

Abstract
The ubiquitin-proteasome system (UPS) promotes the timely degradation of short-lived proteins with key regulatory roles in a vast array of biological processes, such as cell cycle progression, oncogenesis and genome integrity. Thus, abnormal regulation of UPS disrupts the protein homeostasis and causes many human diseases, particularly cancer. Indeed, the FDA approval of bortezomib, the first class of general proteasome inhibitor, for the treatment of multiple myeloma, demonstrated that the UPS can be an attractive anti-cancer target. However, normal cell toxicity associated with bortezomib, resulting from global inhibition of protein degradation, promotes the focus of drug discovery efforts on targeting enzymes upstream of the proteasome for better specificity. E3 ubiquitin ligases, particularly those known to be activated in human cancer, become an attractive choice. Cullin-RING Ligases (CRLs) with multiple components are the largest family of E3 ubiquitin ligases and are responsible for ubiquitination of ~20% of cellular proteins degraded through UPS. Activity of CRLs is dynamically regulated and requires the RING component and cullin neddylation. In this review, we will introduce the UPS and CRL E3s and discuss the biological processes regulated by each of eight CRLs through substrate degradation. We will further discuss how cullin neddylation controls CRL activity, and how CRLs are being validated as the attractive cancer targets by abrogating the RING component through genetic means and by inhibiting cullin neddylation via MLN4924, a small molecule indirect inhibitor of CRLs, currently in several Phase I clinical trials. Finally, we will discuss current efforts and future perspectives on the development of additional inhibitors of CRLs by targeting E2 and/or E3 of cullin neddylation and CRL-mediated ubiquitination as potential anti-cancer agents.
Keywords: Anticancer targets, autophagy, cullins, CRL/SCF E3 ligase, MLN4924, NEDD8, neddylation, protein degradation, ubiquitin, UPS
THE UBIQUITIN-PROTEASOME SYSTEM: AN INTRODUCTION
The ubiquitin-proteasome system (UPS) is a major clearance system for the maintenance of protein homeostasis [1]. A protein doomed for degradation via UPS contains a poly-ubiquitin tag, resulting from a biochemical process known as polyubiquitination, which involves a three-step enzymatic reaction [2]. First, ubiquitin is activated through an ATP-dependent reaction catalyzed by ubiquitin-activating enzyme (E1). Activated ubiquitin is then transferred to an ubiquitin-conjugating enzyme (E2). Finally, a ubiquitin ligase (E3), which recognizes and recruits a target protein, designated as substrate, catalyzes the transfer of ubiquitin from E2 to the substrate via covalent attachment of ubiquitin onto its lysine residue (K). Ubiquitin itself contains seven lysine residues, which serve as the acceptors for second ubiquitin molecule, leading to polyubiquitination of the substrate after multiple runs of this reaction. However, some proteins can be ubiquitinated with only a single ubiquitin on a single lysine residue (mono-ubiquitination) or on multiple lysine residues (multi-mono-ubiquitination). Depending upon the nature of ubiquitin attachment and the type of isopeptide linkage of the polyubiquitin chain, the fate of ubiquitinated proteins varies. The K48/K11-linked poly-ubiquitination predominantly targets the protein for degradation after being recognized by the 26S proteasome, whereas the K63-linked poly-ubiquitination or mono-ubiquitination normally alters protein function and subcellular localization. These ubiquitin-modified proteins, instead of being degraded, play important roles in protein-protein interactions, DNA-damage responses and signal transduction [3-8] (see Fig. 1).
Fig. (1). The ubiquitin-proteasome system.

Ubiquitin is activated through an ATP-dependent reaction catalyzed by ubiquitin-activating enzyme (E1). Activated ubiquitin is then transferred to a ubiquitin-conjugating enzyme (E2), and finally attached to substrates catalyzed by ubiquitin ligase (E3). The fate of ubiquitinated proteins is determined on the nature of ubiquitin attachment and the type of isopeptide linkage of the polyubiquitin chain. The K48/K11-linked polyubiquitination predominantly targets protein for degradation by the 26S proteasome, whereas the K63-linked polyubiquitination and monoubiquitination normally alters protein functions.
Organization of the poly-ubiquitination enzymatic cascade is in a hierarchical order. The human genome encodes two E1 ubiquitin-activating enzymes, 38 E2 ubiquitin-conjugating enzymes and more than 600 E3 ubiquitin ligases [9-13] (Fig. 2). E3 ubiquitin ligases can be further subdivided into four major classes: 1) N-end rule E3s (E3α), 2) HECT (Homologous to E6-AP C-terminus) domain-containing E3s, 3) a RING (Really Interesting New Gene) finger-containing E3s, including its derivatives, the U-box domain-containing E3s and 4) APC/C (Anaphase Promoting Complex/Cyclosome) E3, which consists of 12 subunits [1,14-17]. The HECT E3s contain a conserved cysteine that can form a thioester intermediate with ubiquitin from ubiquitin-charged E2 and then pass it on to the substrate, whereas the RING-finger containing E3s promote the direct transfer of ubiquitin from the ubiquitin-charged E2 to the substrate [1,18]. RING-finger E3s can be farther divided into two subclasses: single peptide E3s, in which the RING-finger and substrate binding motif are different domains within the same protein molecule (e.g. MDM2) [19,20], and multi-component E3s, in which RING-finger and substrate binding units are assembled together from different individual proteins (such as CRLs) [21,22].
Fig. (2). The UPS cascade is pyramidal in design.
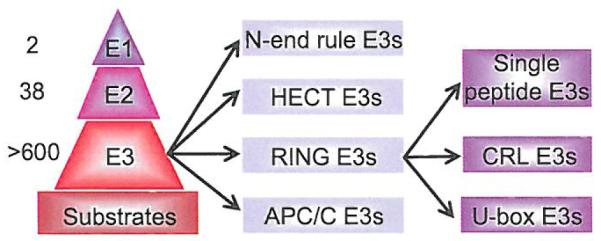
In human cells, there are two E1 ubiquitin-activating enzymes, 38 E2 ubiquitin-conjugating enzymes, more than 600 E3 ubiquitin ligases and thousands of substrates. The E3 ubiquitin ligases, based upon their structure, can be further categorized into 4 classes, including the N-end rule E3s, HECT-E3s, RING-E3s, and APC/C E3s. The RING-E3s can further be subdivided into RING-containing single peptide E3s. Cullin-RING ligases (CRLs) and its derivatives. U-box E3s.
THE COMPOSITION OF CRL E3s
Cullin-RING ligases (CRLs) are the largest family of E3 ubiquitin ligases that promote the ubiquitination of about 20% of cellular proteins doomed for degradation through UPS [23]. CRLs consist of, in general, four components: cullins, RINGs, adaptor proteins and substrate recognition receptors/proteins, catalyzing the transfer of E2-loaded ubiquitin to a substrate (Fig. 3A). In the human genome there are 1) two RING components (RBX1 and RBX2, also known as SAG), which bind to two zinc atoms via a C3H2C3 motif to form the RING finger domain, required for the activity of CRLs (Fig. 3B); 2) eight members of cullins (Cul-1-3, 4A, 4B, 5, 7, and Cul-9, also known as PARC) with an evolutionarily conserved cullin homology domain (CH domain with ~150 amino acids) at the c-terminus (Fig. 3C), responsible for their interaction with RBX1 or RBX2 [24]; 3) four adaptor proteins with Skp1 for Cul-1/7; Elongin B/C for Cul-2/5, and DDB1 for Cul-4A/B (Fig. 3D); and 4) many substrate recognition receptors, including 69 F-box proteins [25] for CRL-1, 80 SOCS proteins [26] for CRL-2/5, ~180 BTB proteins [27] for CRL-3, and 90 DCAF proteins [28] for CRL4A/B (Fig. 3E). Thus, CRLs have a total of more than 400 components, forming 8 cullin-based RING-dependent E3 ubiquitin ligases for targeted ubiquitination and degradation of thousands of substrates [9,29].
Fig. (3). The composition of CRL E3 ubiquitin ligases.

(A) A typical composition of CRL E3, consisting of a cullin, a RING protein, an adaptor protein, and a substrate recognition receptor. (B) The C3H2C3 RING structure of two RING proteins, RBX1 and RBX2. Numbers represent the amino acid codons and X stands for any amino acid. (C) The domain structure of cullin family members. Numbers represent the amino acid codons. The orange line indicates the position of the neddylation site. (D) Known adaptor proteins in CRLs. (E) The number of the family members in each type of receptor proteins found in CRLs. (The color version of the figure is available in the electronic copy of the article).
Fig. (4). Modularity of cullin-RING E3 ligases.

(A) CRL1, (B) CRL2, (C) CRL3, (D) CRL4, (E) CRL5, (F) CRL7, (G) CRL9. Representative and selective substrates of each individual CRL are shown, along with the biological processes they regulate. Note that substrates in red are tumor suppressors, whereas in green are oncoproteins. (The color version of the figure is available in the electronic copy of the article).
All CRLs share a similar core architecture with a cullin protein acting as a molecular scaffold that binds to an adaptor protein and a substrate receptor protein at the N-terminus, and a RING protein, RBX1 or RBX2 at the C-terminus [24]. It is well established that the substrate specificity of CRLs is determined by substrate recognition receptors, such as F-box proteins [25], whereas the core ligase activity is possessed by the cullins-RBX1/2 complex [30]. Furthermore, each individual cullin also contains a key lysine residue at its C-terminus for targeted NEDD8 modification (Fig. 3C). Cullin neddylation is also required for CRL activity (see below for details) [31], Cullins, therefore, act as a “center” player to bring together other members of CRL to form an active multi-unit “holoenzyme” for substrate ubiquitination and subsequent degradation (Fig. 3A). Below are brief discussions of each CRL E3, including their respective components, important targeting substrates and affected biologic processes.
EIGHT CULLIN-BASED RING E3 LIGASES AND THE BIOLOGIC PROCESSES THEY REGULATE
1). CRL1
Among all CRLs, CRL1, also known as SCF (Skp1-Cullin 1-F box protein), is the best studied founding member of CRLs [32,33], Cullin-1 recruits SKP1 adaptor protein, F-box receptor proteins and RJBX1 or RBX2 RING protein to form SCF E3 complex that promotes ubiquitin transfers from RBX1/RBX2-bound E2 to a substrate, recognized/bound by a F-box protein (Fig. 4A). Although the human genome encodes 69 F-box proteins [25], which are characterized by a 40-amino-acid F-box domain for SKP1/cullin-1 binding and a WD40 or LRR region for substrate recognition [34], only three F-box proteins, namely SKP2, FBXW7 and β-TrCP, are well studied [35,36], An interesting phenomenon in CRL1 action is that an F-box protein can recognize several substrates for targeted ubiquitination and degradation, whereas the same substrate can also be recognized and targeted by different F-box proteins. For instance, both SCFFbxw7 [37-39] and SCFSkp2 [40-43] control the turnover of Myc and Cyclin E. Another unique feature for substrate degradation mediated by CRL1/SCF E3s is that a substrate has to be phosphorylated by a kinase or a group of kinases acting in a coordinated manner prior to being recognized by an F-box protein [9,22]. Thus, ubiquitination and degradation of a given substrate by CRL1/SCF E3 is precisely regulated in a cell type and context dependent manner, which would rely at any given time on the availability of a signal trigger to activate kinase(s) for substrate phosphorylation, a “nearby” F-box protein for substrate recognition, free unbound cullin-1 and an E2-Ub-loaded RBX1 or RBX2 to form the E3 complex. These features may explain why the same critical substrate can be recognized by multiple F-box proteins to ensure its timely degradation, when needed.
Among key substrates of CRL1/SCF E3 are oncoproteins (e.g. Myc, c-Jun, β-catenin, Notch), tumor suppressors (p21, p27, NF1) [44,45], cell cycle promoters (cyclin D/E), apoptosis regulators (Mcl-1, BimEL) [46,47], and signaling molecules (IκB, DEPTOR) [48], just to name a few (Fig. 4A) (for more complete list, see [40]). Thus CRL1 E3 regulates many biologically significant processes such as tumorigenesis, cell cycle progression, apoptosis, signal transduction and genome integrity. Since abnormal activation of CRL1/SCF E3 was found in a number of human cancers, it was considered as an attractive anti-cancer target [33,49,50]. However, our understanding of this E3 is still at the infant stage, given the fact that only less than 5% of the entire F-box family has been well studied. Future study will be directed to functional characterization of all 69 F-box proteins and their corresponding substrates to thoroughly elucidate the biological processes regulated by this E3.
2) CRL2
CRL2 is a complex of cullin-2, REX1, adaptor protein Elongin B/C and substrate receptor protein VHL. The best known substrate of CRL2 is HIF1α (Fig. 4B). Analogous to a CRL1 substrate with a requirement for phosphorylation by a kinase before being recognized by a F-box protein, HIF1α has to be hydroxylated at conserved proline residues by the PHD family of prolyl-4-hydroxylases before being recognized by VHL for targeted ubiquitination [51,52]. Activity of prolyl-4-droxylases requires oxygen and therefore, under hypoxic conditions, the enzymes are inactivated, leading to HIF1α accumulation [53], Consistently, HIF1α accumulation can also be caused by mutational inactivation of VHL tumor suppressor, which is responsible for the development of von Hippel-Lindau disease, an inherited human cancer syndrome with high Incidence of tumors in eye, brain, kidney, lung and liver in affected individuals [54]. Accumulated HIF1α then binds to HIF1β, also known as aryl hydrocarbon receptor nuclear translocator (ARNT), to form an active HIF1 transcription factor, which promotes angiogenesis, proliferation and cell survival by transactivating many of its target genes involved in regulation of these processes [55], Other oncogenic substrates of CRL2 include β2-adrenergic receptor (β2-AR) [56], atypical PKCλ [57], and the RNA polymerase II subunit hsRPB7 [58]. Thus, CRL2 mainly regulates hypoxic responses, gene transcription and signaling transduction. Given the tumor suppressive nature of CRL2 E3s, selective targeting of CRL2 may benefit cancer cell growth.
3) CRL3
CRL3 consists of cullin-3, RBX1, and BTB (Bric-a-brac, Tramtrack, Broad-complex domain) proteins as substrate receptors (Fig. 4C). The best characterized substrate of CRL3 is Nrf2 (NF-E2 related factor 2) [59], a transcription factor which, upon activation in response to oxidative stress, binds to antioxidant response elements found in several antioxidant genes and phase II detoxification genes to transactivate their expression [60]. Again, CRL3 mediated Nrf2 ubiquitination and degradation is subjected to stress regulation. Under unstressed conditions, Nrf2 is mainly localized in the cytoplasm and bound to receptor protein Keap1 (Kelch-like ECH-associated protein 1) for targeted degradation. Upon oxidative stresses, Keap1-CRL3-mediated Nrf2 ubiquitination is impeded and spared Nrf2 is rapidly translocated into the nucleus to execute its function as an antioxidant transcription factor [61], The facts that Keap1 is mutated in human lung cancer [62] and Nrf2 is frequently overexpressed in many human cancers [63] suggest that CRL3 may act as a tumor suppressor, which is further supported by its ubiquitination and degradation of BMI1, an oncoprotein [64], and DAXX, a transcriptional repressor of p53 [65], Taken together, CRL3 regulates antioxidant responses as well as gene transcription with tumor suppressive properties, and like CRL2, selective targeting of CRL3 should be avoided.
4) CRL4A/B
CRL4 consists of CRL4A and CRL4B, dependent on either cullin-4A or cullin-4B acting as the scaffold protein. The adaptor protein for CRL4 is DDB1 (DNA damage-binding protein 1), whereas the substrate receptor is DCAF (DDB1-CUL4 associated factor) (Fig. 4D). CRL4 promotes ubiquitination and degradation of a number of DNA damage responsive proteins, such as CDT1 [66,67] and XPC [68] as well as proteins which regulate histone methylation such as PR-Set7/Set8 [69] and WDR5 (for CRL4B) [70]. CRL4, therefore, regulates cellular responses to DNA damage and chromatin remodeling. Since the Cul4A gene is amplified or overexpressed in a number of human cancers (for review see [71,72]), which is associated with poor prognosis of cancer patients [73], while Cul4B mutations are associated with X-linked mental retardation (XLMR) [74-77], selective targeting of CRL4A E3 might be an ideal strategy for cancer therapy.
5) CRL5
CRL5 E3 consists of cullin-5, RING protein RBX2, adaptor proteins Elongin B/C, and receptor proteins SOCS (Suppressors of cytokine signaling) (Fig. 4E). Progress on identification and characterization of specific endogenous CRL5 substrates is lacking, although it has been suggested that active Src oncoprotein might be subjected to degradation by CRL5 [78,79], On the other hand, it has been relatively well established that several viral proteins can hijack cellular CRL5 E3 to promote degradation of several key host proteins. Examples include HIV-1 viral infectivity factor (Vif), which contains a SOCS-box domain, assembles a cellular CRL5 E3 to promote ubiquitination and degradation of the host anti-viral factor APOBEC3G [80]. Adenovirus proteins E4orf6 and E1B55K were also found to assemble an active CRL5 to promote degradation of p53 [81]. Likewise, latency-associated nuclear antigen (LANA) from Kaposi’s sarcoma-associated herpes virus (KSHV) also targets the degradation of tumor suppressors, VHL and p53 via CRL5 E3 [82], Taken together, CRL5 appeal’s to be utilized by these viruses to damage the host. Selective inhibitors of CRL5 E3 might, therefore, be useful as anti-virus agents.
6) CRL7 and CRL9
CRL7 E3 consists of cullin-7, adaptor protein Skp1, RING protein RBX1, and receptor protein FBXW8; whereas very little is known about CRL9 E3 except the founding component cullin-9, also known as PARC (p53-associated parkin-like cytoplasmic protein). A well-characterized substrate of CRL7 E3 is IRS-1 (Insulin receptor substrate 1) [83], which is a signaling molecule positively regulating the PI3K/AKT pathway. Furthermore, mutations of the Cul7 gene are associated with 3-M syndrome, which is characterized by severe pre- and postnatal growth retardation [84], Thus, CRL7 targets the IRS-PI3K-AKT axis, and selective inhibition of CRL7 E3 may confer survival.
CRL E3s AS ANTI-CANCER TARGETS
Given the fact that CRL E3s play a fundamental role in regulating various biological processes including cell cycle progression, gene transcription, apoptosis, signal transduction and DNA replication among others [9,85,86], it is anticipated that deregulation of CRLs is associated with uncontrolled proliferative diseases such as cancer [85]. Among hundreds of components of CRLs, only a few components are well studied for their involvement in cancer. Cancer-associated CRL components can be generally classified as oncogenes (e.g. SKP2 and Cul4A) which are frequently amplified and/or overexpressed in cancers or contact-dependent oncogenes (e.g. β-TrCP), and tumor suppressors which are mutated and inactivated in human cancers (e.g. FBXW7 and VHL) [35,36,50]. Although the oncogenic properties of some CRLs make them potential targets for therapeutic intervention, the tumor suppressive properties of other CRLs may, however, negate them as cancer targets. However, a particular CRL, which promotes the degradation of some dominant tumor suppressor substrates in a specific cellular context during tumorigenesis, would qualify it as a candidate anticancer target. Nevertheless, the overall validation of CRL E3s as candidate cancer targets is mainly based upon the following: 1) enzymatic activity of CRL E3 ligases requires a) the RING component, RBX1 or RBX2 and b) cullin neddylation, 2) both RING components, RBX1 and RBX2 are overexpressed in a number of human cancers [72], and 3) the availability of MLN4924, a small molecule inhibitor of Nedd8-Activating Enzyme (NAE), which indirectly inhibits CRLs by abrogation of cullin neddylation [23].
1) Validation by Targeting the RING Component, RBX1 or RBX2
Several studies from our laboratory showed that either RBX1 or RBX2 is frequently overexpressed in several types of human cancer, including lung, breast, colon, stomach, and liver [87-90], To determine if overexpressed RBX1 or RBX2 is required for tumor cell growth, we used siRNA-based genetic approach and found that cancer cells are “addicted” to high levels of RBX1 or RBX2, since knockdown of either RBX1 or RBX2 suppresses cancer cell growth by inducing three types of cell death, namely apoptosis, senescence and autophagy in sequential as well as simultaneous orders [88-90], Our mechanistic study revealed that apoptosis induced upon RBX1 knockdown is mediated by enhanced DNA damage as a result of accumulation of CDT1 and ORCI [88,91], two DNA licensing proteins and substrates of CRL4 [67] and CRL1 [92], respectively. Senescence induced by RBX1 knockdown is associated at least in part with accumulation of a CRL1 substrate, p21 [90], whereas autophagy induction is attributable to mTORC1 inactivation, resulting from accumulation of DEPTOR, a naturally occurring inhibitor of mTORCs [93] and a newly characterized substrate of CRL1βTrCP [48,94,95]. On the other hand, RBX2 silencing-induced apoptosis is associated with accumulation of procasapse-3 and NOXA, two pro-apoptotic proteins, which are also the substrates of CRLs [89,96]. Furthermore, Rbx2 knockout sensitizes mouse embryonic stem cells to radiation via inactivation of NFκB due to IκBα accumulation [97]. Consistently, overexpression of RBX2 protects cancer cells from apoptosis induced by a variety of stresses in many in vitro cell culture models (for review, see [72]), and promotes skin proliferation and carcinogenesis induced by UV and DMBA-TPA in in vivo transgenic mouse models, through the modulation of CRL substrates, including p27, c-JUN/AP1, and IκBα/NFκB [98,99]. Thus, increased dependence of cancer cells on overexpressed RBX1/RBX2 makes them ideal cancer targets. Furthermore, since either RBX1 or RBX2 is required for the ligase activity of each individual CRL (see Fig. 4), targeted inactivation of RBX1/RBX2 would lead to broad inhibition of CRLs and might be useful approach for the treatment of human cancer with activated CRLs. These studies, therefore, provide validating evidence that CRLs could be selective anti-cancer targets.
2) Validation by Targeting Cullin Neddylation
a) Control of CRL E3 Ubiquitin Ligases by Neddylation
A key feature of CRLs E3 ligases is that the cullin protein must be covalently modified by one NEDD8 molecule, a reaction known as neddylation, which disrupts inhibitory binding by CAND1 and retains the CRLs E3 ligases in an active conformation [100], Like ubiquitination, neddylation requires E1 NEDD8-activating enzyme (NAE), which activates NEDD8; E2 NEDD8-conjugating enzyme, which carries NEDD8; and E3 NEDD8 ligase, which catalyzes the transfer of NEDD8 from an E2 to a target molecule (e.g. cullin) [101-103] (Fig. 5). Each cullin protein contains a conserved lysine at its C-terminus, which is readily neddylated [31] (Fig. 3C).
Fig. (5). Dynamic regulation of CRLs activity by neddylation.
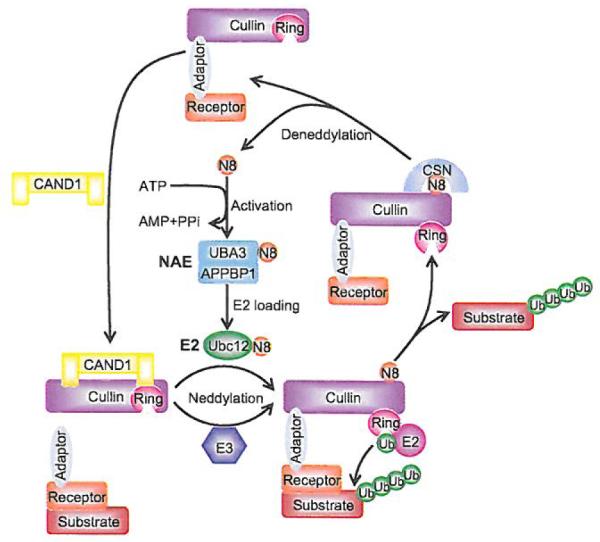
The binding of unmodified cullin to CAND1 inhibits the binding of the substrate receptor-adaptor module to the N-terminus of cullin. Neddylation of cullin disrupts the inhibitory binding by CAND1 and retains the CRLs in an active conformation to promote substrate ubiquitination. Like ubiquitination, neddylation requires E1 NEDDS-activating enzyme (NAE), E2 NEDD8-conjugating enzyme (UBC12), and E3 NEDD8 ligase, which catalyzes the transfer of NEDD8 to cullin. After dissociation of polyubiquitinated substrate from CRL complex, CSN binds to neddylation site of cullin and removes NEDD8 from cullin for recycle. CAND1 then binds to cullin and inactivates the CRLs.
Cullin neddylation has been reported to have a range of functional consequences for the activity of CRLs E3 ligases. First of all, CAND1 (Cullin-associated and neddylation-dissociated-1), a large sequestration factor, binds to unmodified cullin, leading to inhibition of the binding of the substrate receptor-adaptor module to the N-terminus. Conjugation of cullin with NEDD8 disrupts the inhibitory binding by CAND1 and retains the CRLs in an active conformation [100,104-108] (Fig. 5). Second, neddylation enhances the recruitment of ubiquitin charged E2 to CRLs and also stabilizes E2 recruitment [109]. Third, the crystal structure of the Cul1–Rbx1–Skp1–F boxSkp2 SCF complex (unmodified Cul1) suggested the existence of an ~ 50 Å gap between the substrate docking site and the E2 active site [24], which, if bridged by neddylation of Cul1 [109], would greatly facilitate the initial ubiquitin transfer. Fourth, neddylation also increases the rate of ubiquitin chain elongation, indicating that the conformation change also improves the activated E2 access to the end of the nascent polyubiquitin chain [34,109].
The mechanism by which neddylation enhances the transfer of Ub from ubiquitin charged E2 to the substrate is fully illuminated by crystal structures of the C-terminal domain (CTD) of Cul5 bound to Rbx1 in the unmodified and NEDD8-conjugated states [100], Neddylation induces striking conformational rearrangements in Cul5CTD-Rbx1, which results in elimination of the CAND1 binding site and release of Rbx1 RING domain from its tight cullin-binding into an “open” conformation. Although the RING domain remains tethered to Cul5CTD, the linker is more flexible and readily bridges the ~ 50 Å gap between the bound E2 and naked substrate present in the original CRLs/SCFs complex structure, providing a sound explanation why neddylation promotes the initiation of ubiquitin transfer. This flexibility also helps the activated E2 to access to the end of nascent chain to allow efficient polyubiquitination [9,101,102] (Fig. 5).
In a reverse reaction, NEDD8 is removed from cullins by the isopeptidase activity of the COP9 signalosome complex (CSN), a reaction known as deneddylation [110]. Thus, neddylation promotes the assembly of active CRLs E3 complex and stimulates the ubiquitination of substrates, whereas deneddylation promotes the dissociation of CRLs E3 complex and binding of the cullin-RING core with inhibitory CAND1. Dynamic neddylation and deneddylation of CRLs represent an important mechanism by which the activation cycle of CRLs is regulated for the maintenance of cellular homeostasis. Furthermore, this cycle of neddylation/deneddylation facilitates the recycling of the cullin-RING core, which is made available for assembly of other CRLs to allow the ubiquitination of many different substrates as required by the cell [111], Accumulating evidence suggests that mutations in CSN components lead to defects in cell cycle progression, signal transduction and development [112-114], Thus, it is equally important to understand the regulation and mechanism of deneddylation. Understanding of cullin neddylation and its biological significance has been advanced by the discovery of a chemical inhibitor of neddylation, designated as MLN4924 [23].
b) MLN4924, a Cullin Neddylation Inhibitor, as a Novel Class of Anticancer Agent
MLN4924 is a newly discovered potent small molecule inhibitor of NEDD8-activating enzyme (NAE) [23] (Fig. 6A). MLN4924 binds to NAE and creates a covalent NEDD8-MLN4924 adduct, which cannot be further utilized in subsequent intraenzyme reactions, thus blocking NAE enzymatic activity [115]. Since cullin neddylation, a process catalyzed by NAE, is required for the activity of CRL E3 ligases, by blocking cullin neddylation, MLN4924 inactivates the entire CRLs and serves as a very attractive pharmaceutical approach to validate CRLs as the cancer targets.
Fig. (6). MLN4924, an inhibitor of NEDD8-activating enzyme for cancer therapy.

(A) Chemical structure of MLN4924. (B) MLN4924 blocks neddylation of all cullins tested and cause the accumulation of CRL substrates. MCF7 cells were treated with 1.0 μM MLN4924 or DMSO vehicle control for indicated time periods, followed by Western blotting with indicated antibodies. (C) MLN4924 induces apoptosis. MCF7 cells were treated with 1.0 μM MLN4924 for indicated time periods, followed by Western blotting with indicated antibodies. (D) MLN4924 induces autophagy. HeLa cells stably expressing EGFP-LC3 were treated with 1.0 μM MLN4924 or DMSO control for 24 h prior to photography under a fluorescent microscope. (E) A model for suppression of cancer cell growth by MLN4924.
The first study, which reported the discovery of MLN4924, clearly demonstrated that MLN4924, by inactivating CRLs, causes accumulation of CRLs substrates and suppresses tumor cell growth both in vitro and in vivo [23], Indeed, we found that MLN4924 is a potent inhibitor of cullin neddylation, as evidenced by complete deneddylation of all cullins tested, including Cul1, Cul2, Cul4A and Cul5 after 6 hrs of treatment in MCF7 breast cancer cells. Consequently and expectedly, p21, p27 and HIF1α, three well-known substrates of CRLs, were accumulated substantially (Fig. 6B).
Early mechanistic studies of MLN4924 against tumor cell growth revealed that MLN4924 effectively induced apoptosis [23,116-119] and senescence [120-122], Indeed, we found that apoptosis was induced significantly in MCF cells after MLN4924 treatment, as evidenced by increased cleaved-PARP and cleaved-caspase-7 (Fig. 6C). Recently, we further found that MLN4924 effectively induced autophagy in both dose- and time-dependent manners in multiple human cancer cell lines [123,124]. As an example, shown here, MLN4924 significantly induced punctuate structures, a well-defined autophagy marker [125], in EGFP-LC3-expressing HeLa cells (Fig, 6D). Our detailed mechanistic studies revealed that MLN4924-induced autophagy is mainly mediated by inactivation of mTORC1, resulting from 1) accumulation of DEPTOR, a naturally occurring inhibitor of mTORCs [93] and a newly characterized substrate of CRL1βTrCP [48,94,95], and 2) accumulation of HEF1α, a well-known substrate of CRL2 [51,52], followed by activation of HIF1-REDD1-TSC1 axis. We Anther elucidated that mTORC1 inactivation and subsequent autophagy induction act as an overall survival signal, since abrogation of autophagy via genetic and pharmaceutical means led to an increased suppression of tumor growth by enhancing apoptosis induction [123], Our study therefore provides a strong piece of proof-of-concept evidence for a rational drug combination of MLN4924 with an autophagy inhibitor for more effective cancer therapy [126].
Taken together, the data show that MLN4924 effectively suppresses tumor cell growth by inducing all three common types of death, namely senescence, apoptosis, and autophagy. Induction of senescence, largely seen in solid tumor cell lines [120], is mainly mediated by p21 accumulation as a result of inactivation of CRL1 [127] and CRL4 [128,129], Induction of apoptosis, as often seen in both leukemia/lymphoma and solid tumor lines [23,116-119,124], is mainly caused by NFκB inactivation, resulting from IκBα accumulation due to CRL1 inactivation [130]. Finally, induction of autophagy, mainly seen in solid tumor lines [123,124], is largely mediated by inactivation of mTORC1, resulting from accumulation of DEPTOR and HIF1α due to inactivation of CRL1 and CRL2 (Fig. 6E).
The MLN4924 studies provide us two pieces of convincing evidence. First, CRLs are attractive anti-cancer targets. By inactivation of the entire CRLs E3, an overall anticancer activity is achieved. Second, targeting CRLs (by MLN4924) is less toxic than targeting proteasomes (by bortezomib), since MLN4924 selectively blocks degradation of a specific set of cellular proteins regulated by CRL E3s, whereas bortezomib inhibits degradation of all proteins through the 26S proteasome, leading to greater general cytotoxicity [131], In fact, MLN4924 was well-tolerated when administrated to mice [23,132], With promising preclinical efficacy, MLN4924 has been advanced to several Phase I clinical trials against a number of human malignancies [103,133].
CONCLUSION AND FUTURE PERSPECTIVES
In summary, CRLs are validated as attractive anticancer targets via genetic (siRNA targeting RBX1 or RBX2) and small molecule (MLN4924 blocking cullin neddylation) approaches. The fact that complete inactivation of CRLs causes growth suppression of tumor cells indicates that the major function of CRLs in cancer cells is to target the degradation of tumor suppressor proteins in order to sustain an unlimited proliferation. This unique feature renders cancer cells much more vulnerable to targeted therapy against CRLs.
It should be clearly noted that although MLN4924 demonstrated some anti-cancer activity in both in vitro cell culture and in vivo animal studies, like bortezomib, but to a lesser extent, MLN4924 bears a specificity issue. First of all, there are 8 CRLs and, by mechanism, MLN4924 would inhibit all. Given the fact that some CRLs have a tumor suppressor function, their inhibition might be detrimental to normal cells. Secondly, MLN4924 is a NAE inhibitor and would likely inhibit other cellular neddylation reactions (e.g. neddylation of p53 or MDM2) [134,135], although cullins are the major physiological substrates. Third, it has been reported recently that cancer cells can develop resistance to MLN4924 via selection of rare clones with heterozygous mutations in the targeting enzyme NAEβ [136,137], Despite these facts, MLN4924, which targets multiple signaling pathways by inactivating CRLs, would be likely more efficacious than targeted therapy using a single kinase inhibitor, since human cancers harbor multiple mutations with alterations in multiple signaling pathways [138,139].
To overcome the limitations of MLN4924. future discovery efforts should be directed to identify specific inhibitors against neddylation E2 and E3 for more specific modulation of cullin neddylation of CRLs. Some drug discovery efforts have been made in the ubiquitin pathway with discovery of PYR-41 as an inhibitor of ubiquitin E1 [140], CC0651 as an E2 inhibitors [141], and compound A, SCF-I2 and SMER3 as inhibitors of various CRL E3s [142-144] (Fig. 7), although anti-cancer activity of these inhibitors awaits further testing. With validation of CRLs as attractive anticancer targets, inhibitors targeting a specific CRL E3, known to be activated in human cancer, could be eventually discovered and developed as a novel class of anticancer agents for the treatment of human cancers with selective activation of this particular CRL.
Fig. (7).
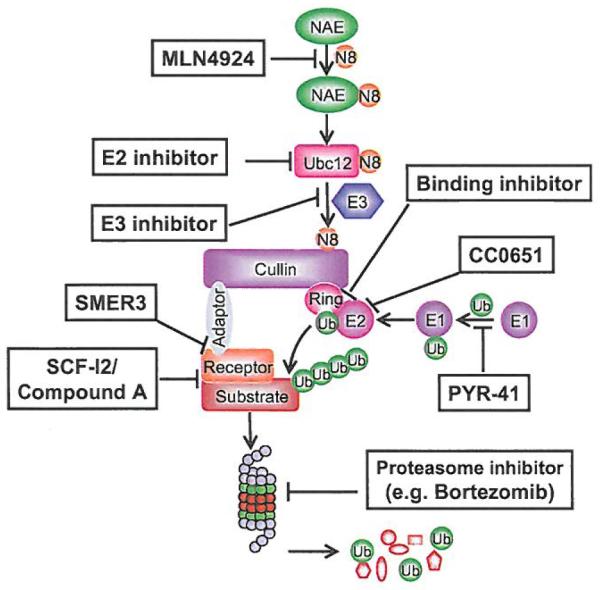
Schematic presentation of neddylation and CRL-mediated ubiquitination and potential points of targeting.
ACKNOWLEDGEMENTS
This work was supported by the NCI grants (CA118762, CA156744, CA171277 and CA170995) to Yi Sun and Susan G. Komen for the Cure PDF Grant (PDF12230424) to Yongchao Zhao.
LIST OF ABBREVIATIONS
- APC/C
Anaphase Promoting Complex/Cyclosome
- ARNT
Aryl hydrocarbon Receptor Nuclear Translocater
- BTB
Bric-a-brac, Tramtrack, Broad-complex domain
- β2 -AR
β2-adrenergic receptor
- βTrCP
Beta-Transducin Repeat Containing Protein
- CAND1
Cullin-Associated and Neddylation-Dissociated-1
- CDT1
Chromatin licensing and DNA replication factor 1
- CRLs
Cullin-RING Ligases
- DAXX
Death domain-Associated protein
- DCAF
DDB1-CUL4 Associated Factor
- DDB1
DNA Damage-Binding protein 1
- DEPTOR
DEP domain containing mTOR-interacting protein
- EGFP
Enhanced Green Fluorescent Protein
- FBXW7
F-box/WD repeat-containing protein 7
- HECT
Homologous to E6-AP C-terminus
- HIF1
Hypoxia-inducible Factor-1
- IRS-1
Insulin Receptor Substrate 1
- Keap1
Kelch-like ECH-associated protein 1
- LC-3
Microtubule-associated protein light chain 3
- MDM2
Murine Double Minute-2
- mTOR
Mammalian target of rapamycin
- mTORC
Mammalian target of rapamycin complex
- NAE
NEDD8-Activating enzyme
- NEDD8
Neural precursor cell Expressed, Developmentally Down-regulated 8
- NF1
Neurofibromatosis-1
- Nrf2
NF-E2 related factor 2
- ORC1
Origin Recognition Complex-1
- PARK
p53-Associated Parkin-like cytoplasmic protein
- PHD
prolyl-4-hydroxylase domain
- RBX1
RING box protein-1
- REDD1
Regulated in Development and DNA damage responses-1
- RING
Really Interesting New Gene
- ROC1
Regulator of Cullins 1
- SAG
Sensitive to Apoptosis Gene
- SCF
Skp1, Cullin and F-box protein
- SKP1
S-phase Kinase-associated Protein 1
- SKP2
S-phase kinase-associated protein 2
- SOCS
Suppressors of Cytokine Signaling
- UPS
Ubiquitin-proteasome system
- VHL
Von Hippel–Lindau
- XLMR
X-linked mental retardation
Footnotes
CONFLICT OF INTEREST The authors confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- [2].Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–60. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–86. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- [4].Deng L, Wang C, Spencer E, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–61. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- [5].Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–65. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pickart CM. Ubiquitin in chains. Trends Biochem Sci. 2000;25:544–8. doi: 10.1016/s0968-0004(00)01681-9. [DOI] [PubMed] [Google Scholar]
- [7].Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol. 1995;15:1265–73. doi: 10.1128/mcb.15.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu P, Duong DM, Seyfried NT, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–45. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- [10].Michelle C, Vourc’h P, Mignon L, Andres C. What Was the Set of Ubiquitin and Ubiquitin-Like Conjugating Enzymes in the Eukaryote Common Ancestor? J Mol Evol. 2009;68:616–28. doi: 10.1007/s00239-009-9225-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Markson G, Kiel C, Hyde R, et al. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 2009;19:1905–11. doi: 10.1101/gr.093963.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Groettrup M, Pelzer C, Schmidtke G, Hofmann K. Activating the ubiquitin family: UBA6 challenges the field. Trends Biochem Sci. 2008;33:230–7. doi: 10.1016/j.tibs.2008.01.005. [DOI] [PubMed] [Google Scholar]
- [13].Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10:755–64. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pickart CM. Ubiquitin enters the new millennium. Mol Cell. 2001;8:499–504. doi: 10.1016/s1097-2765(01)00347-1. [DOI] [PubMed] [Google Scholar]
- [15].Aravind L, Koonin EV. The U box is a modified RING finger - a common domain in ubiquitination. Curr Biol. 2000;10:R132–4. doi: 10.1016/s0960-9822(00)00398-5. [DOI] [PubMed] [Google Scholar]
- [16].Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell. 2007;27:3–16. doi: 10.1016/j.molcel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- [17].van Leuken R, Clijsters L, Wolthuis R. To cell cycle, swing the APC/C. Biochim Biophys Acta. 2008;1786:49–59. doi: 10.1016/j.bbcan.2008.05.002. [DOI] [PubMed] [Google Scholar]
- [18].Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer. 2011;11:629–43. doi: 10.1038/nrc3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- [20].Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- [21].Sun Y. Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol Therapy. 2003;2:623–9. [PubMed] [Google Scholar]
- [22].Willems AR, Schwab M, Tyers M. A hitchhiker’S guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta. 2004;1695:133–70. doi: 10.1016/j.bbamcr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- [23].Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- [24].Zheng N, Schulman BA, Song LZ, et al. Structure of the Cul1-Rbx1-Skp1-F box(Skp2) SCF ubiquitin ligase complex. Nature. 2002;416:703–9. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- [25].Jin J, Cardozo T, Lovering RC, et al. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–80. doi: 10.1101/gad.1255304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life. 2012;64:316–23. doi: 10.1002/iub.1011. [DOI] [PubMed] [Google Scholar]
- [27].Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDSI functions as a linker to recruit receptor WD40 proteins to CVL4-ROCI ubiquitin ligases. Genes Dev. 2006;20:2949–54. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12:220. doi: 10.1186/gb-2011-12-4-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wu K, Fuchs SY, Chen A, et al. The SCFHOS/beta-TRCP-ROC1 E3 ubiquitin ligase utilizes two distinct domains within CUL1 for substrate targeting and ubiquitin ligation. Molecular and Cellular Biology. 2000;20:1382–93. doi: 10.1128/mcb.20.4.1382-1393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985–97. doi: 10.1038/sj.onc.1207414. [DOI] [PubMed] [Google Scholar]
- [32].Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–67. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- [33].Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11:347–56. doi: 10.2174/156800911794519734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Skaar JR, Pagano M. Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr Opin Cell Biol. 2009;21:816–24. doi: 10.1016/j.ceb.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–49. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- [37].Strohmaier H, Spruck CH, Kaiser P, et al. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316–22. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- [38].Yada M, Hatakeyama S, Kamura T, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–25. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Skaar JR, D’Angiolella V, Pagan JK, Pagano M. SnapShot: F Box Proteins II. Cell. 2009;137:1358, 1358–1. doi: 10.1016/j.cell.2009.05.040. [DOI] [PubMed] [Google Scholar]
- [41].Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11:1177–88. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- [42].von der Lehr N, Johansson S, Wu S, et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189–200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- [43].Yeh KH, Kondo T, Zheng J, et al. The F-box protein SKP2 binds to the phosphorylated threonine 380 in cyclin E and regulates ubiquitin-dependent degradation of cyclin E. Biochem Biophys Res Commun. 2001;281:884–90. doi: 10.1006/bbrc.2001.4442. [DOI] [PubMed] [Google Scholar]
- [44].Tan M, Zhao Y, Kim SJ, et al. SAG/RBX2/ROC2 E3 Ubiquitin Ligase Is Essential for Vascular and Neural Development by Targeting NF1 for Degradation. Dev Cell. 2011;21:1062–76. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yan Y, Zhang X, Legerski RJ. Artemis interacts with the Cul4A-DDB1DDB2 ubiquitin E3 ligase and regulates degradation of the CDK inhibitor p27. Cell Cycle. 2011;10:4098–109. doi: 10.4161/cc.10.23.18227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Inuzuka H, Shaik S, Onoyama I, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471:104–9. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dehan E, Bassermann F, Guardavaccaro D, et al. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol Cell. 2009;33:109–16. doi: 10.1016/j.molcel.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR Inhibitor, Is a Physiological Substrate of SCFβTrCp E3 Ubiquitin Ligase and Regulates Survival and Autophagy. Molecular Cell. 2011;44:304–16. doi: 10.1016/j.molcel.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sun Y. E3 ubiquitin ligases as cancer targets and biomarkers. Neoplasia. 2006;8:645–54. doi: 10.1593/neo.06376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- [51].Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- [52].Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- [53].Safran M, Kaelin WG., Jr. HIF hydroxylation and the mammalian oxygen-sensing pathway. J Clin Invest. 2003;111:779–83. doi: 10.1172/JCI18181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pugh CW, Ratcliffe PJ. The von Hippel-Lindau rumor suppressor, hypoxia-inducible factor-1 (HIF-1) degradation, and cancer pathogenesis. Semin Cancer Biol. 2003;13:83–9. doi: 10.1016/s1044-579x(02)00103-7. [DOI] [PubMed] [Google Scholar]
- [55].Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- [56].Xie L, Xiao K, Whalen EJ, et al. Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2:ra33. doi: 10.1126/scisignal.2000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Okuda H, Saitoh K, Hirai S, et al. The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J Biol Chem. 2001;276:43611–7. doi: 10.1074/jbc.M107880200. [DOI] [PubMed] [Google Scholar]
- [58].Na X, Duan HO, Messing EM, et al. Identification of the RNA polymerase II subunit hsRPB7 as a novel target of the von Hippel-Lindau protein. EMBO J. 2003;22:4249–59. doi: 10.1093/emboj/cdg410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 10 a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–56. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- [61].Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48:91–104. doi: 10.1002/mc.20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- [64].Hernandez-Munoz I, Lund AH, van der Stoop P, et al. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc Natl Acad Sci USA. 2005;102:7635–40. doi: 10.1073/pnas.0408918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kwon JE, La M, Oh KH, et al. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem. 2006;281:12664–72. doi: 10.1074/jbc.M600204200. [DOI] [PubMed] [Google Scholar]
- [66].Higa LA, Mihaylov Is, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003;5:1008–15. doi: 10.1038/ncb1061. [DOI] [PubMed] [Google Scholar]
- [67].Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cu14-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–21. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- [68].Sugasawa K, Okuda Y, Saijo M, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;12l:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- [69].Abbas T, Shibata E, Park J, et al. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nakagawa T, Xiong Y. X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyItransferase component WDR5 and regulates neuronal gene expression. Mol Cell. 2011;43:381–91. doi: 10.1016/j.molcel.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lee J, Zhou P. Cullins and cancer. Genes Cancer. 2010;1:690–9. doi: 10.1177/1947601910382899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wei D, Sun Y. Small RING finger proteins RBXI and RBX2 of SCF E3 ubiquitin ligases: the role in cancer and as cancer targets. Genes & Cancer. 2010;1:700–7. doi: 10.1177/1947601910382776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Schindl M, Gnant M, Schoppmann SF, Horvat R, Birner P. Overexpression of the human homologue for Caenorhabditis elegans cul-4 gene is associated with poor outcome in node-negative breast cancer. Anticancer Res. 2007;27:949–52. [PubMed] [Google Scholar]
- [74].Liu L, Yin Y, Li Y, Prevedel L, Lacy EH, Ma L, Zhou P. Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Research. 2012 doi: 10.1038/cr.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhao Y, Sun Y. CUL4B ubiquitin ligase in mouse development: A model for human X-linked mental retardation syndrome? Cell Res. 2012;22:1224–6. doi: 10.1038/cr.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tarpey PS, Raymond FL, O’Meara S, et al. Mutations in CUL4B, Which Encodes a Ubiquitin E3 Ligase Subunit, Cause an X-linked Mental Retardation Syndrome Associated with Aggressive Outbursts, Seizures, Relative Macrocephaly, Central Obesity, Hypogonadism, Pes Cavus, and Tremor. The American Journal of Human Genetics. 2007;80:345–52. doi: 10.1086/511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zou Y, Liu Q, Chen B, et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet. 2007;80:561–6. doi: 10.1086/512489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Laszlo GS, Cooper JA. Restriction of Src activity by Cullin-5. Curr Biol. 2009;19:157–62. doi: 10.1016/j.cub.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pan Q, Qiao F, Gao C, et al. Cdk5 targets active Src for ubiquitin-dependent degradation by phosphorylating Src(S75) Cell Mol Life Sci. 2011;68:3425–36. doi: 10.1007/s00018-011-0638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yu X, Yu Y, Liu B, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- [81].Querido E, Blanchette P, Yan Q, et al. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15:3104–17. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cai QL, Knight JS, Verma SC, Zald P, Robertson ES. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 rumor suppressors. PLoS Pathog. 2006;2:e116. doi: 10.1371/journal.ppat.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xu X, Sarikas A, Dias-Santagata DC, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30:403–14. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huber C, Dias-Santagata D, Glaser A, et al. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005;37:1119–24. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- [85].Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- [86].Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- [87].Huang Y, Duan H, Sun Y. Elevated expression of SAG/ROC2/Rbx2/Hrt2 in human colon carcinomas: SAG does not induce neoplastic transformation, but its antisense transfection inhibits rumor cell growth. Mol Carcinog. 2001;30:62–70. [PubMed] [Google Scholar]
- [88].Jia L, Soengas MS, Sun Y. ROC1/RBX1 E3 ubiquitin ligase silencing suppresses tumor cell growth via sequential induction of G2-M arrest, apoptosis, and senescence. Cancer Res. 2009;69:4974–82. doi: 10.1158/0008-5472.CAN-08-4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Jia L, Yang J, Hao X, et al. Validation of SAG/RBX2/ROC2 E3 Ubiquitin Ligase as an Anticancer and Radiosensitizing Target. Clin Cancer Res. 2010;16:814–24. doi: 10.1158/1078-0432.CCR-09-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yang D, Li L, Liu H, et al. Induction of autophagy and senescence by knockdown of ROC1 E3 ubiquitin ligase to suppress the growth of liver cancer cells. Cell Death Differ. 2012 doi: 10.1038/cdd.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jia L, Bickel JS, Wu J, et al. RBX1 (RING-box protein 1) E3 ubiquitin ligase is required for genomic integrity by modulating DNA replication licensing proteins. J Biol Chem. 2011;286:3379–86. doi: 10.1074/jbc.M110.188425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mendez J, Zou-Yang XH, Kim SY, et al. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol Cell. 2002;9:481–91. doi: 10.1016/s1097-2765(02)00467-7. [DOI] [PubMed] [Google Scholar]
- [93].Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gao D, Inuzuka H, Tan M-Kwang M, et al. mTOR Drives Its Own Activation via SCFβTrCP-Dependent Degradation of the mTOR Inhibitor DEPTOR. Molecular Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Duan S, Skaar Jeffrey R, Kuchay S, et al. mTOR Generates an Auto-Amplification Loop by Triggering the βTrCP- and CK1α-Dependent Degradation of DEPTOR. Molecular Cell. 2011;44:317–24. doi: 10.1016/j.molcel.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tan M, Gallegos JR, Gu Q, et al. SAG/ROC-SCFbeta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006;8:1042–54. doi: 10.1593/neo.06568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Tan M, Zhu Y, Kovacev J, et al. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kB activation in mouse embryonic stem cells. Free Radic Biol Med. 2010;49:976–83. doi: 10.1016/j.freeradbiomed.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gu Q, Bowden GT, Normolle D, Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol. 2007;178:1009–23. doi: 10.1083/jcb.200612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].He H, Gu Q, Zheng M, Normolle D, Sun Y. SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis. 2008;29:858–65. doi: 10.1093/carcin/bgn021. [DOI] [PubMed] [Google Scholar]
- [100].Duda DM, Borg LA, Scott DC, et al. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Deshaies RJ, Emberley ED, Saha A. Groettrup M, editor. Control of cullin-RING ubiquitin ligase activity by Nedd8. Conjugation and Deconjugation of Ubiquitin Family Modifiers: Subcellular Biochemistry. 2010;54:41–56. doi: 10.1007/978-1-4419-6676-6_4. [DOI] [PubMed] [Google Scholar]
- [102].Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–76. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1:708–16. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kamura T, Conrad MN, Yan Q, Conaway RC, Conaway JW. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. 1999;13:2928–33. doi: 10.1101/gad.13.22.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Goldenberg SJ, Cascio TC, Shumway SD, et al. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119:517–28. doi: 10.1016/j.cell.2004.10.019. [DOI] [PubMed] [Google Scholar]
- [106].Yamoah K, Oashi T, Sarikas A, et al. Autoinhibitory regulation of SCF-mediated ubiquitination by human cullin l’s C-terminal tail. Proc Natl Acad Sci USA. 2008;105:12230–5. doi: 10.1073/pnas.0806155105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Liu J, Furukawa M, Matsumoto T, Xiong Y. NEDD8 modification of CUL1 dissociates p120(CAND1), an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell. 2002;10:1511–8. doi: 10.1016/s1097-2765(02)00783-9. [DOI] [PubMed] [Google Scholar]
- [108].Zheng J, Yang X, Harrell JM, et al. CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol Cell. 2002;10:1519–26. doi: 10.1016/s1097-2765(02)00784-0. [DOI] [PubMed] [Google Scholar]
- [109].Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Lyapina S, Cope G, Shevchenko A, et al. Promotion of NEDD8-CUL1 Conjugate Cleavage by COP9 Signalosome. Science. 2001;292:1382–5. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- [111].Lo S-C, Hannink M. CANO1-Mediated Substrate Adaptor Recycling Is Required for Efficient Repression of Nrf2 by Keap1. Molecular and Cellular Biology. 2006;26:1235–44. doi: 10.1128/MCB.26.4.1235-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Bjorklund M, Taipale M, Varjosalo M, et al. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature. 2006;439:1009–13. doi: 10.1038/nature04469. [DOI] [PubMed] [Google Scholar]
- [113].Bech-Otschir D, Seeger M, Dubiel W. The COP9 signalosome: at the interface between signal transduction and ubiquitin-dependent proteolysis. Journal of Cell Science. 2002;115:467–73. doi: 10.1242/jcs.115.3.467. [DOI] [PubMed] [Google Scholar]
- [114].Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends in Biochemical Sciences. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- [115].Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37:102–11. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- [116].Milhollen MA, Traore T, Adams-Duffy J, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–23. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- [117].Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- [l18].Tan M, Li Y, Yang R, Xi N, Sun Y. Inactivation of SAG E3 Ubiquitin Ligase Blocks Embryonic Stem Cell Differentiation and Sensitizes Leukemia Cells to Retinoid Acid. PLoS One. 2011;6:e27726. doi: 10.1371/journal.pone.0027726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Milhollen MA, Narayanan U, Soucy TA, et al. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71:3042–51. doi: 10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- [120].Jia L, Li H, Sun Y. Induction of p21-Dependent Senescence by an NAE Inhibitor, MLN4924, as a Mechanism of Growth Suppression. Neoplasia. 2011;13:561–9. doi: 10.1593/neo.11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Lin HK, Chen Z, Wang G, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–9. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70:10310–20. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zhao Y, Xiong X, Jia L, Sun Y. Targeting cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Disease. 2012;3 doi: 10.1038/cddis.2012.125. doi:10.1038/cddis.2012.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Luo Z, Yu G, Lee HW, et al. The Nedd8-Activating Enzyme Inhibitor MLN4924 Induces Autophagy and Apoptosis to Suppress Liver Cancer Cell Growth. Cancer Res. 2012;72:3360–71. doi: 10.1158/0008-5472.CAN-12-0388. [DOI] [PubMed] [Google Scholar]
- [125].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhao Y, Sun Y. Targeting the mTOR-DEPTOR Pathway by CRL E3 Ubiquitin Ligases: Therapeutic Application. Neoplasia. 2012;14:360–7. doi: 10.1593/neo.12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci USA. 1998;95:11324–29. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Abbas T, Sivaprasad U, Terai K, et al. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22:2496–506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507–19. doi: 10.1101/gad.1703708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Winston JT, Strack P, Beer-Romero P, et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dey. 1999;13:270–83. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- [132].Wei D, Li H, Yu J, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282–93. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15:3912–6. doi: 10.1158/1078-0432.CCR-09-0343. [DOI] [PubMed] [Google Scholar]
- [134].Xirodimas DP. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem Soc Trans. 2008;36:802–6. doi: 10.1042/BST0360802. [DOI] [PubMed] [Google Scholar]
- [135].Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- [136].Milhollen MA, Thomas MP, Narayanan U, et al. Treatment-Emergent Mutations in NAEbeta Confer Resistance to the NEDD8-Activating Enzyme Inhibitor MLN4924. Cancer Cell. 2012;21:388–401. doi: 10.1016/j.ccr.2012.02.009. [DOI] [PubMed] [Google Scholar]
- [137].Toth JI, Yang L, Dahl R, Petroski MD. A gatekeeper residue for NEDD8-activating enzyme inhibition of MLN4924. Cell Reports. 2012;1:309–16. doi: 10.1016/j.celrep.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Yang Y, Kitagaki J, Dai RM, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–81. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- [141].Ceccarelli DF, Tang X, Pelletier S, et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell. 2011;145:1075–87. doi: 10.1016/j.cell.2011.05.039. [DOI] [PubMed] [Google Scholar]
- [142].Aghajan M, Jonai N, Flick K, et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotechnol. 2010;28:738–42. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Chen Q, Xie W, Kuhn DJ, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood. 2008;111:4690–9. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Orlicky S, Tang X, Neduva V, et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol. 2010;28:733–7. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]