are relatively synchronous, in contrast to those of the natural infection (Edungbola and Schiller, 1979). Studies have appeared allowing a detailed dissection of the granulomatous process in Schistosoma spp. by examining the contribution of serum components and cell types involved at different stages of the granulomatous reaction (Cheever, 1987; Oswald et al., 1994; Sher et al., 1996). When using this model, however, it is important for each laboratory to experimentally define and standardize the system rigorously. For example, changing the percentage of the NaCl used to isolate eggs (from 1.7% to 1.2%) can have a drastic effect on the volume of the granulomas after egg injection.
With practice, procedures for collecting most other schistosome life stages from the mammalian host will present few problems. Techniques have been established for many years for most efficient methods of collecting schistosomules from the lungs, adult worms from the portal tract, and eggs from various tissues.
Anticipated Results
Many biotic and abiotic factors influence cercarial production in infected snails. Most of these are mentioned in Support Protocol 4. It is not unusual, however, for an individual infected B. glabrata snail to shed over 2,000 cercariae at any single collection time. For most S. mansoni-mammalian exposures therefore, one can obtain sufficient numbers of cercariae from a few snails, although using larger numbers will more readily ensure an approximately equal male-to-female ratio in the cercarial pool. Since Bulinus spp. and Oncomelania hupensis ssp. produce less cercariae per snail than the S. mansoni/B. glabrata system, it is necessary to shed or crush (Oncomelania hupensis ssp.) more snails in comparison.
Exposing mammals to active S. mansoni cercariae should result in >90% of the cercariae penetrating skin. On the peak day of migration through the lungs (5–7 days post-exposure), one can obtain ~40% of the cercariae used for exposure as schistosomules by the lung-chop method (Alternate Protocol 4). Later, from 35% to 50% of the invading cercariae will usually mature to the adult worm stage in both mice and hamsters, although the percentages may vary with the strain and age of the animal used (Stirewalt et al., 1965).
S. japonicum schistosomules can be detected in mouse lungs before S. mansoni (Gobert et al., 2007) but less is known about recovery of S. haematobium schistosomules. Gui et al. (1995) found that more S. japonicum cercariae can be recovered as adults in comparison to S. mansoni, but in the authors’ experience, this has not been observed regularly. In the authors’ experience, ~30% of invading S. haematobium cercariae in a hamster infection model (Basic Protocol 3) can be recovered as adult worms. The decreased recovery is a result of poorer yields from venous perfusion (as compared to S. mansoni and S. japonicum) mainly due to the fact that many adult worms stay sequestered in the mesenteric veins and must be removed by dissection. Moore and Meleney (1954) reported that ~17% adult S. haematobium worms were recovered from hamsters by perfusion, but there was no mention of dissected worms.
The yield of eggs from the tissues will depend on a number of factors, most notably the worm burden and the length of infection. As a point of reference, one can usually expect to obtain around 20,000 purified eggs from the liver and 20,000 eggs from the intestines of a single Swiss outbred mouse infected 7 weeks with 50 S. mansoni worm pairs. Comparable egg yields have been reported from livers of S. japonicum (Warren et al., 1975);
When preparing SEA from the isolated eggs, the yield will depend both on the concentration of eggs used for initial homogenization, as well as on the extent of homogenization. Under the conditions described in this unit, purified S. mansoni SEA at a protein concentration of ~1.3 mg/ml is regularly achieved.
Time Considerations
For each of the components of this section, approximate time allocations for performing the various tasks are given in Table 19.1.2.
Table 19.1.2.
Time Considerations
| Procedure | Quantity | Time |
|---|---|---|
| Collecting and counting cercariae | From 200 snails | 1.5–2 hr |
| Exposing mammals to cercariae | 30 mice or 10 hamsters | 1.5–2 hr |
| Preparation of in vitro schistosomules | From 2 × 105 cercariae | 2–2.5 hr |
| Collection of lung schistosomules | From 10 mice | 4 hr |
| Collection of adult worms | From 15 mice, 2 hamsters | 1 hr |
| Counting eggs in tissues (including overnight incubation) | From 10 mice | 24 hr |
| Isolating eggs for injection or antigen preparation | From 20 livers | 4 hr |
| Preparation of crude SEA | From 2 × 106 eggs | 2 hr |
| Rearing newly hatched snails to exposable size (5–8 mm diameter [B. glabrata]; 2–3 mm length [Bulinus spp.]; 4–6 mm in length [O. hupensis ssp.]) | Indeterminate | 1–2 months (depending on species, water temperature) |
| Exposing snails to miracidia | 500 snails | 2–3 hr |
| Cryopreserving schistosomules | 20,000 cercariae | 4–5 hr |
| Thawing cryopreserved schistosmules for injection into mice | <20,000 schistosomules | < 1 hr |
Acknowledgments
The authors wish to acknowledge NIH-NIAID Contract No. HHSN272201000005I.
Footnotes
Juvenile snails are very small and transparent and may be easily overlooked, particularly if mud and debris have accumulated in one area of the petri dish. One may use the Pasteur pipette to very gently swirl the water to move the solid material around the petri dish, thus revealing any newly hatched juvenile snails.
Contributed by Matthew S. Tucker, Laksiri B. Karunaratne, and Fred A. Lewis, Biomedical Research Institute, Rockville, Maryland
Literature Cited
- Basch PF. Cultivation of Schistosoma mansoni in vitro. I Establishment of cultures from cercariae and development until pairing. J Parasitol. 1981;67:179–185. [PubMed] [Google Scholar]
- Boros DL, Warren KS. Delayed hypersensitivity granuloma formation and dermal reaction induced and elicited by a soluble factor isolated from Schistosoma mansoni eggs. J Exp Med. 1970;132:488–507. doi: 10.1084/jem.132.3.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce JI, Liang YS. Cultivation of schistosomes and snails for researchers in the United States of America and other countries. J Med Appl Malacol. 1992;4:13–30. [Google Scholar]
- Bruce JI, Radke MG, Davis GM. Biomedical Report No 19,406th Medical Laboratory. U.S. Army; 1971. Culturing Biomphalaria and Oncomelania (Gastropoda) for large-scale studies of schistosomiasis. [Google Scholar]
- Cai Y, Langley JG, Smith DI, Boros DL. A cloned major Schistosoma mansoni egg antigen with homologies to small heat shock proteins elicits Th1 responsiveness. Infect Immun. 1996;64:1750–1755. doi: 10.1128/iai.64.5.1750-1755.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WC, Cuckler AC. The prophylactic effect of topically applied cedarwood oil on infection with Schistosoma mansoni in mice. Am J Trop Med Hyg. 1961;10:712–715. doi: 10.4269/ajtmh.1961.10.712. [DOI] [PubMed] [Google Scholar]
- Cheever AW. Relative resistance of the eggs of human schistosomes to digestion in potassium hydroxide. Bull WHO. 1970;43:601–603. [PMC free article] [PubMed] [Google Scholar]
- Cheever AW. Comparison of pathologic changes in mammalian hosts infected with Schistosoma mansoni, S. japonicum and S. haematobium. Mem Inst Oswaldo Cruz. 1987;82 (suppl 4):39–45. doi: 10.1590/s0074-02761987000800008. [DOI] [PubMed] [Google Scholar]
- Cheever AW, Yap GS. Immunologic basis of disease and disease regulation in schistosomiasis. In: Freedman DO, editor. Immunopathogenetic Aspects of Disease Induced by Helminth Parasites. S. Karger; Basel: 1997. pp. 159–176. [DOI] [PubMed] [Google Scholar]
- Chernin E, Michelson EH. Studies on the biological control of schistosome-bearing snails. III The effects of population density on the growth and fecundity in Australorbis glabratus. Am J Hyg. 1957;65:57–70. doi: 10.1093/oxfordjournals.aje.a119856. [DOI] [PubMed] [Google Scholar]
- Cohen LM, Neimark H, Eveland LK. Schistosoma mansoni: Response of cercariae to a thermal gradient. J Parasitol. 1980;66:363–364. [PubMed] [Google Scholar]
- Colley DG, Wikel SK. Schistosoma mansoni: Simplified method for the production of schistosomules. Exp Parasitol. 1974;35:44–51. doi: 10.1016/0014-4894(74)90005-8. [DOI] [PubMed] [Google Scholar]
- Cooper LA, Lewis FA, File-Emperador S. Re-Establishing a life cycle of Schistosoma mansoni from cryopreserved larvae. J Parasitol. 1989;75:353–356. [PubMed] [Google Scholar]
- Cousin CE, Stirewalt MS, Dorsey CH. Schistosoma mansoni: ultrastructure of early transformation of skin- and shear-pressure-derived schistosomules. Experimental Parasitol. 1981;51:341–365. doi: 10.1016/0014-4894(81)90122-3. [DOI] [PubMed] [Google Scholar]
- Damian RT. Immunity in schistosomiasis: A holistic view. In: Marchalonis JJ, editor. Contemporary Topics in Immunobiology. Plenum; New York: 1984. pp. 359–420. [DOI] [PubMed] [Google Scholar]
- deWalick S, Tielens AG, van Hellemond JJ. Schistosoma mansoni: The egg, biosynthesis of the shell and interaction with the host. Exp Parasitol. 2012;132:7–13. doi: 10.1016/j.exppara.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Duvall RH, DeWitt WB. An improved perfusion technique for recovering adult schistosomes from laboratory animals. Am J Trop Med Hyg. 1967;16:483–486. doi: 10.4269/ajtmh.1967.16.483. [DOI] [PubMed] [Google Scholar]
- Edungbola LD, Schiller EL. Histopathology of hepatic and pulmonary granulomata experimentally induced with eggs of Schistosoma mansoni. J Parasitol. 1979;65:253–261. [PubMed] [Google Scholar]
- Everts B, Perona-Wright G, Smits HH, Hokke CH, van der Ham AJ, Fitzsimmons CM, Doenhoff MJ, van der Bosch J, Mohrs K, Haas H, Mohrs M, Yazdanbakhsh M, Schramm G. Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J Exp Med. 2009;206:1673–1680. doi: 10.1084/jem.20082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert GN, Chai M, McManus DP. Biology of the schistosome lung-stage schistosomulum. Parasitology. 2007;134:453–460. doi: 10.1017/S0031182006001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui M, Kusel JR, Shi YE, Ruppel A. Schistosoma japonicum and S. mansoni: comparison of larval migration patterns in mice. J Helminthol. 1995;69:19–25. doi: 10.1017/s0022149x0001378x. [DOI] [PubMed] [Google Scholar]
- James ER, Farrant J. Recovery of infective Schistosoma mansoni schistosomula from liquid nitrogen. A step towards storage of a live schistosomiasis vaccine. Trans R Soc Trop Med Hyg. 1977;71:498–500. doi: 10.1016/0035-9203(77)90143-2. [DOI] [PubMed] [Google Scholar]
- James ER. Schistosoma mansoni: cryopreservation of schistomules by two-step addition of ethanediol and rapid cooling. Exp Parasitol. 1981;52:105–116. doi: 10.1016/0014-4894(81)90066-7. [DOI] [PubMed] [Google Scholar]
- Lazdins JK, Stein MJ, David JR, Sher A. Schistosoma mansoni: rapid isolation and purification of schistosomula of different developmental stages by centrifugation on discontinuous density gradients of percoll. Exp Parasitol. 1982;53:39–44. doi: 10.1016/0014-4894(82)90090-x. [DOI] [PubMed] [Google Scholar]
- Lewis FA, Colley DG. Modification of the lung recovery assay for schistosomula and correlations with worm burdens in mice infected with Schistosoma mansoni. J Parasitol. 1977;63:413–417. [PubMed] [Google Scholar]
- Lewis FA, Stirewalt MA, Souza CP, Gazzinelli G. Large-scale laboratory maintenance of Schistosoma mansoni, with observations on three schistosome/snail host combinations. J Parasitol. 1986;72:813–829. [PubMed] [Google Scholar]
- Lewis FA, White-Ziegler CA, Ball JE, Niemann GM. Schistosoma mansoni larvacidal activity of murine bronchoalveolar lavage cells. Infect Immun. 1990;58:3903–3908. doi: 10.1128/iai.58.12.3903-3908.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis FA. Schistosomiasis. In: Coico R III, editor. Current Protocols in Immunology. John Wiley and Sons, Inc; 1999. pp. 19.1.1–19.1.28. [Google Scholar]
- Liang YS, van der Schalie H, Berry EG. Transmission of ostracods in snails. Malacol Rev. 1973;6:66. [Google Scholar]
- Liang YS. Cultivation of Bulinus (Physopsis) globosus (Morelet) and Biomphalaria pfeifferi pfeifferi (Krauss), snail hosts of schistosomiasis. Sterkiana. 1974;54:7–73. [Google Scholar]
- Liang YS, Bruce JI, Boyd DA. Laboratory cultivation of schistosome vector snails and maintenance of schistosome life cycles. Proc First Sino-Am Sym. 1987;1:34–48. [Google Scholar]
- Lo CT. The inhibiting action of ostracods on snail cultures. Trans Am Microsc Soc. 1967;86:402–405. [Google Scholar]
- Lukacs NW, Boros DL. Splenic and granuloma T-lymphocyte responses to fractionated soluble egg antigens of Schistosoma mansoni–infected mice. Infect Immun. 1991;59:941–948. doi: 10.1128/iai.59.3.941-948.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustigman S, Mahmoud AAF, Hamburger J. Glycoproteins in soluble egg antigen of Schistosoma mansoni: Isolation, characterization, and elucidation of their immunochemical and immunopathological relation to the major egg glycoprotein (MEG) J Immunol. 1985;134:1961–1971. [PubMed] [Google Scholar]
- McMullen DB, Beaver P. Studies on schistosome dermatitis. IX The life cycles of three dermatitis-producing schistosomes from birds and a discussion of the subfamily Bilharziellinae (Trematoda: Schistosomatidae) Am J Trop Med Hyg. 1945;42:128–154. [Google Scholar]
- Michaelson EH. The protective action of Chaetogaster limnaei on snails exposed to Schistosoma mansoni. J Parasitol. 1964;50:441–444. [PubMed] [Google Scholar]
- Meevissen MH, Yazdanbakhsh M, Hokke CH. Schistosoma mansoni egg glycoproteins and C-type lectins of host immune cells: molecular partners that shape immune responses. Exp Parasitol. 2012;132:14–21. doi: 10.1016/j.exppara.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Moloney NA, Hinchcliffe P, Webbe G. The simple laboratory maintenance of a highly productive Schistosoma japonicum life cycle. Trans R Soc Trop Med Hyg. 1987;81:67–68. doi: 10.1016/0035-9203(87)90286-0. [DOI] [PubMed] [Google Scholar]
- Moore DV, Thillet CJ, Carney DM, Meleney HE. Experimental infection of Bulinus truncatus with Schistosoma haematobium. J Parasitol. 1953;39:215–221. [PubMed] [Google Scholar]
- Moore DV, Meleney HE. Comparative susceptibility of common laboratory animals to experimental infection with Schistosoma haematobium. J Parasitol. 1954;40:392–97. [PubMed] [Google Scholar]
- Najarian HH. Biological studies on the snail, Bulinus truncatus, in central Iraq. Bull World Health Organ. 1961;25:435–46. [PMC free article] [PubMed] [Google Scholar]
- Oswald IP, Caspar P, Jankovic D, Wynn TA, Pearce EJ, Sher A. IL-12 inhibits Th2 cytokine responses induced by eggs of Schistosoma mansoni. J Immunol. 1994;153:1707–1713. [PubMed] [Google Scholar]
- Pearce EJ, James SL. Post lung-stage schistosomula of Schistosoma mansoni exhibit transient susceptibility to macrophage-mediated cytotoxicity in vitro that may relate to late phase killing in vivo. Parasite Immunol. 1986;8:513–527. doi: 10.1111/j.1365-3024.1986.tb00866.x. [DOI] [PubMed] [Google Scholar]
- Pearce EJ. Priming of the immune response by schistosome eggs. Parasite Immunol. 2005;27:265–70. doi: 10.1111/j.1365-3024.2005.00765.x. [DOI] [PubMed] [Google Scholar]
- Perez H, Clegg JA, Smithers SR. Acquired immunity to Schistosoma mansoni in the rat: Measurement of immunity by the lung recovery technique. Parasitol. 1974;69:99–116. doi: 10.1017/s0031182000063046. [DOI] [PubMed] [Google Scholar]
- Ramalho-Pinto FJ, Gazzinelli G, Howells RE, Monta-Santos TA, Figueiredo EA, Pellegrino J. Schistosoma mansoni: A defined system for the stepwise transformation of a cercaria to schistosomule in vitro. Exp Parasitol. 1974;36:360–372. doi: 10.1016/0014-4894(74)90076-9. [DOI] [PubMed] [Google Scholar]
- Richards CS, Shade PC. The genetic variation of compatibility in Biomphalaria glabrata and Schistosoma mansoni. J Parasitol. 1987;73:1146–1151. [PubMed] [Google Scholar]
- Rowan WB. Mass cultivation of Australorbis glabratus, intermediate host of Schistosoma mansoni in Puerto Rico. J Parasitol. 1958;44:247. [Google Scholar]
- Sher A, Jankovic D, Cheever A, Wynn T. An IL-12 based vaccine approach for preventing immunopathology in schistosomiasis. Interleukin. 1996;12:288–301. doi: 10.1111/j.1749-6632.1996.tb52669.x. [DOI] [PubMed] [Google Scholar]
- Sher FA, MacKenzie P, Smithers SR. Decreased recovery of invading parasites from the lungs as a parameter of acquired immunity to schistosomiasis in the laboratory mouse. J Infect Dis. 1974;130:626–634. doi: 10.1093/infdis/130.6.626. [DOI] [PubMed] [Google Scholar]
- Smithers SR, Terry RJ. The infection of laboratory hosts with cercariae of Schistosoma mansoni and the recovery of the adult worms. Parasitol. 1965;55:695–700. doi: 10.1017/s0031182000086248. [DOI] [PubMed] [Google Scholar]
- Sodeman WA, Jr, Dowda M. Laboratory maintenance of Bulinus vectors of Schistosoma haematobium. Am J Trop Med Hyg. 1973;22:678–681. doi: 10.4269/ajtmh.1973.22.678. [DOI] [PubMed] [Google Scholar]
- Standen OD. Some observations upon the maintenance of Australorbis glabratus in the laboratory. Ann Trop Med Parasitol. 1951;45:80–83. doi: 10.1080/00034983.1951.11685473. [DOI] [PubMed] [Google Scholar]
- Steinfelder S, Andersen JF, Cannons JL, Feng CG, Joshi M, Dwyer D, Caspar P, Schwartzberg PL, Sher A, Jankovic D. The major component in schistosome eggs responsible for conditioning dendritic cells for Th2 polarization is a T2 ribonuclease (omega-1) J Exp Med. 2009;206:1681–90. doi: 10.1084/jem.20082462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirewalt MA. Effect of snail maintenance temperatures on development of Schistosoma mansoni. Exp Parasitol. 1954;3:504–516. doi: 10.1016/0014-4894(54)90046-6. [DOI] [PubMed] [Google Scholar]
- Stirewalt MA. Schistosoma mansoni: Cercaria to schistosomule. In: Dawes B, editor. Advances in Parasitology. Academic Press; London: 1974. pp. 115–182. [DOI] [PubMed] [Google Scholar]
- Stirewalt MA, Lewis FA. Schistosoma mansoni: Effect of rotifers on cercarial output, motility and infectivity. Int J Parasitol. 1981;11:301–308. doi: 10.1016/0020-7519(81)90040-0. [DOI] [PubMed] [Google Scholar]
- Stirewalt MA, Shepperson JR, Lincicome DR. Comparison of penetration and maturation of Schistosoma mansoni in four strains of mice. Parasitol. 1965;55:227–235. doi: 10.1017/s0031182000068724. [DOI] [PubMed] [Google Scholar]
- Stirewalt MA, Cousin CE, Dorsey CH. Schistosoma mansoni: Stimulus and transformation of cercariae into schistosomules. Exp Parasitol. 1983;56:358–368. doi: 10.1016/0014-4894(83)90081-4. [DOI] [PubMed] [Google Scholar]
- Stirewalt M, Lewis FA, Cousin CE, Leef JL. Cryopreservation of schistosomules of Schistosoma mansoni in quantity. Am J Trop Med Hyg. 1984;33:116–124. doi: 10.4269/ajtmh.1984.33.116. [DOI] [PubMed] [Google Scholar]
- Warren KS, Boros DL, Hang LM, Mahmoud AA. The Schistosoma japonicum egg granuloma. Am J Pathol. 1975;80:279–294. [PMC free article] [PubMed] [Google Scholar]
- Weiss JB, Magnani JL, Strand M. Identification of Schistosoma mansoni glycolipids that share immunogenic carbohydrate epitopes with glycoproteins. J Immunol. 1986;136:4275–4282. [PubMed] [Google Scholar]
- Wilson RA, Coulson PS. Lung-phase immunity to schistosomes: A new perspective on an old problem. Parasitol Today. 1989;5:274–278. doi: 10.1016/0169-4758(89)90017-3. [DOI] [PubMed] [Google Scholar]
- Wright CA. The Crowding Phenomenon in Laboratory Colonies of Freshwater Snails. Ann Trop Med Parasitol. 1960;54:224–32. doi: 10.1080/00034983.1960.11685978. [DOI] [PubMed] [Google Scholar]