

Abstract
Objectives: To understand if there exists a functional interaction between arsenic trioxide and paclitaxel in vitro.
Materials and methods: HeLa and HCT116 (ρ53+/+ and ρ53−/−) cells were treated with As2O3 and/or paclitaxel for various times. Treated cells were collected for analyses using a combination of flow cytometry, fluorescence microscopy and Western blotting.
Results: Because As2O3 is capable of inhibiting tubulin polymerization and inducing mitotic arrest, we examined whether there existed any functional interaction between As2O3 and paclitaxel, a well‐known microtubule poison. Flow cytometry and fluorescence microscopy revealed that although As2O3 alone caused a moderate level of mitotic arrest, it greatly attenuated paclitaxel‐induced mitotic arrest in cells with p53 deficiency. Western blot analysis showed that As2O3 significantly blocked phosphorylation of BubR1, Cdc20, and Cdc27 in cells treated with paclitaxel, suggesting that arsenic compromised the activation of the spindle checkpoint. Our further studies revealed that the attenuation of paclitaxel‐induced mitotic arrest by As2O3 resulted primarily from sluggish cell cycle progression at S phase but not enhanced mitotic exit.
Conclusion: The observations that As2O3 has a negative impact on the cell cycle checkpoint activation by taxol should have significant clinical implications because the efficacy of taxol in the clinics is associated with its ability to induce mitotic arrest and subsequent mitotic catastrophe.
Introduction
Arsenic compounds have been used for medicinal purposes for thousands of years (1, 2). Recently, arsenic trioxide (As2O3) has been approved by the US Food and Drug Administration for treatment of acute promyelocytic leukaemia (APL). It is especially effective for treating leukaemia resistant to all‐trans retinoic acid (3, 4, 5). Extensive in vitro and in vivo studies show that in APL cells arsenic trioxide induces partial differentiation at low doses (0.1–0.5 µm) and apoptosis at high doses (1.0–2.0 µm) (3, 6, 7). Arsenic compounds also induce apoptosis in a variety of solid tumour cells, as well as leukaemic cells other than APL (8, 9, 10, 11, 12); also, As2O3 exhibits promising therapeutic properties in inhibiting tumour growth in an orthotopic prostate cancer model (13). For the past a few years, clinical trials have been initiated to test efficacy of arsenic compounds in treatment of solid tumours as well as lymphoid malignancies (14).
The mechanism by which arsenic compounds, including As2O3 and sodium arsenite [As(III)], induce apoptosis appears to be complicated. As(III) can activate c‐Jun NH2‐terminal kinases (15), perturb mitochondrial transmembrane potential, and activate caspase 3 (16, 17). Also, it is capable of producing reactive oxygen species, eliciting DNA damage responses, and slowing down cell‐cycle progression (18). Notably, As(III) can induce mitotic arrest and mitotic catastrophe in a variety of cells, including leukaemia cells and cells derived from solid tumours (1, 12, 19, 20), strongly suggesting its ability to target a common signalling pathway(s) in these cells. Supporting this notion, it has been shown that As(III) directly interferes with the function of tubulins (21, 22, 23); this potentially affects integrity of microtubules and mitotic spindles. Indeed, As(III) inhibits GTP‐induced formation of microtubules in vitro by acting as a non‐competitive inhibitor (21). It has been hypothesized that As(III) is capable of cross‐linking two vicinal cysteine residues (Cys‐12 and Cys‐213), which inactivates the GTP binding site (21).
Taxol is one of the most effective anti‐tumour drugs used in the clinic. It has been approved for treatment of a variety of human malignancies, including breast, ovarian, and non‐small cell lung cancers (24). Taxol stabilizes microtubules by binding to the β‐subunit of tubulin; this prevents the dynamic instability of mitotic spindles and leads to mitotic arrest and prolonged mitotic arrest often results in mitotic catastrophe (25). Because of their common properties in induction of mitotic arrest and apoptosis of cancer cells, As2O3 and taxol have been used in combination in clinical trials for treating stage III osteosarcoma and Ewing sarcoma (1, 26). Intriguingly, a previous study has suggested that when they are used in combination in a cell culture system, As(III) and paclitaxel behave antagonistically (27).
Given the clinical importance of As2O3 and taxol, we have carefully examined the effect of these compounds on cell‐cycle progression as well as on spindle checkpoint activation. We demonstrated that As2O3 suppressed mitotic arrest induced by paclitaxel and interfered with paclitaxel‐induced activation of BubR1, suggesting antagonism between As2O3 and taxol. The compromised mitotic arrest resulted from sluggish cell‐cycle progression rather than accelerated mitotic exit. We also observed that the antagonistic effect on mitotic arrest and cell death was exacerbated by p53 deficiency.
Materials and methods
Cell culture and treatment
HeLa cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in dishes or on Lab‐Tek II chamber slides (Fisher Scientific, Pittsburgh, PA, USA) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS) with 5% CO2 at 37 °C. HCT116 cell lines (p53+/+ and p53−/–) generously provided by Dr Bert Vogelstein (Johns Hopkins University, Baltimore, MD, USA) were grown in McCoy's 5a Medium Modified supplemented with 10% FBS at 37 °C under 5% CO2 atmosphere.
As2O3 (Sigma Chemical Co., St. Louis, MO, USA) was dissolved in 1.0 mol/l NaOH and diluted in growth medium without FBS to make stock solution at a concentration of 50 mm. Stock solutions of paclitaxel and nocodazole (Sigma Chemical Co.) were prepared in dimethyl sulphoxide (DMSO). Maximal final concentration of DMSO used in cell culture never exceeded 0.1%. To follow cell‐cycle progression, HeLa cells were pulsed with 0.1 mm (final concentration) bromodeoxyuridine (BrdU, Sigma) in the presence of various drugs, overnight. To synchronize cells in mitosis, they were cultured in medium containing 2 mm thymidine for 18 h, then released into fresh medium with no thymidine for 9 h; these cells were again cultured in medium with 2 mm thymidine for 18 h before releasing them into medium containing 50 ng/ml nocodazole for 9 h. Mitotic cells were collected by shake‐off.
Fluorescence microscopy
Cells after various treatments, were fixed in 4% paraformaldehyde in phosphate‐buffered saline (PBS) for 5 min on ice. Following fixation, they were permeabilized at room temperature for 10 min. Following blocking with 2% bovine serum albumin (BSA), the cells were incubated with anti‐human phosphorylated histone H3 (p‐histone H3) monoclonal antibody (PharMingen, BD Bioscience, San Jose, CA, USA) in PBS containing 2% BSA for 1 h at room temperature. They were then washed and stained with Alexa Fluor 488‐labelled secondary antibody (Invitrogen) at room temperature for 1 h in the dark; cells were finally stained with 4′,6‐diamidino‐2‐phenylindole (1 µg/ml, Fluka, St Louis, MO, USA). Fluorescence microscopy was performed on a Nikon microscope, and images were captured using a digital camera (Optronics MacroFire, Optronics, Goleta, CA, USA), using Image‐Pro Plus software.
Flow cytometry analysis
Cells were first fixed in 75% ethanol. They were then incubated with anti‐BrdU‐FITC antibody (Becton Dickinson, Franklin Lakes, NJ, USA) in a staining solution [PBS with 0.5% Tween 20 (v/v), 1% BSA (w/v), 0.5 mg/ml RNAse] at 4 °C overnight. DNA was subsequently stained with propidium iodide. Cell‐cycle distributions of various treatments were analysed on a Beckman Coulter® Epics XL‐MCL™ Flow Cytometer (Fullerton, CA, USA). Cell‐cycle distributions were analysed using Muticycle software (Phoenix Flow System, San Diego, CA, USA).
Western blot analysis
Cells were harvested and lysed in lysis buffer as previously described (28); lysates were centrifuged at 12 000 g for 15 min at 4 °C and supernatants were collected. Approximately equal amounts of protein were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS‐PAGE) followed by electro‐transfer to PVDF membranes. Protein blots were probed with antibodies to BubR1, securin (Novocastra, Newcastle, UK), Cdc27, cyclin B, and β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Specific signals were detected using horseradish peroxidase‐conjugated goat‐anti‐rabbit (or anti‐mouse) secondary antibodies (Sigma) and enhanced chemiluminescence reagents (Amersham Pharmacia Biotech, Pittsburgh, PA, USA).
Cell viability
Cell viability was assayed using the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay method. HCT116 cells (5 × 103 cells/well) were seeded in a 96‐well plate in sextuplicates. Arsenic trioxide was added to cell cultures at indicated concentrations, for 24 h. MTT(15 µl) was supplied to each well. After incubation at 37 °C for additional 4 h, the medium was removed. MTT formazan precipitates were dissolved in 100 µl of an SDS/dimethyl formamide solution for at least 1 h at 37 °C. Optical density of dissolved samples was measured at 570 nm using a plate reader.
Results
Taxol is capable of inducing mitotic arrest because of its ability to disrupt microtubule dynamics. Given that both taxol and arsenic trioxide [As(III)] are widely used in the clinic for cancer treatment, we examined whether these two compounds had a synergistic effect on causing mitotic arrest, as well as causing mitotic catastrophe. HeLa cells treated with or without paclitaxel and/or As(III) overnight were first examined for their morphology. After overnight treatment with paclitaxel, almost all treated cells changed their appearance. They detached themselves from the bottom of the culture plate and rounded‐up (Fig. 1a), consistent with the established role of taxol in induction of mitotic arrest. Cells treated with As(III) at high concentration (5 µm) also induced the rounded‐up phenotype in a small but significant fraction of cells (Fig. 1a) compared to that of untreated control cells. Interestingly, paclitaxel‐induced detachment of HeLa cells was partially blocked by co‐treatment with As(III); this effect was more pronounced when high concentration of As(III) was included in the taxol‐treated culture (Fig. 1a).
Figure 1.
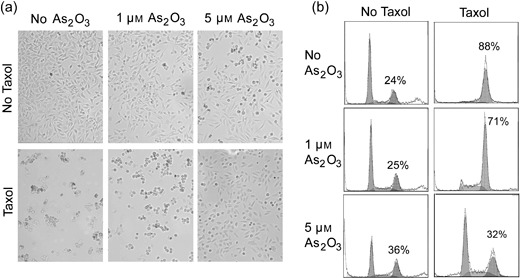
Arsenic trioxide (As2O3) blocks paclitaxel‐induced cell rounding and G2/M arrest. (a) HeLa cells were treated with 10 nm paclitaxel (taxol) and/or As2O3 at indicated concentrations for 24 h. Images were captured by light microscopy. Rounded‐up cells and adherent cells are morphologically distinguishable. (b) Flow cytometry analysis of cells treated with 10 nm paclitaxel and/or As2O3 at indicated concentrations for 16 h. Percentage of G2/M cells was indicated.
The fewer rounded‐up cells after co‐treatment with paclitaxel and As(III) suggests that arsenic may attenuate the effect of paclitaxel‐induced cell‐cycle arrest. To test this possibility, we stained HeLa cells that had been treated with paclitaxel and/or As(III) with propidium iodide. The stained cells were then analysed by flow cytometry to determine their cell‐cycle distributions. We observed that paclitaxel alone arrested a majority of cells at the G2/M stage. Paclitaxel‐induced mitotic arrest was partially suppressed when the cells were co‐treated with As(III), and inhibition of paclitaxel‐induced G2/M arrest by As(III) was dose‐dependent (Fig. 1b). Whereas around 70% of cells were in the G2/M phase when paclitaxel‐treated HeLa cells were supplemented with 1 µm of As(III), around 32% of paclitaxel‐treated cells were in G2/M when they were supplemented with 5 µm of As(III) (Fig. 1b).
Analysis of cell‐cycle distribution by flow cytometry, as shown in Fig. 1b, could not differentiate G2 cells from mitotic ones. Thus, we analysed HeLa cells treated with paclitaxel and/or As(III), for presence of p‐histone H3, a specific mitotic marker. Fluorescent microscopy and flow cytometry revealed that paclitaxel‐treated cells highly expressed p‐histone H3 (Fig. 2a–c). Treatment with As(III) also induced increased number, albeit much smaller than that of paclitaxel alone, of p‐histone H3‐positive cells (Fig. 2a–c). However, when HeLa cells were treated with paclitaxel in the presence of As(III), the p‐histone H3‐positive cell population was significantly reduced compared to that of paclitaxel treated cells (Fig. 2a–c). These observations thus indicate that As(III) suppressed paclitaxel‐induced mitotic arrest.
Figure 2.
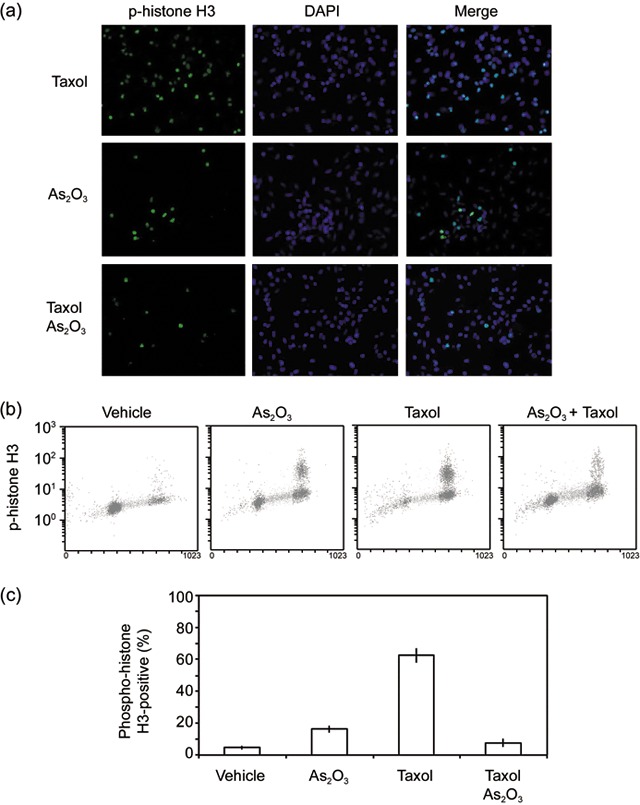
Arsenic trioxide (As2O3) inhibits paclitaxel‐induced mitotic arrest. (a) HeLa cells treated with or without 10 nm paclitaxel and/or 5 µm As(III) for 16 h were stained with the antibody to phorphorylated histone H3 (p‐histone H3, green), DAPI (blue) stained DNA. Representative cell images shown. (b) HeLa cells treated with or without 10 nm paclitaxel and/or 5 µm As(III) for 16 h were fixed and labelled with the antibody to p‐histone H3 for flow cytometry analysis. (c) Flow cytometry data as shown in B were quantified from three independent experiments.
We then analysed spindle checkpoint status in cells exposed to paclitaxel and/or As(III). Consistent with its known effect in induction of mitotic arrest, paclitaxel treatment alone caused activation of the spindle checkpoint, manifested as accumulation of phospho‐BubR1 (p‐BubR1) and phospho‐Cdc27 (p‐Cdc27) (Fig. 3a). Paclitaxel also significantly increased cyclin B and securin levels (Fig. 3a). On the other hand, co‐treatment of HeLa cells with paclitaxel and As(III) significantly compromised induction of p‐BubR1, p‐Cdc27, securin, and cyclin B, indicating weakened mitotic arrest and spindle checkpoint activation. At high concentration (5 µm), As(III) completely suppressed paclitaxel‐induced phosphorylation of BubR1 and Cdc27. Interestingly, treatment with As(III) alone also weakly induced accumulation of phospho‐BubR1 and securin, consistent with its ability to induce mitotic arrest.
Figure 3.
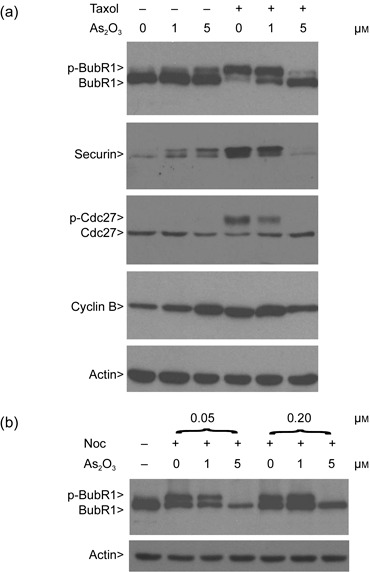
Arsenic trioxide (As2O3) compromises the spindle checkpoint activation. (a) HeLa cells were treated with or without paclitaxel and/or As(III) as indicated for 24 h. Equal amounts of protein lysates prepared from treated cells were blotted for BubR1, Cdc27, securin, cyclin B, and β‐actin. Arrows p‐BubR1 and p‐Cdc27 denote phosphorylated BubR1 and phosphorylated Cdc27, respectively. (b) HeLa cells were treated with or without nocodazole (Noc, 0.05 and 0.2 µm) and/or As(III) for 24 h. Equal amounts of protein lysates prepared from treated cells were blotted for BubR1 and β‐actin.
As nocodazole activates spindle checkpoint by destabilizing microtubules, we treated HeLa cells with both As(III) and nocodazole (Noc) overnight. Western blot analysis revealed that As(III) at high concentration (5 µm) was capable of suppressing nocodazole‐induced activation of BubR1 (Fig. 3b). Combined, the above‐described studies indicate that arsenic compromises mitotic arrest and spindle checkpoint activation, induced by microtubule poisons.
One simple and straightforward interpretation of above results is that As(III) either impeded mitotic entry or accelerated mitotic exit when cells were treated with paclitaxel. To differentiate between these two possibilities, we pulsed cells overnight with BrdU in the presence of As(III) and/or paclitaxel, followed by examining BrdU‐positive population using flow cytometry. This experimental approach would allow both use of asynchronous cell populations and determination of alterations in cell‐cycle progression, specific to cells treated with As(III) and/or paclitaxel. As expected, paclitaxel treatment blocked almost all cells (either BrdU‐positive or ‐negative) in mitosis (Fig. 4a). Co‐treatment with As(III) and paclitaxel greatly enriched mitotic cells with no increase in BrdU‐positive G1 population; on the other hand, a significant fraction of S‐phase cells remained (Fig. 4a), suggesting that As(III) treatment did not promote mitotic exit and slowed down cell‐cycle progression. To further confirm that As(III) delayed cell‐cycle progression, we pulsed HeLa cells for 45 min with BrdU in the presence of As(III) and/or paclitaxel. Flow cytometry analysis revealed that compared with that of vehicle‐treated control, magnitude of BrdU incorporation was significantly reduced in cells treated with both As(III) and paclitaxel (Fig. 4b,c).
Figure 4.
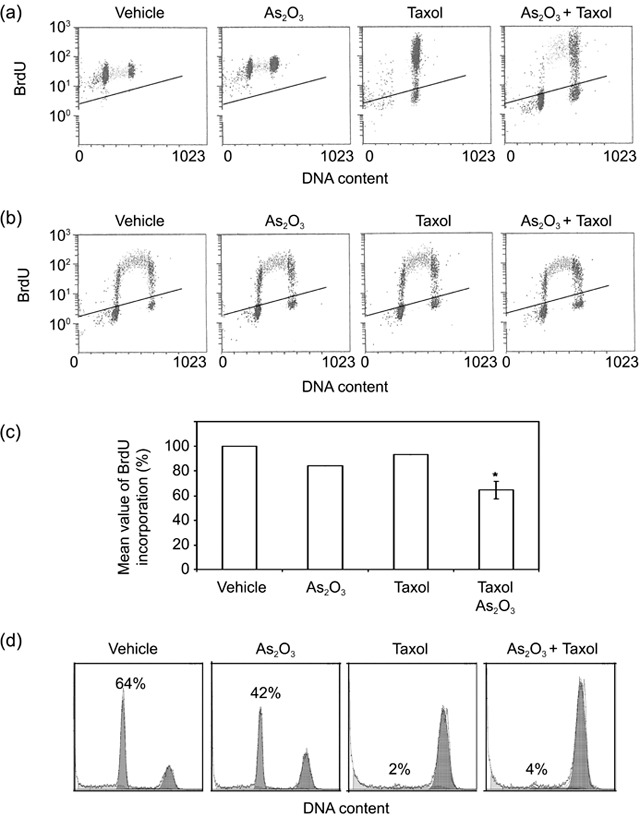
Arsenic trioxide (As2O3) delays cell‐cycle progression. (a) HeLa cells were pulsed with bromodeoxyuridine (BrdU) in the presence of vehicle, 5 µm As2O3, and/or 10 nm paclitaxel for 16 h. Cells were then collected and stained with fluorescein isothiocyanate (FITC)‐conjugated antibody to BrdU and propidium iodide (PI) followed by flow cytometry analysis. (b) HeLa cells were pulsed with BrdU for 45 min in the presence of vehicle, 5 µm As2O3, and/or 10 nm paclitaxel. After that, cells were collected and stained with the FITC‐conjugated BrdU antibody and PI followed by flow cytometry analysis. (c) The data presented in B were quantified from three independent experiments. *denotes the difference between the treatment and the controls (vehicle, As2O3, or paclitaxel alone) is statistically significant (P < 0.01). (d) HeLa cells synchronized at M phase by double‐thymidine block followed by release into nocodazole‐containing medium were incubated in the presence of As(III) and/or paclitaxel for 90 min. Cell‐cycle distributions were analysed by flow cytometry.
To directly study the effect of As2O3 on mitotic exit, HeLa cells were synchronized at the G1/S junction by double thymidine treatment and then released into the medium containing nocodazole, which arrested cells at mitosis. Subsequently, cells were cultured in fresh, nocodazole‐free medium containing As(III) and/or paclitaxel, for 90 min. Flow cytometry analysis revealed that whereas the majority of control cells (64% G1 cells) had exited from mitosis, As(III) treatment significantly attenuated mitotic exit (42% G1 cells) (Fig. 4d). As expected, paclitaxel completely blocked mitotic exit with few G1 cells; co‐treatment with As(III) did not significantly accelerate mitotic exit (Fig. 4d). These results thus indicate that As(III) slows down cell‐cycle progression at all stages and that suppression of paclitaxel‐induced mitotic arrest by As(III) is not due to enhanced mitotic exit.
Extensive studies in the past have indicated that p53 status significantly affects the response of cells to arsenic in vitro (29, 30). As HeLa cells do not have functional p53, we asked whether As(III)‐mediated suppression of paclitaxel‐induced mitotic arrest and if checkpoint activation was p53‐dependent. To this end, we selected a pair of isogenic cell lines [HCT116 (p53+/+) and HCT116 (p53−/–)] for our studies. We first measured survival rate of paired HCT116 cells treated with various concentrations of As(III) for 24 h. MTT assays revealed that HCT116 (p53+/+) cells did not suffer significant cytotoxicity in the presence of As(III) with IC50 about 40 µm (Fig. 5a). Interestingly, isogenic HCT116 (p53−/–) cells were slightly more resistant to As(III) treatment, especially at concentrations below 20 µm (Fig. 5a). We then treated the paired HCT116 cell lines with As(III) and/or paclitaxel and examined their gross morphologies. As expected, treatment with paclitaxel caused significant rounding‐up in both cell lines (Fig. 5b). Co‐treatment with As(III) had a little effect on modulating the detached (rounded‐up) phenotype of HCT116 (p53+/+) cells caused by paclitaxel; however, As(III) greatly attenuated paclitaxel‐induced cell rounding/detachment in HCT116 (p53−/–) cells (Fig. 5b). Our subsequent flow cytometric analyses revealed that As(III) significantly suppressed paclitaxel‐induced mitotic arrest in HCT116 (p53−/–) cells but not in HCT116 (p53+/+) cells (Fig. 5c). Combined, our studies strongly suggest that p53‐deficiency cooperates with As(III) in compromising spindle checkpoint function and suppressing mitotic arrest induced by paclitaxel.
Figure 5.
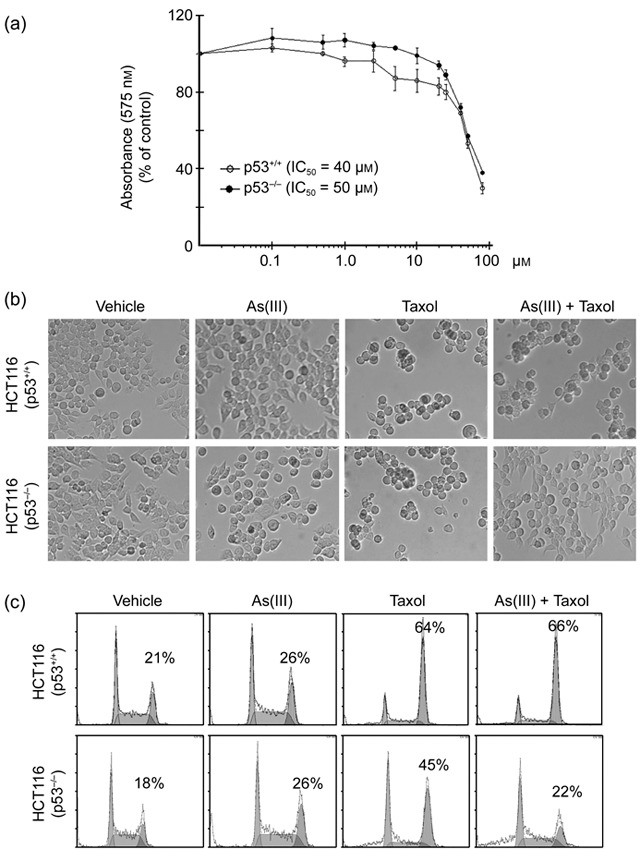
Suppression of paclitaxel‐induced mitotic arrest by arsenic trioxide (As2O3) is p53‐dependent. (a) Isogenic HCT116 (p53+/+) and HCT116 (p53−/–) cells were treated with various concentrations of As(III) for 24 h. Cell survival was measured using the MTT assay. Data were summarized from three independent experiments. (b) Paired HCT116 cells were cultured in the presence or absence of As(III) (5 µm) and/or paclitaxel (10 nm) for 24 h. Representative cell images were shown. (c) Paired HCT116 cells were cultured in the presence or absence of As(III) (5 µm) and/or paclitaxel (10 nm) for 24 h. Cells of various treatments were then processed for flow cytometry analysis. Representative data were shown. Percent of G2/M cell population in each treatment was indicated.
Discussion
Our current study showed that As(III) interferes with paclitaxel‐induced activation of the spindle checkpoint as well as mitotic arrest in vitro. We also demonstrate that attenuation of paclitaxel‐induced mitotic arrest by As(III) is primarily due to slowed cell‐cycle progression through S phase, but not enhanced mitotic exit. It is intriguing to observe that As(III) attenuates paclitaxel‐induced mitotic arrest. Clinical efficacy of paclitaxel is associated with its ability to induce mitotic arrest and subsequent mitotic catastrophe. Given the wide clinical applications of these two compounds in cancer treatment, it is essential for us to understand the mechanism by which As(III) suppresses spindle checkpoint activation and attenuates mitotic arrest induced by paclitaxel.
As(III) directly interferes with function of tubulins (21, 22, 27, 31), thus potentially affecting integrity of microtubules and mitotic spindles. We and others have documented that As(III) treatment alone induces features often found in cells treated with microtubule‐stabilizing or destabilizing agents, such as taxol and nocodazole (1, 19, 22). Indeed, As(III) inhibits GTP‐induced formation of microtubules in vitro as a non‐competitive inhibitor (21). It has been hypothesized that As(III) is capable of cross‐linking two vicinal cysteine residues (Cys‐12 and Cys‐213), which inactivates the GTP binding site (21). It has also been shown that As(III) antagonizes the effect of taxol on tubulin and microtubules and that it binds to SH groups blocking stoichiometric interaction of paclitaxel with tubulin (27). Despite differences in its exact mode of action, As(III) significantly affects the dynamics of microtubules in vivo, thus sharing similar properties with many well‐known microtubule disrupting agents. This may account for the fact that As(III) blocks cell‐cycle progression and induces mitotic arrest, frequently followed by mitotic catastrophe (1, 18).
It remains unclear how As(III) interferes with the action of paclitaxel. Despite that As(III) and taxol affect microtubule dynamics, they exhibit no synergistic effect on blocking cell proliferation and inducing apoptosis. An early study has shown that when they are used in combination, As(III) and paclitaxel behave antagonistically (27). As(III) suppresses paclitaxel‐induced perturbation of microtubule structures and mitotic arrest; likewise, paclitaxel reduces the inhibitory effect of As(III) on tubulin polymerization (27). Interestingly, the interaction between As(III) with SH groups of tubulin is not affected by binding of paclitaxel to tubulin (27), suggesting the antagonism is not simply due to stereo‐hindrance between these compounds blocking access of each of them to tubulin.
p53 status is known to affect arsenite‐induced genomic instability and apoptosis and p53 is induced or activated upon treatment with As(III) (32, 33). A series of in vitro studies has revealed that cell lines with p53 mutation or p53 deficiency are more sensitive to arsenite‐induced apoptosis than those with wild‐type p53 (29, 33). A recent study demonstrates that activation of cell‐cycle checkpoints, including G1 and G2, by arsenite does not require p53; however, arsenite‐induced mitotic catastrophe occurs preferentially in cells without functional p53 (29), suggesting that p53 is involved in guarding certain aspects of normal mitotic progression and initiating mitotic catastrophe when they are deregulated. Supporting this, p53 appears to influence mitotic exit induced by arsenite; different from p53‐positive cells, p53‐negative cells exit from mitosis more slowly when they are exposed to arsenite (30), consistent with the notion that persistent mitotic arrest can result in mitotic catastrophe. In the current study, we have shown that p53 deficiency somewhat compromises mitotic arrest induced by paclitaxel (Fig. 5c). This may partly explain why As(III) has a preferential effect on attenuating paclitaxel‐induced mitotic arrest.
The observation that p53 deficiency greatly facilitates suppression of paclitaxel‐induced spindle checkpoint activation and mitotic arrest by As(III), should have profound clinical implication. For example, mutations in or inactivation of TP53, the gene encoding p53, are found in at least 50% of all human cancers. Moreover, deficiency in, or haploinsufficiency of, spindle check point genes often results in enhanced tumourigenesis (34). In fact, many tumour cells harbour deficiencies in spindle checkpoint control (35). If the observed antagonism between As(III) and paclitaxel also occurs in vivo, it is imperative for us to take into consideration spindle checkpoint status/integrity and p53 status of tumour cells, when patients undergo chemotherapy with arsenic compounds.
Acknowledgements
We thank Katherine Xie for her assistance in the current study and co‐workers in the laboratory for discussions. This work was supported in part by grants from the National Institutes of Health to W.D. (CA074229, CA090658).
References
- 1. Halicka HD, Smolewski P, Darzynkiewicz Z, Dai W, Traganos F (2002) Arsenic trioxide arrests cells early in mitosis leading to apoptosis. Cell Cycle 1, 201–209. [PubMed] [Google Scholar]
- 2. Huang X (2003) The revival of the ancient drug – arsenic. Chin. Med. J. (Engl.) 116, 1637–1638. [PubMed] [Google Scholar]
- 3. Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY et al (1997b) Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89, 3354–3360. [PubMed] [Google Scholar]
- 4. Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ et al (1998) Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 339, 1341–1348. [DOI] [PubMed] [Google Scholar]
- 5. Zhang TD, Chen GQ, Wang ZG, Wang ZY, Chen SJ, Chen Z (2001) Arsenic trioxide, a therapeutic agent for APL. Oncogene 20, 7146–7153. [DOI] [PubMed] [Google Scholar]
- 6. Kitamura K, Yoshida H, Ohno R, Naoe T (1997) Toxic effects of arsenic (As3+) and other metal ions on acute promyelocytic leukemia cells. Int. J. Hematol. 65, 179–185. [DOI] [PubMed] [Google Scholar]
- 7. Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY et al (1996) In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with downregulation of Bcl‐2 expression and modulation of PML‐RAR α/PML proteins. Blood 88, 1052–1061. [PubMed] [Google Scholar]
- 8. Zhu XH, Shen YL, Jing YK, Cai X, Jia PM, Huang Y et al (1999) Apoptosis and growth inhibition in malignant lymphocytes after treatment with arsenic trioxide at clinically achievable concentrations. J. Natl. Cancer Inst. 91, 772–778. [DOI] [PubMed] [Google Scholar]
- 9. Rousselot P, Labaume S, Marolleau JP, Larghero J, Noguera MH, Brouet JC et al (1999) Arsenic trioxide and melarsoprol induce apoptosis in plasma cell lines and in plasma cells from myeloma patients. Cancer Res. 59, 1041–1048. [PubMed] [Google Scholar]
- 10. Siu KP, Chan JY, Fung KP (2002) Effect of arsenic trioxide on human hepatocellular carcinoma HepG2 cells: inhibition of proliferation and induction of apoptosis. Life Sci. 71, 275–285. [DOI] [PubMed] [Google Scholar]
- 11. Shen ZY, Shen J, Cai WJ, Hong C, Zheng MH (2000) The alteration of mitochondria is an early event of arsenic trioxide induced apoptosis in esophageal carcinoma cells. Int. J. Mol. Med. 5, 155–158. [DOI] [PubMed] [Google Scholar]
- 12. Uslu R, Sanli UA, Sezgin C, Karabulut B, Terzioglu E, Omay SB et al (2000) Arsenic trioxide‐mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines. Clin. Cancer Res. 6, 4957–4964. [PubMed] [Google Scholar]
- 13. Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y et al (2001) Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen‐independent prostate cancer. Cancer Res. 61, 5432–5440. [PubMed] [Google Scholar]
- 14. Murgo AJ (2001) Clinical trials of arsenic trioxide in hematologic and solid tumors: overview of the National Cancer Institute Cooperative Research and Development Studies. Oncologist. 6(Suppl. 2), 22–28. [DOI] [PubMed] [Google Scholar]
- 15. Huang C, Li J, Ding M, Wang L, Shi X, Castranova V et al (2001) Arsenic‐induced NFκB transactivation through Erks‐ and JNKs‐dependent pathways in mouse epidermal JB6 cells. Mol. Cell. Biochem. 222, 29–34. [PubMed] [Google Scholar]
- 16. Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L et al (2000) Arsenic trioxide‐induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemia 14, 262–270. [DOI] [PubMed] [Google Scholar]
- 17. Larochette N, Decaudin D, Jacotot E, Brenner C, Marzo I, Susin SA et al (1999) Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp. Cell. Res. 249, 413–421. [DOI] [PubMed] [Google Scholar]
- 18. Kligerman AD, Tennant AH (2007) Insights into the carcinogenic mode of action of arsenic. Toxicol. Appl. Pharmacol. 222, 281–288. [DOI] [PubMed] [Google Scholar]
- 19. Cai X, Yu Y, Huang Y, Zhang L, Jia PM, Zhao Q et al (2003) Arsenic trioxide‐induced mitotic arrest and apoptosis in acute promyelocytic leukemia cells. Leukemia 17, 1333–1337. [DOI] [PubMed] [Google Scholar]
- 20. Park JW, Choi YJ, Jang MA, Baek SH, Lim JH, Passaniti T et al (2001) Arsenic trioxide induces G2/M growth arrest and apoptosis after caspase‐3 activation and Bcl‐2 phosphorylation in promonocytic U937 cells. Biochem. Biophys. Res. Commun. 286, 726–734. [DOI] [PubMed] [Google Scholar]
- 21. Li YM, Broome JD (1999a) Arsenic targets tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res. 59, 776–780. [PubMed] [Google Scholar]
- 22. Ling YH, Jiang JD, Holland JF, Perez‐Soler R (2002b) Arsenic trioxide produces polymerization of microtubules and mitotic arrest before apoptosis in human tumor cell lines. Mol. Pharmacol. 62, 529–538. [DOI] [PubMed] [Google Scholar]
- 23. Ramirez P, Eastmond DA, Laclette JP, Ostrosky‐Wegman P (1997) Disruption of microtubule assembly and spindle formation as a mechanism for the induction of aneuploid cells by sodium arsenite and vanadium pentoxide. Mutat. Res. 386, 291–298. [DOI] [PubMed] [Google Scholar]
- 24. Jordan MA, Wilson L (2004) Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265. [DOI] [PubMed] [Google Scholar]
- 25. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G (2004) Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825–2837. [DOI] [PubMed] [Google Scholar]
- 26. Guo W, Tang XD, Tang S, Yang Y (2006) [Preliminary report of combination chemotherapy including Arsenic trioxide for stage III osteosarcoma and Ewing sarcoma]. Zhonghua Wai Ke. Za Zhi. 44, 805–808 (in Chinese). [PubMed] [Google Scholar]
- 27. Carre M, Carles G, Andre N, Douillard S, Ciccolini J, Briand C et al (2002a) Involvement of microtubules and mitochondria in the antagonism of arsenic trioxide on paclitaxel‐induced apoptosis. Biochem. Pharmacol. 63, 1831–1842. [DOI] [PubMed] [Google Scholar]
- 28. Ouyang B, Pan H, Lu L, Li J, Stambrook P, Li B et al (1997) Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J. Biol. Chem. 272, 28646–28651. [DOI] [PubMed] [Google Scholar]
- 29. Taylor BF, McNeely SC, Miller HL, Lehmann GM, McCabe MJ Jr, States JC (2006) p53 Suppression of arsenite‐induced mitotic catastrophe is mediated by p21CIP1/WAF1. J. Pharmacol. Exp. Ther. 318, 142–151. [DOI] [PubMed] [Google Scholar]
- 30. McNeely SC, Xu X, Taylor BF, Zacharias W, McCabe MJ Jr, States JC (2006) Exit from arsenite‐induced mitotic arrest is p53 dependent. Environ. Health Perspect. 114, 1401–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ochi T, Nakajima F, Nasui M (1999) Distribution of gamma‐tubulin in multipolar spindles and multinucleated cells induced by dimethylarsinic acid, a methylated derivative of inorganic arsenics, in Chinese hamster V79 cells. Toxicology 136, 79–88. [DOI] [PubMed] [Google Scholar]
- 32. Yih LH, Lee TC (2000) Arsenite induces p53 accumulation through an ATM‐dependent pathway in human fibroblasts. Cancer Res. 60, 6346–6352. [PubMed] [Google Scholar]
- 33. Salazar AM, Ostrosky‐Wegman P, Menendez D, Miranda E, Garcia‐Carranca A, Rojas E (1997) Induction of p53 protein expression by sodium arsenite. Mutat. Res. 381, 259–265. [DOI] [PubMed] [Google Scholar]
- 34. Wassmann K, Benezra R (2001) Mitotic checkpoints: from yeast to cancer. Curr. Opin. Genet. Dev. 11, 83–90. [DOI] [PubMed] [Google Scholar]
- 35. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD et al (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303. [DOI] [PubMed] [Google Scholar]