

Abstract
A concise total synthesis of the complex indole alkaloid (±)-actinophyllic acid was accomplished by a sequence of reactions requiring only 10 steps from readily-available, known starting materials. The approach featured a Lewis acid-catalyzed cascade of reactions involving stabilized carbocations that delivered the tetracyclic core of the natural product in a single chemical operation. Optimal conversion of this key intermediate into (±)-actinophyllic acid required judicious selection of a protecting group strategy.
Keywords: Total Synthesis, Cascade Reaction, Iminium Ion, Mannich-type Reaction
1. Introduction
It has been estimated that as many as 600,000 people suffer annually from thrombotic diseases in the United States.1 The mortality rate associated with thrombosis is exceedingly high, necessitating an improvement in the current standard of care. One risk factor for thrombosis is unregulated fibrinolysis, or the breakdown of fibrin clots by plasmin. Fibrinolysis is in part regulated by carboxypeptidase U (CPU), alternatively called activated thrombin-activatable fibrinolysis inhibitor (TAFIa), which catalyzes the cleavage of C-terminal arginine and lysine residues from fibrin and fibrin degradation products.2 These basic residues are essential for the activation of plasmin via the formation of a ternary complex between fibrin, plasminogen, and tissue-type plasminogen activator (tPA). Because plasmin promotes the breakdown of fibrin, CPU acts as an endogenous inhibitor of the fibrinolysis process.3 Accordingly, CPU has attracted considerable attention from the scientific community as a potential target for the treatment of thrombosis,4–7 although there is currently no CPU inhibitor in the clinic.8 In 2005, Carroll and co-workers reported the isolation of the indole alkaloid actinophyllic acid (1) from the leaves of Alstonia actinophylla, and using a CPU/hippuricase coupled enzyme assay they discovered that 1 was a potent CPU inhibitor (IC50 = 0.84 µM) (Scheme 1).9
Scheme 1.

Retrosynthetic analysis
The key structural features of the bridged architecture of 1, which is unique among known natural products, comprise a pyrrolidine ring, a cyclic hemiketal, a carboxylic acid appended to a quaternary carbon atom, an indol-3-ylmethanamine moiety, and five contiguous stereocenters. Indeed, the compact and highly functionalized hexacyclic framework embodied in 1 has prompted several total synthesis campaigns. Overman and co-workers completed the first total syntheses of (±)- and (–)-1 through the elegant application of the venerable aza-Cope/Mannich sequence.10 Importantly, the synthesis of (–)-1 allowed for the determination of the absolute configuration of the natural product via analysis of its methyl ester derivatives through electronic circular dichroism (ECD).11 Other reports from the groups of Wood,12 Taniguchi,13 Maldonado,14 and Coldham15 have revealed a diverse array of approaches toward 1.
Our plan for the synthesis of (±)-actinophyllic acid (1) is outlined retrosynthetically in Scheme 1 and commences with the oxidation of the neopentyl alcohol in 2, which would result from deprotection of the primary alcohols in 3 and subsequent hemiketalization. The pyrrolidine ring in 3 would be installed via deprotection of the nitrogen atom in 4, followed by reductive alkylation of N(2) and cyclization at C(19) by intramolecular enolate alkylation. We envisioned that the tetracyclic core of the natural product embodied in 4 would arise from the union of electrophile 6 and π-nucleophile 7. In particular, we reasoned that capture of the stabilized carbocation generated upon Lewis-acid initiated ionization of 6 with the π-nucleophile 7 via a vinylogous Mannich-like reaction16 would produce the N-acyliminium ion 5 that would then undergo attack by the 3-position of the indole in a Pictet-Spengler type reaction.17
It is notable that the pivotal reaction cascade proposed for the transformation of 6 and 7 into the tetracyclic intermediate 4 was inspired by chemistry developed during efforts directed toward our syntheses of the welwitindolinone alkaloids.18 Indeed, we had developed this reaction into a general method to couple stabilized carbocations derived from heteroaromatic carbinols with various π-nucleophiles to deliver β-heteroaryl propionates as exemplified by the preparation of 10 by the TMSOTf-promoted reaction of 8 and 9 (Equation 1).19
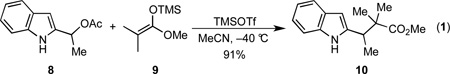 |
(1) |
We thus sought to further demonstrate the utility of this powerful transformation by applying it to the total synthesis of (±)-actinophyllic acid (1), and selected aspects of our work in the area and a total synthesis of (±)-1 were recently disclosed.20 Herein, we provide a full account of those studies.
2. Results and Discussion
In order to probe the feasibility of the key cascade sequence, we sought an efficient route to the requisite starting materials. Accordingly, carboxylate-directed deprotonation at the 2-position of indole (11) produced a dianion that underwent reaction with 1,3-dibenzyloxyacetone21 to afford an alkoxide intermediate that was acetylated with acetic anhydride to deliver acetate 12 in 83% yield.22 The structure of compound 12 was confirmed through single crystal X-ray analysis. Deprotonation of the known hydroazepinone 13, which was prepared in a single step from the photolysis of N-vinylpyrrolidinone,23 followed by reaction with several alkyl chloroformates gave carbamates 14–16, which were then converted into their corresponding triethylsilyl enol ethers 17–19.
With the desired electrophile precursor 12 and the π-nucleophiles 17–19 in hand, we explored the pivotal cascade sequence, beginning with conditions similar to those shown in Equation 1. However, upon treatment of a mixture of 12 and 17 with TMSOTf in CH2Cl2 at −78 °C, the formation of the desired tetracycle 20 was not observed, and the α,β-unsaturated ketone 21 and tetracycle 22 were isolated in 15% and 18% yields, respectively (Scheme 3). The ratio of 21:22 was found to be temperature dependent with tricycle 21 being the sole product (isolated in 63% yield) when the temperature was maintained at −78 °C. We found that compound 21 was converted completely to tetracycle 22 upon resubjection to the reaction conditions at room temperature. Although this transformation did not provide the desired bridged compound 20, the observation that the two key C-C bonds had been successfully formed encouraged us to pursue the reaction.
Scheme 3.

Failed cascade reaction
Based on this unexpected result, we reasoned that 21 and 22 were formed by degradation of the initially formed product 20 via two potential pathways (Scheme 4). In one scenario, β-elimination of the carbamate group from the silyl enol ether 20 would give the α,β-unsaturated ketone 21 (Path A), which upon cyclization and dehydration would produce tetracycle 22. Alternatively, 20 could suffer a gramine-type fragmentation to give intermediate 23 (Path B).24 Loss of the silyl group would then furnish 21, which may then cyclize to 22.
Scheme 4.

Proposed fragmentation pathways
These plausible degradation pathways suggested that the initially formed tetracycle 20 was unstable to the acidic reaction conditions, so we queried whether adding a suitable base might suppress any deleterious side reactions. In an initial experiment, we found that the presence of Hünig's base precluded any reaction, presumably due to coordination of TMSOTf with the basic nitrogen atom of the amine. In order to prevent such coordination, we employed 2,6-di-tert-butylpyridine as the base, but upon the usual aqueous workup, we again isolated a mixture of 21 and 22. We eventually discovered that quenching the reaction at –78 °C with NaOMe in MeOH, followed by an aqueous work-up, avoided the formation of any fragmentation products. In the event, treatment of a mixture of indole 12 and the π-nucleophiles 17–19 with TMSOTf in the presence of 2,6-ditert-butylpyridine, followed by addition of NaOMe in MeOH at −78 °C delivered the silyl enol ethers 20, 24, and 25 in 53–80% yields (Equation 2). The structure of methyl carbamate 24 was confirmed by single crystal X-ray analysis (Figure 1). To our dismay, we found that 20, 24, and 25 were rather unstable to further synthetic manipulations, a fact that severely limited their utility as intermediates en route to (±)-actinophyllic acid (1).
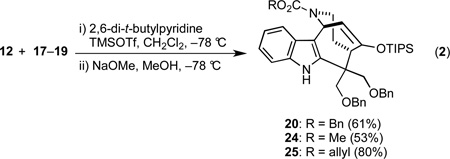 |
(2) |
Given that the enol ether moieties of 20, 24, and 25 are poised to undergo facile β-elimination (Scheme 4, Path A), we hypothesized that the corresponding ketones might be more stable. Recognizing that N-Alloc groups can be easily removed under mild conditions, we prepared the ketone 26 in 92% yield by treating 25, which was formed in situ by the reaction of 12 with 19 in the presence of TMSOTf and 2,6-di-tert-butylpyridine, with TBAF (Scheme 5). Palladium-catalyzed removal of the N-Alloc group25 gave the secondary amine 27, but this intermediate proved to be rather labile and difficult to isolate in pure form. Although we did not fully characterize any of the side-products derived from 27, 1H NMR spectra of product mixtures suggested that fragmentation pathways similar to those outlined in Scheme 4 were operative.
Figure 1.

X-ray structure of 24; hydrogen atoms omitted for clarity.
Scheme 5.

Setting the stage to form a pentacyclic intermediate
Two possible strategies to increase the stability of intermediates derived from 26 were thus considered: We could either reduce the ketone, a tactic with some precedent,13,26 or place an electron withdrawing group on the nitrogen atom of the indole ring. Because we had encountered some difficulties in preliminary attempts to introduce an electron withdrawing group on the indole nitrogen atom of 26, we elected instead to reduce the ketone function in 26 with lithium borohydride and obtained 28 as a single diastereomer in 82% yield (Scheme 5). The structure of 28 was unambiguously assigned via single crystal X-ray analysis (Figure 2). Alcohol 28 underwent facile palladium-catalyzed Alloc cleavage using phenylsilane as the π-allyl cation scavenger to yield the amino alcohol 29 in 73% yield.25
Figure 2.

X-ray structure of 28; hydrogen atoms omitted for clarity.
With 29 in hand, we turned our attention to forming the pyrrolidine ring, thereby establishing the pentacyclic core of actinophyllic acid (1). In an exploratory experiment, the secondary amine group in 29 was reductively alkylated with chloroacetaldehyde, but several attempts to oxidize the alcohol group in 30 and induce a base-catalyzed ring closure of the resultant ketone to give 31, even under the conditions described by Taniguchi for a similar conversion,13 were unavailing; the tetracyclic alcohol 34 was obtained in 41% yield instead (Scheme 6). Isolation of the primary alcohol 34 may be rationalized by the formation of an aziridinium ion such as 32 that underwent subsequent fragmentation (cf., Scheme 4) and nucleophilic ringopening with water to give 33, which could then provide 34 via sequential cyclization and dehydration.27
Scheme 6.

First generation route to a pentacyclic intermediate.
At this juncture, the main issue to be confronted was whether 26 or a compound derived therefrom was a viable intermediate in the synthesis of (±)-actinophyllic acid (1). Because step-count was a secondary consideration in establishing proof-of-principle, we developed a strategy that relied upon installing an electron withdrawing group on the indole nitrogen atom to mitigate fragmentation via a gramine-type pathway. Toward this end, protection of the hydroxyl group in 28 as its trimethylsilyl ether, followed by treatment with neat Boc-anhydride in the presence of a catalytic amount of DMAP at 100 °C and then TBAF-mediated deprotection of the alcohol furnished the N-protected indole 35 in 88% overall yield (Scheme 7). Utilizing the previously developed conditions for palladium-catalyzed deprotection and reductive amination, 36 was obtained in 56% overall yield. Although oxidation of 36 with IBX gave mixtures of the desired ketone 37 together with side products that appeared to arise from fragmentation, oxidation of 36 under Swern conditions cleanly delivered 37 in 86% yield. Treatment of 37 with sodium tert-butoxide resulted in intramolecular enolate alkylation and formation of the pyrrolidine ring, giving the pentacycle 38 in 94% yield.
Scheme 7.

Second generation route to a pentacyclic intermediate.
Toward completing the total synthesis of (±)-actinophyllic acid (1), our initial plan was to remove the 1,3-diol and indole protecting groups in 38 by hydrogenolysis in the presence of protic acid to give 2, but these conditions led exclusively to the cyclic carbamate 39 (Scheme 8). Although 39 was converted into 2 upon standing in wet methanol, we developed a facile two-step, one-pot procedure whereby the Boc group was first removed by treating 38 with HCl in methanol at 65 °C. The solution was simply cooled, Pd/C (10%) was added, and the mixture was stirred under an atmosphere of hydrogen to furnish the hexacyclic alcohol 2 as its hydrochloride salt in 86% yield. The structure of 2 was confirmed through single crystal X-ray analysis (Figure 3).
Scheme 8.

Synthesis of hexacyclic alcohol 2.
Figure 3.

X-ray structure of 2 with chloride ion (green); hydrogen atoms omitted for clarity.
The oxidation of the neopentyl alcohol 2 to give (±)-actinophyllic acid (1) proved surprisingly difficult. The insolubility of 2 precluded use of many standard reaction conditions, and numerous attempts to oxidize 2 led to poor material balances and low yields of 1.28,29 We surmised that oxidation of the indole ring and/or ketal opening and oxidation of both primary alcohols were complicating factors, but we were not able to isolate products that support either of these hypotheses. After considerable experimentation, we discovered that oxidation of 2 with TPAP/NMO30 or PDC in DMF31 produced (±)-actinophyllic acid (1), albeit in irreproducible yields. Finally, we found that oxidation of 2 with IBX in DMSO at 50 °C produced an intermediate aldehyde, which upon treatment with N-hydroxysuccinimide and further oxidation yielded an activated ester that was readily hydrolyzed under basic conditions to deliver (±)-actinophyllic acid (1) in 31% yield (Equation 3).32 The spectral data for 1 matched that of a synthetic sample graciously provided by Professor Larry E. Overman.
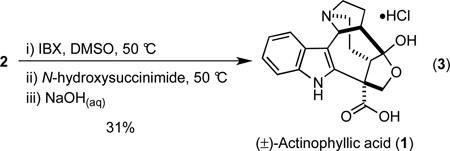 |
(3) |
This first generation synthesis of (±)-actinophyllic acid (1) was completed in 14 steps and 5% overall yield from known materials. However, the sequence suffers from far too many protection/deprotection and refunctionalization steps; avoiding so many "unproductive" steps was clearly desirable in order to enhance the overall efficiency of the synthesis. After considering various possibilities, we decided to revisit the tactic of placing an electron withdrawing group on the indole nitrogen atom of 26. This seemed a risky undertaking because 26 had previously proven difficult to handle (vide supra), and the forcing conditions required to introduce the Boc group onto a precursor of 35 did not augur well for such a protective move. However, despite our reservations, we discovered that treating 26 with Boc-anhydride and DMAP in toluene at room temperature delivered the desired N-protected compound 40 in 83% yield (Scheme 9). Under these milder reaction conditions no products resulting from fragmentation of 26 were observed in the 1H NMR spectra of the crude reaction mixture. The subsequent Alloc deprotection was optimized through the use of N,N-dimethylbarbituric acid as the π-allyl cation scavenger,33 and the amine 41 was obtained in 92% yield. Reductive amination of 41 with chloroacetaldehyde, followed by base-induced cyclization as before furnished pentacycle 38 in 83% yield over the two steps. This improved second generation synthesis delivered (±)-1 in only 10 total steps and 10% overall yield from known materials.
Scheme 9.

Second generation synthesis
3. Summary
Inspired by previous work in our laboratories, a novel cascade of reactions involving stabilized carbocations and π-nucleophiles was developed to deliver the tetracyclic core of (±)-actinophyllic acid (1) in a single chemical operation from readily-available starting materials. Although several synthetic intermediates exhibited a proclivity to suffer a troublesome fragmentation, consideration of possible degradation pathways enabled us to solve the underlying problems and complete the synthesis of (±)-actinophyllic acid (1) via two different routes. The first of these required 14 steps and proceeded in 5% overall yield from readily-available starting materials, whereas the second generation approach proceeded in 10% overall yield and required only 10 steps from the same starting materials. Toward identifying novel, biologically-active compounds, several intermediates were diverted to prepare analogs for biological evaluation,34 and a number of tetracyclic derivatives of 28 were found to exhibit significant anticancer activity as reported elsewhere.20 Notably, these compounds are not readily accessible by known synthetic pathways, including those that have been reported for the synthesis of actinophyllic acid. Accordingly, this discovery of promising anticancer leads underscores the value, which is not universally acknowledged, of developing alternative entries to complex natural products as a strategy to generate compounds for biological screening that would not be otherwise accessible. Details of our biological studies and other applications of cascade reactions to address problems in alkaloid synthesis will be reported in due course.
4. Experimental
4.1. General
Tetrahydrofuran (THF) and toluene were passed through two columns of activated neutral alumina prior to use.35 Dichloromethane (CH2Cl2), acetic anhydride (Ac2O), and allyl chloroformate (Alloc-Cl) were freshly distilled over CaH2. Trimethylsilyl trifluoromethanesulfonate (TMSOTf) was distilled over P2O5. All other reagents and solvents were reagent grade and were used as received unless otherwise noted. Reactions were performed under a nitrogen or argon atmosphere, and moisture sensitive reactions were performed with flame- or oven-dried glassware. Reaction temperatures are reported as the temperatures of the bath surrounding the vessel.
Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were acquired in CDCl3 unless otherwise noted. Chemical shifts are reported in parts per million (ppm, δ), downfield from tetramethylsilane (TMS, δ = 0.00 ppm) and are referenced to residual solvent (CDCl3, δ = 7.26 ppm (1H) and 77.16 ppm (13C)). Coupling constants (J) are reported in hertz (Hz) and the resonance multiplicity abbreviations used are: s, singlet; d, doublet; t, triplet; q, quartet; dt, doublet of triplets; dd, doublet of doublets; ddd, doublet of doublet of doublets; dddd, doublet of doublet of doublet of doublets; m, multiplet; comp, overlapping multiplets of magnetically non-equivalent protons. The abbreviations br and app stand for broad and apparent, respectively. Infrared (IR) spectra were obtained as thin films on sodium chloride plates. Thin-layer chromatography (TLC) was performed on EMD 60 F254 glass-backed pre-coated silica gel plates. Flash chromatography was performed using Silicycle® SiliaFlash F60® (40–63 µm) silica gel eluting with the solvents indicated according to the procedure of Still.36 Reverse phase chromatography was performed on a CombiFlash® Companion instrument utilizing a RediSep® Rf 43 g C18 Reverse Phase column.
4.2. Selected Experimental Procedures
1,3-Bis(benzyloxy)-2-(1H-indol-2-yl)propan-2-yl acetate (12)
A solution of n-BuLi (6.8 mL of a 2.17 M solution in hexanes, 14.8 mmol) was added dropwise to a solution of indole (11) (1.6 g, 13.5 mmol) in dry THF (39 mL) at −78 °C. The reaction was stirred for 30 min, and dried CO2 gas (passed through a short column of anhydrous CaSO4) was bubbled through the reaction for 15 min. The reaction was stirred for an additional 15 min at −78 °C, the bath was removed, and the reaction was stirred for 1 h at room temperature. Excess CO2 was removed via freeze-pump-thaw (3 cycles), and then the reaction was evacuated for 30 sec and sparged with Ar for 20 min. The solution was cooled to −78 °C, whereupon t-BuLi (10.0 mL of a 1.47 M solution in pentane, 14.8 mmol) was added dropwise. The reaction was stirred for 80 min and transferred via cannula to a solution of 1,3-dibenzyloxyacetone (4.0 g, 14.8 mmol) in dry THF (39 mL) at −78 °C. The reaction was stirred for 2.5 h, acetic anhydride (3.8 mL, 40.5 mmol) was added dropwise, and the reaction was allowed to warm to room temperature over 7 h. Saturated aqueous NaHCO3 (50 mL) was added, and the reaction was stirred for 30 min, whereupon saturated brine (80 mL) and Et2O (200 mL) were added. The layers were separated, and the aqueous layer was extracted with Et2O (2 × 100 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (90 : 10 → 85 : 15 → 80 : 20 → 75 : 25 → 70 : 20) containing 1% Et3N to give 5.0 g (85%) of 12 as a pale yellow solid: mp 78–79 °C; 1H NMR (400 MHz, CD3CN) δ 9.55 (br s, 1 H), 7.58 (d, J = 8.0 Hz, 1 H), 7.45 (d, J = 8.0 Hz, 1 H), 7.39–731 (comp, 10 H), 7.21-7.17 (m, 1 H), 7.11-7.07 (m, 1 H), 6.46 (dd, J = 2.4, 0.8 Hz, 1 H), 4.58 (s, 4 H), 4.21 (s, 4 H), 2.08 (s, 3 H); 13C NMR (100 MHz, CD3CN) δ 169.8, 138.6, 137.3, 136.2, 128.6, 128.0, 127.9, 122.0, 120.5, 119.8, 111.4, 99.9, 80.7, 73.4, 70.7, 21.5; IR (neat) 3416, 2864, 1741, 1454, 1367, 1237, 1103, 1018, 738, 698 cm−1; mass spectrum (CI) m/z 430.2010 [C27H28NO4 (M+1) requires 430.2018].
General procedure for the acylation of hydroazepinone 13
A solution of n-BuLi (1.2 equiv of a 2.5 M solution in hexanes) was added dropwise to a solution of 13 (1 equiv) in THF (0.11 M) at −78 °C. After stirring for 0.5 h, the chloroformate (1.3 equiv) was added dropwise, and the reaction was stirred for 1.5 h. The bath was removed, and the reaction was stirred for 1.5 h at room temperature. The reaction was concentrated and partitioned between a mixture of saturated aqueous NaHCO3, saturated brine and CH2Cl2. The layers were separated, and the aqueous layer was extracted twice with CH2Cl2.The combined organic layers were washed with saturated brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography.
Benzyl 5-oxo-2,3,4,5-tetrahydro-1H-azepine-1-carboxylate (14)
Purification: hexanes/EtOAc (4 : 1 → 3 : 2) to give 1.94 g (84%) of 14 as a colorless amorphous solid: 1H NMR (400 MHz, CD2Cl2) δ 7.47 (d, J = 10.6 Hz, 1 H), 7.41-7.34 (comp, 5 H), 5.24 (s, 2 H), 5.19 (d, J = 10.6 Hz, 1 H), 3.97-3.94 (m, 2 H), 2.62 (app t, J = 6.5 Hz, 2 H), 2.06-2.00 (m, 2 H); 13C NMR (100 MHz, CD2Cl2) δ 200.7, 135.8, 129.0, 128.9, 128.6, 110.1, 110.0, 69.4, 48.5, 43.5, 22.9; IR (neat) 1722, 1613, 1315, 1250, 1221, 1112 cm−1; mass spectrum (CI) m/z 246.1131 [C14H16NO3 (M+1) requires 246.1130].
Methyl 5-oxo-2,3,4,5-tetrahydro-1H-azepine-1-carboxylate (15)
Purification: hexanes/EtOAc (4 : 1 →1 : 1) to give 1.27 g (79%) of 15 as a pale yellow oil: 1H NMR (400 MHz, CD2Cl2) δ 7.44 (d, J = 10.6 Hz, 1 H), 5.18 (d, J = 10.6 Hz, 1 H), 3.94-3.92 (m, 2 H), 3.83 (s, 3 H), 2.62 (app t, J = 6.5 Hz, 2 H), 2.05-2.00 (m, 2 H); IR (neat) 2956, 1729, 1614, 1441, 1351, 1319, 1256, 1116, 943, 767 cm−1; mass spectrum (CI) m/z 170.0815 [C8H12NO3 (M+1) requires 170.0817].
Allyl 5-oxo-2,3,4,5-tetrahydro-1H-azepine-1-carboxylate (16)
Purification: hexanes/EtOAc (7 : 3 →8 : 4 →1 : 1) to give 3.7 g (84%) of 16 as a colorless solid: 1H NMR (400 MHz) δ 7.46 (d, J = 10.6 Hz, 1 H), 5.99-5.89 (m, 1 H), 5.37-5.23 (comp, 3 H), 4.70 (app dt, J = 1.4, 5.8 Hz, 2 H), 3.96-3.93 (comp, 2 H), 2.65 (app t, J = 6.5 Hz, 2 H), 2.07-2.01 (comp, 2 H); 13C NMR (100 MHz) δ 200.9, 166.0, 137.2, 131.4, 119.3, 109.9, 68.0, 48.1, 42.7, 22.7; IR (neat) 3492, 3088, 2945, 1730, 1614, 1434, 1114, 943, 766 cm−1; mass spectrum (CI) m/z 196.0974 [C10H13NO3 (M+1) requires 196.0974].
Allyl 5-((triisopropylsilyl)oxy)-2,3-dihydro-1H-azepine-1-carboxylate (19)
Triisopropylsilyl chloride (7.2 g, 8.0 mL, 37.4 mmol) was added to a solution of azepine 16 (6.0 g, 30.9 mmol) in THF (330 mL) at −78 °C and stirred for 12 min. Sodium hexamethyldisilazide (20.0 mL of a 2.04 M solution in THF, 40.1 mmol) was added to the mixture dropwise, and the solution was stirred at −78 °C for 1.5 h. Saturated NaHCO3 (100 mL) was added, the bath was removed, and stirring was continued for 15 min. Once at room temperature, the reaction was partitioned between a mixture of saturated aqueous NaHCO3 (100 mL), saturated brine (150 mL), and Et2O (200 mL). The layers were separated, and the aqueous layer was extracted with Et2O (2 × 200 mL). The combined organic layers were washed with saturated brine (300 mL), dried (Na2SO4), filtered and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (92 : 8 → 93 : 7) containing 1% Et3N to give 11.3 g (92%) of 16 as a pale yellow oil: 1H NMR (400 MHz, CD3CN) δ 6.84 (d, J = 8.9 Hz, 1 H), 6.03-5.93 (m, 1 H), 5.36-5.19 (comp, 3 H), 5.06 (d, J = 9.9 Hz, 1 H), 4.64 (app dt, J = 1.4, 5.5 Hz, 2 H), 3.74 (app t, J = 4.1 Hz, 2 H), 2.35 (app q, J = 5.5 Hz, 2 H), 1.23-1.14 (comp, 3 H), 1.10-1.09 (comp, 18 H); 13C NMR (125 MHz, CD3CN) 153.9, 148.9, 133.7, 128.6, 118.2, 110.4, 109.6, 67.6, 46.2, 28.6, 18.5, 13.3; IR (neat) 3060, 3030, 2944, 2866, 1691, 1461, 1411, 1366, 1233, 1199, 1106, 885, 743.6, 697.9 cm−1; mass spectrum (CI) m/z 351.2225 [C19H33NO3Si (M+1) requires 351.2230].
General procedure for the formation of silyl enol ethers 20, 24, and 25
Trimethylsilyl triflate (1.1 equiv) was added dropwise to a solution of 12 (1.0 equiv), 17–19 (1.1 equiv) and 2,6-di-tert-butylpyridine (2.0 equiv) in CH2Cl2 (0.1 M in 12) at −78 °C under argon. The reaction was stirred for 3.5 h, and sodium methoxide (14.5 equiv of a 3.20 M solution in dry methanol) was added. After 1 h, saturated aqueous NaHCO3 was added, the dry-ice bath was removed, and the reaction was stirred for 1 h. Additional portions of saturated aqueous NaHCO3 and saturated brine were added, and the reaction was extracted three-times with CH2Cl2. The combined organic layers were washed with saturated brine, dried (Na2SO4), filtered and concentrated. The crude material was purified by flash column chromatography.
Benzyl (1S/R,5S/R)-6,6-bis((benzyloxy)methyl)-12-((triisopropylsilyl)oxy)-1,3,4,5,6,7-hexahydro-2H-1,5-ethenoazocino[4,3-b]indole-2-carboxylate (20)
Purification: pentane/Et2O (4 : 1) containing 1% Et3N to give 33 mg (61%) of 20 as a colorless amorphous solid: 1H NMR (600 MHz, CD2Cl2, as a mixture of rotamers) δ 9.17 (s, 0.5 H), 9.16 (s, 0.5 H), 7.56 (d, J = 7.8 Hz, 0.5 H), 7.49 (d, J = 7.4 Hz, 1 H), 7.43 (app t, J = 7.4 Hz, 2 H), 7.40-7.27 (comp, 13.5 H), 7.13-7.10 (m, 1 H), 7.08-7.03 (m, 1 H), 5.92 (d, J = 8.6 Hz, 0.5 H), 5.81 (d, J = 8.4 Hz, 0.5 H), 5.76 (dd, J = 2.4, 8.6 Hz, 0.5 H), 5.68 (dd, J = 2.4, 8.4 Hz, 0.5 H), 5.25 (d, J = 12.5 Hz, 0.5 H), 5.20 (d, J = 12.5 Hz, 0.5 H), 5.18 (d, J = 12.5 Hz, 0.5 H), 5.09 (d, J = 12.5 Hz, 0.5 H), 4.69-4.64 (comp, 2 H), 4.60-4.59 (d, J = 11.9 Hz, 1 H), 4.49-4.47 (dd, J = 3.5, 11.8 Hz, 1 H), 4.11-3.97 (m, 1 H), 3.98-3.96 (m, 1 H), 3.89-3.86 (m, 1 H), 3.83-3.80 (comp, 1H), 3.72-3.69 (m, 1 H), 3.33-3.28 (app dt, J = 2.4, 15.6 Hz, 0.5 H), 3.25-3.20 (app dt, J = 2.6, 14.0 Hz, 0.5 H), 2.86-2.84 (comp 1H), 2.13-2.05 (m, 1 H), 1.82-1.78 (m, 1 H), 1.26-1.15 (comp, 3 H), 1.10-1.06 (comp, 18 H); 13C NMR (150 MHz, CD2Cl2, as a mixture of rotamers) δ 159.0, 158.7, 115.7, 155.5, 139.5, 139.4, 139.0, 138.52, 138.50, 138.0, 137.9, 135.0, 134.9, 128.9, 128.8, 128.7, 128.6, 128.2, 128.1, 128.1, 128.1, 128.0, 128.0, 127.9, 127.8, 126.9, 126.7, 121.9, 119.8, 119.7, 118.3, 117.9, 111.1, 111.0, 109.7, 109.57, 109.55, 109.5, 74.2, 73.8, 72.5, 72.4, 72.2, 72.1, 67.2, 67.1, 60.6, 45.6, 45.4, 45.0, 41.5, 41.3, 27.5, 27.0, 18.3, 13.0; IR (neat) 2943, 2866, 1691, 1460, 1419, 1199, 1106, 884, 744, 697 cm−1; mass spectrum (CI) m/z 771.4195 [C48H59N2O5Si (M+1) requires 771.4188].
(1S/R,5S/R)-Methyl 6,6-bis((benzyloxy)methyl)-12-((triisopropylsilyl)oxy)-3,4,5,6-tetrahydro-1H-1,5-ethenoazocino[4,3-b]indole-2(7H)-carboxylate (24)
Purification: hexanes/EtOAc (9 : 1 →7 : 3) containing 1% Et3N to give 324 mg (53%) of 24 a colorless amorphous solid: 1H NMR (600 MHz, CD2Cl2, as a mixture of rotamers) δ 9.16-9.13 (comp, 1H), 7.52 (d, J = 30.7 Hz, 0.5 H), 7.47 (d, J = 30.7 Hz, 0.5 H), 7.37-7.25 (comp, 11 H), 7.10 (app dt, J = 2.9, 7.2 Hz, 1 H), 7.05 (app q, J = 6.9, 1 H), 5.85 (d, J = 8.5 Hz, 0.5 H), 5.72 (dd, J = 2.3, 8.6 Hz, 0.5 H), 5.67 (app s, 1 H), 4.65 (dd, J = 3.7, 11.9 Hz, 2 H), 4.58 (d, J = 11.9 Hz, 1 H), 4.46 (d, J = 11.8 Hz, 1 H), 4.01 (app dt, J = 3.7, 14.7 Hz, 0.5 H), 3.95 (d, J = 8.7 Hz, 1 H), 3.86 (app dd, J = 3.5, 12.0 Hz, 1 H), 3.88-3.84 (comp, 0.5 H), 3.79 (app dd, J = 2.3, 8.6 Hz, 1 H), 3.75 (s, 1.5 H), 3.68 (app t, 7.7 Hz, 1 H), 3.64 (s, 1.5 H), 3.25 (app t, J = 12.8 Hz, 1 H), 2.82 (d, J = 2.1 Hz, 1 H), 2.10-2.03 (comp, 0.5 H), 2.06-2.00 (comp, 0.5 H), 1.77-1.76 (comp, 0.5 H), 1.75-1.74 (comp, 0.5 H), 1.25-1.17 (comp, 3 H), 1.10-1.04 (comp, 18 H); 13C NMR (150 MHz, CD2Cl2 as a mixture of rotamers) δ 158.8, 158.6, 156.40, 156.36, 139.4, 139.0, 138.5, 135.0, 134.9, 128.9, 128.6, 128.2, 128.1, 128.0, 127.8, 126.9, 126.8, 121.9, 119.7, 118.3, 117.9, 111.1, 111.0, 110.0, 109.7, 109.6, 109.5, 74.2, 73.8, 72.43, 72.40, 72.2, 72.1, 52.7, 52.6, 45.5, 45.4, 45.3, 45.02, 45.00, 41.5, 41.2, 27.4, 26.8, 18.3, 13.0, 12.9; IR (neat) 3030, 2945, 2866, 1692, 1460, 1401, 1367, 1233, 1198, 1106, 1073, 885, 744, 698 cm−1; mass spectrum (CI) m/z 694.3805 [C42H54N2O5Si (M+1) requires 694.3802].
Allyl (1S/R,5S/R)-6,6-bis((benzyloxy)methyl)-12-((triisopropylsilyl)oxy)-1,3,4,5,6,7-hexahydro-2H-1,5-ethenoazocino[4,3-b]indole-2-carboxylate (25)
Purification: hexanes/EtOAc (9 : 1 →7 : 3) containing 1% Et3N to give 915 mg (80%) of 25 of an amorphous, colorless solid: 1H NMR (600 MHz, CD3CN, as a mixture of rotamers) 9.49-9.47 (m, 1 H), 7.41 (d, J = 7.9 Hz, 1 H), 7.38-7.21 (comp, 11 H), 7.11-710 (m, 1 H), 7.02 (dd, J = 7.6, 15.1 Hz, 1 H), 6.08-6.00 (m, 0.5 H), 5.95-5.88 (m, 0.5 H), 5.81 (d, J = 8.6 Hz, 0.5 H), 5.74 (d, J = 8.4 Hz, 0.5 H), 5.67 (dd, J = 2.3, 8.3 Hz, 1 H), 5.43-5.20 (m, 1 H), 5.40 (dd, J = 1.5, 17.3 Hz, 0.5 H), 5.13 (d, J = 10.5 Hz, 0.5 H), 4.63 (d, J = 12.0 Hz, 1 H), 4.58 (d, J = 12.0 Hz, 1 H), 4.64-4.58 (m, 1 H), 4.50–4.58 (m, 1 H), 4.53 (d, J =12.3 Hz, 1 H), 4.45 (d, J =12.3 Hz, 1 H), 4.04 (d, J = 9.0 Hz, 1 H), 3.93 (app dt, J = 3.1, 13.9 Hz, 0.5 H), 3.88 (app dt, J = 3.1, 13.9 Hz, 0.5 H), 3.73 (d, J = 9.2 Hz, 1 H), 3.71 (d, J = 9.8 Hz, 1 H), 3.64 (d, J =8.9 Hz, 1 H), 3.20 (dt, J = 2.3, 12.8, Hz, 0.5 H), 3.11 (dt, J = 2.3, 12.8, Hz, 0.5 H), 2.70 (dd, J = 2.8, 5.4 Hz, 1 H), 1.97-1.91 (m, 1 H), 1.87-1.79 (m, 1 H), 1.19-1.12 (comp, 3 H), 1.04-1.01 (comp, 18 H); 13C NMR (150 MHz, CD3CN, as a mixture of rotamers) 159.51, 159.46, 156.0, 155.9, 139.9, 139.8, 139.5, 139.4, 135.84, 135.81, 134.8, 129.4, 129.2, 128.8, 128.6, 128.4, 127.24, 127.22, 122.4, 120.2, 117.14, 117.05 112.0, 111.9, 110.4, 110.1, 109.5, 74.2, 74.1, 73.2, 71.9, 71.8, 66.5, 66.3, 46.3, 46.0, 45.72, 45.68, 42.0, 41.8, 27.9, 27.3, 18.6, 18.5, 13.3; IR (neat) 3030, 2943, 2866, 1691, 1461, 1411, 1366, 1233, 1199, 1106, 885, 744 cm−1; mass spectrum (CI) m/z 720.3954 [C44H56N2O5Si (M+1) requires 720.3959].
Allyl (1S/R,5S/R)-6,6-bis((benzyloxy)methyl)-12-oxo-1,3,4,5,6,7-hexahydro-2H-1,5-ethanoazocino[4,3-b]indole-2-carboxylate (26)
A solution of TMSOTf (0.9 g, 0.7 mL, 3.7 mmol) in CH2Cl2 (1.5 mL) was added dropwise to a solution of 19 (1.3 g, 3.6 mmol), 12 (1.5 g, 3.5 mmol), and 2,6-di(tert-butyl) pyridine (1.4 g, 1.3 mL, 7.0 mmol) in dry CH2Cl2 (13.5 mL) at −78 °C. The reaction was stirred for 3 h at −78 °C, whereupon a solution of TBAF·3H2O (5.5 g, 17.5 mmol) in CH2Cl2 (27.0 mL) was added via cannula. The bath was removed, and the reaction was stirred at room temperature for 1 h. The reaction was quenched with saturated aqueous NaHCO3 (30 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 30 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (8 : 2 → 7 : 3 → 6 : 4) containing 1% Et3N to give 1.8 g (92%) of 26 as an amorphous yellow solid: 1H NMR (600 MHz, CD3CN, as a mixture of rotamers) δ 9.60 (s, 1 H), 7.45 (d, J = 7.81 Hz, 1 H), 7.36-7.23 (comp 9 H), 7.38 (d, J = 8.1 Hz, 1 H), 7.17-7.15 (m, 1 H), 7.16-7.12 (m, 1 H), 7.07 (app t, J = 7.9 Hz, 1 H), 6.10-5.89 (m, 1 H), 5.81-5.76 (m, 1 H), 5.40-5.15 (comp, 2 H), 4.66-4.54 (comp, 2 H), 4.62 (d, J = 11.0 Hz, 1 H), 4.57 (d, J = 11.0 Hz, 1 H), 4.38 (d, J = 11.9 Hz, 1 H), 4.31 (d, J = 11.9 Hz, 1 H), 3.98-3.80 (m, 1 H), 3.96 (d, J = 9.3 Hz, 1 H), 3.75 (d, J = 9.3 Hz, 1 H), 3.70 (d, J = 9.3 Hz, 1 H), 3.63 (d, J = 9.3 Hz, 1 H), 3.07 (dd, J = 2.6, 5.9 Hz, 1 H), 3.04 (app dd, J = 4.4, 18.8 Hz, 1 H), 3.03-2.87 (m, 1 H), 2.98-2.84 (m, 1 H), 2.13-2.01 (m, 1 H), 1.80-1.70 (m, 1 H); 13C NMR (150 MHz, CD3CN, as a mixture of rotamers) 211.9, 155.6, 139.13, 139.09, 136.4, 134.6, 129.4, 129.2, 128.9, 128.8, 128.7, 128.5, 127.4, 123.0, 120.5, 117.3, 112.0, 110.4, 75.9, 74.2, 74.0, 73.0, 66.6, 58.1, 50.4, 49.6, 46.1, 45.7, 40.5, 28.8, 28.4; IR (neat) 3031, 2863, 1694, 1414, 1344, 1305, 1228, 1103, 1207, 983, 941, 861, 744, 699 cm−1; mass spectrum (CI) m/z 564.2625 [C35H36N2O5 (M+1) requires 564.2725].
Allyl (1S/R,5S/R,12R/S)-6,6-bis((benzyloxy)methyl)-12-hydroxy-1,3,4,5,6,7-hexahydro-2H-5,1-ethanoazocino[4,3-b]indole-2-carboxylate (28)
Lithium borohydride (4 mg, 0.18 mmol) was added to a solution of 26 (20 mg, 0.04 mmol) in MeOH (0.4 mL) at 0 °C. The reaction was stirred for 18 h at 0 °C and then partitioned between saturated brine (3 mL) and CH2Cl2 (3 mL). The layers were separated, and the aqueous layer was extracted (2 × 3 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant yellow solid was purified by flash column chromatography, eluting with hexanes/EtOAc (1 : 1) containing 1% Et3N to give 16 mg (82%) of 28 as a colorless solid: mp 167–169 °C; 1H NMR (500 MHz, DMSO-d6, 120 °C) δ 10.23 (br s, 1 H), 7.40 (d, J = 8.0 Hz, 1 H), 7.36-7.31 (comp, 5 H), 7.29-7.21 (comp, 6 H), 7.07-7.04 (m, 1 H), 7.00-6.97 (m, 1 H), 6.05-5.97 (m, 1 H), 5.56 (d, J = 6.5 Hz, 1 H), 5.34 (d, J = 17.5 Hz, 1 H), 5.22 (d, J = 10.0 Hz, 1 H), 4.68-4.59 (comp, 4 H), 4.48 (dd, J = 14.5, 13.0 Hz, 2 H), 4.41 (br s, 1 H), 4.29 (d, J = 9.0 Hz, 1 H), 4.26-4.23 (m, 1 H), 4.08 (d, J = 10.0 Hz, 1 H), 4.02 (d, J = 10.0 Hz, 1 H), 3.76-3.73 (comp, 2 H), 2.97-2.91 (m, 1 H), 2.68-2.63 (comp, 2 H), 1.93 (app dt, J = 14.5, 3.5 Hz, 1 H), 1.85 (dd, J = 13.5, 9.5 Hz, 1 H), 1.76-1.68 (m, 1 H); 13C NMR (125 MHz, DMSO-d6, 120 °C) δ 154.0, 138.9, 138.2, 138.0, 134.9, 133.3, 127.5, 127.4, 126.74, 126.7, 126.6, 126.4, 125.5, 120.2, 118.2, 116.0, 115.97, 110.8, 110.5, 73.3, 73.2, 72.4, 72.2, 71.8, 64.4, 46.7, 46.4, 45.4, 39.9, 39.8, 29.4; IR (neat) 3408, 2943, 1680, 1454, 1413, 1104, 739 cm−1; mass spectrum (ESI) m/z 566.2823 [C35H39N2O5 (M+1) requires 566.2853].
(1S/R,5S/R,12R/S)-6,6-bis((Benzyloxy)methyl)-2,3,4,5,6,7-hexahydro-1H-5,1-ethanoazocino[4,3-b]indol-12-ol (29)
A solution of tetrakis(triphenylphosphine)palladium(0) (92 mg, 0.08 mmol) in CH2Cl2 (2.0 mL) was added dropwise to a solution of alcohol 28 (0.9 g, 1.59 mmol) and PhSiH3 (688 mg, 0.8 mL, 6.36 mmol) in CH2Cl2 (7.4 mL) at room temperature, and the reaction was stirred for 0.5 h. Saturated aqueous NaHCO3 (20 mL) was added, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 25 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant brown gum was purified by flash column chromatography eluting with CH2Cl2/MeOH (75 : 25) containing 1% Et3N to give 0.56 g (73%) of 29 as a tan amorphous solid foam:1H NMR (600 MHz, CD2Cl2) δ 9.40 (br s, 1 H), 7.65 (d, J = 7.8 Hz, 1 H), 7.42-7.22 (comp, 11 H), 7.13 (ddd, J = 7.2, 7.2, 1.2 Hz, 1 H), 7.09 (ddd, J = 8.4, 8.4, 1.2 Hz, 1 H), 4.96 (d, J = 6.0 Hz, 1 H), 4.71-4.60 (comp, 3 H), 4.52-4.48 (m, 1 H), 4.09-4.00 (comp, 3 H), 3.93 (d, J = 9.0 Hz, 1 H), 3.71 (d, J = 9.0 Hz, 1 H), 3.05 (ddd, J = 14.4, 8.4, 6.6 Hz, 1 H), 2.87 (dd, J = 13.8, 4.8 Hz, 1 H), 2.66-2.55 (comp, 2 H), 2.07 (dd, J = 13.8, 9.0 Hz, 1 H), 2.01-1.85 (comp, 2 H); 13C NMR (150 Hz, CD2Cl2) δ 140.0, 138.42, 138.38, 135.5, 128.97, 128.94, 128.7, 128.3, 128.2, 128.1, 126.5, 122.2, 119.9, 118.5, 111.0, 110.3, 74.7, 74.2, 74.0, 72.8, 64.5, 47.8, 46.5, 41.8, 40.6, 31.0, 29.0; mass spectrum m/z (ESI) 483.2642 [C31H35N2O3 (M+1) requires 483.2642].
Allyl (1S/R,5S/R,12R/S)-6,6-bis((benzyloxy)methyl)-12-((trimethylsilyl)oxy)-1,3,4,5,6,7-hexahydro-2H-5,1-ethanoazocino[4,3-b]indole-2-carboxylate
TMSCl (2.2 mL, 1.8 g, 17.4 mmol) was added to a solution of 28 (3.3 g, 5.8 mmol) and imidazole (2.0 g, 29.0 mmol) in anhydrous DMF (62 mL), and the reaction was stirred 2 h. The mixture was partitioned between saturated aqueous NaHCO3 (100 mL) and Et2O (150 mL), and the layers were separated. The aqueous layer was extracted with Et2O (2 × 100 mL), and the combined organic layers were washed with brine (1 × 100 mL). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. The resultant clear oil was purified by flash column chromatography eluting with hexanes/EtOAc (9 : 1) containing 1% Et3N to provide 3.6 g (97%) of the trimethylsilyl protected alcohol as a colorless amorphous solid foam: mp 115–116 °C (colorless crystals from i-PrOH/H2O); 1H NMR (500 MHz, DMSO-d6, 120 °C) δ 10.23 (br s, 1 H), 7.39 (d, J = 8.0 Hz, 1H), 7.32-7.29 (comp, 5 H), 7.27-7.22 (comp, 6 H), 7.05 (t, J = 8.0 Hz, 1 H), 6.97 (t, J = 8.0 Hz, 1 H), 6.04-5.97 (m, 1 H), 5.51 (d, J = 6.5 Hz, 1 H), 5.33 (d, J = 17.0 Hz, 1 H), 5.21 (d, J = 10.5 Hz, 1 H), 4.67-4.58 (comp, 4 H), 4.48 (d, J = 12.5 Hz, 1 H), 4.45 (d, J = 12.5 Hz, 1 H), 4.32-4.27 (comp, 2 H), 3.98 (d, J = 9.5 Hz, 1 H), 3.92 (d, J = 9.5 Hz, 1 H), 3.75-3.71 (comp, 2 H), 2.93-2.85 (m, 1 H), 2.67-2.61 (comp, 2 H), 1.92 (ddd, J = 14.5, 3.5, 3.5 Hz, 1 H), 1.86 (dd, J = 13.5, 9.0 Hz, 1 H), 1.75-1.67 (m, 1 H), 0.05 (s, 9 H); 13C NMR (125 MHz, DMSO-d6, 120 °C) δ 154.0, 138.8, 138.1, 138.0, 134.9, 133.2, 127.4, 127.3, 126.6, 126.6, 126.5, 126.4, 125.3, 120.2, 118.1, 116.0, 116.0 110.6, 110.5, 74.8, 72.5, 72.3, 72.1, 71.5, 64.4, 46.7, 46.2, 45.5, 40.5 39.7, 29.1, −0.8; IR (neat) 3030, 2951, 2871, 1693, 1454, 1412, 1251, 1072, 873, 842, 744, 698 cm−1; mass spectrum (ESI) m/z 661.3066 [C38H46N2NaO5Si (M+1) requires 661.3068].
2-Allyl 7-(tert-butyl) (1S/R,5S/R,12R/S)-6,6-bis((benzyloxy)methyl)-12-((trimethylsilyl)oxy)-3,4,5,6-tetrahydro-1H-5,1-ethanoazocino[4,3-b]indole-2,7-dicarboxylate
DMAP (0.2 g, 1.64 mmol) was added to a solution of the trimethylsilyl protected alcohol (3.5 g, 5.5 mmol) from the previous step in Boc2O (16 mL) at 100 °C, and the reaction was stirred for 30 min at 100 °C. The mixture was partitioned between H2O (100 mL) and Et2O (150 mL), and the layers were separated. The aqueous layer was extracted with Et2O (2 × 150 mL), and the combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The resultant brown oil was purified by flash column chromatography eluting with hexanes/EtOAc (1 : 0 → 95 : 5 → 9 : 1) to provide 3.8 g (93%) of the Boc protected indole as a colorless amorphous solid foam: mp 139–140 °C (colorless crystals from i-PrOH/H2O); 1H NMR (400 MHz, as a mixture of rotamers) δ 7.72 (dd, J = 8.4, 2.4 Hz, 1 H), 7.45 (d, J = 7.6 Hz, 0.5 H), 7.36-7.27 (comp, 4 H), 7.24-7.19 (comp, 2.5 H), 7.16-7.08 (comp, 4 H), 7.00-6.96 (comp, 2 H), 6.06-5.96 (m, 0.5 H), 5.93-5.84 (m, 0.5 H), 5.66 (d, J = 6.8 Hz, 0.5 H), 5.54 (d, J = 6.8 Hz, 0.5 H), 5.37 (dd, J = 17.2, 1.6 Hz, 0.5 H), 5.26-5.20 (m, 1 H), 5.13 (dd, J = 10.4, 1.2, 0.5 H), 4.77-4.72 (comp, 1.5 H), 4.67-4.49 (comp, 3.5 H), 4.45-4.41 (comp, 2 H), 4.35-4.32 (m, 1 H), 4.28-4.15 (comp, 2 H), 3.80 (dd, J = 14.4, 6.4 Hz, 0.5 H), 3.58 (dd, J = 14.4, 6.4 Hz, 0.5 H), 3.58 (d, J = 10.0 Hz, 1 H), 2.94-2.87 (comp, 1.5 H), 2.83-2.76 (m, 0.5 H), 2.69-2.57 (m, 1 H), 2.08-1.99 (m, 1 H), 1.91-1.65 (comp, 2 H), 1.50 (s, 9 H); 0.01 (s, 9 H); 13C NMR (100 MHz, as a mixture of rotamers) δ 155.4, 155.3, 151.0, 139.5, 138.9, 138.8, 138.2, 138.1, 136.3, 136.2, 133.31, 133.28, 127.2, 127.1, 126.9, 126.8, 126.7, 126.4, 126.3, 124.1, 121.74, 121.71, 121.3, 121.1, 118.0, 117.6, 117.4, 116.9, 114.1, 114.0, 83.7, 83.6, 78.0, 77.8, 77.2, 75.5, 75.4, 72.5, 72.4, 72.3, 72.0, 65.9, 65.8, 52.7, 52.6, 49.5, 47.0, 46.8, 41.1, 41.0, 39.8, 38.8, 31.5, 30.7, 28.0, 27.4, −0.08; IR (neat) 2952, 1739, 1696, 1456, 1316, 1250, 1112, 845, 737 cm−1; mass spectrum (ESI) m/z 739.3765 [C43H55N2O7Si (M+1) requires 739.3773].
2-Allyl 7-(tert-butyl) (1S/R,5S/R,12R/S)-6,6-bis((benzyloxy)methyl)-12-hydroxy-3,4,5,6-tetrahydro-1H-5,1-ethanoazocino[4,3-b]indole-2,7-dicarboxylate (35)
TBAF·3H2O (289 mg, 0.915 mmol) was added to a solution of the Boc protected indole (135 mg, 0.183 mmol) from the previous step in CH2Cl2 (1.8 mL), and the reaction was stirred for 2 h. The mixture was partitioned between saturated aqueous NaHCO3 (10 mL) and CH2Cl2 (10 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant yellow gum was purified by flash column chromatography eluting with hexanes/EtOAc (70 : 30) containing 1% Et3N to provide 118 mg (97%) of 35 as a colorless amorphous solid foam: mp 182–183 °C (colorless crystals from i-PrOH/H2O); 1H NMR (400 MHz, as a mixture of rotamers) δ 7.75 (dd, J = 8.4, 4.0 Hz, 1 H), 7.50 (d, J = 8.4 Hz, 0.5 H), 7.39 (d, J = 7.6 Hz, 0.5 H), 7.31-7.12 (comp, 12 H), 6.09-5.99 (m, 0.5 H), 5.95-5.87 (m, 0.5 H), 5.74 (d, J = 6.4 Hz, 0.5 H), 5.60 (d, J = 6.4 Hz, 0.5 H), 5.43-5.15 (comp, 2 H), 4.73-4.70 (m, 1 H), 4.63-4.25 (comp, 9 H), 4.12 (d, J = 9.2 Hz, 0.5 H), 4.05 (d, J = 9.2 Hz, 0.5 H), 3.88-3.66 (comp, 2 H), 3.09-2.95 (m, 1 H), 2.84 (br s, 1 H), 2.61-2.48 (m, 1 H), 2.14-2.05 (m, 1 H), 1.97-1.73 (comp, 2 H), 1.58 (s, 9 H); 13C NMR (100 MHz, as a mixture of rotamers) δ 155.2, 155.1, 151.4, 151.3, 137.8, 137.75, 137.66, 137.6, 137.4, 137.3, 136.4, 133.2, 133.1, 128.4, 128.2, 128.1, 128.0, 127.8, 127.5, 127.4, 127.3, 127.2, 126.8, 126.6, 124.5, 122.2, 122.1, 118.3, 117.9, 117.4, 117.1, 113.9, 84.2, 84.1, 77.2, 76.2, 75.4, 75.1, 74.8, 74.7, 73.54, 73.48, 72.9, 72.8, 66.1, 65.9, 53.7, 53.4, 49.5, 49.4, 47.1, 47.0, 40.7, 40.6, 39.2, 38.2, 31.4, 30.8, 28.0; IR (neat) 2941, 1739, 1693, 1456, 1316, 1112, 738 cm−1; mass spectrum (ESI) m/z 667.3374 [C40H47N2O7 (M+1) requires 667.3378].
tert-Butyl (1S/R,5S/R,12R/S)-6,6-bis((benzyloxy)methyl)-12-hydroxy-1,2,3,4,5,6-hexahydro-7H-5,1-ethanoazocino[4,3-b]indole-7-carboxylate
A solution of tetrakis-(triphenylphosphine)palladium(0) (10.4 mg, 0.009 mmol) in CH2Cl2 (0.1 mL) was added to a solution of 35 (118 mg, 0.177 mmol) and PhSiH3 (0.44 mL, 383 mg, 3.54 mmol) in CH2Cl2 (1.7 mL), and the reaction was stirred for 0.5 h at room temperature. The mixture was partitioned between saturated aqueous NaHCO3 (10 mL) and CH2Cl2 (10 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with EtOAc/MeOH (85 : 15) containing 1% Et3N to provide 75 mg (73%) of the secondary amine as a tan amorphous solid foam: 1H NMR (400 MHz) δ 7.76 (d, J = 8.0 Hz, 1 H), 7.46 (d, J = 7.2 Hz, 1 H), 7.30-7.20 (comp, 10 H), 7.17-7.14 (comp, 2 H), 4.64-4.60 (comp, 2 H), 4.56 (s, 2 H), 4.51-4.46 (comp, 3 H), 4.42 (d, J = 12.4 Hz, 1 H), 4.05 (d, J = 9.2 Hz, 1 H), 3.74 (d, J = 9.2 Hz, 1 H), 2.86-2.78 (comp, 2 H), 2.58 (dd, J = 12.8, 5.2 Hz, 1 H), 2.36 (ddd, J = 12.8, 12.8, 3.6 Hz, 1 H), 2.13 (dd, J = 13.2, 9.2 Hz, 1 H), 1.97-1.90 (m, 1 H), 1.80-1.71 (m, 1 H), 1.58 (s, 9 H); 13C NMR (100 MHz) δ 151.6, 138.0, 137.5, 137.3, 136.8, 128.3, 128.2, 127.9, 127.7, 127.2, 126.8, 124.1, 122.8, 121.8, 117.7, 114.0, 83.8, 77.2, 76.3, 75.6, 75.0, 73.5, 72.9, 53.4, 49.5, 47.5, 42.3, 41.7, 31.8, 28.0; IR (neat) 2931, 1736, 1456, 1316, 1135, 738 cm−1; mass spectrum (ESI) m/z 583.3170 [C36H43N2O5 (M+1) requires 583.3166].
tert-Butyl (1S/R,5S/R,12R/S)-6,6-bis((benzyloxy)methyl)-2-(2-chloroethyl)-12-hydr-oxy-1,2,3,4,5,6-hexahydro-7H-5,1-ethanoazocino[4,3-b]indole-7-carboxylate (36)
Sodium triacetoxyborohydride (15 mg, 0.068 mmol) was added to a solution of the secondary amine (20 mg, 0.034 mmol) from the previous step and chloroacetaldehyde (11 µL of a 50% by weight solution in H2O, 0.068 mmol) in 1,2-dichloroethane (0.4 mL) at 0 °C, and the reaction was stirred for 0.5 h at 0 °C. The mixture was partitioned between saturated aqueous NaHCO3 (3 mL) and CH2Cl2 (3 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 3 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (60 : 40) to provide 17 mg (77%) of 36 as a yellow gum: 1H NMR (400 MHz, CD3CN) δ 7.69 (dd, J = 8.0, 0.4 Hz, 1 H), 7.48 (d, J = 8.0 Hz, 1 H), 7.28-7.12 (comp, 12 H), 4.48-4.28 (comp, 8 H), 4.20 (d, J = 9.6 Hz, 1 H), 3.69 (d, J = 6.4 Hz, 1 H), 3.64-3.59 (comp, 2 H), 3.10-3.04 (m, 1 H), 2.79-2.72 (m, 1 H), 2.70-2.62 (comp, 2 H), 2.38-2.23 (comp, 2 H), 2.15 (br s, 1 H), 1.89-1.85 (m, 1 H), 1.79-1.71 (m, 1 H), 1.56 (s, 9 H); 13C NMR (400 MHz, CD3CN) δ 152.6, 139.3, 138.4, 137.3, 129.3, 129.2, 129.0, 128.7, 128.5, 128.2, 128.1, 124.9, 122.6, 122.4, 119.5, 114.6, 85.3, 77.9, 75.6, 73.6, 73.5, 60.0, 54.4, 54.3, 50.2, 49.4, 44.4, 44.3, 42.0, 32.2, 31.7, 28.2; IR (neat) 3450, 2934, 1737, 1456, 1315, 1142, 738 cm−1; mass spectrum (ESI) m/z 645.3081 [C38H46 35ClN2O5 (M+1) requires 645.3090].
tert-Butyl (1S/R,5S/R)-6,6-bis((benzyloxy)methyl)-2-(2-chloroethyl)-12-oxo-1,2,3,4,5,6-hexahydro-7H-1,5-ethanoazocino[4,3-b]indole-7-carboxylate (37)
Anhydrous DMSO (56 µL, 62 mg, 0.792 mmol) was added to a solution of oxalyl chloride (35 µL, 50 mg, 0.397 mmol) in CH2Cl2 (1.1 mL) at –78 °C. The reaction was stirred for 10 min at –78 °C, whereupon a solution of 36 (128 mg, 0.198 mmol) in CH2Cl2 (1.1 mL) was added, and the reaction was stirred an additional 15 min at –78 °C. Et3N (220 µL, 160 mg, 1.58 mmol) was added, and the reaction was stirred for 15 min at –78 °C. The bath was removed, and the reaction was stirred for 1 h at room temperature. The mixture was partitioned between H2O (15 mL) and CH2Cl2 (15 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 15 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (8 : 2) to provide 109 mg (86%) of 37 as a yellow amorphous solid foam: 1H NMR (400 MHz, CD2Cl2) δ 7.78 (d, J = 8.4 Hz, 1 H), 7.53 (d, J = 7.6 Hz, 1 H), 7.35-7.20 (comp, 12 H), 4.56-4.52 (comp, 2 H), 4.46-4.43 (comp, 2 H), 4.40 (d, J = 11.2 Hz, 1 H), 4.30 (d, J = 11.2 Hz, 1 H), 4.03 (d, J = 9.2 Hz, 1 H), 3.83-3.78 (comp, 2 H), 3.63-3.59 (comp, 2 H), 3.24 (dd, J = 5.6, 3.2 Hz, 1 H), 3.13 (ddd, J = 13.2, 6.8, 6.8 Hz, 1 H), 3.06-3.94 (comp, 2 H), 2.81-2.74 (m, 1 H), 2.66 (ddd, J = 12.8, 12.8, 2.8 Hz, 1 H), 2.51-2.45 (m, 1 H), 2.02-1.96 (m, 1 H), 1.84-1.76 (m, 1 H), 1.66 (s, 9 H); 13C NMR (100 MHz, CD2Cl2) δ 212.2, 151.9, 138.6, 138.3, 137.0, 136.9, 129.0, 128.7, 128.5, 128.3, 127.9, 127.8, 127.7, 124.5, 122.4, 118.9, 118.8, 114.4, 84.9, 76.2, 73.7, 73.6, 73.2, 63.0, 60.0, 53.0, 49.2, 49.0, 48.2, 43.3, 28.2, 28.0; IR (neat) 2978, 2934, 2859, 1739, 1699, 1455, 1312, 1145, 1097, 740, 699 cm−1; mass spectrum (ESI) m/z 643.2931 [C38H44 35ClN2O5 (M+1) requires 643.2933].
tert-Butyl (1R/S,4R/S,7S/R,13cS)-8,8-bis((benzyloxy)-methyl)-14-oxo-1,2,3,5,6,7,8,13c-octahydro-9H-1,7-methanopyrrolo[1',2':1,2]azocino[4,3-b]indole-9-carboxylate (38) Method A
Sodium tert-butoxide (81 mg, 0.847 mmol) was added to a solution of 37 (109 mg, 0.169 mmol) in THF/t-BuOH (4.2 mL/1.4 mL) at 0 °C. The bath was removed, and the reaction was stirred for 1 h at room temperature. The mixture was partitioned between saturated aqueous NH4Cl (15 mL) and Et2O (15 mL), and the layers were separated. The aqueous layer was extracted with Et2O (2 × 15 mL), and the combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with CH2Cl2/MeOH (95 : 5) to provide 96 mg (94%) of 38 as a pale yellow amorphous solid foam: 1H NMR (400 MHz) δ 7.82 (d, J = 7.2 Hz, 1 H), 7.77 (dd, J = 7.6, 1.2 Hz, 1 H), 7.37-7.22 (comp, 10 H), 7.18-7.16 (comp, 2 H), 4.73 (d, J = 4.8 Hz, 1 H), 4.53 (d, J = 12.4 Hz, 1 H), 4.45- 4.39 (comp, 2 H), 4.37-4.32 (comp, 2 H), 4.14 (d, J = 9.6 Hz, 1 H), 3.95 (d, J = 5.2 Hz, 1 H), 3.78 (d, J = 9.6 Hz, 1 H), 3.67-3.60 (m, 1 H), 3.29-3.28 (m, 1 H), 3.23 (dd, J = 7.2, 5.2 Hz, 1 H), 3.19-3.11 (comp, 2 H), 2.67 (dd, J = 14.4, 3.2 Hz, 1 H), 2.45-2.29 (comp, 2 H), 2.01-1.93 (m, 1 H), 1.88-1.80 (m, 1 H), 1.64 (s, 9 H); 13C NMR (100 MHz) δ 214.2, 151.0, 137.8, 137.7, 137.2, 136.2, 128.4, 128.1, 127.9, 127.6, 127.4, 127.1, 127.0, 124.4, 122.3, 118.6, 115.0, 113.7, 84.3, 74.4, 73.0, 72.6, 72.5, 61.6, 58.0, 54.3, 49.2, 49.1, 47.5, 32.3, 27.8, 23.8; IR (neat) 2929, 2873, 1740, 1695, 1314, 1147, 742 cm−1; mass spectrum (ESI) m/z 607.3169 [C38H43N2O5 (M+1) requires 607.3166].
(1R/S,4R/S,7S/R,8R/S,13cS/R,16S/R)-8-(Hydroxymethyl)-2,3,5,6,7,8,9,13c-octahydro-1H-1,7,8-(epimethanetriyloxy-methano) pyrrolo[1',2':1,2]azocino[4,3-b]indol-16-ol hydrochloride (2)
A solution of 38 (1.0 g, 1.65 mmol) in a mixture of MeOH (23 mL) and 5 M HCl(aq) (23 mL) was stirred at 65 °C for 1 h. The reaction was cooled to room temperature, palladium on carbon (250 mg of 10% loading) was added, and the reaction was stirred under a balloon of H2 for 4 h. The reaction was filtered through a plug of cotton and concentrated under reduced pressure. The resultant yellow residue was purified by reverse phase chromatography eluting with MeOH/H2O (0 : 100 → 20 : 80 over 15 mins) containing 0.1% TFA to give 513 mg (86%) of 2 as a pale yellow amorphous solid foam: 1H NMR (600 MHz, CD3OD) δ 7.61 (d, J = 8.0 Hz, 1 H), 7.44 (d, J = 8.0 Hz, 1 H), 7.16 (ddd, J = 8.0, 7.0, 1.0 Hz, 1 H), 7.11 (ddd, J = 8.0, 7.0, 1.0 Hz, 1 H), 5.45 (d, J = 7.4 Hz, 1 H), 4.17 (d, J = 11.3 Hz, 1 H), 4.13 (d, J = 11.3 Hz, 1 H), 3.97 (d, J = 7.8 Hz, 1 H), 3.70 (ddd, J = 12.4, 12.4, 8.0 Hz, 1 H), 3.60-3.55 (m, 1 H), 3.56 (d, J = 7.8 Hz, 1 H), 3.21-3.14 (comp, 2 H), 2.95 (ddd, J = 13.7, 3.2, 3.2 Hz, 1 H), 2.74 (dddd, J = 12.8, 10.3, 8.0, 2.4 Hz, 1 H), 2.64-2.63 (m, 1 H), 2.53-2.46 (m, 1 H), 2.40-2.33 (m, 1 H), 2.23 (dddd, J = 12.2, 9.0, 3.0, 3.0 Hz, 1 H); 13C NMR (150 MHz, CD3OD) δ 143.1, 137.2, 128.4, 123.3, 121.1, 118.0, 112.5, 108.4, 102.5, 75.3, 64.3, 62.8, 55.3, 53.4, 53.1, 50.5, 47.3, 26.2, 19.7; IR (neat) 3395, 3285, 1441, 1233, 1086, 997, 738, 721 cm−1; mass spectrum (ESI) m/z 327.1704 [C19H23N2O3 (M+1) requires 327.1703].
(±)-Actinophyllic acid hydrochloride (1)
A solution of IBX (115 mg, 0.41 mmol) and alcohol 2 (30 mg, 0.08 mmol) in DMSO (0.33 mL) was stirred at 50 °C for 2 hours. N-hydroxysuccinimide (67 mg, 0.58 mmol) was added, and the reaction was stirred at 50 °C for 2 h. The reaction was cooled to room temperature, whereupon a solution of 1 M NaOH was added until the reaction was basic (~pH 10) according to pH paper. The reaction mixture was dry-loaded onto SiO2 and purified by flash column chromatography eluting with CHCl3/MeOH/NH4OH (6 : 3 : 0.5), followed by reverse phase chromatography eluting with MeOH/H2O (0 : 100 → 20 : 80 over 15 mins) to give 9.8 mg (31%) of 1 as a colorless amorphous solid: 1H NMR (600 MHz, D2O, internal reference to MeOH at 3.34 ppm) δ 7.64 (d, J = 8.0 Hz, 1 H), 7.54 (d, J = 8.1 Hz, 1 H), 7.33-7.30 (m, 1 H), 7.25-7.22 (m, 1 H), 5.52 (d, J = 7.5 Hz, 1 H), 4.40 (d, J = 8.2 Hz, 1 H), 3.79 (d, J = 8.2 Hz, 1 H), 3.80-3.74 (m, 1 H), 3.64-3.60 (m, 1 H), 3.30-3.25 (comp, 2 H), 3.09-3.05 (comp, 2 H), 2.70-2.63 (m, 1 H), 2.61-2.54 (m, 1 H), 2.45-2.40 (m, 1 H), 2.37-2.33 (m, 1 H); 13C NMR (150 MHz, D2O, internal reference to MeOH at 49.5 ppm) δ 173.8, 138.5, 136.1, 127.2, 123.7, 121.2, 117.8, 112.4, 107.7, 102.3, 74.6, 61.2, 58.3, 54.4, 51.8, 50.2, 46.2, 25.3, 18.7; IR (neat) 3321, 1589, 1460, 1372, 1108, 1035, 738 cm−1; mass spectrum (ESI) m/z 341.1497 [C19H21N2O4 (M+1) requires 341.1496].
2-Allyl 7-(tert-butyl) (1S/R,5S/R)-6,6-bis((benzyloxy)methyl)-12-oxo-3,4,5,6-tetrahydro-1H-1,5-ethanoazocino[4,3-b]indole-2,7-dicarboxylate (40)
4-Dimethylaminopyridine (1.5 g, 12.3 mmol) was added to a solution of 26 (4.6 g, 8.2 mmol) in a mixture of di-tert-butyl dicarbonate (23 mL, 22 g, 102.5 mmol) and toluene (18 mL), and the reaction was stirred for 3 h at room temperature. The reaction was partitioned between H2O (100 mL) and Et2O (100 mL). The layers were separated, and the aqueous layer was extracted with Et2O (2 × 100 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with hexanes/EtOAc (8 : 2) to give 4.6 g (83%) of 40 as a yellow amorphous solid: 1H NMR (400 MHz, as a mixture of rotamers) δ 7.80 (d, J = 8.0 Hz, 1 H), 7.58 (d, J = 7.6 Hz, 0.5 H), 7.43 (d, J = 8.4 Hz, 0.5 H), 7.34-7.15 (comp, 12 H), 6.09-5.76 (comp, 2 H), 5.44-5.18 (comp, 2 H), 4.75-4.34 (comp, 7 H), 4.06-3.93 (comp, 1.5 H), 3.83-3.71 (comp, 2.5 H), 3.33-3.28 (m, 1 H), 3.17-3.03 (comp, 2 H), 2.95-2.78 (m, 1 H), 2.07-1.95 (m, 1 H), 1.90-1.75 (m, 1 H), 1.64 (s, 9 H); 13C NMR (100 MHz, as a mixture of rotamers) δ 211.7, 211.5, 154.8, 154.5, 151.1, 137.7, 137.5, 137.3, 137.2, 136.3, 132.9, 132.8, 128.2, 128.0, 127.9, 127.6, 127.5, 127.4, 127.1, 127.0, 124.6, 122.3, 119.4, 119.2, 118.3, 117.7, 117.3, 114.1, 114.0, 84.4, 84.3, 75.5, 75.2, 73.2, 73.1, 73.0, 72.6, 66.3, 66.1, 63.3, 63.1, 48.8, 47.5, 46.7, 45.0, 39.7, 28.8, 28.3, 27.9; IR (neat) 2979, 2936, 2862, 1784, 1698, 1456, 1306, 1145, 1111, 767, 741, 700 cm−1; mass spectrum (ESI) m/z 665.3215 [C40H45N2O7 (M+1) requires 665.3221].
tert-Butyl (1S/R,5S/S)-6,6-bis((benzyloxy)methyl)-12-oxo-1,2,3,4,5,6-hexahydro-7H-1,5-ethanoazocino[4,3-b]indole-7-carboxylate (41)
A solution of Pd2(dba)3 (8.5 mg, 0.009 mmol) and 1,4-bis(diphenylphosphino)butane (dppb) (3.9 mg, 0.009 mmol) in THF (3.1 mL) was stirred for 5 min at room temperature, and the resultant solution was transferred to a solution of 40 (614 mg, 0.92 mmol) and N,N-dimethylbarbituric acid (NMDBA) (1.44 g, 9.24 mmol) in THF (6.2 mL). The reaction was stirred for 1 h at room temperature, whereupon Et2O (20 mL) and saturated aqueous NaHCO3 (30 mL) were slowly added. The layers were separated, and the aqueous layer was extracted with Et2O (2 × 15 mL). The combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with EtOAc containing 1% Et3N to provide 495 mg (92%) of 41 as a pale yellow amorphous solid: 1H NMR (400 MHz) δ 7.82 (d, J = 8.8 Hz, 1 H), 7.49 (d, J = 7.6 Hz, 1 H), 7.35-7.20 (comp, 12 H), 4.67-4.65 (m, 1 H), 4.60 (d, J = 8.8 Hz, 1 H), 4.53 (d, J = 12.0 Hz, 1 H), 4.43 (d, J = 11.6 Hz, 1 H), 4.35 (d, J = 12.0 Hz, 1 H), 3.97 (d, J = 8.8 Hz, 1 H), 3.85- 3.81 (comp, 2 H), 3.37-3.34 (m, 1 H), 3.18 (dd, J = 2.4, 18.0 Hz, 1 H), 2.97 (dd, J = 4.4, 18.0 Hz, 1 H), 2.72-2.65 (comp, 2 H), 2.07-1.97 (comp, 2 H), 1.80-1.73 (m, 1 H), 1.64 (s, 9 H); 13C NMR (100 MHz) δ 213.2, 151.7, 138.4, 138.0, 137.1, 136.9, 128.6, 128.4, 128.2, 128.1, 127.8, 127.6, 127.4, 124.6, 122.4, 120.6, 117.9, 114.5, 84.5, 75.9, 73.5, 73.4, 73.0, 62.9, 50.3, 49.2, 46.0, 41.4, 29.0, 28.3; IR (neat) 2920, 2857, 1737, 1698, 1456, 1311, 1143, 1098, 740, 699 cm−1; mass spectrum (ESI) m/z 581.3012 [C36H41N2O5 (M+1) requires 581.3010].
tert-Butyl (1R/S,4R/S,7S/R,13cS)-8,8-bis((benzyloxy)methyl)-14-oxo-1,2,3,5,6,7,8,13c-octahydro- 9H-1,7-methanopyrrolo[1',2':1,2]azocino[4,3-b]indole-9-carboxylate (38) Method B
Sodium triacetoxyborohydride (0.69 g, 3.27 mmol) was added to a mixture of 41 (0.95 g, 1.64 mmol) and chloroacetaldehyde (0.51 mL of a 50% by weight solution in H2O, 3.27 mmol) in 1,2-dichloroethane (18 mL) at 0 °C, and the reaction was stirred for 15 min at 0 °C. The reaction was partitioned between saturated aqueous NaHCO3 (20 mL) and CH2Cl2 (10 mL), and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 20 mL), and the combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure to give 1.16 g of yellow amorphous solid. The crude material was dissolved in a mixture of THF (38 mL) and t-BuOH (11.7 mL) and cooled to 0 °C. Sodium tert-butoxide (0.79 g, 8.20 mmol) was added at 0 °C, the bath was removed, and the reaction was stirred for 1.5 h at room temperature. The reaction was partitioned between saturated aqueous NH4Cl (30 mL) and Et2O (30 mL), and the layers were separated. The aqueous layer was extracted with Et2O (2 × 30 mL), and the combined organic layers were dried (MgSO4), filtered, and concentrated under reduced pressure. The resultant yellow oil was purified by flash column chromatography eluting with CH2Cl2/MeOH (95 : 5) to provide 820 mg (83% over the two steps) of 38 as a pale yellow amorphous solid. The spectra for 38 prepared via this procedure were identical to that prepared via the procedure listed as Method A above.
Supplementary Material
Scheme 2.

Preparation of starting materials
Acknowledgments
We thank the National Institutes of Health (GM 25439) and the Robert A. Welch Foundation (F-652) for their generous support of this research. B.A.G thanks the Cancer Prevention Research Institute of Texas for a training fellowship and for their support of this research. We are grateful to Dr. Vincent Lynch (The University of Texas at Austin) for performing X-ray crystallography, Dr. Chi-Li Chen for helpful discussions, and Prof. Larry E. Overman (University of California, Irvine) for supplying an authentic sample of 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beckman MG, Hooper WC, Critchley SE, Ortel TL. Am. J. Prev. Med. 2010;38:S495–S501. doi: 10.1016/j.amepre.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 2.Willemse JL, Hendriks DF. Clin. Chem. 2006;52:30–36. doi: 10.1373/clinchem.2005.055814. [DOI] [PubMed] [Google Scholar]
- 3.Dubis J, Witkiewicz W. Adv. Clin. Exp. Med. 2010;19:379–387. [Google Scholar]
- 4.(a) Barrow JC, Nantermet PG, Stauffer SR, Ngo PL, Steinbeiser MA, Mao SS, Carroll SS, Bailey C, Colussi D, Bosserman M, Burlein C, Cook JJ, Stiko G, Tiller PR, Miller-Stein CM, Rose M, McMasters DR, Vacca JP, Selnick HG. J. Med. Chem. 2003;46:5294–5297. doi: 10.1021/jm034141y. [DOI] [PubMed] [Google Scholar]; (b) Nantermet PG, Barrow JC, Lindsley SR, Young M, Mao SS, Carroll S, Bailey C, Bosserman M, Colussi D, McMasters DR, Vacca JP, Selnick HG. Bioorg. Med. Chem. Lett. 2004;14:2141–2145. doi: 10.1016/j.bmcl.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 5.Bunnage ME, Blagg J, Steele J, Owen DR, Allerton C, McElroy AB, Miller D, Ringer T, Butcher K, Beaumont K, Evans K, Gray AJ, Holland SJ, Feeder N, Moore RS, Brown DG. J. Med. Chem. 2007;50:6095–6103. doi: 10.1021/jm0702433. [DOI] [PubMed] [Google Scholar]
- 6.Islam I, Bryant J, May K, Mohan R, Yuan S, Kent L, Morser J, Zhao L, Vergona R, White K, Adler M, Whitlow M, Buckman BO. Bioorg. Med. Chem. Lett. 2007;17:1349–1354. doi: 10.1016/j.bmcl.2006.11.078. [DOI] [PubMed] [Google Scholar]
- 7.Bunnage ME, Owen DR. Curr. Opin. Drug Disc. Dev. 2008;11:480–486. [PubMed] [Google Scholar]
- 8.Willemse JL, Heylen E, Nesheim ME, Hendriks DF. J. Thromb. Haemost. 2009;7:1962–1971. doi: 10.1111/j.1538-7836.2009.03596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carroll AR, Hyde E, Smith J, Quinn RJ, Guymer G, Forster PI. J. Org. Chem. 2005;70:1096. doi: 10.1021/jo048439n. [DOI] [PubMed] [Google Scholar]
- 10.(a) Martin CL, Overman LE, Rohde JM. J. Am. Chem. Soc. 2008;130:7568–7569. doi: 10.1021/ja803158y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin CL, Overman LE, Rohde JM. J. Am. Chem. Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniguchi T, Martin CL, Monde K, Nakanishi K, Berova N, Overman LE. J. Nat. Prod. 2009;39:806–810. doi: 10.1021/np800665s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaswani RG, Day JJ, Wood JL. Org. Lett. 2009;11:4532–4535. doi: 10.1021/ol901746c. [DOI] [PubMed] [Google Scholar]
- 13.Zaimoku H, Taniguchi T, Ishibashi H. Org. Lett. 2012;14:1656–1658. doi: 10.1021/ol300280s. [DOI] [PubMed] [Google Scholar]
- 14.Galicia IZ, Maldonado LA. Tetrahedron Lett. 2013;54:2180–2182. [Google Scholar]
- 15.Mortimer D, Whiting M, Harrity JPA, Jones S, Coldham I. Tetrahedron Lett. 2014;55:1255–1257. [Google Scholar]
- 16.For reviews of the vinylogous Mannich reaction, see: Martin SF. Acc. Chem. Res. 2002;35:895–904. doi: 10.1021/ar950230w. Casiraghi G, Battistini L, Curti C, Rassu G, Zanardi F. Chem. Rev. 2011;111:3076–3154. doi: 10.1021/cr100304n.
- 17.For a review of the Pictet-Spengler reaction, see: Stöckigt J, Antonchick AP, Wu F, Waldmann H. Angew. Chem. Int. Ed. 2011;50:8538–8564. doi: 10.1002/anie.201008071.
- 18.(a) Heidebrecht RW, Jr, Gulledge B, Martin SF. Org. Lett. 2010;12:2492–2495. doi: 10.1021/ol1006373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu T–H, McElroy WT, Shamszad M, Heidebrecht RW, Jr, Gulledge B, Martin SF. Org. Lett. 2012;14:3834–3837. doi: 10.1021/ol301424h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fu T–H, McElroy WT, Shamszad M, Heidebrecht RW, Jr, Gulledge B, Martin SF. Tetrahedron. 2013;69:5588–5603. doi: 10.1016/j.tet.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu T–H, Bonaparte A, Martin SF. Tetrahedron Lett. 2009;50:3253–3257. doi: 10.1016/j.tetlet.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granger BA, Jewett IT, Butler JD, Hua B, Knezevic CE, Parkinson EI, Hergenrother PJ, Martin SF. J. Am. Chem. Soc. 2013;135:12984–12986. doi: 10.1021/ja4070206. [DOI] [PubMed] [Google Scholar]
- 21.Choi M–H, Kim H–D. Arch. Pharm. Res. 2003;26:990–996. doi: 10.1007/BF02994747. [DOI] [PubMed] [Google Scholar]
- 22.Katritzky AR, Akutagawa K. Tetrahedron Lett. 1985;26:5935–5938. [Google Scholar]
- 23.(a) Buhr G. 1971 DE2013761, [Google Scholar]; (b) Shizuka H, Ogiwara T, Morita T. Bull. Chem. Soc. Jpn. 1977;50:2067–2073. [Google Scholar]
- 24.For a review on gramine chemistry, see: Semenov BB, Granik VG. Pharm. Chem. J. 2004;38:287–310.
- 25.Minami I, Ohashi Y, Shimizu I, Tsuji J. Tetrahedron Lett. 1985;26:2449–2452. [Google Scholar]
- 26.Martin CL, Nakamura S, Otte R, Overman LE. Org. Lett. 2011;13:138. doi: 10.1021/ol102709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reactions of 1,2-aminochloroethanes with water are known to proceed through an aziridinium ion intermediate: Kerwin JF, Ullyot GE, Fuson RC, Zirkle CL. J. Am. Chem. Soc. 1974;69:2961–2965.
- 28.For a review on the oxidation of primary alcohols to carboxylic acids, see: Tojo G, Fernández M. Oxidation of Primary Alcohols to Carboxylic Acids: A Guide to Current Common Practices. New York: Springer; 2007.
- 29.For attempted procedures, see: Jefford CW, Wang Y. J. Chem. Soc. Chem. Commun. 1988:634–635. Zhao M, Li J, Song Z, Desmond R, Tschaen DM, Grabowski EJJ, Reider PJ. Tetrahedron Lett. 1998;39:5323–5326. Epp JB, Widlanski TS. J. Org. Chem. 1999;64:293–295. doi: 10.1021/jo981316g. Ravi G, Lee K, Ji X–D, Kim HS, Soltysiak KA, Marquez VE, Jacobson KA. Bioorg. Med. Chem. Lett. 2001;11:2295–2300. doi: 10.1016/s0960-894x(01)00450-4. Thottumkara AP, Bowsher MS, Vinod TK. Org. Lett. 2005;7:2933–2936. doi: 10.1021/ol050875o. Grill JM, Ogle JW, Miller SA. J. Org. Chem. 2006;71:9291–9296. doi: 10.1021/jo0612574. An G, Anh H, de Castro KA, Rhee H. Synthesis. 2010:477–485. Das R, Chakraborty D. Appl. Organometal. Chem. 2011;25:437–442.
- 30.Schmidt A-KC, Stark CBW. Org. Lett. 2011;13:4164–4167. doi: 10.1021/ol2014335. [DOI] [PubMed] [Google Scholar]
- 31.Corey EJ, Schmidt G. Tetrahedron Lett. 1979;5:399–402. [Google Scholar]
- 32.Mazitschek R, Mülbaier M, Giannis A. Angew. Chem. Int. Ed. 2002;41:4059–4061. doi: 10.1002/1521-3773(20021104)41:21<4059::AID-ANIE4059>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 33.Kunz H, März J. Angew. Chem. Int. Ed. 1988;127:1375–1377. [Google Scholar]
- 34.For some lead references on diverted total synthesis, see: Gaul C, Njardarson JT, Shan D, Dorn DC, Wu K–D, Tong WP, Huang X–Y, Moore MAS, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:11326–11337. doi: 10.1021/ja048779q. Wilson RM, Danishefsky SJ. J. Org. Chem. 2006;71:8329–8351. doi: 10.1021/jo0610053. Szpilman AM, Carreira EM. Angew. Chem. Int. Ed. 2010;49:9592–9628. doi: 10.1002/anie.200904761.
- 35.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 36.Still WC, Kahn M, Mitra A. J. Org. Chem. 1978;43:2923–2925. [Google Scholar]
- 37.CCDC 990247
- 38.CCDC 990248
- 39.CCDC 990249
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.