

Abstract
Quantitation of low-abundance protein modifications involves significant analytical challenges, especially in biologically important applications, such as studying the role of post-translational modifications in biology and measurement of the effects of reactive drug metabolites. 14C labeling combined with accelerator mass spectrometry (AMS) provides exquisite sensitivity for such experiments. Here, we demonstrate real-time 14C quantitation of high-performance liquid chromatography (HPLC) separations by liquid sample accelerator mass spectrometry (LS-AMS). By enabling direct HPLC-AMS coupling, LS-AMS overcomes several major limitations of conventional HPLC-AMS, where individual HPLC fractions must be collected and converted to graphite before measurement. To demonstrate LS-AMS and compare the new technology to traditional solid sample AMS (SS-AMS), reduced and native bovine serum albumin (BSA) was modified by 14C-iodoacetamide, with and without glutathione present, producing adducts on the order of 1 modification in every 106 to 108 proteins. 14C incorporated into modified BSA was measured by solid carbon AMS and LS-AMS. BSA peptides were generated by tryptic digestion. Analysis of HPLC-separated peptides was performed in parallel by LS-AMS, fraction collection combined with SS-AMS, and (for peptide identification) electrospray ionization and tandem mass spectrometry (ESI-MS/MS). LS-AMS enabled 14C quantitation from ng sample sizes and was 100 times more sensitive to 14C incorporated in HPLC-separated peptides than SS-AMS, resulting in a lower limit of quantitation of 50 zmol 14C/peak. Additionally, LS-AMS turnaround times were minutes instead of days, and HPLC trace analyses required 1/ 6th the AMS instrument time required for analysis of graphite fractions by SS-AMS.
Carbon-14 accelerator mass spectrometry (14C-AMS) has been used for a wide variety of biological experiments including measurement of drugs and their metabolites,1,2 bioavailability determination of dietary components,3 and detection of protein4–6 and DNA adducts7,8 formed by reactive compounds. Experiments using 14C-AMS have proven useful in predicting pharmacokinetic properties of several drugs.9–11 In addition, measurement of protein therapeutics in experimental animals using 14C-AMS has recently been reported.12,13, 14C-AMS is an attractive approach for quantitation of therapeutic proteins or low-abundance post-translational modifications displayed on proteins because its sensitivity allows the use of doses that impose negligible chemical and radioactive risk to humans, while providing robust quantitative data.14,15
Historically, 14C-AMS has required that all samples be processed into solid carbon in the form of graphite before measurement.16 Quantitation of 14C by AMS in HPLC separations has required individual fractions to be collected and processed to graphite prior to AMS analysis. This process can be time-consuming and typically involves processing 30 to 60 fractions for each HPLC run. Since the carbon contained in an individual HPLC fraction is less than that required for successful conversion to graphite, 14C-depleted carrier carbon must be carefully added to provide sufficient material for AMS analysis. Small amounts of 14C in the carrier carbon decrease sensitivity and, in some instances, potentially compromise 14C quantitation.
As an alternative to solid carbon sample preparation, we have developed a real-time liquid sample AMS method (LS-AMS). LS-AMS enables the measurement of liquid samples through an ultra high-efficiency moving wire interface (MWI)17 coupled to an AMS instrument through a CO2 gas-accepting ion source. 18 LS-AMS accepts samples of nonvolatile material dissolved or suspended in a solvent or buffer solution, evaporates the solvent or buffer, combusts the remaining analyte, and directs the resulting CO2 to a gas-accepting ion source, generating C− ions and enabling normal AMS 14C quantitation of the sample (Figure 1). As many materials analyzed by BioAMS laboratories are already suspended in a compatible solvent, very little sample handling is required. Such a sample may be measured in minutes, with results generated in real time. LS-AMS is also capable of accepting a continuous flow of fluid directly from an HPLC instrument, which can lead to increased sensitivity, better temporal peak resolution, and substantial reduction of AMS instrument time. Other approaches to couple biochemical separation instrumentation for the analysis of nonvolatile compounds to AMS have been developed, including systems that use a chemical agent19 or a laser20,21 to oxidize the sample. However, these systems are either more restrictive or not compatible with direct coupling of HPLC to AMS, either due to mobile phase limitations or an off-line solvent removal step.
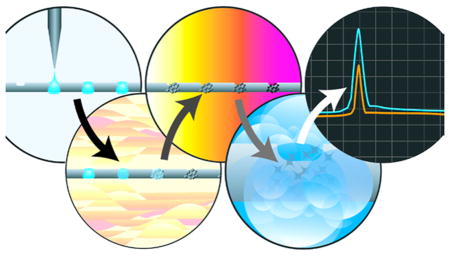
Figure 1.

Schematic layout of LS-AMS. (1) Wire indenter generates periodic indentations on the wire. (2) Surface carbon is removed and the wire is oxidized at high temperature. (3) A stream of effluent or single droplet is placed on the wire. (4) Solvent is evaporated at elevated temperature in an atmosphere of helium. (5) Nonvolatile analyte is combusted at high temperature in a helium and oxygen atmosphere. (6) The flow of gas through the combustion oven directs all combustion products to the gas accepting ion source through a narrow i.d. fused silica capillary. (7) The 14C and 12C content of the CO2 is measured using a gas accepting cesium sputter ion source followed by AMS analysis. (8) Used wire is collected on a separate spool for disposal, preventing cross contamination.
Here the LS-AMS method is combined with liquid chromatography-mass spectrometry (LC-MS/MS), to quantify 14C chemical adducts of bovine serum albumin (BSA) and characterize peptide modification patterns under several reaction conditions. This model system was chosen to explore the application of LS-AMS for quantitation of low-abundance protein modifications by real-time measurement of bulk protein samples and of HPLC-separated peptides.
EXPERIMENTAL SECTION
Materials
BSA, dithiothreitol (DTT), iodoacetamide (IAC), HPLC-grade acetonitrile, HPLC-grade water, formic acid, acetic acid, and proteomics-grade trypsin were purchased from Sigma (St. Louis, MO). Iodoacetamide [1-14C] with a specific activity of 56 mCi/mmol was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO).
14C-Iodoacetamide Adduct Quantitation and Competitive Binding Experiments
Reaction of 14C-iodoacetamide with BSA was performed under several experimental conditions.
Initial range-finding experiments were performed with the goal of generating protein adducts measuring between 2000 and 20000 amol 14C/mg protein. These experiments used a range of concentrations of native and reduced BSA and 14C-iodoacetamide. A selection of these samples were measured by the solid sample (carbon)-AMS (SS-AMS) method for comparison with LS-AMS.
For competitive binding experiments, 2 mg/mL BSA was suspended in a solution of 100 mM ammonium bicarbonate with 5% acetonitrile. Solutions of 50 mM dithiothreitol, 500 mM reduced glutathione (GSH), and 100 mM unlabeled iodoacetamide were prepared in water. For labeling experiments, a dosing solution of 100 pCi/mL of [1-14C] iodoacetamide was prepared in water. Activity of the source material was verified by liquid scintillation counting. Each reaction was performed in triplicate. For control (no label) experiments, BSA disulfide bonds were reduced by incubation with 5 mM DTT for 5 min at 65 °C in the dark. The reaction was cooled, and unlabeled iodoacetamide was added to a final concentration of 10 mM and cysteine alkylation was allowed to proceed for 30 min at 30 °C in the dark. For native albumin 14C-iodoacetamide binding experiments, 1 pCi of 14C-iodoacetamide was added to each 1 mL of BSA and reacted at 30 °C for 30 min in the dark. Disulfides were then reduced, and the remaining free cysteine residues were alkylated with unlabeled iodoacetamide as described for the control samples. For reduced albumin experiments, albumin disulfides were reduced with DTT as in the control experiments, followed by reaction with 1 pCi of 14C-iodoacetamide and subsequent alkylation of the remaining free cysteine residues with 10 mM of unlabeled iodoacetamide. For competitive binding experiments, GSH was added to native or reduced BSA to a final concentration of 5 mM, after which BSA was reacted with 1 pCi of 14C-iodoacetamide, with subsequent alkylation of free cysteine (Cys) with unlabeled iodoacetamide or reduction of disulfides followed by alkylation with unlabeled cysteine, as described for the control samples. Following protein modification, all samples were desalted by filtration through 50 kDa molecular weight cutoff spin tubes. Protein was washed three times with 0.5 mL of 100 mM ammonium bicarbonate with 5% acetonitrile by spinning for 30 min at 14000 rpm. Washed protein was collected in 1 mL of 100 mM ammonium bicarbonate with 5% acetonitrile and stored at −80 °C until analysis.
Adducted Peptide Analysis Experiments
For characterization of modified peptides, unlabeled BSA was reduced and alkylated with unlabeled iodoacetamide as above, except that the BSA concentration was 5 mg/mL and reactions were performed in quadruplicate. Alkylation of native and reduced BSA was likewise performed using 5 mg/mL BSA. Reactions used 4 mL of BSA solution per reaction, and labeling experiments used 4 pCi of 14C-iodoacetamide per reaction. After reactions were completed, protein was precipitated by addition of 6 mL of ice-cold methanol and centrifugation for 10 min at 4400 rpm in a swinging bucket centrifuge. The precipitating solution was collected and stored at −80 °C for subsequent analysis. Precipitated protein was washed four times with 10 mL of methanol, after which the protein was resuspended in 4 mL of 100 mM ammonium bicarbonate with 5% acetonitrile. Samples were stored at −80 °C until analysis or digestion.
Trypsin Digestion of Modified BSA
Modified protein samples were vortex-mixed and 100 μL aliquots were collected for trypsin digestion. Trypsin was resuspended to a concentration of 1 mg/mL in 1 mM HCl. Reconstituted trypsin was then added to each protein at a 1:20 trypsin:protein ratio (mg/mL). Digestion was allowed to proceed overnight with incubation at 37 °C. Following digestion, the reaction was stopped by addition of 10 μL of 0.5% acetic acid.
Analysis of Intact Protein Adducts by AMS
Intact proteins were analyzed for 14C-iodoacetamide incorporation by LS-AMS and SS-AMS analysis. For LS-AMS analysis, 1 μL (2 μg/μL) samples were directly applied to the MWI as droplets at 1 min intervals by pipet. Each sample was measured three times. For comparison, standards of known isotopic ratio and carbon content were applied before and after each series of replicates. The design of the MWI was as detailed previously,17 with the addition that the exhaust of the drying oven is now collected by a sample pump equipped with carbon traps. The instrument configuration is shown in Figure 1. The wire was set to move at 2.5 cm/s with indentations every 0.6 cm. The cleaning furnace was held at 900 °C under atmosphere. The drying oven was held at 140 °C with a helium flow rate of 400 mL/min. The combustion oven was held at 750 °C with an atmosphere of 97.5% helium and 2.5% oxygen. The capillary from the combustion oven to the gas-accepting ion source was 180 μm in diameter and 4.5 m long. From the ion beam generated by the gas accepting ion source, the AMS instrument directed 12C ions to a Faraday cup and 14C ions to a solid-state particle detector. The charge collected by the Faraday cup and number of 14C atoms measured by the particle detector were summed over 1 s intervals. Isotopic ratios of samples were determined by the background subtracted integral of the resulting 14C and 12C signals from the combusted samples in comparison to that of the standards.
For SS-AMS analysis, 300 μL (600 μg C) of each sample was converted to graphite and measured by AMS, following standard biochemical AMS sample preparation procedures.16
HPLC Analysis of Peptide Adducts
Prior to HPLC analysis, tubes containing labeled peptides were centrifuged at 14000g for 5 min and the supernatant was loaded into HPLC analysis vials. HPLC separation of peptides: an Agilent 300 Extend C18 3.5 μm 2.1 × 100 mm column; mobile phases: A, water and B, acetonitrile; flow rate = 100 μL/minute; time: 0, 5% B; 25 min, 30% B; 35 min, 60% B; 35.1 min, 90% B; 38 min, 90% B; 38.1 min, 5% B; stop time: 45 min, 5% B. Experiments were initially performed with mobile phases containing 0.1% formic acid to improve peak shape and enhance peptide ionization for MS/MS, but an elevated 12C background attributable to the formic acid was observed. Formic acid was therefore omitted from the HPLC mobile phases without a significant reduction in the quality of peptide separations. LC-MS/MS analysis was performed on a Thermo LTQ XL linear ion-trap instrument. Files were converted to profile.mzXML files using the MassMatrix file conversion utility. Peptides were identified using the MassMatrix search server.22,23 In addition to LC-MS/MS analysis, digests were separated using identical chromatographic conditions and fractions were collected to allow for comparison of peptide adduction quantitation by directly coupled liquid chromatography LS-AMS (LC-LS-AMS) and SS-AMS. SS-AMS HPLC fractions were prepared for graphite analysis by the standard biochemical AMS protocol.16 The LC-LS-AMS and SS-AMS traces were aligned with the total ion chromatograms generated by LC-MS/MS analysis to identify 14C-modified peptides.
RESULTS AND DISCUSSION
Cys residues in proteins play important roles in maintaining protein structure and function.24 Numerous biological modifications of protein Cys residues are known to modulate protein activity. In addition, Cys residues are important sites of modification by reactive chemicals, potentially resulting in impairment of protein function.25,26 Bovine serum albumin is a well-characterized protein containing 35 cysteine residues, 34 of which participate in disulfide bonds. Iodoacetamide readily alkylates free Cys residues to form carbamidomethylated cysteine in proteins and peptides but cannot react with disulfide cysteines. Because the reaction of iodoacetamide with BSA is predictable, this reaction was selected to explore the utility of LS-AMS for absolute quantitation of protein and peptide modifications. In principle, this method of protein analysis can be extended to measure protein therapeutics or other proteins synthesized to contain a 14C label.
To determine appropriate labeling procedures and to enable comparison of measurements made by LS-AMS analysis with solid sample AMS analysis, several alkylation experiments involving modification of BSA by 14C-iodoacetamide were performed with native BSA or BSA with chemically reduced disulfide bonds. Aliquots of each sample were measured by LS-AMS and by graphitization followed by AMS. Results are given in Table 1 and a typical LS-AMS signal is depicted in Figure 2. Quantification of binding is computed by the difference between 14C incorporation in the control and the sample. To enable routine solid carbon AMS measurement, samples for this comparison were designed to yield 14C concentrations less than 5000 amol/mg protein.
Table 1.
Comparison of Solid Sample and Liquid Sample AMS
| sample | amol 14C/mg BSA
|
|
|---|---|---|
| graphite | LS-AMS | |
| control BSA | 57.2 ± 0.8 | 57.2 ± 1.1 |
| modified native BSA | 3074 ± 35 | 2900 ± 130 |
| modified reduced BSA | 2342 ± 21 | 2313 ± 59 |
Figure 2.
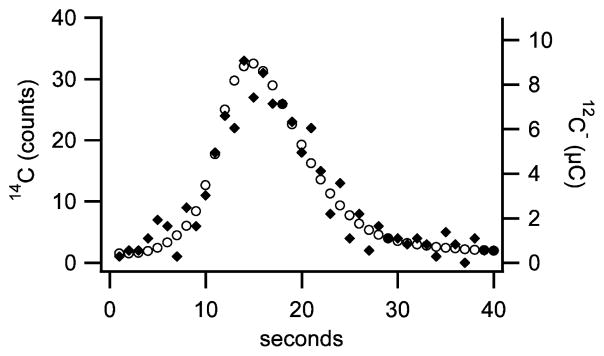
Typical LS-AMS peak generated from a 1 μL droplet containing 0.7 μg of unlabeled BSA; the symbols ◆ represents 14C and ○ represents 12C, in microCoulombs. Integrated signals from this sample were 94 ± 10 counts 14C and 334.8 ± 0.1 μC 12C. A comparison to a standard of known isotopic ratio indicated a 14C concentration of 59 ± 6 amol 14C/mg BSA. Precision was limited by the number of 14C counts detected.
Comparison of SS-AMS and LS-AMS analysis of selected controls and samples modified with 14C-iodoacetamide. Native and reduced albumin were prepared at 5 mg/mL and alkylated with various preparations of 14C-iodoacetamide to achieve 14C incorporation in the optimal range of SS-AMS. The control was prepared by alkylation of reduced BSA with unlabeled iodoacetamide. 14C incorporation of each sample was computed from the mean and standard deviation of the mean of 4 replicate reactions.
The LS-AMS measurements produced equivalent values to the SS-AMS measurements, albeit with 2–4% precision instead of 1%, using only 1 to 2 μg of material for each measurement (Table 1). For routine SS-AMS quantitation of biomedical materials, a sample carbon mass of 500 μg 12C (~1000 μg protein) is required.16 14C-labeled biological samples containing as little as 50 μg 12C are possible to measure by SS-AMS but require substantially more time and effort to measure.27
A typical peak resulting from LS-AMS measurement of unlabeled BSA is depicted in Figure 2. The precision of individual peak 14C/C quantitation is primarily limited by 14C content, requiring ~40 zmol 14C for 10% precision. Uncertainties of 12C peak integrations indicate that such samples could contain as little as 10 ng 12C with little change to precision.
After appropriate labeling procedures were determined, competitive binding experiments were performed with BSA and 14C-iodoacetamide in the presence or absence of the thiol-containing peptide glutathione (GSH). The molar ratio of albumin to 14C-iodoacetamide in these alkylation experiments was approximately 1.6 million to 1. As expected, measurements of label incorporation demonstrated increased binding of 14C-iodoacetamide to reduced BSA (6468 ± 85 amol/mg protein, indicating 81% of added label bound to BSA) compared to native BSA (4100 ± 200 amol/mg protein, indicating 51% of added label bound to BSA) due to the availability of additional free Cys residues (Figure 3). Competitive binding experiments with GSH show decreased binding of 14C-iodoacetamide to both native (623 ± 25 amol/mg protein, indicating 7.8% of added label bound to BSA) and reduced BSA (3780 ± 75 amol/mg protein, indicating 47% of added label bound to BSA) compared to native and reduced BSA in the absence of glutathione (Figure 3). In all cases, measurement of intact protein allowed absolute quantitation of protein modification by 14C-iodoacetamide.
Figure 3.

Absolute quantitation of an albumin 14C-iodoacetamide modification competitive binding experiment. Treated and untreated BSA samples were exposed to 1 pCi 14C-iodoacetamide and described as follows: 14C-iodoacetamide-modified native BSA (14C-IAC-native):native BSA with 1 free Cys residue; 14C-IAC-native + GSH:native BSA and 5 mM GSH; 14C-iodoacetamide-modified reduced BSA (14C-IAC-reduced):disulfide bonds reduced; 14C-IAC-reduced + GSH:disulfide bonds reduced and 5 mM GSH. Results were background subtracted from the control, which showed no excess 14C above the endogenous level expected for a contemporary biological sample. Error bars represent the standard error of the mean of three replicate samples.
In order to determine specific sites of BSA modification, quadruplicates of 14C-iodoacetamide-modified native and reduced BSA (respectively 14C-IAC-native and 14C-IAC-reduced) were subjected to tryptic digestion and analysis. Peptide identification was accomplished by LC-MS/MS, and the 14C tracer was detected by LC-LS-AMS. Additionally, fractions collected from identical separations were measured by solid carbon AMS. Representative traces from these separations, including peptide total ion chromatograms and peptide 14C and 12C traces are depicted in Figures 4 and 5.
Figure 4.

Modification of native bovine serum albumin by 14C-iodoacetamide. (A) MS/MS relative peptide abundance total ion chromatogram. (B) Peptide fraction collection and SS-AMS analysis. (C) LS-AMS 12C trace in units of ng 12C/measurement interval (top) and LS-AMS 14C trace in units of amol 14C/measurement interval (bottom). 14C peak 3 is due to the peptide containing Cys 58, GLVLIAFSQYLQQCPFDEHVK. Minor 14C peaks 1 and 2 contain multiple Cys-containing peptides.
Figure 5.

Modification of reduced bovine serum albumin by 14C-labeled iodoacetamide. (A) MS/MS relative peptide abundance total ion chromatogram. (B) Peptide fraction collection and SS-AMS analysis. (C) 12C (top) and 14C (bottom) traces from LS-AMS. The following numbered 14C peaks were found by MS/MS to correspond to the following Cys containing peptides: 2, ETYGDMADCCEK; 3, TCVADESHAGCEK and QNCDQFEK; 4, ETYGDMADCCEK and SHCIAEVEK; 7, LKPDPNTLCDEFK and YNGVFQECCQAEDK; 8, RPCFSALTPDETYVPK and SLHTLFGDELCK; 10, GLVLIAFSQYLQQCPFDEHVK and LFTFHADICTLPDTEK. All other numbered peaks have elution times corresponding to more than two Cys-containing peptides.
For LS-AMS analysis, effluent from the HPLC was applied directly to the interface. For solid carbon AMS, as the HPLC separations contained only ~15 μg C, it was necessary to add 1 μL of tributyrin carrier (containing 0.61 mg C with an isotopic ratio of 11.2 ± 0.3 amol 14C/mg 12C) to each fraction, to provide sufficient carbon for graphitization and AMS analysis. Under this regime, the amount of 12C from the carrier dominates the 12C from the sample and 1 μL of tributyrin carrier adds 6.8 ± 0.4 amol 14C to each fraction. 14C from the HPLC eluate in each fraction was determined by subtracting the measured isotopic ratio of the carrier from the measured isotope ratio of each fraction and then multiplying by the carbon mass of the carrier.28 This process adds a significant uncertainty to the 14C measurement. As a result, the uncertainty of individual solid carbon AMS fraction measurements is approximately ±1 amol 14C/fraction, while the lower limit of quantitation (LLOQ) is 10 amol 14C/fraction. In contrast, LC-LS-AMS 12C backgrounds were between 2 to 4 ng/second and the 14C background was less than 10−3 amol 14C/second with a LLOQ of 50 zmol 14C/peak.
Although the magnitude of the 12C signal measured by LC-LS-AMS was sufficient for quantitation, peptide coelution made it impossible to assign measured 12C signals to individual peptides (Figures 4C and 5C), thus preventing measurement of an isotope ratio that would easily enable absolute quantitation of each modified peptide. The 14C content in the separation was estimated by comparison to a standard of known 14C content measured before and after the separation. With improved separations, it should be possible to obtain 12C quantitation of individual peptides.
The HPLC traces in Figures 4 and 5 indicate that LC-LS-AMS analysis is more sensitive for quantifying 14C-containing peaks than conventional solid carbon HPLC-AMS. Figure 4C reveals 3 distinct 14C peaks from separation of the digested 14C-IAC-native by LC-LS-AMS. The 14C peak eluting at 33.2 min and containing ~75% (17 amol) of 14C incorporated into the 14C-IAC-native is consistent with the elution time of the peptide containing Cys 58, the only free Cys residue in BSA, as detected by LC-MS/MS (Figure 4C). The other 14C peaks in Figure 4C eluting at 3.8 and 20.3 min, respectively, contained 1.9 and 2.9 amol 14C and correspond to a number of Cys-containing peptides. Conversely, with solid carbon AMS, the only 14C peak detected eluted at fraction 65, corresponding to an elution time of 34 min and was near the practical limit of quantitation for solid carbon AMS (Figure 4B). Additionally, graphitization of 3 fractions, including fraction 66, failed. Without repeating the experiment, it cannot be known if some portion of the 14C elevation detected by fraction 65 carried over to fraction 66.
From separation of the digested 14C-IAC-reduced, LC-LS-AMS detected 13 distinct 14C peaks, corresponding to additional Cys-containing peptides (Figure 5C). 14C peaks 1, 8, and 9 have elution times within a few seconds of 14C peaks 1, 2, and 3 of the digested 14C-IAC-native separation shown in Figure 4C. Peak overlap limits precise quantitation of individual peaks in the 14C trace in Figure 5C. Conversely, no substantially elevated fractions were detected by solid carbon AMS from the 14C-IAC-reduced separation in Figure 5B. Failure to detect elevated fractions from the 14C-IAC-reduced separation is due to a lack of sensitivity, arising from the combination of extremely low-level labeling of numerous peptides and the presence of 14C from the tributyrin present in all fractions. Overall, the data in Figures 4 and 5 suggest that LC-LS-AMS is 100 times more sensitive in quantifying 14C incorporated in HPLC-separated peptides than SS-AMS.
LC-LS-AMS also provides superior temporal resolution compared to fraction collection combined with solid carbon AMS. For LC-LS-AMS, the integrated 12C charge and 14C counts are recorded at 1 s intervals. Peak maximum locations could be determined to the nearest second and varied by less than 5 s between replicates. Typical peak widths, measured by full width at half-maximum, were in the range of 35 s and varied by less than 2 s between replicates. Without employing complex peak fitting algorithms, the temporal resolution of solid carbon AMS analysis is limited by the fraction collection time. Increasing the number of fractions can improve temporal resolution, but this would require more carrier carbon and thus decrease sensitivity.
In addition to improved sensitivity, improved temporal resolution and reduced sample preparation, traces measured by LC-LS-AMS took significantly less AMS instrument time to measure: ~1 h per trace for Figures 4C and 5C. In contrast, the low 14C content and dilution by carrier carbon of the solid carbon fractions (resulting in an average isotopic ratio of ~15 amol 14C/mg C) required ~6 h of AMS instrument time per one separation to measure the 75 fractions collected from each of the two separations shown in Figures 4B and 5B.
CONCLUSIONS AND FUTURE DIRECTIONS
Recent advances in the application of proteins as therapeutics have revealed an urgent need for improved methods for tracking and quantifying proteins and protein modifications in a variety of chemical and biological matrices.29,30 Sensitive, specific methods for protein quantitation are essential for determining the biological fates and pharmacokinetic characteristics of protein therapeutics in experimental animals and in humans.31 Measurement of protein therapeutics in animals and humans is difficult, as analytical methods such as immunoassays frequently lack sensitivity and are subject to interferences.12,30 Analysis is further complicated by the fact that many protein drugs are nearly indistinguishable from proteins that occur naturally in the body.12,31
Detection of a modified protein is difficult using conventional methods, due to the low frequency of occurrence of the modification relative to the total amount of a given protein present (i.e., only a small fraction of the target protein is expected to be modified). In contrast, the approach described here enables sensitive, rapid characterization of minimally modified proteins (in the range of 1 modification for every 106–108 proteins) using a 14C-labeled modifying agent. We envision that application of this technology can be extended to a variety of classes of biological molecules of interest and to HPLC separations of greater complexity.
Our analyses show that LS-AMS can measure submicrogram protein samples containing tens of zeptomoles of 14C with good accuracy and acceptable precision compared to solid carbon AMS analysis, which requires 100 to 1000 times more 12C and 100 times more 14C per sample. We further demonstrate the utility of LC-LS-AMS for characterization of modified peptides by 14C-labeled compounds of interest. Compared to solid carbon AMS, analysis of HPLC separations by LC-LS-AMS provides around 100 times more 14C sensitivity, the capability to measure HPLC-relevant 12C quantities, superior temporal resolution, greater reliability, results in real-time instead of days, and a 6-fold reduction in AMS instrument time.
The improvements offered by the LS-AMS approach should find ready application to essential tracer experiments in biology and medicine, especially in studies requiring protein or post-translational modification quantitation, in the development of protein-based drugs, and in the broader field of protein functional characterization.
Acknowledgments
The authors thank Kurt Haack for assistance preparing solid carbon samples and Drs. Alma Burlingame and Giselle Knudsen for technical advice.
The work was performed at the Research Resource for Biomedical AMS, which is operated at LLNL under the auspices of the U.S. Department of Energy under contract DE-AC52-07NA27344. The Research Resource is supported by the National Institutes of Health, National Institute of General Medical Sciences under Grant P41 GM103483.
Footnotes
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval of the final version of the manuscript. A.T.T. and B.J.S. contributed equally.
The authors declare no competing financial interest.
References
- 1.Boddy AV, Sludden J, Griffin MJ, Garner C, Kendrick J, Mistry P, Dutreix C, Newell DR, O’Brien SG. Clin Cancer Res. 2007;13(14):4164–9. doi: 10.1158/1078-0432.CCR-06-2179. [DOI] [PubMed] [Google Scholar]
- 2.Xu XS, Dueker SR, Christopher LJ, Lohstroh PN, Keung CF, Cao KK, Bonacorsi SJ, Cojocaru L, Shen JX, Humphreys WG, Stouffer B, Arnold ME. Bioanalysis. 2012;4(15):1855–70. doi: 10.4155/bio.12.171. [DOI] [PubMed] [Google Scholar]
- 3.Ross AB, Vuong le T, Ruckle J, Synal HA, Schulze-Konig T, Wertz K, Rumbeli R, Liberman RG, Skipper PL, Tannenbaum SR, Bourgeois A, Guy PA, Enslen M, Nielsen IL, Kochhar S, Richelle M, Fay LB, Williamson G. Am J Clin Nutr. 2011;93(6):1263–73. doi: 10.3945/ajcn.110.008375. [DOI] [PubMed] [Google Scholar]
- 4.Dingley KH, Freeman SP, Nelson DO, Garner RC, Turteltaub KW. Drug Metab Dispos. 1998;26(8):825–8. [PubMed] [Google Scholar]
- 5.Williams KE, Carver TA, Miranda JJ, Kautiainen A, Vogel JS, Dingley K, Baldwin MA, Turteltaub KW, Burlingame AL. Mol Cell Proteomics. 2002;1(11):885–95. doi: 10.1074/mcp.m200067-mcp200. [DOI] [PubMed] [Google Scholar]
- 6.Yamazaki H, Kuribayashi S, Inoue T, Tateno C, Nishikura Y, Oofusa K, Harada D, Naito S, Horie T, Ohta S. Chem Res Toxicol. 2010;23(1):152–8. doi: 10.1021/tx900323a. [DOI] [PubMed] [Google Scholar]
- 7.Malfatti MA, Dingley KH, Nowell-Kadlubar S, Ubick EA, Mulakken N, Nelson D, Lang NP, Felton JS, Turteltaub KW. Cancer Res. 2006;66(21):10541–7. doi: 10.1158/0008-5472.CAN-06-1573. [DOI] [PubMed] [Google Scholar]
- 8.Xie Q, Sun H, Liu Y, Ding X, Fu D, Liu K. Toxicol Lett. 2006;163(2):101–8. doi: 10.1016/j.toxlet.2005.09.041. [DOI] [PubMed] [Google Scholar]
- 9.Sandhu P, Vogel JS, Rose MJ, Ubick EA, Brunner JE, Wallace MA, Adelsberger JK, Baker MP, Henderson PT, Pearson PG, Baillie TA. Drug Metab Dispos. 2004;32(11):1254–9. doi: 10.1124/dmd.104.000422. [DOI] [PubMed] [Google Scholar]
- 10.Lappin G, Kuhnz W, Jochemsen R, Kneer J, Chaudhary A, Oosterhuis B, Drijfhout WJ, Rowland M, Garner RC. Clin Pharmacol Ther (NY, NY, US) 2006;80(3):203–15. doi: 10.1016/j.clpt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Gao L, Li J, Kasserra C, Song Q, Arjomand A, Hesk D, Chowdhury SK. Anal Chem. 2011;83(14):5607–16. doi: 10.1021/ac2006284. [DOI] [PubMed] [Google Scholar]
- 12.Lappin G, Garner RC, Meyers T, Powell J, Varley P. J Pharm Biomed Anal. 2006;41(4):1299–302. doi: 10.1016/j.jpba.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Salehpour M, Ekblom J, Sabetsky V, Hakansson K, Possnert G. Rapid Commun Mass Spectrom. 2010;24(10):1481–9. doi: 10.1002/rcm.4544. [DOI] [PubMed] [Google Scholar]
- 14.Buchholz BA, Sarachine Falso MJ, Stewart BJ, Haack KW, Ognibene TJ, Bench G, Salazar Quintero GA, Malfatti MA, Kulp KS, Turteltaub KW, Lyubimov AV. In: Encyclopedia of Drug Metabolism and Interactions. Lyubimov A, editor. John Wiley & Sons, Inc; Hoboken, NJ: 2011. [Google Scholar]
- 15.Stewart BJ, Bench G, Buchholz BA, Haack KW, Malfatti MA, Ognibene TJ, Turteltaub KW. In: Mass Spectrometry Handbook. Lee MS, editor. John Wiley & Sons, Inc; Hoboken, NJ: 2012. pp. 259–269. [Google Scholar]
- 16.Ognibene TJ, Bench G, Vogel JS, Peaslee GF, Murov S. Anal Chem. 2003;75(9):2192–2196. doi: 10.1021/ac026334j. [DOI] [PubMed] [Google Scholar]
- 17.Thomas AT, Ognibene T, Daley P, Turteltaub K, Radousky H, Bench G. Anal Chem. 2011;83(24):9413–7. doi: 10.1021/ac202013s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ognibene TJ, Salazar GA. Nucl Instrum Methods Phys Res, Sect B. 2013;294(0):311–314. doi: 10.1016/j.nimb.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krummen M, Hilkert AW, Juchelka D, Duhr A, Schluter HJ, Pesch R. Rapid Commun Mass Spectrom. 2004;18(19):2260–2266. doi: 10.1002/rcm.1620. [DOI] [PubMed] [Google Scholar]
- 20.Liberman RG, Tannenbaum SR, Hughey BJ, Shefer RE, Klinkowstein RE, Prakash C, Harriman SP, Skipper PL. Anal Chem. 2004;76(2):328–334. doi: 10.1021/ac030181y. [DOI] [PubMed] [Google Scholar]
- 21.Daniel R, Mores M, Kitchen R, Sundquist M, Hauser T, Stodola M, Tannenbaum S, Skipper P, Liberman R, Young G, Corless S, Tucker M. Nucl Instrum Methods Phys Res, Sect B. 2013;294:291–295. [Google Scholar]
- 22.Xu H, Freitas MA. BMC Bioinf. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu H, Freitas MA. Proteomics. 2009;9(6):1548–55. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Netto LE, de Oliveira MA, Monteiro G, Demasi AP, Cussiol JR, Discola KF, Demasi M, Silva GM, Alves SV, Faria VG, Horta BB. Comp Biochem Physiol, Part C: Toxicol Pharmacol. 2007;146(1–2):180–93. doi: 10.1016/j.cbpc.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Nagahara N, Matsumura T, Okamoto R, Kajihara Y. Curr Med Chem. 2009;16(33):4419–44. doi: 10.2174/092986709789712880. [DOI] [PubMed] [Google Scholar]
- 26.Spadaro D, Yun BW, Spoel SH, Chu C, Wang YQ, Loake GJ. Physiol Plant. 2010;138(4):360–71. doi: 10.1111/j.1399-3054.2009.01307.x. [DOI] [PubMed] [Google Scholar]
- 27.Salehpour M, Forsgard N, Possnert G. Rapid Commun Mass Spectrom. 2008;22(23):3928–3934. doi: 10.1002/rcm.3808. [DOI] [PubMed] [Google Scholar]
- 28.Brown K, Dingley KH, Turreltaub KW. Biol Mass Spectrom. 2005;402:423–443. doi: 10.1016/S0076-6879(05)02014-8. [DOI] [PubMed] [Google Scholar]
- 29.Weng Z, DeLisi C. Trends Biotechnol. 2002;20(1):29–35. doi: 10.1016/s0167-7799(01)01846-7. [DOI] [PubMed] [Google Scholar]
- 30.Bronsema KJ, Bischoff R, van de Merbel NC. J Chromatogr, B. 2012;893–894:1–14. doi: 10.1016/j.jchromb.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Nat Rev Drug Discovery. 2012;11(7):527–40. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]