

Significance
Hippocampal hyperactivity predicts psychosis and may disrupt aspects of cognition in schizophrenia. Here, we use interneuron precursor transplants in mice lacking cyclin D2 (Ccnd2) to test links between hippocampal GABAergic interneurons and psychosis-relevant phenotypes. Ccnd2-null mice show parvalbumin interneuron deficits and increased in vivo hippocampal excitatory neuron spiking and metabolic activity. This hippocampal disinhibition is associated with cognitive deficits and excess dopamine activity. Transplanting interneuron progenitors derived from the embryonic medial ganglionic eminence into adult hippocampus mitigates these abnormalities. This study thus provides a paradigm for elucidating mechanisms by which limbic cortical interneuron hypofunction may contribute to cognitive deficits and dopamine dysregulation in psychosis. The sustained efficacy of the transplants supports a rationale for targeting hippocampal GABA interneurons with novel therapies for psychosis.
Keywords: parvalbumin, temporal lobe-dependent cognition, neural stem cell therapy, functional magnetic resonance imaging, contextual fear conditioning
Abstract
GABAergic interneuron hypofunction is hypothesized to underlie hippocampal dysfunction in schizophrenia. Here, we use the cyclin D2 knockout (Ccnd2−/−) mouse model to test potential links between hippocampal interneuron deficits and psychosis-relevant neurobehavioral phenotypes. Ccnd2−/− mice show cortical PV+ interneuron reductions, prominently in hippocampus, associated with deficits in synaptic inhibition, increased in vivo spike activity of projection neurons, and increased in vivo basal metabolic activity (assessed with fMRI) in hippocampus. Ccnd2−/− mice show several neurophysiological and behavioral phenotypes that would be predicted to be produced by hippocampal disinhibition, including increased ventral tegmental area dopamine neuron population activity, behavioral hyperresponsiveness to amphetamine, and impairments in hippocampus-dependent cognition. Remarkably, transplantation of cells from the embryonic medial ganglionic eminence (the major origin of cerebral cortical interneurons) into the adult Ccnd2−/− caudoventral hippocampus reverses these psychosis-relevant phenotypes. Surviving neurons from these transplants are 97% GABAergic and widely distributed within the hippocampus. Up to 6 mo after the transplants, in vivo hippocampal metabolic activity is lowered, context-dependent learning and memory is improved, and dopamine neuron activity and the behavioral response to amphetamine are normalized. These findings establish functional links between hippocampal GABA interneuron deficits and psychosis-relevant dopaminergic and cognitive phenotypes, and support a rationale for targeting limbic cortical interneuron function in the prevention and treatment of schizophrenia.
Precursors of most γ-aminobutyric acid (GABA)-releasing interneurons of the cerebral cortex and the hippocampus originate in the embryonic medial ganglionic eminence (MGE) (1–3). A subpopulation of MGE-derived cells differentiates into fast-spiking, parvalbumin-expressing (PV+) interneurons that tightly regulate the activity and synchronization of cortical projection neurons (2, 4). Structural and functional deficits in PV+ interneurons are hypothesized as a pathophysiological mechanism in schizophrenia and psychotic disorders (4–6).
Although psychotic disorders are clearly heterogeneous in etiology, disinhibition within temporolimbic cortical circuits is postulated as a core pathophysiology underlying positive symptoms (e.g., delusions and hallucinations) and a subset of cognitive disturbances that manifest with psychosis (4, 5, 7). Postmortem studies of brains from individuals with psychotic disorders show reduced molecular markers of the number and/or function of PV+ interneurons in the hippocampus (6, 8). Consistent with these observations, basal metabolic activity in the hippocampus, as measured with functional magnetic resonance imaging (fMRI), is increased in schizophrenia, a phenotype that predicts psychosis and positive symptom severity (5, 7). This abnormal resting activity is postulated to underlie abnormal recruitment of hippocampal circuits during cognitive performance (5, 9). Striatal dopamine (DA) release capacity is also increased and correlated with positive symptoms in schizophrenia and its risk states (10, 11). Importantly, hippocampal hyperactivity may contribute to DA dysregulation (12), because rodent studies show that caudoventral hippocampal (in the primate, anterior hippocampal) efferents regulate the activity of DA neurons and medial striatal DA release (13, 14).
Thus, converging evidence implicates hippocampal disinhibition in the abnormal striatal DA transmission and cognitive impairment in schizophrenia. However, the role of hippocampal inhibitory interneurons in psychosis-relevant circuitry remains to be established. To this end, we used the cyclin D2 (Ccnd2) knockout mouse model (15), which displays a relatively selective deficit in cortical PV+ interneurons, and transplantation of interneuron precursors from the MGE to elucidate relationships between reduced hippocampal GABA interneuron function and multiple psychosis-relevant phenotypes, and to explore a novel treatment strategy for psychosis.
Results
PV+ Cell Density and Functional Deficits in Ccnd2−/− Hippocampus.
CCND2 is a G1-phase active cell cycle protein expressed within the subventricular zone of the MGE. A loss-of-function mutation of Ccnd2 leads to reduced proliferation in the MGE and lower PV+ interneuron density in the cortex, while not affecting densities of glutamatergic projection neurons or other interneuron subtypes of the cerebral cortex (15, 16). To determine the impact of the PV+ neuron deficit on hippocampal output, we compared PV+ interneuron density, GABA-mediated synaptic inhibition, and in vivo activity of presumptive projection neurons in the hippocampus of Ccnd2−/− mice and their wild-type (Ccnd2+/+) littermates. Stereology-based quantification showed a significant reduction in PV+ interneuron density in hippocampus, particularly in CA1 (Fig. 1 A and B and Fig. S1). The apparent PV+ neuron deficit in these Ccnd2−/− mice showed regional heterogeneity, with no significant differences between genotypes in the medial prefrontal cortex (mPFC) (Fig. S1). In parallel, GABA-mediated miniature inhibitory postsynaptic currents (mIPSC) frequencies at projection neurons were decreased in the Ccnd2−/− hippocampus (Fig. 1 C and D) but not in mPFC (Fig. S1B). Indices of postsynaptic GABA function (e.g., mIPSC amplitude and kinetics) and excitatory synaptic transmission appeared unchanged in Ccnd2−/− mice (Fig. S2). Consistent with the loss of inhibitory input observed in vitro, a reduction in the density of spontaneously active presumptive inhibitory interneurons was observed in Ccnd2−/− mice in vivo, a deficit corresponding with increased in vivo spike activity of excitatory projection neurons (Fig. 1 E–G and Fig. S3). Overall, these data suggest regional heterogeneity in the deficits in PV+ interneuron number and function in the Ccnd2−/− cortex, with a relatively greater deficit in hippocampus leading to disinhibition of hippocampal outputs in vivo.
Fig. 1.

Impaired inhibition in the hippocampus of Ccnd2−/− mice. (A) Representative sections of the rostral hippocampus showing PV+ neuron distribution. Right shows higher-magnification of the CA1 region outlined in yellow on Left. (B) PV+ interneuron density in CA1 is reduced in Ccnd2−/− mice (t6 = 3.8, P < 0.05; n = 4). (C) Representative traces from whole-cell voltage-clamp recordings showing GABA-mediated mIPSCs in CA1 pyramidal neurons in Ccnd2+/+ (Upper) and Ccnd2−/− (Lower) mice. (D) mIPSC frequency in CA1 pyramidal cells is reduced in Ccnd2−/− mice (t19 = 2.7, P < 0.05; n = 9–12). (E) Schematic showing starting track for in vivo single-unit recordings in caudal hippocampus. (F) In vivo recording data showing fewer hippocampal units with spike characteristics of GABAergic (including PV+) interneurons in Ccnd2−/− mice (t11 = 3.9, P < 0.005; n = 6–7). (G) Increased in vivo spike activity (average firing rate) of hippocampal excitatory projection neurons in Ccnd2−/−mice (t11 = 4.1, P < 0.005). (H) MRI images of horizontal brain sections showing the hippocampal ROI. (I) Increased hippocampal CBV in Ccnd2 −/− mice (t14 = 3.3, P < 0.01; n = 8). Box plots show interquartile range; whiskers extend to the outermost data points within 1.5x of the interquartile range. Means are marked by dotted lines and squares, medians by solid lines, and outliers by solid circles. *P < 0.05, independent t tests.
fMRI Shows Increased in Vivo Basal Metabolic Activity in Ccnd2−/− Hippocampus.
Whether reduced GABA-mediated inhibition increases hippocampal metabolic activity in vivo was assessed by using basal cerebral blood volume (CBV) maps of the mouse brain from gadodiamide-contrast fMRI (Fig. 1H and Fig. S4A). This method is used to characterize hippocampal hypermetabolism as a biomarker for psychosis (7). Relative to Ccnd2+/+ littermates, Ccnd2−/− mice showed increased basal CBV in the hippocampus (Fig. 1I). Basal CBV in the cerebellum was not different (Fig. S4B), despite an apparent deficit in cerebellar stellate neurons in the Ccnd2−/− mouse (17). We thus used the cerebellum as a reference region for analyses of additional brain regions and hippocampal subregions. Consistent with the regional variation in the cortical PV+ interneuron deficit, Ccnd2−/− mice showed increased basal CBV in the hippocampus but not in the mPFC or sensorimotor cortex (Fig. S4). Thus, whereas loss of Ccnd2 function has multiple developmental effects, decreases in PV+ interneuron function and increased hippocampal activity comprise a prominent phenotype cluster associated with this mutation.
Impaired Hippocampus-Mediated Cognition in Ccnd2−/− Mice.
The increased basal or resting activity in hippocampus in schizophrenia patients may compromise recruitment of this region in response to cognitive demands (5). Thus, we evaluated contextual fear conditioning (18), a cognitive process that recruits and depends on the hippocampus in rodents. Conditioning took place in one distinct context (training context) by using five pairings of tones (conditioned stimulus, CS+) and shock (unconditioned stimulus, US+). Task parameters, including the use of highly contrasting contexts, were adjusted to produce robust context-dependent conditioned behavior in the wild-type mice (Fig. S5) and minimize contribution of the adult neurogenesis deficit in the dentate gyrus of Ccnd2−/− mice (19, 20). Twenty-four hours later, we measured freezing to the tone CS+ (without shock), an amygdala-dependent, largely hippocampus-independent response (18). During subsequent CS+ presentations in the novel context, we monitored posttone freezing, a behavior that is sensitive to hippocampal lesions (21). Six hours after the tone CS+ test, we tested memory for the training context. Ccnd2−/− mice showed robust and selective deficits in the hippocampus-dependent components of learning and memory in this task (Fig. 2). This impairment was not associated with qualitative changes in sensory or motor functions or shock sensitivity (Table S1).
Fig. 2.
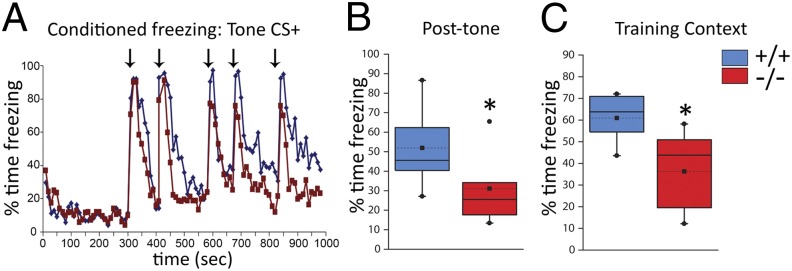
Impaired hippocampus-dependent cognition in Ccnd2−/− mice. Responses to tone CS+ and training context 24–30 h following aversive conditioning. (A) Conditioned behavioral freezing to the tone CS+ presented in a novel context (arrows indicate CS+ presentation). Both Ccnd2+/+ (blue line) and Ccnd2−/− (red line) mice show minimal baseline freezing (<300 s) and robust conditioned freezing to the first tone CS+ (t16 = 1.3, P > 0.25; n = 9 per genotype). On subsequent tone CS+ presentations, Ccnd2−/− mice show less posttone freezing, a hippocampus-dependent behavior. (B) Average posttone freezing in the novel context (t15.8 = 2.3, P < 0.05). (C) Ccnd2−/− mice show reduced context-conditioned freezing (t16 = 2.3, P < 0.05). The mixed repeated measures ANOVA showed a significant effect of genotype (F1,16 = 14.4, P < 0.01). Box plot parameters are described in Fig. 1. *P < 0.05, planned comparisons with independent t tests.
Increases in Mesolimbic DA Neuron Population Activity and Responsiveness to Amphetamine in Ccnd2−/− Mice.
Projections from the ventral hippocampus to the basal ganglia regulate ventral tegmental area (VTA) DA neuron activity and responses to amphetamine (AMPH). Ventral hippocampal activation increases DA neuron population activity via disinhibitory circuits involving GABA neurons in the nucleus accumbens, ventral pallidum, and VTA (14, 22). We thus predicted that the hippocampal disinhibition in Ccnd2−/− mice would lead to increased VTA DA neuron activity. In vivo recordings within the VTA of anesthetized mice (Fig. 3A and Fig. S6 A and B) revealed increases in spontaneously active DA neurons in Ccnd2−/− mice (Fig. 3B).
Fig. 3.
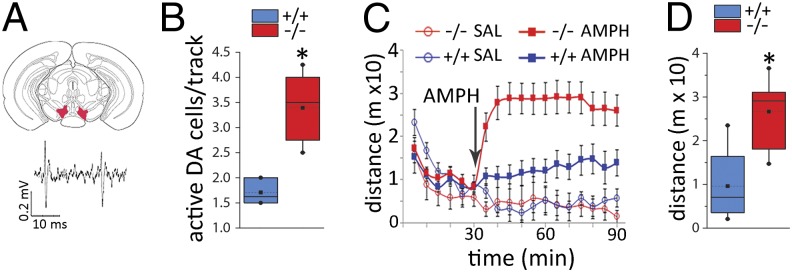
Increased VTA dopamine neuron population activity and hyperresponsiveness to AMPH in Ccnd2−/− mice. (A) Schematic of coronal section through the VTA recording region (Upper) and representative extracellular trace of VTA DA neuron spiking patterns in vivo (Lower) (see also Fig. S6). (B) The number of spontaneously active DA cells is increased in Ccnd2−/− mice (t8.8 = 6.3, P < 0.01; n = 6–7). (C) Locomotor activity before and after injection of saline or AMPH (2.0 mg/kg, i.p.) (n = 4 and 13 per genotype for saline and AMPH, respectively; arrow indicates time of injection). The pattern shows an interaction between genotype and dose (F1,29 = 6.0, P < 0.05), genotype and time (F1,29 = 11.2, P < 0.01), and a trend for a three-way interaction (P = 0.078) (n = 4 per genotype for saline; 10–14 per genotype for AMPH). Planned comparisons show no differences between genotypes before injection (t31 = 0.2, P > 0.25) or following saline (t6 = 0.13, P > 0.25, drug groups combined). (D) Average post-AMPH locomotion is increased in Ccnd2−/− mice (t23 = 4.5, P < 0.001). Box plot parameters are described in Fig. 1. *P < 0.05, independent t tests.
By modulating activity within the limbic basal ganglia, ventral hippocampal activity can drive medial striatal DA release (13, 14) and behaviors mediated by this system, including the locomotor response to AMPH (23, 24). Consistent with this function of the hippocampus, Ccnd2−/− mice showed a dose-dependent increase in AMPH-induced locomotion (Fig. 3 C and D and Fig. S6C), a difference blocked by systemic administration of a DA D2 receptor antagonist (Fig. S6D). Importantly, this augmented response in the Ccnd2−/− mice was eliminated by partial lesions of the caudal (including caudoventral) hippocampus, but not by lesions of the overlying parietal cortex (Fig. S6E). Together these data indicate that the augmented response to AMPH in the Ccnd2−/− mice is DA mediated and requires hippocampal activity.
Normalizing Effects of MGE-Derived Interneuronal Precursors Transplanted into Adult Hippocampus.
The data above support the hypothesis that decreased PV+ interneuron function in the hippocampus contributes to the augmented DA system activity and cognitive impairment in Ccnd2−/− mice. However, because the Ccnd2 mutation exerts its effects early in development, one must manipulate hippocampal GABA interneuron population in adulthood to test this hypothesis. Previous studies have shown that MGE-derived cells transplanted into the postnatal cortex differentiate into GABA+ neurons with interneuron-like morphology. These new GABA neurons enhance synaptic inhibition of the surrounding neurons and attenuate seizures and related memory deficits (3, 25). We thus quantified the distribution and phenotypes of neurons derived from embryonic MGE cells transplanted into the hippocampus of adult Ccnd2−/− mice and examined their impact on hippocampal metabolic activity, hippocampus-mediated cognition, and VTA DA neuron activity. Cells were dissociated from the ventral MGE of green fluorescent protein (GFP)-expressing mouse embryos at embryonic day 15.5 (Fig. 4A) and transplanted bilaterally into the ventral hippocampus of 6- to 8-wk-old Ccnd2−/− mice. Control Ccnd2−/− mice received ventral hippocampal injections of killed (freeze-thawed) MGE cells.
Fig. 4.

MGE-derived progenitor cells transplanted into the adult hippocampus mature into GABAergic interneurons. (A) Summary of the transplant approach. From GFP+ embryos at embryonic day 15.5 (Top), cells were dissociated from the ventral MGE (Middle, solid white arrow shows dissected area) and transplanted bilaterally into the hippocampi of adult Ccnd2−/− mice. (B) Representative coronal section through the hippocampus of an adult host 6 mo after transplantation of active MGE cells showing surviving GFP+ cells with interneuron-like morphology. Images show progressive magnifications of boxed regions. (C and D) Transplanted GFP+ cells in the mature hippocampus showing neurons coexpressing GABA (97% of GFP+ neurons) (C) and PV (D). (E) Percentage of total GFP+ cells colabeled with PV or somatostatin (SST). Colabeling was assessed separately for PV, SST, and GABA. Remaining portion (green slice) includes a mixture of GABAergic neuron subtypes.
At 4–6 mo after transplantation, brains with active (live-cell) MGE transplants contained an average of 3,706 [±573 (SEM); n = 4] surviving (GFP+) cells. These cells showed features of mature interneurons (Fig. 4 B–D) and were dispersed throughout the longitudinal axis of the hippocampus. Nearly all of the GFP+ cells in the hippocampus were GABAergic (97%), with ∼56% and 35% expressing PV and somatostatin, respectively (Fig. 4E). The new neurons were seen primarily in strata oriens and pyramidale of CA fields. Remarkably, MGE transplants normalized multiple aspects of the Ccnd2−/− phenotype. Ccnd2−/− mice with MGE transplants showed reduced hippocampal CBV, to levels similar to those observed in wild-type littermates (Fig. 5A). Ccnd2−/− mice with active MGE transplants also showed improved hippocampus-dependent cognition (Fig. 5 B and C), and normalization of DA neuron population activity (Fig. 5D) and the response to AMPH (Fig. 5 E and F).
Fig. 5.

Active MGE transplants into adult hippocampus normalize multiple psychosis-relevant neural, behavioral, and cognitive phenotypes in Ccnd2−/− mice. (A) Hippocampal CBV (expressed relative to cerebellum) in Ccnd2−/− mice with control (killed cells; red stipple) or active (live cells; gray stipple) transplants derived from the ventral MGE. Active transplants in Ccnd2−/− mice lower CBV (t4 = 20.2, P < 0.01; n = 3 per transplant condition) to CBV values similar to Ccnd2+/+ mice (blue reference line; data from Fig. S4C). (B and C) Hippocampus-dependent cognition assessed using the fear conditioning paradigm described for Fig. 2, showing a significant effect of transplant condition (F1,11 = 5.3, P < 0.05; n = 3–8). Active MGE transplants lead to increased conditioned posttone freezing (tested in the novel context) (t9 = 3.1, P < 0.01) (B) and improved learning and/or memory of the training context (t9 = 2.8, P < 0.05) (C). (D–F) DA neuron VTA population activity and locomotor responses to AMPH are normalized (reduced) in Ccnd2−/− mice with active MGE cell transplants. (D) Spontaneously active DA neurons in the VTA (active < control: t3 = 4.6, P < 0.01; n = 2–3). (E) Time course of locomotion in Ccnd2−/− mice with control (red squares; red dashed line) or active (gray squares; gray dashed line) MGE-derived transplants. Also shown are reference data for nontransplanted Ccnd2+/+ mice (solid blue line, triangles) and Ccnd2−/− mice (solid red line, triangles); vertical arrow indicates time of AMPH injection. (F) Average post-AMPH locomotor activity (active < control; t12 = 2.4, P < 0.05; n = 5–9). Data in A–D and F are box plots as described for Fig. 1; solid blue horizontal lines show expected mean values for Ccnd2+/+ mice (derived from data in Figs. 1–3 and Fig. S4C). *P < 0.05, one-tailed independent t tests.
Previous studies reported a neurogenesis deficit in the adult dentate gyrus of Ccnd2−/− mice (26, 27). We thus examined the effects of the transplanted interneurons on newly born neurons in the adult Ccnd2−/− dentate by quantifying cells expressing doublecortin (DCX+), a marker of immature neurons. Relative to wild-type colonymates, both control- and active MGE-transplanted Ccnd2−/− mice showed a similarly marked reduction in DCX+ dentate gyrus neurons (Fig. S7). Thus, the effects of MGE-derived cells transplanted into the caudoventral hippocampus are not likely mediated by a change in adult hippocampal neurogenesis.
Discussion
The PV+ interneuron deficit in Ccnd2−/− mice is associated with adult neurobehavioral phenotypes relevant to psychosis, including increased hippocampal basal metabolic activity as assessed with fMRI, increased midbrain DA neuron activity, augmented response to AMPH, and disruption of cognitive processes that recruit and depend on the hippocampus. Partial restoration of GABAergic interneurons to the adult hippocampus of Ccnd2−/− mice normalizes these interrelated in vivo phenotypes, thus establishing the plausibility of causal links between hippocampal interneuron hypofunction and specific psychomotor and cognitive disturbances in psychotic disorders.
Cortical PV+ interneuron pathology, particularly in the hippocampus, is a frequent feature of rodent models of schizophrenia pathogenesis and may be an outcome common to multiple susceptibility pathways in this disorder (6, 7, 12, 28). The current study examines the impact of the loss of a major regulator of cortical PV+ interneuron progenitor proliferation. Our findings are consistent with postmortem studies implicating altered development of cortical interneurons in schizophrenia patients (6, 29, 30). Cyclin D2 expression in the MGE subventricular zone is important for its selective regulation of PV+ interneuron precursors (15) and raises the possibility of a CCND2-related mechanism underlying the regional heterogeneity in cortical PV+ interneurons, and greater vulnerability of the hippocampal population. CCND2 is robustly expressed in human fetal brain (16), and its impacts on neural structure and function are of potential relevance to developmental brain disorders. Whereas loss of Ccnd2 leads to a decrease in PV+ interneuron numbers across the cerebral cortex, the present quantitative neuroanatomical study suggests a greater impact of this deficit in the hippocampus. Taken together with the effects of the MGE transplants, the data indicate that hippocampal GABAergic interneurons are important for a subset of the behavioral and neurophysiological effects of the Ccnd2 mutation that is particularly relevant to psychosis and point to the potential importance of mechanisms regulating cortical interneuron development in the pathogenesis of schizophrenia (6).
Although the Ccnd2−/− mutation has quite selective effects on brain structure and function, the potential contributions of neurophenotypes other than the hippocampal PV+ interneuron deficit must be considered. Perhaps most relevant to the cognitive domain tested in this study is the neurogenesis deficit in the adult Ccnd2−/− dentate gyrus (26, 27) (Fig. S7). The significance of “adult neurogenesis” to behavioral health remains under intense investigation (20, 26, 31), and we do not rule out that this deficit may impact cognition in Ccnd2−/− mice (e.g., ref. 32). However, the present study indicates that adding new MGE-derived GABA interneurons to hippocampal CA fields of adult Ccnd2−/− mice can improve cognition without stimulating neurogenesis. Ccnd2−/− mice also show reduced cerebral cortical volume (16) and a deficit in cerebellar stellate neurons (17). Although we expect these abnormalities to have effects on cognition and behavior, we and others have observed no evidence for qualitative sensory or motor (including cerebellar) phenotypes that would confound psychomotor or cognitive assessments used in this study. Notably, in schizophrenia, and in several rodent models of genetic susceptibility, hippocampal interneuron deficits occur in the context of cerebral cortical thinning, cerebellar pathology, and cell metabolic abnormalities (6, 28, 29, 33–35). We thus consider the effects of the hippocampus-targeted transplants demonstrated in the Ccnd2−/− model to be of particular relevance to schizophrenia in part because they occur on a background of other schizophrenia-relevant neuropathologies (see also ref. 36).
Increased resting metabolic activity in the hippocampus and adjacent medial temporal cortex is characteristic of schizophrenia and its risk states (7). Interneuron dysfunction has been hypothesized to be both a potential driver and consequence of this abnormal state, in part through dysregulation of glutamatergic signaling (4, 5, 7, 37). Our findings are consistent with a deficit in PV+ interneurons driving abnormal hippocampal activity, and highlight a need for additional studies on mechanisms linking interneuron function to local cortical metabolism and hemodynamics. Our in vitro and in vivo electrophysiological data indicate that a disinhibition of hippocampal projection neurons may contribute to the fMRI hypermetabolism phenotype in the Ccnd2−/− mouse. Although the potential effects of the Ccnd2 mutation on basal ganglia interneurons should also be studied, our current data argue for a model in which a deficit in the number or function in MGE-derived GABAergic interneurons in the hippocampus dysregulates hippocampal projections to limbic structures and the basal ganglia, leading to multiple phenotypes associated with schizophrenia including increased basal or resting hippocampal activity, increased striatal DA neurotransmission, and related psychomotor and cognitive disturbances (5–7, 9, 12) (Fig. S8).
In the Ccnd2−/− mouse model, psychosis-relevant phenotypes derived in part from a developmental PV+ interneuron deficit can be reversed in adulthood with transplantation of MGE-derived cells into the hippocampus. We hypothesize that the remarkable, sustained impact of the transplants observed here is related to the source and developmental stage of the transplanted cells: the ventral MGE at mouse embryonic day 15.5. Our findings extend previous studies showing that transplanted MGE-derived GABA neurons establish a high degree of synaptic connectivity and induce plasticity in the host cortex, and have procognitive effects in disease models (3, 25, 38, 39). The impacts of the MGE transplants on DA system function observed here and in a recent report (36) further illustrate the potential of GABA interneuron precursor-rich transplants in experimental therapeutics in schizophrenia (3, 6). Indeed, determining the molecular and physiological changes induced by these transplants, GABA-related and otherwise, will lead to discovery of novel treatment targets for schizophrenia.
In conclusion, the current findings support the hypothesis that hippocampal PV+ interneurons play an important role in regulating DA neurotransmission and specific aspects of cognition, and provide a rationale for developing GABA interneuron transplant therapies for treatment-resistant psychosis associated with elevated hippocampal activity. Of equal or greater importance for experimental therapeutics in schizophrenia will be to elucidate the mechanisms mediating the differentiation, survival, migration, connectivity, and function of MGE-derived neuronal precursors transplanted into the adult cortex.
Materials and Methods
Animals and Treatments.
All experiments were approved by the New York State Psychiatric Institute Animal Care and Use Committee in accordance with standards set by the National Institutes of Health Office of Laboratory Animal Welfare. Ccnd2 knockout mice (cf. ref. 15) were maintained on a C57BL/6J background; sex- and age-matched wild-type littermates (or colony-mates) were used as controls.
Immunohistochemistry and Neuron Quantification.
Histological and cell quantification protocols are fully described in SI Materials and Methods. Briefly, paraformadehyde-fixed brains were cryosectioned into 40-μm sections and collected serially into five equal sets according to the principles of systematic random sampling. Sets were processed with primary antibodies for parvalbumin (anti-mouse α-PV-235), somatostatin (anti-rat polyclonal; Chemicon/Millipore), GABA (anti-rabbit; Sigma-Aldrich), doublecortin (anti-goat; Santa Cruz Biotechnology), and/or GFP (anti-rabbit or -chicken; Molecular Probes/Invitrogen). Fluorescent secondary antibodies were used (Rhodamine Red, DyLight 488, Dylight 405, and DyLight 647; Jackson ImmunoResearch, diluted to 1:250 or 1:200). A subset of sections were stained for PV by using goat anti-mouse IgG (1:200) and a standard avidin-biotin-peroxidase reaction method. Region of interest (ROI) boundaries for the hippocampus, medial prefrontal cortex, and somatosensory and motor cortices were based on standard anatomical landmarks as described in SI Materials and Methods. The number of labeled neurons in the entirety of each ROI was estimated by using a modified fractionator method. Density was calculated by dividing the total cells counted by the ROI volume as determined with the Cavalieri estimator.
In Vitro Slice Electrophysiology.
Brains extracted from ketamine/xylazine-anesthetized Ccnd2−/− and sex-matched Ccnd2+/+ littermates, ages 3–6 wk, were processed for slice electrophysiology by using a standard protocol detailed in SI Materials and Methods. Whole-cell voltage-clamp recordings of identified pyramidal neurons used glass capillary pipettes (3-5 MΩ impedance) filled with a buffered CsCl solution to record GABAA currents or a buffered potassium gluconate to record excitatory currents. GABAergic mIPSCs, at a holding potential of −70 mV, were isolated by including tetrodotoxin (TTX, 1 µM), 2-amino-5-phosphonopentanoic acid (AP5, 50 µM) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 40 µM) to the perfusion solution and confirmed via blockade by gabazine (SR95531, 20 µM). Glutamatergic miniature excitatory postsynaptic currents were recorded at −80 mV, isolated by adding TTX and gabazine to the perfusion solution, and confirmed via blockade with CNQX and AP5. Data were filtered at 5 kHz, digitized at 10 kHz, and filtered offline by using a low-pass elliptic filter (1 kHz cutoff) to remove high frequency noise. Amplitude threshold for mPSC detection was set to three times the square root of mean of noise. Averages of the frequency, amplitude, 10–90% rise time, and 10–90% decay time of synaptic currents were calculated from at least 100 s of recording for each cell and averaged across cells for each subject.
In Vivo fMRI.
Design and procedures of imaging experiments were based on methods developed by Small and colleagues (cf. ref. 7). Briefly, four sets of axial T2-weighted images were acquired sequentially to generate 86 µm × 86 µm CBV maps, each set consisting of 24 images of 16 min each. Gadodiamide was injected (13 mmol/kg i.p.) after a precontrast set was acquired. CBV was mapped as the change in the transverse relaxation rate (R2) induced by the contrast agent. CBV maps were measured from steady-state T2-weighted images as CBV R2 = ln(Spre/Spost)/TE, where TE is the effective echo time, Spre is the signal before the contrast administration, and Spost is the signal after the contrast agent reaches steady state. The derived maps were normalized to the maximum four-pixel signal value of the posterior cerebral artery. Standard atlases were used to identify anatomical landmarks and define ROIs (SI Materials and Methods). The hippocampal ROI included the CA fields, subiculum, and dentate gyrus. For additional genotype comparisons of CBV across brain regions and of active versus control MGE transplants, CBV in the target region was expressed as a ratio to basal cerebellar CBV (Fig. S4).
In Vivo Hippocampal and VTA DA Neuron Recordings.
Stereotaxic surgery and single-unit extracellular recording and neuron sampling methods were performed in chloral hydrate- or urethane-anesthetized mice by adapting published methods (14) as described in detail in SI Materials and Methods. Briefly, glass electrodes (4–10 MΩ) filled with 2 M NaCl were used to sample four tracks spaced 150 µm apart within the VTA (Fig. 3A and Fig. S6A) or six tracks at 200 µm apart in the caudal hippocampus (Fig. 1E). VTA DA neurons and hippocampal pyramidal and nonpyramidal neurons and the spontaneous activity thereof were quantified as described in SI Materials and Methods (Figs. S3 and S6).
Behavioral Experiments.
Ccnd2−/− mice and sex-matched Ccnd2+/+ littermates 2.5–4 mo of age were used for behavioral testing. Mice were habituated to handling but were otherwise behaviorally naïve for each experiment except for some transplanted mice, for which contextual fear conditioning followed locomotor testing by a few days. See SI Materials and Methods for full details. Spontaneous and amphetamine-induced locomotor activity was measured in 17 × 17-inch open field boxes under standard lighting conditions. Mice were placed in open field for 30 min, after which, amphetamine (2 mg/kg dissolved in isotonic saline at 0.2 mg/mL) or saline was injected i.p. Activity (distance traveled) was measured for another 60 min. A mixed ANOVA design with genotype and drug as factors, and time (before or after injection) as the repeated measure, was used. This analysis was followed with planned Student t test comparisons of genotypes within drug condition separately for baseline and postinjection locomotion. Contextual fear conditioning methods were adapted from previous studies (e.g., refs. 19 and 21 as detailed in SI Materials and Methods. Briefly, mice were acclimated to the testing room 1 h before training. The training/testing apparatus was a chamber with shock grid floors placed within a sound-attenuating chamber. The inner chamber featured a distinctive combination of visuospatial, tactile, and odor cues, which together defined the context. On the day of training, mice were placed in one context (“training context”) and the CS+ consisting of a tone (85 dB, 20 s duration, 4.5 kHz) was presented at 300, 470, 580, 670, and 840 s. During the last second of each tone, a 0.7-mA scrambled current was delivered through the floor grid (US+). Mice were removed from the training context 140 s following the last CS-US presentation. Twenty-four hours later, mice were placed in a novel context and the tone CS+ was presented without shock at 300, 410, 580, 670, and 830 s. Six hours after the tone CS+ retrieval test, mice were placed in the training context for 600 s. Conditioned freezing, defined as absence of movement except for respiration, was quantified for the following epochs: (i) during the first presentation of the tone-CS+, (ii) for the 40–100 s following the offset of CS+ presentations 2–5 (posttone freezing; averaged for all five tones), and (iii) in the training context. Data were analyzed with a mixed ANOVA with retrieval phase as the repeated measure and genotype as the between-subjects factor. Independent t tests were used for planned comparisons between genotypes.
Transplantation of Progenitor Cells from the MGE.
Methods were adapted from those of Tyson and Anderson (3) as detailed in SI Materials and Methods. Wild-type dams impregnated by transgenic male mice expressing GFP (FVB.Cg-Tg (CAG-EGFP) B5Nagy/J Jackson Laboratories) were killed on embryonic day 15.5. GFP+ embryos, identified by fluorescence under 488-nm light, were placed in Hanks' balanced salt solution. Brains were removed and embedded in 4% (wt/vol) low melting point agarose (Invitrogen) and sliced to produce 250-μm sections from which the ventral two-thirds of the MGE was dissected (Fig. 4A, Middle). Donor cells were collected as described in SI Materials and Methods. For control transplants, cells were killed by repeated freeze-thaw cycles. Active (live-cell) (average density of 30,000 live cells per microliter) or control (killed-cell) suspensions were injected bilaterally into the caudoventral hippocampal CA1 of 6- to 8-wk-old mice by using a glass pipette (50-μm outer-tip diameter) connected to a nano-injector.
Statistics.
Except as noted, effects of genotype or transplants were tested with independent t tests. For experiments with multiple outcomes, multivariate analyses were also run to corroborate t tests. Results are reported as “significant” if P < 0.05 and as “not significant (n.s.)” if P > 0.25; trends are reported individually.
Supplementary Material
Acknowledgments
We thank Sara Steinfeld Tsantes, Bozena Slowinska-Castaldo, Karin H. Krueger, Alexei Shemyakin, John Castrillon, E. De La Cruz, Kenneth Hess, and Fang Hua for critical contributions, and Drs. Stephen G. Rayport and Helen Scharfman for sharing their laboratories. The New York State Psychiatric Institute Rodent Neurobehavioral Analysis Core provided assistance. This work was supported by Public Health Service Awards P01 NS048120 (to M.E.R., S.A.A., and H.M.) and K23MH090563 (to S. A. Schobel); the Fulbright (to A.I.G.), Sidney R. Baer, Jr. (H.M.), and Broitman (S. A. Small) Foundations; Brain & Behavior Research Foundation (to R.M. and S. A. Schobel); and New York State Office of Mental Health.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316488111/-/DCSupplemental.
References
- 1.Ma T, et al. Subcortical origins of human and monkey neocortical interneurons. Nat Neurosci. 2013;16(11):1588–1597. doi: 10.1038/nn.3536. [DOI] [PubMed] [Google Scholar]
- 2.Tricoire L, et al. A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J Neurosci. 2011;31(30):10948–10970. doi: 10.1523/JNEUROSCI.0323-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyson JA, Anderson SA. GABAergic interneuron transplants to study development and treat disease. Trends Neurosci. 2014;37(3):169–177. doi: 10.1016/j.tins.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakazawa K, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62(3):1574–1583. doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heckers S, Konradi C. In: Hippocampal Pathology in Schizophrenia. Behavioral Neurobiology of Schizophrenia and Its Treatment. Swerdlow NR, editor. Berlin: Springer; 2010. pp. 529–553. [DOI] [PubMed] [Google Scholar]
- 6.Inan M, Petros TJ, Anderson SA. Losing your inhibition: Linking cortical GABAergic interneurons to schizophrenia. Neurobiol Dis. 2013;53:36–48. doi: 10.1016/j.nbd.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schobel SA, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78(1):81–93. doi: 10.1016/j.neuron.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konradi C, et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res. 2011;131(1-3):165–173. doi: 10.1016/j.schres.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suazo V, Díez A, Tamayo P, Montes C, Molina V. Limbic hyperactivity associated to verbal memory deficit in schizophrenia. J Psychiatr Res. 2013;47(6):843–850. doi: 10.1016/j.jpsychires.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Howes OD, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69(8):776–786. doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillin O, Abi-Dargham A, Laruelle M. Neurobiology of dopamine in schizophrenia. Int'l Rev Neurobiology. 2007;78:1–39. doi: 10.1016/S0074-7742(06)78001-1. [DOI] [PubMed] [Google Scholar]
- 12.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32(9):507–513. doi: 10.1016/j.tips.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Legault M, Rompré PP, Wise RA. Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J Neurosci. 2000;20(4):1635–1642. doi: 10.1523/JNEUROSCI.20-04-01635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6(9):968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- 15.Glickstein SB, et al. Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development. 2007;134(22):4083–4093. doi: 10.1242/dev.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME. Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci. 2009;29(30):9614–9624. doi: 10.1523/JNEUROSCI.2284-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang G, Huard JM, Beitz AJ, Ross ME, Iadecola C. Stellate neurons mediate functional hyperemia in the cerebellar molecular layer. J Neurosci. 2000;20(18):6968–6973. doi: 10.1523/JNEUROSCI.20-18-06968.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256(5057):675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 19.Saxe MD, et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci USA. 2006;103(46):17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaholkowski P, et al. New hippocampal neurons are not obligatory for memory formation; cyclin D2 knockout mice with no adult brain neurogenesis show learning. Learn Mem. 2009;16(7):439–451. doi: 10.1101/lm.1459709. [DOI] [PubMed] [Google Scholar]
- 21.Quinn JJ, Wied HM, Ma QD, Tinsley MR, Fanselow MS. Dorsal hippocampus involvement in delay fear conditioning depends upon the strength of the tone-footshock association. Hippocampus. 2008;18(7):640–654. doi: 10.1002/hipo.20424. [DOI] [PubMed] [Google Scholar]
- 22.Xia Y, et al. Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J Neurosci. 2011;31(21):7811–7816. doi: 10.1523/JNEUROSCI.1504-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bardgett ME, Henry JD. Locomotor activity and accumbens Fos expression driven by ventral hippocampal stimulation require D1 and D2 receptors. Neuroscience. 1999;94(1):59–70. doi: 10.1016/s0306-4522(99)00303-6. [DOI] [PubMed] [Google Scholar]
- 24.White IM, Whitaker C, White W. Amphetamine-induced hyperlocomotion in rats: Hippocampal modulation of the nucleus accumbens. Hippocampus. 2006;16(7):596–603. doi: 10.1002/hipo.20189. [DOI] [PubMed] [Google Scholar]
- 25.Hunt RF, Girskis KM, Rubenstein JL, Alvarez-Buylla A, Baraban SC. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal behavior. Nat Neurosci. 2013;16(6):692–697. doi: 10.1038/nn.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ansorg A, Witte OW, Urbach A. Age-dependent kinetics of dentate gyrus neurogenesis in the absence of cyclin D2. BMC Neurosci. 2012;13:46. doi: 10.1186/1471-2202-13-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowalczyk A, et al. The critical role of cyclin D2 in adult neurogenesis. J Cell Biol. 2004;167(2):209–213. doi: 10.1083/jcb.200404181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlson GC, et al. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci USA. 2011;108(43):E962–E970. doi: 10.1073/pnas.1109625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gandal MJ, Nesbitt AM, McCurdy RM, Alter MD. Measuring the maturity of the fast-spiking interneuron transcriptional program in autism, schizophrenia, and bipolar disorder. PLoS ONE. 2012;7(8):e41215. doi: 10.1371/journal.pone.0041215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fung SJ, et al. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am J Psychiatry. 2010;167(12):1479–1488. doi: 10.1176/appi.ajp.2010.09060784. [DOI] [PubMed] [Google Scholar]
- 31.Groves JO, et al. Ablating adult neurogenesis in the rat has no effect on spatial processing: Evidence from a novel pharmacogenetic model. PLoS Genet. 2013;9(9):e1003718. doi: 10.1371/journal.pgen.1003718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song J, et al. Parvalbumin interneurons mediate neuronal circuitry-neurogenesis coupling in the adult hippocampus. Nat Neurosci. 2013;16(12):1728–1730. doi: 10.1038/nn.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeganeh-Doost P, Gruber O, Falkai P, Schmitt A. The role of the cerebellum in schizophrenia: From cognition to molecular pathways. Clinics (Sao Paulo) 2011;66(Suppl 1):71–77. doi: 10.1590/S1807-59322011001300009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Amelsvoort T, et al. Brain anatomy in adults with velocardiofacial syndrome with and without schizophrenia: Preliminary results of a structural magnetic resonance imaging study. Arch Gen Psychiatry. 2004;61(11):1085–1096. doi: 10.1001/archpsyc.61.11.1085. [DOI] [PubMed] [Google Scholar]
- 35.Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49(1-2):1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18(11):1193–1198. doi: 10.1038/mp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moghaddam B, Javitt D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37(1):4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Southwell DG, Froemke RC, Alvarez-Buylla A, Stryker MP, Gandhi SP. Cortical plasticity induced by inhibitory neuron transplantation. Science. 2010;327(5969):1145–1148. doi: 10.1126/science.1183962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka DH, Toriumi K, Kubo K, Nabeshima T, Nakajima K. GABAergic precursor transplantation into the prefrontal cortex prevents phencyclidine-induced cognitive deficits. J Neurosci. 2011;31(40):14116–14125. doi: 10.1523/JNEUROSCI.2786-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.