

Abstract
SQSTM1 mutations are common in patients with Paget disease of bone (PDB), with most affecting the C-terminal ubiquitin-associated (UBA) domain of the SQSTM1 protein. We performed structural and functional analyses of two UBA domain mutations, an I424S mutation relatively common in UK PDB patients, and an A427D mutation associated with a severe phenotype in Southern Italian patients. Both impaired SQSTM1's ubiquitin-binding function in pull-down assays and resulted in activation of basal NF-κB signalling, compared to wild-type, in reporter assays. We found evidence for a relationship between the ability of different UBA domain mutants to activate NF-κB signalling in vitro and number of affected sites in vivo in 1152 PDB patients from the UK and Italy, with A427D-SQSTM1 producing the greatest level of activation (relative to wild-type) of all PDB mutants tested to date. NMR and isothermal titration calorimetry studies were able to demonstrate that I424S is associated with global structural changes in the UBA domain, resulting in 10-fold weaker UBA dimer stability than wild-type and reduced ubiquitin-binding affinity of the UBA monomer. Our observations provide insights into the role of SQSTM1-mediated NF-κB signalling in PDB aetiology, and demonstrate that different mutations in close proximity within loop 2/helix 3 of the SQSTM1 UBA domain exert distinct effects on protein structure and stability, including indirect effects at the UBA/ubiquitin-binding interface.
Keywords: Sequestosome 1, SQSTM1, p62, Paget disease of bone, NF-κB, Ubiquitin
Highlights
-
•
The I424S and A427D SQSTM1 mutations affect ubiquitin-binding and NF-κB signalling.
-
•
Mutant SQSTM1's ability to activate NF-κB signalling may be related to disease extent in PDB.
-
•
A427D-SQSTM1 produces the greatest activation (relative to wild-type) of all PDB mutants.
-
•
The I424S mutant destabilises the UBA dimer causing unfolding of the monomer.
-
•
UBA domain mutations of SQSTM1 exert distinct effects on protein structure and stability.
1. Introduction
Paget disease of the bone (PDB; OMIM 60208) is a chronic disorder in which increased osteoclast activity, accompanied by secondary increases in osteoblast activity, leads to focal increases in bone turnover at different sites throughout the skeleton [1]. There is a wide variation in disease severity within PDB patients; whilst some remain asymptomatic, others present with a range of symptoms including bone pain, susceptibility to fracture and deformity. PDB aetiology is thought to involve a complex interplay between genetic and environmental factors. The most important genetic link to PDB identified so far came from positional cloning studies of patients with familial PDB, where mutations affecting the C-terminal ubiquitin-associated (UBA) domain of the SQSTM1 protein were identified [2,3]. Numerous truncating and missense SQSTM1 mutations have now been discovered [2–16] and are PDB specific; patients with mutations in SQSTM1 are on average diagnosed with PDB 5 years earlier than patients without [15]. Further, the skeletal phenotype of a mouse model of PDB carrying a P394L missense mutation, equivalent to the most common PDB-associated P392L human mutation, supports the causal role of SQSTM1 mutations in PDB aetiology [17]. However, recent genome-wide association studies (GWAS) have identified variants close to or within other genes (CSF1, OPTN, PML, RIN3, NUP205, TM7SF4, and TNFRSF11A) which also significantly act as risk factors for PDB [18,19] and disease severity [20].
The SQSTM1 protein is involved in several distinct cellular pathways but of particular relevance to PDB is its role in receptor activator of nuclear factor κB (RANK)-NF-κB signalling, which is important for osteoclast differentiation and function [21]. A number of studies indicate that UBA domain mutations of SQSTM1 impair its ability to bind to ubiquitin and result in increased NF-κB activity [7,11,22,23] and we have provided structural insights into some of these effects [24]. In particular we noted that different PDB mutations impact on ubiquitin recognition by the UBA domain via distinct mechanisms that either affect UBA domain structural integrity and/or UBA dimer stability, or perturb key contacts at the UBA–ubiquitin interface [24]. In addition we and others [7,22] noted that changes in SQSTM1's ability to regulate NF-κB signalling may be related to disease severity in PDB patients. Notably, truncating mutants of SQSTM1, which earlier genotype–phenotype analyses suggested to be associated with more severe PDB than common missense mutants [6,7,25] were found to be associated with a greater activation of basal NF-κB activity in reporter assays than the missense mutants analysed [22]. Further support for such a relationship came from observations that a non-synonymous variant (T575C, V192A) of RANK (encoded by TNFRSF11A) associated with PDB severity in Italian patients, in reporter assays synergises with wild-type and PDB-mutant SQSTM1 to produce a level of activation of NF-κB signalling greater than with RANK-V192 [26]. Whilst a recent study confirms that risk alleles previously found to predispose to PDB by GWAS are associated (in an additive manner with SQSTM1 mutations) with disease extent and severity in several populations, notably SQSTM1 mutation status alone plays a major role in determining the disease phenotype in PDB patients [20].
Here, we present structural and functional analyses of the PDB-associated I424S and A427D UBA domain mutants of SQSTM1 [14,15]. Both mutations were recently identified in independent studies and are localised close to the site of the PDB-associated G425R missense mutation, which maps to a solvent exposed site in loop 2 of the three-helix bundle UBA domain, and which we have previously characterised in structural detail [24]. The UBA domain of SQSTM1 forms a highly stable dimer involving residues largely along helix 2, but also at the C-terminus of helix 3 [27,28]. The latter also forms part of the ubiquitin-binding surface such that dimerisation of the UBA partially occludes the ubiquitin-binding surface making UBA dimerisation and ubiquitin-binding mutually exclusive processes [27]. Of interest, we previously reported that the G425R mutation exerts only local effects on UBA domain tertiary structure but is associated with an increase in dimer stability which may in part rationalise the inhibitory effects of the mutation observed in ubiquitin-binding assays [29].
The I424S mutation, which is immediately adjacent to the G425 site, but is buried in the hydrophobic core of the protein, was identified in the PRISM study from UK patients who consented to provide DNA samples for analysis. The mutation was identified in a randomised trial comparing the effects of symptomatic treatment with intensive bisphosphonate therapy in a cohort of 1324 patients with PDB [15,30]. Within this cohort, 80 PDB patients were found to carry SQSTM1 mutations, 5 of which carried the I424S substitution alone and one with an I424S/G425R double mutation. I424S therefore constitutes a relatively common PDB-associated SQSTM1 mutation in the UK, occurring in 7.5% of the patients in the PRISM study with SQSTM1 mutations. The A427D mutation was identified in two individuals of a southern Italian family [14]. This mutation is located in close proximity to I424/G425, within helix 3 of the UBA domain, and notably is associated with a high number of affected sites, with both reported cases presenting as polyostotic PDB with 7.00 ± 2.8 affected sites compared to 3.60 ± 2.6 sites in SQSTM1 mutation carriers collectively within the cohort [14]. Although located in close proximity in the primary and tertiary structure of the SQSTM1 UBA domain within the loop 2/helix 3 region, we show that the three PDB-associated mutations I424S, G425R and A427D, exert very different effects on protein structure and function.
2. Materials and methods
2.1. Plasmids
The plasmids for expression of the full-length wild-type and G425R mutant SQSTM1 protein (residues 1–440) and UBA domain only (residues 387–436) as GST fusion proteins (pGEX-4T-1, GE Healthcare) in Escherichia coli were described previously [22,29,31]. The I424S and A427D mutants were produced from the wild-type plasmids by site-directed mutagenesis (QuikChange kit; Stratagene) and subsequently verified by DNA sequencing. Mammalian expression vectors (pcDNA3.1 construct; Invitrogen) for wild-type and selected PDB-mutant SQSTM1 proteins (His-FLAG tagged) were generated previously [22]. The I424S and A427D mutations (as well as other SQSTM1 mutations) were again introduced into the wild-type SQSTM1 construct by site-directed mutagenesis and confirmed by DNA sequencing.
2.2. Ubiquitin pull-down assays
Ubiquitin pull-down assays of GST-SQSTM1 proteins were performed as described previously [31]. Briefly, the proteins were expressed in 10 ml cultures of E. coli and cells were lysed by sonicating in 1 ml TBS-T buffer (10 mM Tris, 150 mM NaCl, 0.1% v/v Triton X-100, pH 7.5). The lysed cells were centrifuged at 13,000 rpm for 10 min at room temperature and the cleared supernatants were diluted 1:10 in TBS-T buffer. 1 ml of each diluted lysate was incubated at 37 °C with either excess glutathione-Sepharose (GE Healthcare), ubiquitin-Sepharose (10 mg/ml bovine ubiquitin immobilised on CNBr-activated Sepharose 4B) or control-Sepharose (CNBr-activated Sepharose prepared without ubiquitin). The unbound proteins were then removed and the beads washed with 3 × 1 ml TBS-T buffer at 37 °C. Bound proteins were eluted from the beads with 50 μl of SDS PAGE loading buffer. Bound proteins were revealed by western blotting with the mouse anti-SQSTM1 lck ligand antibody (BD Biosciences Pharmingen). Each assay was repeated on a minimum of 4 independent occasions.
2.3. Cell transfection and NF-κB reporter assays
The same protocol as described previously using a NF-κB reporter that contained the − 415/− 93 fragment of the human IL8 promoter cloned into the KpnI and HindIII restriction sites of the pGL4.10 basic plasmid (Promega) was followed [22]. Briefly, HEK293T cells were cultured in 24-well plates. 24 h after the cells were seeded they were co-transfected with 0.2 ng/well of control Renilla luciferase plasmid (pRL0CMV; Promega), 100 ng/well of NF-κB Firefly luciferase reporter plasmid and 700 ng/well of His-FLAG-SQSTM1 construct (either wild-type or PDB-mutant). 2 μl of the transfection reagent Lipofectamine 2000 (Invitrogen) and 50 μl of Opti-MEM medium (Invitrogen) were used per well and then cells were incubated at 37 °C, 5.5% CO2. The cells were lysed 30 h after transfection and luciferase activity was determined using the Steady-Glo luciferase assay system (Promega) according to the manufacturer's instructions. A GloMax-96 Microplate Luminometer (Promega) was used to measure Firefly and Renilla activities. Data were normalised to Renilla activity and for each mutant expressed relative to wild-type activity. For each experiment the normalised values of the quadruplicates were averaged, and data are presented as the mean of the average values from the four separate experiments ± SD. Statistical analyses used a one-way ANOVA (Prism5) and a Dunnett's test to calculate the level of significance between wild-type SQSTM1 and PDB-mutant values. Significance was set at p < 0.05. Western blotting verified that the expression levels of the different SQSTM1 constructs were similar as well as the integrity of the transfected SQSTM1 proteins.
2.4. Protein stability experiments
The stability of transfected His-FLAG-SQSTM1 proteins was assessed essentially as described previously [7] except using HEK293 cells. Briefly, cells were transfected with wild-type, P392L or A427D mutant SQSTM1 constructs and 48 h after transfection, were either lysed (time 0) or treated with 2 μg/ml cycloheximide to prevent further protein synthesis. Samples were collected after incubation for a further 24, 48, or 72 h and protein extracts analysed by western blotting with anti-FLAG or anti-α-tubulin antibodies. After densitometric analysis, intensities of the FLAG-SQSTM1 bands were normalised to those of the α-tubulin bands with values at time zero set to 1.0.
2.5. Patients and clinical assessments
The relation between SQSTM1 mutations, disease extent and age at diagnosis was assessed in 1156 PDB patients of whom 766 were from the UK and had participated in the PRISM study [30] and 390 were recruited from secondary referral centres in Tuscany and Campania in Italy as previously described [20]. Disease extent was assessed in all patients by radionuclide bone scan in which the number of affected sites was counted. Age at diagnosis was obtained by a review of medical records. Clinical data for patients with truncating mutations (E396X (n = 6) and Y383X (n = 5)), which both result in complete loss of the UBA domain, were similar and these genotypes were combined for statistical analysis. We excluded four compound heterozygotes from the analysis leaving a final study population of 1152.
2.6. Statistical analysis
The relation between mutation status, disease extent and age at first diagnosis was assessed by ANOVA. Two analyses were performed; in the first we analysed disease extent and age at first diagnosis in relation to the presence or absence of SQSTM1 mutations (Fig. 3a/b, first two bars); in the second we analysed the same variables in relation to individual mutations by ANOVA, and assessed differences between wild-type and individual mutations by pairwise testing using Tukey's test. The relation between the ability of different SQSTM1 mutants to activate basal NF-κB signalling, disease extent and age at first diagnosis was assessed by comparing the mean value of each clinical variable with the mean value of NF-κB activation using Pearson's correlation.
Fig. 3.
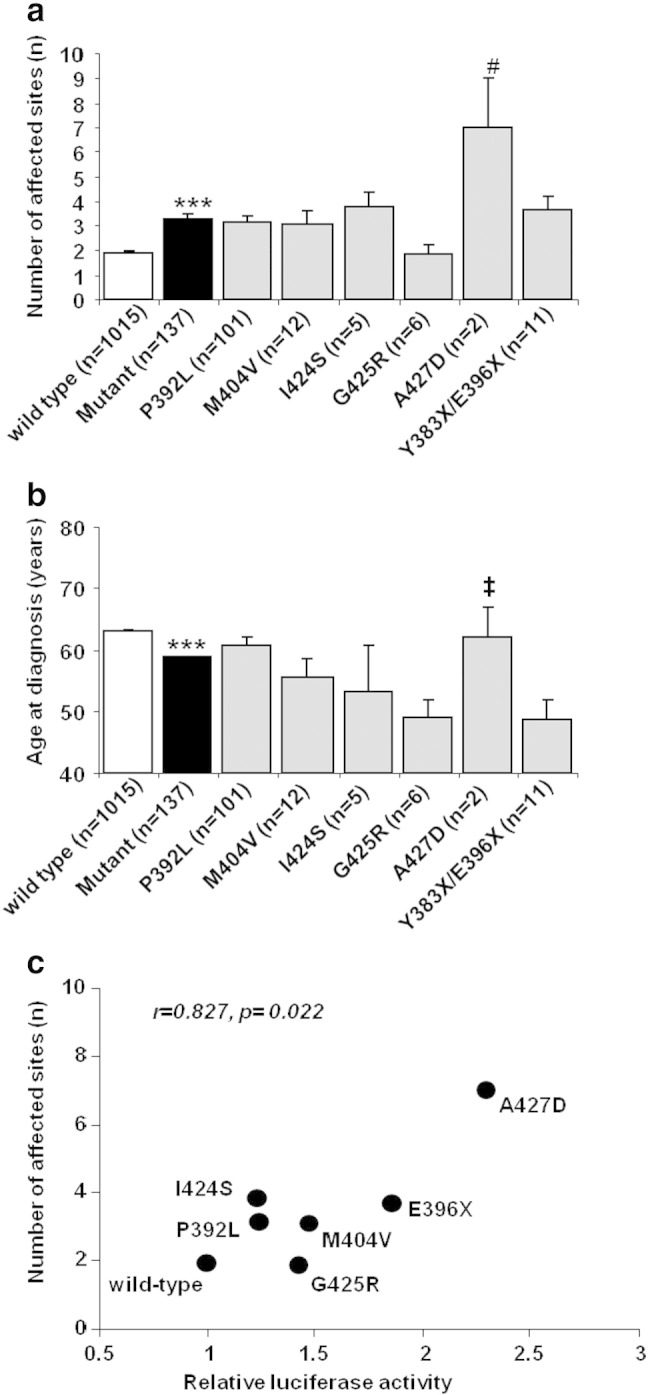
Clinical data for UK and Italian PDB patients in this study. a. Mean number of affected sites +/− SEM and b. mean age at first diagnosis for PDB patients with the indicated genotypes +/− SEM. Wild-type refers to PDB patients with no SQSTM1 mutations; mutant refers to all SQSTM1 mutations combined. The number in brackets refers to number of PDB patients with the given mutation. ***p < 0.001 from wild-type, #p < 0.05 from all other mutants except I424S, and ‡p < 0.05 from P392L mutant. c. Pearson's correlation analysis was used to investigate the relationship between the ability of different SQSTM1 mutants to activate basal NF-κB signalling in reporter assays (relative luciferase activity) and disease extent (number of affected sites) in 1152 PDB patients with the corresponding mutations. Clinical data for “E396X” represents that from combining data for patients with truncating mutations (E396X and Y383X).
2.7. Isothermal titration calorimetry (ITC) and circular dichroism (CD)
The unlabelled and 15N-labelled GST-UBA mutants, untagged ubiquitin and 15N-labelled untagged ubiquitin, were expressed and purified as described previously [27,32]. Thrombin cleavage was used to remove the GST-tag from the glutathione-Sepharose-immobilised UBA domain fusions. Purification of the eluted proteins by gel filtration (Superdex75; GE Healthcare) in 30 mM potassium phosphate, 100 mM NaCl, pH 7, allowed fractions of purified UBA domains to be concentrated by lyophilisation. Fractions were then subsequently desalted on a 5 × 5 ml HiTrap™ desalting column (GE Healthcare) before lyophilisation.
Methods for isothermal titration calorimetry (ITC) have been described previously [32]. In brief, ITC was carried out using a MicroCal VP-ITC instrument. 5 μl of wild-type UBA or I424S-UBA stock solution (250 mM) in 50 mM potassium phosphate buffer, pH 7.0, 50 mM NaCl, was sequentially injected into the ITC cell at 298 K, and the endothermic heat pulse was measured. Data were corrected for the effects of buffer mixing by including an enthalpy of mixing as an iterated variable. The data were analysed using the MicroCal Origin software to determine Kdim using a standard nonlinear least-squares regression analysis [27].
Far UV circular dichroism (CD) spectra were collected on an Applied PhotoPhysics Pi-Star™ spectrophotometer at 298 K using a 0.5 mm (80–110 μM solutions) path length quartz cuvette over a wavelength range from 180 to 260 nm.
2.8. Protein NMR titration experiments
NMR experiments were performed on an AVANCE800 Bruker spectrometer using a TXI cryoprobe and standard pulse sequences with watergate solvent suppression on a cryoprobe. Protein buffer solutions consisted of 25 mM potassium phosphate buffer, 25 mM NaCl, 5% (v/v) D2O, 0.02% (w/v) sodium azide, pH 7.0. NMR titration studies of 15N-labelled ubiquitin with unlabelled I424S-UBA or unlabelled wild-type-UBA domain were performed at 298 K and 1H–15N–HSQC spectra were collected up to a UBA:ubiquitin ratio of 6:1. Chemical shift perturbations (CSPs) were calculated as ΔδHSQC = [(ΔδH)2 + (ΔδN/5)2]1/2 [24], where ΔδH and ΔδN are the observed shift perturbations in the 1H and 15N dimension of the HSQC spectrum. Binding isotherms were constructed and analysed from the dependence of ΔδHSQC on [UBA]TOT. CSPs (> 0.1 ppm) were fitted to a 1:1 binding model using the programme KaleidaGraph to derive an apparent Kobs. From fits shown in Fig. 5c, the Kobs were calculated averaged over 4–5 different residues. Since Kobs = Kd2/Kdim, where Kd is the UBA-Ub dissociation constant and Kdim the dimer dissociation constant, using Kdim values derived from the ITC experiments the Kd values could be calculated.
Fig. 5.

Portion of the 1H–15N-HSQC spectra from titrating ubiquitin (180 μM) with increasing concentrations of a. I424S-UBA and b. wild-type-UBA, at 298 K up to a ubiquitin:UBA ratio of 1:6. The spectra focus on the perturbation to the shift of the peak for V70 showing little movement for binding of I424S-UBA and a more significant shift for wild-type. c. Binding isotherms determined from CSPs (> 0.1 ppm) measured from the titrations of 15N-ubiquitin (180 μM) with either wild-type (wt) UBA or I424S mutant to a final ratio of ubiquitin:UBA 1:6 at 298 K. A 1:1 binding model was used with the programme KaleidaGraph to derive an apparent Kobs. The graph shows four representative residues of which the CSPs were fitted to the 1:1 binding model for both wild-type-UBA (black) and I424S-UBA (white, CSPs are small and overlay). d. Weighted CSP data showing the residues of 15N-ubiquitin (180 μM) that are perturbed by either wild-type (wt) UBA (white bars) or I424S-UBA (black bars) at a final ratio of 1:6 (ubiquitin:UBA) at 298 K. Binding isotherms were constructed and analysed from the dependence of ΔδHSQC on [UBA]TOT. All CSPs above 0.1 are deemed significant. The I424S-UBA mutant resulted in very minor perturbations to the spectra.
3. Results
3.1. Position of mutations and effects on ubiquitin-binding
The positions of the I424, A427 and previously characterised PDB-associated G425 mutation sites are shown mapped on to the SQSTM1 UBA monomer structure in Fig. 1a and are in close proximity within the tertiary structure. I424 and G425 are localised at the C-terminal end of loop 2, and A427 is located at the N-terminus of helix 3 of the UBA domain. The C-terminal end of helix 3 is the common interaction surface for both ubiquitin-binding and UBA domain dimerisation [27]. We first examined the effects of the I424S and A427D mutations on SQSTM1's ubiquitin-binding function in vitro using pull-down assays. In this case recombinant GST-SQSTM1 proteins were incubated with glutathione (g), control (c), and ubiquitin-Sepharose (u) beads at 37 °C, pH 7.5. Here G425R-mutant GST-SQSTM1 acts as a control for complete loss of ubiquitin-binding [29]. Both the I424S and A427D mutants were also associated with a reduced ability to bind to ubiquitin (Fig. 1b). Notably however, although the denatured molecular weight of the A427D-mutant GST-SQSTM1 was consistent with that of the wild-type protein and other (I424S, G425R) mutants, longer exposures of western blots were required to detect protein bands in the glutathione-Sepharose-bound samples, suggesting reduced stability or solubility of the A427D-mutant protein.
Fig. 1.

a. SQSTM1 UBA domain structure (from the dimeric structure PDB accession code: 2knv) with the PDB mutation sites I424, G425 and A427 highlighted. α1-3 refer to the three helices. b. In vitro ubiquitin-binding assays showing the effects of the I424S and A427D mutations on GST-SQSTM1 ubiquitin-binding function. Mutations as indicated (or wild-type, wt) were introduced into the full-length GST-SQSTM1 sequence and pull-down assays were performed at 37 °C. Bacterial lysates containing the GST-SQSTM1 fusions were incubated with glutathione- (G), control- (C) and ubiquitin-Sepharose (Ub) beads and captured proteins were detected by western blotting (anti-SQSTM1 antibodies). The G425R mutation has previously been shown to cause complete loss of ubiquitin-binding function in similar assays. c. Purification of bacterial expressed GST-UBA SQSTM1 proteins or GST only. Bacterial over-expressions of GST-UBA proteins were affinity purified using glutathione-Sepharose beads. The products from the purification were loaded onto a 5-20% gradient SDS-PAGE gel which was subsequently stained with Coomassie blue.
3.2. Effects of mutations on NF-κB signalling
The effects of over-expressing mutant SQSTM1 proteins on the activation of basal NF-κB signalling in HEK293T cells were assessed (Fig. 2a). Previous studies using the same approach have shown that different PDB-associated UBA domain missense mutants of SQSTM1, including G425R, increase basal NF-κB activity relative to wild-type which itself is associated with repression of signalling compared to empty vector control [7]. In addition, we also previously noted that truncating mutants of SQSTM1 (e.g. E396X), suggested to be associated with more severe PDB than missense mutants [6,7], gave the greatest level of activation of basal NF-κB activity in reporter assays [22]. Here we examined the effects of expression of not only the I424S and A427D mutants, but also in parallel several other PDB-associated SQSTM1 UBA domain mutants, on basal NF-κB activity. The other mutants P392L, E396X, M404V and G425R were chosen because they were the most common mutations within the PRISM study [15]. Like other PDB-associated mutants tested, both I424S and A427D activated basal signalling, relative to wild-type SQSTM1 (Fig. 2a), although the values for the P392L mutant did not reach significance by one-way ANOVA and Dunnett's test (p = 0.23); notably A427D-SQSTM1 was associated with the greatest level of activation (relative to wild-type) of all PDB mutants tested to date. In contrast to the observations with the GST-SQSTM1 proteins expressed in E. coli, comparable steady-state levels of all transfected proteins, including the A427D mutant, were detected in these mammalian cells (Fig. 2b). Consistent with these observations, analyses in HEK293 cells demonstrated that SQSTM1 mutations had only minor effects on protein stability over the course of 72 h (see Supplementary data Fig. S1), and importantly these did not correlate with the ability of different mutant SQSTM1 proteins to activate NF-κB activity in our reporter assays. Further, co-immunoprecipitation experiments found no evidence of altered interactions of the transfected SQSTM1 proteins with endogenous aPKC proteins (data not shown), indicating that the effects of the mutations are not mediated through altered interactions with these critical regulators of NF-κB signalling.
Fig. 2.
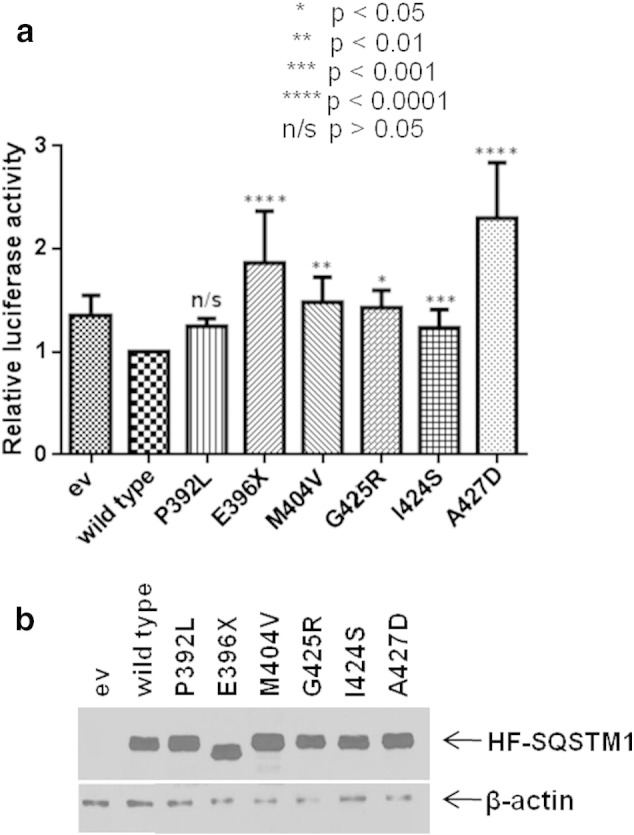
a. Effects of wild-type and PDB mutant SQSTM1 over-expression on basal NF-κB activity. HEK293T cells were co-transfected with His-FLAG-SQSTM1 pcDNA3.1 constructs as indicated or empty vector (ev) pcDNA3.1 control along with NF-κB Firefly luciferase reporter and control Renilla luciferase construct [36]. Cells were harvested 30 h after transfection and luciferase activity was measured. Firefly luciferase values were normalised to Renilla values. Experiments were performed in quadruplicate and assays were repeated on 4 separate occasions. Data is displayed as mean ± SD with p values indicating significance of difference compared with wild-type as indicated. The A427D mutant gave had the highest level of activation of all PDB mutants tested to date. b. Western blotting analysis (anti-SQSTM1, anti-β-actin) showed comparable expression levels of the different His-FLAG-SQSTM1 constructs (HF-SQSTM1) from a representative set of samples from a single NF-κB reporter assay.
Supplementary Fig. S1.
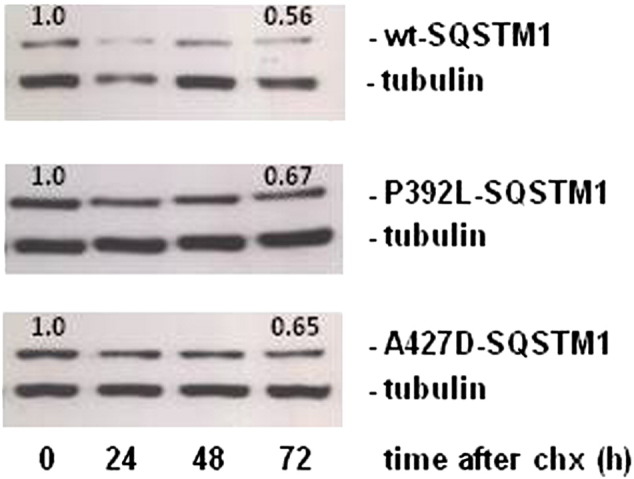
HEK293 cells were transfected with His-FLAG-SQSTM1 pcDNA3.1 constructs as indicated and 48 h after transfection, cells were treated with cycloheximide (chx) to inhibit protein synthesis. After 0, 24, 48, and 72 h of incubation, cells were lysed and proteins were separated on 10% SDS polyacrylamide gels then transferred to nitrocellulose membranes. Proteins were visualised after western blot analyses with anti-FLAG and anti-α-tubulin antibodies (representative blot shown). Numbering on blots relates to SQSTM1 band intensity, normalised to α-tubulin bands and with the value at time zero for each SQSTM1 sequence set to 1.0 and values at the 72 h end-point indicated.
The effects of over-expressing mutant SQSTM1 proteins on the activation of basal NF-κB signalling in HEK293T cells were assessed (Fig. 2a). Previous studies using the same approach have shown that different PDB-associated UBA domain missense mutants of SQSTM1, including G425R, increase basal NF-κB activity relative to wild-type which itself is associated with repression of signalling compared to empty vector control [7]. In addition, we also previously noted that truncating mutants of SQSTM1 (e.g. E396X), suggested to be associated with more severe PDB than missense mutants [6,7], gave the greatest level of activation of basal NF-κB activity in reporter assays [22]. Here we examined the effects of expression of not only the I424S and A427D mutants, but also in parallel several other PDB-associated SQSTM1 UBA domain mutants, on basal NF-κB activity. The other mutants P392L, E396X, M404V and G425R were chosen because they were the most common mutations within the PRISM study [15]. Like other PDB-associated mutants tested, both I424S and A427D activated basal signalling, relative to wild-type SQSTM1 (Fig. 2a), although the values for the P392L mutant did not reach significance by one-way ANOVA and Dunnett's test (p = 0.23); notably A427D-SQSTM1 was associated with the greatest level of activation (relative to wild-type) of all PDB mutants tested to date. In contrast to the observations with the GST-SQSTM1 proteins expressed in E. coli, comparable steady-state levels of all transfected proteins, including the A427D mutant, were detected in these mammalian cells (Fig. 2b). Consistent with these observations, analyses in HEK293 cells demonstrated that SQSTM1 mutations had only minor effects on protein stability over the course of 72 h (see Supplementary data Fig. S1), and importantly these did not correlate with the ability of different mutant SQSTM1 proteins to activate NF-κB activity in our reporter assays. Further, co-immunoprecipitation experiments found no evidence of altered interactions of the transfected SQSTM1 proteins with endogenous aPKC proteins (data not shown), indicating that the effects of the mutations are not mediated through altered interactions with these critical regulators of NF-κB signalling.
3.3. Genotype–phenotype analyses
We went on to investigate possible associations between the ability of different UBA domain mutants of SQSTM1 to activate basal NF-κB signalling and clinical markers of disease severity, including the age at first diagnosis and number of affected sites as determined by radionuclide bone scan. The relation between mutation type, age at first diagnosis and number of affected sites is shown in Fig. 3. Patients with SQSTM1 mutations had significantly more extensive disease (Fig. 3a) and an earlier age at first diagnosis (Fig. 3b) than those without mutations. The number of affected sites and age at first diagnosis also varied by mutation type but pairwise comparisons revealed few significant differences between genotypes probably because of the small sample size for most mutations. Although the A427D mutation was associated with an increase in number of affected sites relative to all other mutants except I424S, it is notable that this mutation is found in only two (related) individuals (with 5 and 9 affected sites, respectively). There was a significant association between the ability of different mutations to activate basal NF-κB signalling in vitro and average number of affected sites (r = 0.827, p = 0.022) (Fig. 3c), but no significant association between NF-κB activation and age at first diagnosis (r = − 0.08, p = 0.86) (data not shown). As the clinical data related to the A427D mutation again came from only two related individuals we also repeated the Pearson's correlation of NF-κB activation and average number of affected sites without the A427D values, and although the correlation remained positive (0.43) the result was no longer significant (p = 0.395), primarily because we are dealing with only limited observations since we correlated mean NF-κB expression with mean number of lesions (data not shown). However, repeating this analysis with every patient in the study population assigned a NF-κB score according to mutation status (which makes use of all study data) and relating this to affected bone numbers, the result was highly significant both when we included all patients (r = 0.277, p < 0.0001) and also when the two patients with A427D mutations were excluded (r = 0.244, p < 0.001) (data not shown).
3.4. Impact of mutations on UBA domain structure and dimerisation
We next sought to understand the structural basis of the effects of the I424S and A427D mutants on SQSTM1's ubiquitin-binding function revealed in the pull-down assays (Fig. 1b), and in particular, given the close proximity of these mutations to the site affected by the previously characterised G425R mutant, whether common or distinct mechanisms are manifested. These structural analyses necessitated that we focussed on the UBA domain sequences alone (residues 387–436) expressed in E. coli. Prior to thrombin cleavage to remove the GST tag, purified GST-A427D-UBA was found by SDS PAGE to be present at much lower levels than the wild-type or I424S/G425R variants (Fig. 1c). In addition, lower molecular weight bands, presumably representing truncated UBA sequences generated by adventitious proteolysis, were evident for the A427D mutant. After thrombin cleavage, although the wild-type and I424S mutant UBA domains yielded a significant amount of purified protein (3–4 mg/l culture), no protein of the expected denatured molecular weight (as determined by SDS PAGE) was recovered for the A427D mutant (not shown). In addition, electrospray ionisation-mass spectrometry (ESI-MS) analyses as previously described [33] confirmed the expected mass values of the wild-type, I424S and G425R-UBA domains, but the A427D-UBA domain could not be detected (not shown). We speculate that this could be due to the A427D mutation causing a reduction in stability or solubility of the SQSTM1-UBA, as was noted for the full-length bacterially-expressed protein (Fig. 1b). Indeed, inspection of the UBA domain NMR structure [27] indicates that A427 is a buried residue that stabilises the UBA domain through hydrophobic packing of helix 3. A427 is highly conserved within the SQSTM1 sequence of different species. The A427D-UBA mutation is likely to be highly destabilising as a consequence of the greater steric bulk of Asp and the large energetic penalty due to burying the polar side chain from access to solvent. This analysis is consistent with the inability to isolate purified material.
Molecular modelling (not shown) also predicted that the I424S mutation is likely to affect the packing of helix 3 within the UBA domain structure, but in contrast to the A427D mutant, the I424S-UBA mutant could be expressed and readily isolated from E. coli. We thus were able to perform detailed structural analyses of this UBA mutant. We first investigated the effects of the I424S mutation on formation of a stable UBA dimer, a feature of the wild-type UBA domain which negatively regulates ubiquitin-binding [27]. ITC was used to determine the Kdim value for dimer dissociation from dilution experiments (see Fig. 4c). Endothermic heat pulses were observed after injection of the protein into buffer solution. Kdim values indicate that the I424S mutant is a 10-fold weaker dimer (Kdim = 19.8 ± 1.0 μM compared to wild-type UBA analysed under identical conditions (Kdim = 2.0 ± 0.3 μM), in contrast with our previous observations that showed that the G425R mutant is a slightly stronger dimer than wild-type [24].
Fig. 4.

Biophysical data demonstrating the structural differences between wild-type and I424S-UBA domains of SQSTM1. a. Weighted chemical shift perturbations (CSPs) for the UBA dimer ([UBA] ~ 750 μM; CSP, weighted average from 1H and 15N shifts) showing the residues that are perturbed when introducing the I424S mutation into the UBA domain, compared to wild-type. All CSPs > 0.1 ppm are deemed significant (with the 0.2 CSP threshold highlighted with a dotted line), indicating extensive structural effects propagated from the mutation site. b. CSPs from (a) mapped on to the UBA surface showing the position of surface residues that are perturbed by the I424S mutation; darker red represents bigger CSP values (PDB structure 2KNV) c. Dimer dissociation isotherms of wild-type (wt) and I424S-UBA derived from the integrated heat absorbance profile of ITC dilution experiments. d. Far-UV CD spectra for the re-folded wild-type-UBA (wt, solid line) and I424S mutant (dotted line) recorded at concentrations between 80 and 110 μM showing a helical spectrum consistent with fully folded dimeric structure.
NMR experiments were performed on the 15N-isotopically labelled I424S-UBA domain mutant to examine the effects on UBA domain structure. Initial analysis at a protein concentration of 750 μM, well above the Kdim, ensured that the mutant was dimeric, permitting close comparison with analogous data for wild-type UBA. The 15N-HSQC NMR ‘footprint’ for each protein provides a sensitive residue-specific probe of changes in protein structure. Comparing the I424S-UBA mutant to the wild-type UBA spectra, 11 out of 50 residues showed significant (> 0.1 ppm) CSPs indicative of structural changes (Fig. 4a). The location of the perturbed residues identified from the CSP analysis was mapped onto the surface of the wild-type UBA dimer structure (Fig. 4b). This structural analysis shows that not only is the I424S change affecting the local environment around the mutation site, but also exerts effects across the whole domain. Although the mutation occurs at the N-terminus of helix 3, large effects (CSPs > 0.2 ppm) are propagated along the helix significantly disrupting many of the critical residues required for both UBA dimerisation and ubiquitin-binding. Residues L428, T430, and I431 all show CSPs over 0.1 ppm and form important contacts across the dimer interface. I431 and L428 are key residues on the ubiquitin-binding surface of the UBA domain. Further away from the mutation site, residues within helix 1 (Q400, M401 and L402), loop 1 (S406 and E409) and helix 2 (E418) all show CSPs over 0.1 ppm; many of these residues are required to stabilise the UBA dimer interface [27]. In contrast, more local effects were previously noted for the G425R mutant (only residues I424-A427, D429 and K435 have CSPs > 0.1 ppm) suggesting only small structural changes adjacent to this mutation site [24], consistent with the location of this residue on the protein surface.
3.5. NMR insights into impact of mutations on UBA dimerisation
We further investigated the dimer–monomer equilibrium of the UBA domain using NMR by recording 2D-HSQC spectra at concentrations below the Kdim values where the dimer is fully dissociated to the biologically active monomeric form [27]. As previously reported, wild-type UBA at < 5 μM concentration shows two sets of well dispersed peaks corresponding to an equilibrium between the stable dimer and a folded monomer (see Supplementary data Fig. S2 part b). In contrast, and as a control, we showed that the engineered T419K-UBA mutant, which has a destabilised dimer interface, retains its structural integrity as a folded monomer at 10 μM [27] (Supplementary data Fig. S2 part c). In spectra of the I424S-UBA mutant at 10 μM concentrations, where the ITC results predict the exclusive presence of monomer, we observed a poorly dispersed spectrum with peaks significantly broadened and clustered around a narrow chemical shift range (Supplementary data Fig. S2 part d). This spectrum is highly indicative of a largely unfolded protein, suggesting that whilst the dimer retains a large part of its structural integrity, stabilised by contacts at the dimer interface, in isolation the monomeric form of the I424S-UBA is unstable and converts to the unfolded state. The process was entirely reversible, with lyophilisation of dilute solutions of I424S-UBA and concentration of samples to > 100 μM showing that the mutant readily reformed the dimer structure. A set of well dispersed peaks in the 15N-HSQC spectrum and a helical CD spectrum (Fig. 4d) was consistent with that of the wild-type UBA dimer at similar sample concentrations (80–110 μM). ESI-MS showed no sign of protein degradation over this period (data not shown), indicative of an equilibrium between different folded and unfolded states.
Supplementary Fig. S2.

1H–15N HSQC NMR spectra demonstrating the structural differences between wild-type (wt), T419K and I424S-UBA domains of SQSTM1 at pH 7.0 and 298 K. The red boxes are expanded below the structural schematic representation of UBA equilibrium of which the position in equilibrium is dependent on UBA concentration and mutation status. Arrows from each spectra point to the species the UBA domains exist in. A. Spectrum of wt-UBA at > 100 μM showing a single species corresponding to a folded dimer (D) with well dispersed cross-peaks. B. Spectrum of wt-UBA at 10 μM showing a doubling of peaks corresponding to dimer and monomer (D/M) in equilibrium. C. Spectrum of the engineered T419K-UBA mutant at 10 μM with a destabilising mutation at the dimer interface leading to the folded helical monomer (M) being fully populated in the absence of dimer. D. Spectrum of the I424S-UBA mutant at 10 μM showing the unfolded monomer (Munf). The well dispersed peaks shown in the red box (expanded below), indicative of a stable folded helical UBA motif, are no longer evident, with peaks confined to a much narrower region of the spectrum and not assigned (?).
We further investigated the dimer–monomer equilibrium of the UBA domain using NMR by recording 2D-HSQC spectra at concentrations below the Kdim values where the dimer is fully dissociated to the biologically active monomeric form [27]. As previously reported, wild-type UBA at < 5 μM concentration shows two sets of well dispersed peaks corresponding to an equilibrium between the stable dimer and a folded monomer (see Supplementary data Fig. S2 part b). In contrast, and as a control, we showed that the engineered T419K-UBA mutant, which has a destabilised dimer interface, retains its structural integrity as a folded monomer at 10 μM [27] (Supplementary data Fig. S2 part c). In spectra of the I424S-UBA mutant at 10 μM concentrations, where the ITC results predict the exclusive presence of monomer, we observed a poorly dispersed spectrum with peaks significantly broadened and clustered around a narrow chemical shift range (Supplementary data Fig. S2 part d). This spectrum is highly indicative of a largely unfolded protein, suggesting that whilst the dimer retains a large part of its structural integrity, stabilised by contacts at the dimer interface, in isolation the monomeric form of the I424S-UBA is unstable and converts to the unfolded state. The process was entirely reversible, with lyophilisation of dilute solutions of I424S-UBA and concentration of samples to > 100 μM showing that the mutant readily reformed the dimer structure. A set of well dispersed peaks in the 15N-HSQC spectrum and a helical CD spectrum (Fig. 4d) was consistent with that of the wild-type UBA dimer at similar sample concentrations (80–110 μM). ESI-MS showed no sign of protein degradation over this period (data not shown), indicative of an equilibrium between different folded and unfolded states.
3.6. Impact of mutations on ubiquitin-binding
Finally, we used NMR to probe the interaction of the I424S-UBA domain with ubiquitin. Titration of the unfolded 15N-labelled I424S-UBA monomer at 10 μM concentration with unlabelled ubiquitin, up to a 40:1 excess of ubiquitin to I424S-UBA, resulted in very minor perturbations to the spectra (not shown). This is indicative of weak binding of the monomeric unfolded mutant UBA to ubiquitin, consistent with pull-down experiments. In contrast, the wild-type UBA monomer underwent significant perturbations (many > 0.2 ppm, as reported [24]) demonstrating strong binding at low concentrations of protein. The effects of the I424S mutation on the UBA domain's ability to bind to ubiquitin were also studied in NMR titrations with 15N-labelled ubiquitin as the spectroscopic probe and unlabelled UBA domains as the ‘silent’ partner. Spectra were recorded up to an excess ratio of 6:1 UBA:ubiquitin, but the I424S-UBA showed only small CSP effects on K48 and L71 (0.05 to 0.1 ppm) of 15N-ubiquitin compared with the larger number of significant perturbations observed for wild-type UBA (Fig. 5d). In attempts to quantify the effects of the I424S mutation on the UBA-binding affinity we constructed binding isotherms for the two NMR titration experiments with 15N-ubiquitin. From fits shown in Fig. 5c, the Kobs were calculated; Kobs = 189 μM (wild-type UBA) and Kobs = 709 μM (I424S-UBA), averaged over 4 different residues. Fitting the data to allow for a two-step process involving UBA dimer dissociation and folding by ubiquitin-binding we estimated that the mutant binds at least 6-fold weaker than the wild-type UBA (wild-type UBA Kd = 19 μM and I424S-UBA Kd = 119 μM). This is likely to be an underestimate of the reduction in affinity since the large differences in the limiting CSPs for residues in the fully bound state for the I424S mutant versus wild-type UBA are consistent with the mutant forming a much weaker binding interface with only a small set of the contacts observed for the wild-type UBA. For example, I13, I44 and V70, which form the widely reported canonical UBA hydrophobic binding surface of ubiquitin, are largely unperturbed, suggesting that the loss of structural integrity in the I424S mutant is no longer compatible with binding to the ubiquitin I44 patch. The free energy of the binding interaction is not sufficient to pull the unfolded monomer into a folded bound form. In contrast, our data show, in the context of the UBA domain dimer, many more contacts are formed along helix 2 making the dimerisation interface much stronger than the UBA/ubiquitin interaction.
4. Discussion
PDB is the second most common metabolic bone disease after osteoporosis. The precise mechanisms that underlie PDB aetiology are yet to be established but there is considerable evidence that SQSTM1 mutations play a central role [3,4,6,7,11,22,23]. Initially all SQSTM1 mutations were thought to contribute to the pagetic phenotype through increases in osteoclastogenesis and osteoclast activity, via activation of RANK-NF-κB signalling [7,11,22,23]. Another common property of the first mutant SQSTM1 proteins analysed was impaired binding to ubiquitin in vitro [7,22,29,31]. Here we found that both the I424S and A427D mutations also impair SQSTM1's ability to bind to ubiquitin, and when over-expressed mutants are associated with activation of basal NF-κB signalling in reporter assays. Indeed all six of the PDB-mutant SQSTM1 sequences tested (covering the most common mutations found within the PRISM study [15]) were able to activate basal NF-κB activity, relative to wild-type SQSTM1, in our HEK293T cell model. We also present evidence for an association between the ability of the different UBA domain mutants to activate basal NF-κB signalling and number of affected sites in 1152 PDB patients. Our analyses did not take into account secular trends indicating a progressive decrease in the number of affected skeletal sites in PDB [34] although we did note that most patients with truncating mutations (E396X and Y383X) had a severe phenotype even in younger patients. Significantly 12/13 of the PDB patients in our study with truncating or A427D mutations, associated with the highest levels of NF-κB activity in reporter assays, exhibited polyostotic PDB and in a previous study a similar phenotype was noted for three patients with the Y383X mutation [10]. Further the A427D mutant, associated with a high number of affected sites in the two reported (albeit related) carriers (7.00 ± 2.8 affected pagetic sites compared to 3.60 ± 2.6 sites in SQSTM1 mutation carriers collectively within the cohort [14]), gave a higher level of activation (relative to wild-type) than any other mutant analysed to date, including the E396X truncating mutant. Thus, despite the caveat that other gene variants can influence severity in PDB with and indeed without SQSTM1 mutations [20] at least in some cases (i.e. for UBA domain mutations) the SQSTM1 status alone may exert significant effects on disease phenotype via changes in RANK-NF-κB signalling. Our findings are however counter to the notion of a direct relationship between SQSTM1 protein function (i.e. ubiquitin-binding activity) and disease severity – in the truncating mutant the UBA domain is completely lost, which would clearly have a more marked effect on protein function than a missense mutation e.g. although weaker, we show that I424S mutation still has a measurable ubiquitin-binding affinity). In fact analyses of other PDB-associated mutants demonstrate that the relationship between SQSTM1 protein function, NF-κB activity and PDB severity is more complex than initially anticipated. Some non-UBA domain mutants have no effect on ubiquitin-binding function but can activate NF-κB activity [11], and we have very recently described a non-UBA domain mutation which affects neither SQSTM1's ubiquitin-binding function nor its ability to regulate NF-κB activity in reporter assays [35]. Clearly further studies are required in this area, including in more physiologically-relevant cell models and including correlations with other aspects of SQSTM1 function.
Our initial goal was to characterise both the I424S and A427D mutant UBA domains in structural detail although notably the latter was not amenable to biophysical analysis. Over-expression of the GST-A427D-UBA sequence led to rapid degradation in E. coli, consistent with the A427D mutation destabilising the three-helix bundle of the UBA monomer rendering the unfolded protein highly susceptible to proteolysis. This is similar to the M404V/T UBA mutants which we have previously reported to be associated with poor expression levels (as UBA only sequences), lower solubility, and aggregation effects consistent with destabilisation of the UBA domain [24]. However, full-length eukaryotic and prokaryotic A427D-SQSTM1 sequences were detectable by western blotting, and we found no overtly detrimental effects of the mutation on protein half-life in mammalian cells, suggesting that there may be an intrinsic stabilisation of the UBA domain of SQSTM1 within the full-length protein.
Although the I424S and previously characterised G425R mutants are located in close proximity within the UBA tertiary fold they exert distinct effects on protein function and structure. As illustrated in Fig. 6, we have established that the SQSTM1 UBA is involved in a complex equilibrium between an inactive dimeric form, an active ubiquitin-binding monomer and an inactive unfolded state. It would appear that PDB mutations can perturb these equilibria in a manner that both directly and indirectly affects the binding affinity of the active monomer. Wild-type SQSTM1 exists in equilibrium where the stability of the UBA dimer regulates ubiquitin-binding by sequestering the biologically active monomeric UBA [27]. The dimer interface protects the ubiquitin-binding surface making UBA dimerisation and ubiquitin-binding competitive recognition events. In contrast to the neighbouring loop 2 mutant (G425R) which stabilised the UBA dimer, the I424S mutation produces a 10-fold reduction in dimer stability with extensive structural perturbations to helix 3 and core packing interactions. In this respect, the PDB-associated I424S mutation appears to result in similar effects on SQSTM1 structure as the S399P mutation which destabilises helix 1 [24]. NMR titrations of 15N-ubiquitin with the I424S-UBA revealed that the mutant also resulted in at least a 6-fold weaker binding affinity for ubiquitin than that of wild-type UBA. Moreover, not only are the limiting CSP effects much smaller for the mutant UBA, indicative of a much weaker binding interface, but the canonical hydrophobic patch on the surface of ubiquitin (I13, I44 and V70) appears to be largely unperturbed by the interaction with the I424S-UBA mutant, also suggesting loss of binding specificity. The I424S mutation involves a key residue in helix 3 that packs against helix 1 and forms part of the stabilising hydrophobic core of the three-helix bundle. NMR studies show that the monomeric UBA domain is largely unfolded in isolation, although it is stabilised at higher concentrations through formation of the dimeric structure. Thus, both I424S and A427D are clear examples of PDB mutations that have such dramatic effects on the stability and structural integrity of the folded UBA domain structure that they are no longer able to directly interact with ubiquitin with any significant binding affinity. In contrast, the G425R mutation affects a surface exposed residue and results in none of the associated effects on the stability of the UBA motif. Substitution of G with a bulky charged R side chain (G425R) appears to affect the steric complementarity of the UBA binding surface on helix 3 [24].
Fig. 6.

Equilibrium showing inter-conversion between the SQSTM1 UBA dimer (left), monomer (centre) and an unfolded state (right), with the equilibrium constants Kdim and Keq defining the relative stabilities of these species. Only the folded monomer is compatible with binding to ubiquitin via the I44 hydrophobic patch with a measurable affinity, Kd. PDB mutations are able to reduce Kd via a number of mechanisms that either directly perturb the UBA:ubiquitin interface (I424S, G425R) or indirectly stabilise either the dimer (G425R) or the unfolded state (I424S). Both reduce the population of the active monomeric UBA for binding to ubiquitin. I424S appears to destabilise both the dimer and monomer, partially favouring the unfolded state under physiological conditions. In contrast, G425R not only increases the stability of the dimer compared with wild-type UBA, but also directly affects the binding interface through steric and electrostatic effects.
In summary we show that both the I424S and A427D PDB-associated SQSTM1 mutants exhibit impaired ubiquitin-binding function in pull-down assays and dysregulated basal NF-κB signalling in luciferase reporter assays, and find that there is a significant correlation between the ability of different UBA domain mutants of SQSTM1 to regulate NF-κB activity in cell models and extent of PDB in patients with the corresponding mutations. Further, we report loss of both structural integrity and stability of the I424S-UBA domain in biophysical experiments. The I424S and A427D mutants exert differing effects on SQSTM1 function relative to other PDB mutants, demonstrating diverse molecular mechanisms operating in the aetiology of PDB.
The following are the Supplementary data to this article.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbadis.2014.03.006.
Acknowledgements
This study was supported in part by grants to RL and MSS from Arthritis Research UK (reference 18585), to SHR from Arthritis Research UK (reference 19799), from the BBSRC (BB/I011420/1) in support of JEL, a National Health and Medical Research Council of Australia grant to SLR (APP1027932), and by a Telethon grant (GGP11119) to LG and FG. Dr. Huw Williams is thanked for help with preliminary modelling studies and Dr. Jennifer Adlington for the HSQC spectrum of T419K-UBA.
References
- 1.Selby P.L., Davie M.W., Ralston S.H., Stone M.D. Guidelines on the management of Paget's disease of bone. Bone. 2002;31:366–373. doi: 10.1016/s8756-3282(02)00817-7. [DOI] [PubMed] [Google Scholar]
- 2.Laurin N., Brown J.P., Morissette J., Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 2002;70:1582–1588. doi: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hocking L.J., Lucas G.J., Daroszewska A., Mangion J., Olavesen M., Cundy T., Nicholson G.C., Ward L., Bennett S.T., Wuyts W., Van Hul W., Ralston S.H. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum. Mol. Genet. 2002;11:2735–2739. doi: 10.1093/hmg/11.22.2735. [DOI] [PubMed] [Google Scholar]
- 4.Johnson-Pais T.L., Wisdom J.H., Weldon K.S., Cody J.D., Hansen M.F., Singer F.R., Leach R.J. Three novel mutations in SQSTM1 identified in familial Paget's disease of bone. J. Bone Miner. Res. 2003;18:1748–1753. doi: 10.1359/jbmr.2003.18.10.1748. [DOI] [PubMed] [Google Scholar]
- 5.Eekhoff E.W., Karperien M., Houtsma D., Zwinderman A.H., Dragoiescu C., Kneppers A.L., Papapoulos S.E. Familial Paget's disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum. 2004;50:1650–1654. doi: 10.1002/art.20224. [DOI] [PubMed] [Google Scholar]
- 6.Hocking L.J., Lucas G.J., Daroszewska A., Cundy T., Nicholson G.C., Donath J., Walsh J.P., Finlayson C., Cavey J.R., Ciani B., Sheppard P.W., Searle M.S., Layfield R., Ralston S.H. Novel UBA domain mutations of SQSTM1 in Paget's disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J. Bone Miner. Res. 2004;19:1122–1127. doi: 10.1359/JBMR.0403015. [DOI] [PubMed] [Google Scholar]
- 7.Rea S.L., Walsh J.P., Ward L., Yip K., Ward B.K., Kent G.N., Steer J.H., Xu J., Ratajczak T. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget's disease of bone with a severe phenotype. J. Bone Miner. Res. 2006;21:1136–1145. doi: 10.1359/jbmr.060405. [DOI] [PubMed] [Google Scholar]
- 8.Beyens G., Wuyts W., Cleiren E., de Freitas F., Tiegs R., Van Hul W. Identification and molecular characterization of a novel splice-site mutation (G1205C) in the SQSTM1 gene causing Paget's disease of bone in an extended American family. Calcif. Tissue Int. 2006;79:281–288. doi: 10.1007/s00223-006-0122-3. [DOI] [PubMed] [Google Scholar]
- 9.Collet C., Michou L., Audran M., Chasseigneaux S., Hilliquin P., Bardin T., Lemaire I., Cornelis F., Launay J.M., Orcel P., Laplanche J.L. Paget's disease of bone in the French population: novel SQSTM1 mutations, functional analysis, and genotype–phenotype correlations. J. Bone Miner. Res. 2007;22:310–317. doi: 10.1359/jbmr.061106. [DOI] [PubMed] [Google Scholar]
- 10.Falchetti A., Di Stefano M., Marini F., Ortolani S., Ulivieri M.F., Bergui S., Masi L., Cepollaro C., Benucci M., Di Munno O., Rossini M., Adami S., Del Puente A., Isaia G., Torricelli F., Brandi M.L., GenePage P. Genetic epidemiology of Paget's disease of bone in Italy: sequestosome1/p62 gene mutational test and haplotype analysis at 5q35 in a large representative series of sporadic and familial Italian cases of Paget's disease of bone. Calcif. Tissue Int. 2009;84:20–37. doi: 10.1007/s00223-008-9192-8. [DOI] [PubMed] [Google Scholar]
- 11.Rea S.L., Walsh J.P., Ward L., Magno A.L., Ward B.K., Shaw B., Layfield R., Kent G.N., Xu J., Ratajczak T. Sequestosome 1 mutations in Paget's disease of bone in Australia: prevalence, genotype/phenotype correlation, and a novel non-UBA domain mutation (P364S) associated with increased NF-kappaB signaling without loss of ubiquitin binding. J. Bone Miner. Res. 2009;24:1216–1223. doi: 10.1359/jbmr.090214. [DOI] [PubMed] [Google Scholar]
- 12.Chung P.Y., Beyens G., Guanabens N., Boonen S., Papapoulos S., Karperien M., Eekhoff M., Van Wesenbeeck L., Jennes K., Geusens P., Offeciers E., Van Offel J., Westhovens R., Zmierczak H., Devogelaer J.P., Van Hul W. Founder effect in different European countries for the recurrent P392L SQSTM1 mutation in Paget's disease of bone. Calcif. Tissue Int. 2008;83:34–42. doi: 10.1007/s00223-008-9137-2. [DOI] [PubMed] [Google Scholar]
- 13.Rendina D., Gianfrancesco F., De Filippo G., Merlotti D., Esposito T., Aloia A., Benvenuto D., Vivona C.L., Annunziata G., Nuti R., Strazzullo P., Mossetti G., Gennari L. Epidemiological, clinical and genetic characteristics of Paget's disease of bone in a rural area of Calabria, Southern Italy. J. Endocrinol. Invest. 2009;33:519–525. doi: 10.1007/BF03346640. [DOI] [PubMed] [Google Scholar]
- 14.Gennari L., Gianfrancesco F., Di Stefano M., Rendina D., Merlotti D., Esposito T., Gallone S., Fusco P., Rainero I., Fenoglio P., Mancini M., Martini G., Bergui S., De Filippo G., Isaia G., Strazzullo P., Nuti R., Mossetti G. SQSTM1 gene analysis and gene–environment interaction in Paget's disease of bone. J. Bone Miner. Res. 2010;25:1375–1384. doi: 10.1002/jbmr.31. [DOI] [PubMed] [Google Scholar]
- 15.Visconti M.R., Langston A.L., Alonso N., Goodman K., Selby P.L., Fraser W.D., Ralston S.H. Mutations of SQSTM1 are associated with severity and clinical outcome in Paget's disease of bone. J. Bone Miner. Res. 2010;25:2368–2373. doi: 10.1002/jbmr.132. [DOI] [PubMed] [Google Scholar]
- 16.Michou L., Morissette J., Gagnon E.R., Marquis A., Dellabadia M., Brown J.P., Siris E.S. Novel SQSTM1 mutations in patients with Paget's disease of bone in an unrelated multiethnic American population. Bone. 2011;48:456–460. doi: 10.1016/j.bone.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Daroszewska A., van 't Hof R.J., Rojas J.A., Layfield R., Landao-Basonga E., Rose L., Rose K., Ralston S.H. A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget's disease-like disorder in mice. Hum. Mol. Genet. 2011;20:2734–2744. doi: 10.1093/hmg/ddr172. [DOI] [PubMed] [Google Scholar]
- 18.Albagha O.M., Visconti M.R., Alonso N., Langston A.L., Cundy T., Dargie R., Dunlop M.G., Fraser W.D., Hooper M.J., Isaia G., Nicholson G.C., Del Pino Montez J., Gonzalez-Sarmiento R., di Stefano M., Tenesa A., Walsh J.P., Ralston S.H. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget's disease of bone. Nat. Genet. 2010;42:520–524. doi: 10.1038/ng.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albagha O.M., Wani S.E., Visconti M.R., Alonso N., Goodman K., Brandi M.L., Cundy T., Chung P.Y., Dargie R., Devogelaer J.P., Falchetti A., Fraser W.D., Gennari L., Gianfrancesco F., Hooper M.J., Van Hul W., Isaia G., Nicholson G.C., Nuti R., Papapoulos S., Montes J.D., Ratajczak T., Rea S.L., Rendina D., Gonzalez-Sarmiento R., Di Stefano M., Ward L.C., Walsh J.P., Ralston S.H. Genome-wide association identifies three new susceptibility loci for Paget's disease of bone. Nat. Genet. 2011;43:685–689. doi: 10.1038/ng.845. [DOI] [PubMed] [Google Scholar]
- 20.Albagha O.M., Visconti M.R., Alonso N., Wani S., Goodman K., Fraser W.D., Gennari L., Merlotti D., Gianfrancesco F., Esposito T., Rendina D., di Stefano M., Isaia G., Brandi M.L., Giusti F., Del Pino-Montes J., Corral-Gudino L., Gonzalez-Sarmiento R., Ward L., Rea S.L., Ratajczak T., Walsh J.P., Ralston S.H. Common susceptibility alleles and SQSTM1 mutations predict disease extent and severity in a multinational study of patients with Paget's disease. J. Bone Miner. Res. 2013;28:2338–2346. doi: 10.1002/jbmr.1975. [DOI] [PubMed] [Google Scholar]
- 21.Duran A., Serrano M., Leitges M., Flores J.M., Picard S., Brown J.P., Moscat J., Diaz-Meco M.T. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev. Cell. 2004;6:303–309. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 22.Najat D., Garner T., Hagen T., Shaw B., Sheppard P.W., Falchetti A., Marini F., Brandi M.L., Long J.E., Cavey J.R., Searle M.S., Layfield R. Characterization of a non-UBA domain missense mutation of sequestosome 1 (SQSTM1) in Paget's disease of bone. J. Bone Miner. Res. 2009;24:632–642. doi: 10.1359/jbmr.081204. [DOI] [PubMed] [Google Scholar]
- 23.Chamoux E., Couture J., Bisson M., Morissette J., Brown J.P., Roux S. The p62 P392L mutation linked to Paget's disease induces activation of human osteoclasts. Mol. Endocrinol. 2009;23:1668–1680. doi: 10.1210/me.2009-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garner T.P., Long J., Layfield R., Searle M.S. Impact of p62/SQSTM1 UBA domain mutations linked to Paget's disease of bone on ubiquitin recognition. Biochemistry. 2011;50:4665–4674. doi: 10.1021/bi200079n. [DOI] [PubMed] [Google Scholar]
- 25.Bolland M.J., Tong P.C., Naot D., Callon K.E., Wattie D.J., Gamble G.D., Cundy T. Delayed development of Paget's disease in offspring inheriting SQSTM1 mutations. J. Bone Miner. Res. 2007;22:411–415. doi: 10.1359/jbmr.061204. [DOI] [PubMed] [Google Scholar]
- 26.Gianfrancesco F., Rendina D., Di Stefano M., Mingione A., Esposito T., Merlotti D., Gallone S., Magliocca S., Goode A., Formicola D., Morello G., Layfield R., Frattini A., De Filippo G., Nuti R., Searle M., Strazzullo P., Isaia G., Mossetti G., Gennari L. A nonsynonymous TNFRSF11A variation increases NFkappaB activity and the severity of Paget's disease. J. Bone Miner. Res. 2012;27:443–452. doi: 10.1002/jbmr.542. [DOI] [PubMed] [Google Scholar]
- 27.Long J., Garner T.P., Pandya M.J., Craven C.J., Chen P., Shaw B., Williamson M.P., Layfield R., Searle M.S. Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-kappaB signalling. J. Mol. Biol. 2010;396:178–194. doi: 10.1016/j.jmb.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 28.Isogai S., Morimoto D., Arita K., Unzai S., Tenno T., Hasegawa J., Sou Y.S., Komatsu M., Tanaka K., Shirakawa M., Tochio H. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J. Biol. Chem. 2011;286:31864–31874. doi: 10.1074/jbc.M111.259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavey J.R., Ralston S.H., Hocking L.J., Sheppard P.W., Ciani B., Searle M.S., Layfield R. Loss of ubiquitin-binding associated with Paget's disease of bone p62 (SQSTM1) mutations. J. Bone Miner. Res. 2005;20:619–624. doi: 10.1359/JBMR.041205. [DOI] [PubMed] [Google Scholar]
- 30.Langston A.L., Campbell M.K., Fraser W.D., MacLennan G.S., Selby P.L., Ralston S.H., Group P.T. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget's disease of bone. J. Bone Miner. Res. 2010;25:20–31. doi: 10.1359/jbmr.090709. [DOI] [PubMed] [Google Scholar]
- 31.Cavey J.R., Ralston S.H., Sheppard P.W., Ciani B., Gallagher T.R., Long J.E., Searle M.S., Layfield R. Loss of ubiquitin binding is a unifying mechanism by which mutations of SQSTM1 cause Paget's disease of bone. Calcif. Tissue Int. 2006;78:271–277. doi: 10.1007/s00223-005-1299-6. [DOI] [PubMed] [Google Scholar]
- 32.Long J., Gallagher T.R., Cavey J.R., Sheppard P.W., Ralston S.H., Layfield R., Searle M.S. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J. Biol. Chem. 2008;283:5427–5440. doi: 10.1074/jbc.M704973200. [DOI] [PubMed] [Google Scholar]
- 33.Strachan J., Roach L., Sokratous K., Tooth D., Long J., Garner T.P., Searle M.S., Oldham N.J., Layfield R. Insights into the molecular composition of endogenous unanchored polyubiquitin chains. J. Proteome Res. 2012;11:1969–1980. doi: 10.1021/pr201167n. [DOI] [PubMed] [Google Scholar]
- 34.Corral-Gudino L., Borao-Cengotita-Bengoa M., Del Pino-Montes J., Ralston S. Epidemiology of Paget's disease of bone: a systematic review and meta-analysis of secular changes. Bone. 2013;55:347–352. doi: 10.1016/j.bone.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 35.Wright T., Rea S.L., Goode A., Bennett A.J., Ratajczak T., Long J.E., Searle M.S., Goldring C.E., Park B.K., Copple I.M., Layfield R. The S349T mutation of SQSTM1 links Keap1/Nrf2 signalling to Paget's disease of bone. Bone. 2013;52:699–706. doi: 10.1016/j.bone.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 36.Ueki K., Kondo T., Tseng Y.H., Kahn C.R. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10422–10427. doi: 10.1073/pnas.0402511101. [DOI] [PMC free article] [PubMed] [Google Scholar]