

Abstract
The concept of the Vanishing Zero, which was first discussed 50 years ago in relation to pesticide residues in foods and food crops, focused on the unintended regulatory consequences created by ever-increasing sensitivity and selectivity of analytical methods, in conjunction with the ambiguous wording of legislation meant to protect public health. In the interim, the ability to detect xenobiotics in most substrates has increased from tens of parts per million to parts per trillion or less, challenging our ability to interpret the biological significance of exposures at the lowest detectable levels. As a result the focus of risk assessment, especially for potential carcinogens, has shifted from defining an acceptable level, to extrapolating from the best available analytical results. Analysis of gene expression profiles in exposed target cells using genomic technologies can identify biological pathways induced or repressed by the exposure as a function of dose and time. This treatise explores how toxicogenomic responses at low doses may inform risk assessment and risk management by defining thresholds for cellular responses linked to modes or mechanisms of toxicity at the molecular level.
Introduction
The Vanishing Zero, as first discussed in relation to pesticide detection and safety in a 1970 essay by Zweig [1] addressed the unanticipated problems created by the ever-increasing sensitivity of analytical methods in conjunction with the imprecise wording of legislation meant to protect the public. The ability to detect the presence of toxicants in foods, clinical samples and environmental samples has continued to the point where it is now possible to detect specific compounds in the low femtomole (10-15) range [2] and in some cases even the attamole (10-18) level [3-5]. Thus the definition of the term “undetectable” has in some cases decreased a billion-fold, challenging the definition of non-detectable, and the interpretation of the biological significance of the lowest detectable levels. Even in 1970, Zweig realized that continued increase in analytical power posed a problem for risk assessment, and applauded government agencies “that had the courage to replace the unrealistic zero tolerance or no residue classification by more realistic terms like ‘inconsequential pharmacologically’ and are placing finite residue tolerances on crops, beyond which the residue chemist is no longer obligated to search for the vanishing zero.”
Zweig correctly predicted that over the next several decades the focus would shift from defining zero to finding more specific methods to measure compounds and then using this information to establish acceptable exposure levels. However, this line of reasoning and its application to risk assessment is not without detractors. For example, a recent online column (http://www.thefreelibrary.com/Journal+of+Environmental+Health/1993/October/1-p5229) referred to this line of reasoning as “the myth of the vanishing zero”. The author questioned the notion that a human risk estimate produced at a concentration two or three orders of magnitude above the detection level would be better than one performed at the detection level. He argues that risk assessment should always be performed with the most sensitive analytical data. His central tenet is that there is no valid scientific argument to support the myth of the vanishing zero. He contends that it is perpetuated to only support the perception that “persons exposed to ‘vanishingly small’ quantities of a potentially carcinogenic substance need have no concerns about the health consequences.” In essence, this statement defines the current concept of setting allowable exposures on the basis of an acceptable increase in cancer risk, such as one excess case per million (10-6) people exposed to the compound. By contrast, one interpretation of the precautionary principle states that in the absence of scientific data to ensure that no harm would ensue, any exposure that has the potential to harm must be mitigated to protect the public or the environment [6, 7]. The burden of proof falls on those who would advocate for use of the compound, while the plausibility of risk is left to the discretion of the decision maker, often without a clear definition of risk. This is a controversial issue with strong arguments on both sides, and pits the concept of acceptable risk against the precautionary principle. The goal of this thesis is to examine how emerging genomic technologies might contribute to scientific arguments on both sides, and better inform risk assessment in the era of unprecedented analytical capabilities.
Biological Endpoints and Risk Assessment
As the sensitivity and specificity of detecting compounds continues to improve, the question of what constitutes a biologically meaningful exposure becomes more relevant [8]. Non-genotoxic and non-carcinogenic compounds often have exposure thresholds for observable toxicity. The latter are defined using dose response data by establishing a NOEL (No Observable Effect Level), or a NOAEL (No Observable Adverse Effect Level) or benchmark modeling of dose response using classical toxicological endpoints, such as changes in organ weight, clinical chemistry or histopathological changes. Default safety factors that take account of individual, species and developmentally-related differences in sensitivity are then applied to develop a reference dose that defines an acceptable exposure for regulatory purposes. For genotoxic and carcinogenic compounds, it is generally assumed that there is no threshold and that risk increases as a function of exposure. For regulatory purposes, this is sometimes dealt with by the establishment of what is considered an acceptable increase in population cancer risk. The Risk Assessment Paradigm [9] assumes that there is no threshold, which leads to a linear dose extrapolation from a point of departure within the lower range of the experimentally observed responses through zero. The point of departure dose is estimated by benchmark modeling of available data. The linear extrapolation is then used in risk management to define a dose that yields an acceptable increase in risk (Figure 1).
Figure 1. Hypothetical Dose Response Curve as Applied to Risk Assessment.
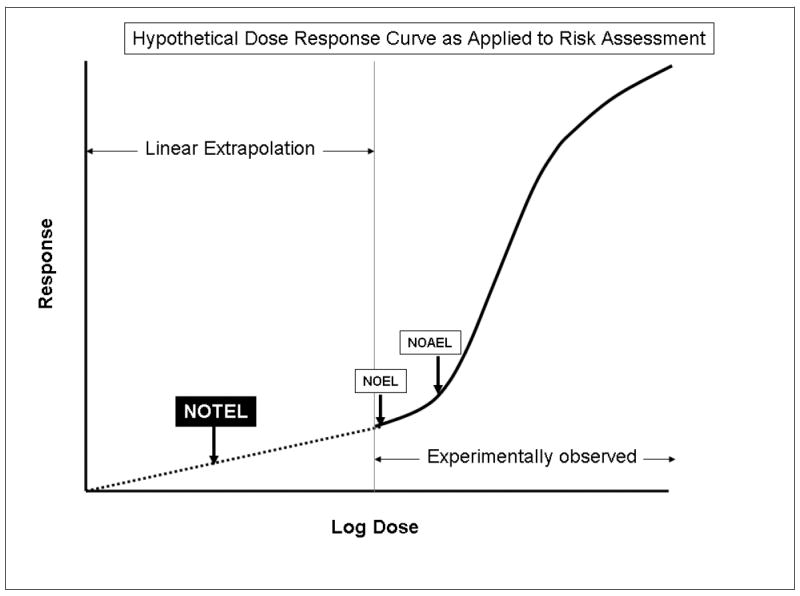
The solid line indicates a typical response curve that shows a threshold for the experimentally measured endpoint. These data are then used to determine a NOEL and/or a NOAEL. Data can also be used for modeling to estimate a benchmark dose. Default safety factors are then used to determine a reference dose. In the case of genotoxic or carcinogenic compounds, linear dose extrapolation is used to set an acceptable risk level. The use of genomics provides the opportunity to define much more sensitive NOTEL (indicated on graph), NOAEL or a mechanistically based benchmark dose (the latter two not indicated) for use in risk assessment.
However, neither of these approaches is completely satisfactory. In the case of non-genotoxic/non-carcinogenic exposures that show a response threshold, the endpoints used often represent overt pathological changes that are complicated by interspecies differences in metabolism, mode of action, dose response and genetic susceptibility. In the case of genotoxicants/carcinogens, increase in cancer risk is difficult to reliably ascertain since sufficiently large cohorts of exposed individuals are rarely available. In animal experiments, exposures even at relatively high doses often generate what appears to be a threshold, but may be an artifact generated by the fact that an insufficient number of animals are at the lower doses or by the dose selection used in the study. Presumably, if a sufficient number of animals were treated and at enough dose levels, one would be able to observe any increase in incidence, but this is not economically, ethically, or logistically feasible. To address these limitations, considerable effort has been made to develop biological response indicators or biomarkers that might be predictors of incipient disease. These include indicators or biomarkers of internal dose (e.g., uptake, metabolic products, DNA or protein adducts) and biologically effective dose (e.g., mutation, changes in cell function or differentiation, and cell death) [10].
Among these indices, DNA adducts have garnered significant attention as exposure biomarkers, owing primarily to the tremendous sensitivity of mass spectrometry based methods, which can now routinely detect a few adducts per cell [2, 11, 12]. Using the most advanced accelerator mass spectrometry methods, it is now possible to demonstrate that adduct formation is in fact linear over the entire low dose range until the signal vanishes into the background [13, 14]. This level of sensitivity is, of course, the ultimate expression of the vanishing zero dilemma. What is the biological relevance of a few additional adducts per cell above a measurable background level? Those who choose to err on the side of caution would argue that no additional adducts are acceptable as they all have the potential to cause mutations [7]. However, arguments can also be made for the opposing view. One could surmise that low adduct levels can be efficiently repaired and hence are not highly mutagenic [15, 16]. In support of such argument, recent studies with large numbers of animals and mass spectrometry approaches that can detect DNA adducts as low as 1 per 109 to 1010 have revealed that adduct formation at low doses is not linear [17]. Moreover, for adducts that can be formed by endogenous processes, the number of adducts induced by the exposure may be small compared to existing background levels, and therefore both endogenous and exogenous DNA adducts must be taken into account [13]. In addition, recent in vitro and in vivo studies [16, 18-20], and an accidental exposure of patients to ethylmethane sulfonate as a drug contaminant [21], have suggested the existence of a genotoxicity threshold for DNA-reactive carcinogens at low dose ranges. Below the threshold, DNA adduct formation is no longer correlated with genotoxic endpoints, presumably due to efficient DNA repair. Even though there are examples of mutagens/genotoxic carcinogens for which the existence of a threshold was convincingly demonstrated, the expert consensus for regulatory purposes is that DNA adduct data should always be trumped by mutational and carcinogenicity data [22]. Nonetheless, the availability of highly sensitive methods for detection of DNA adducts [12, 17] continues to fuel the vanishing zero debate.
Use of genomics in risk assessment
Genomic technologies have the unprecedented ability to assess global cellular responses to exposures as a function of dose, and hence the potential for predicting mechanisms of toxicity and/or mode of action, thereby providing unique opportunities for risk assessment. For example, studies by Paules and coworkers examined the in vivo transcriptional response of blood cells and hepatocytes to acetaminophen [23]. Their results indicated that a gene expression profile indicative of incipient toxicity could be detected in blood cells at doses significantly below the levels required to observe histopathological changes in liver. Similarly, Thomas et al. [24] showed that data from expression profiles obtained after a two week exposure of mice to 26 individual carcinogens could predict tumor formation following long-term exposure with accuracy of 60-80%. The potential use of these approaches in predictive toxicology and risk assessment was the subject of two recent National Academy of Sciences/National Research Council reports. The first report, entitled “Toxicity Testing in the 21st Century: A Vision and a Strategy”, took a long-range view of how pathway-based response to exposures in model organisms might eventually replace current testing paradigms [25, 26]. The second report, entitled “Applications of Toxicogenomic Technologies to Predictive Toxicology and Risk Assessment” looked at the current state of genomic technologies, their application to toxicology, and assessed the impact of these approaches on predictive toxicology and risk assessment in the immediate and near term [27]. Relevant to the vanishing zero debate, the latter report concluded that one of the ways genomic approaches could contribute to risk assessment is by providing mechanistically-based dose response data, especially in the area of low dose extrapolation and mixtures of xenobiotics. This prediction was based primarily on the seminal experiments that looked at the transcriptional response of estrogen receptor positive cells to decreasing estrogen concentrations in vitro [28] and in vivo [29]. In both cases, there was a clear threshold for an estrogen specific transcriptional response which was related to the dissociation constant of estrogen from its receptor. They referred to this threshold as the No Transcriptional Effect Level, or NOTEL [28].
While these studies laid the ground work for the concept of a NOTEL in response to receptor mediated signal transduction, not all toxicants mediate their effects through specific binding to receptors. We and others therefore asked whether cells exposed to decreasing concentrations of nongenotoxic [30] or genotoxic/carcinogenic compounds also showed a NOTEL. We exposed human BEAS-2B bronchial epithelial cells to the genotoxic carcinogens N-hydroxy-PhIP (N-hydroxy-2- amino-1-methyl-6-phenylimidazo[4’5-b] pyridine) and benzo[a]pyrene diolepoxide (BPDE). To minimize the argument that an observed threshold was simply a technical limit of the microarray-based expression profiling, we anchored the transcriptional response to several biological endpoints and response indicators including cell survival, mutagenicity, and DNA adduct levels. In these preliminary studies, BEAS-2B bronchial epithelial cells were dosed in duplicate for 24 hours to N-hydroxy- PhIP, the active metabolite of PhIP, at concentrations ranging from 10-6 to 10-13 M. Total RNA and DNA were simultaneously isolated from each of the exposed cultures using the Qiagen AllPrep™ kit, which uses a series of column-based separations to isolate DNA, RNA and protein from the same sample. RNA preparations were used to generate cRNAs and hybridized to Affymetrix GeneChip Human Genome U133 Plus™ arrays. Microarray data were extracted and analyzed for changes in gene expression as a function of carcinogen dose using Array Assist software. A correlation plot was generated for pair-wise comparison of gene expression patterns among all doses and the controls (Figure 2). These preliminary data indicated that at dose below 10-7 M, the pattern of gene expression of the treated cells was indistinguishable from that of the control. These observations suggested that NOTEL for N-hydroxy-PhIP in BEAS-2B cells occurred at doses below 10-7 M. The observed NOTEL was thus at least two orders of magnitude lower than the lowest dose (10-5 M) that yielded detectable cytotoxicity and mutagenicity in BEAS-2B cells. These initial observations were confirmed in subsequent studies where exposures and microarray analyses were performed in triplicate with a larger number of intermediate doses (data not shown).
Figure 2. Correlation Plot of Gene Expression Profiles as a Function of N-OH-PhIP Dose.
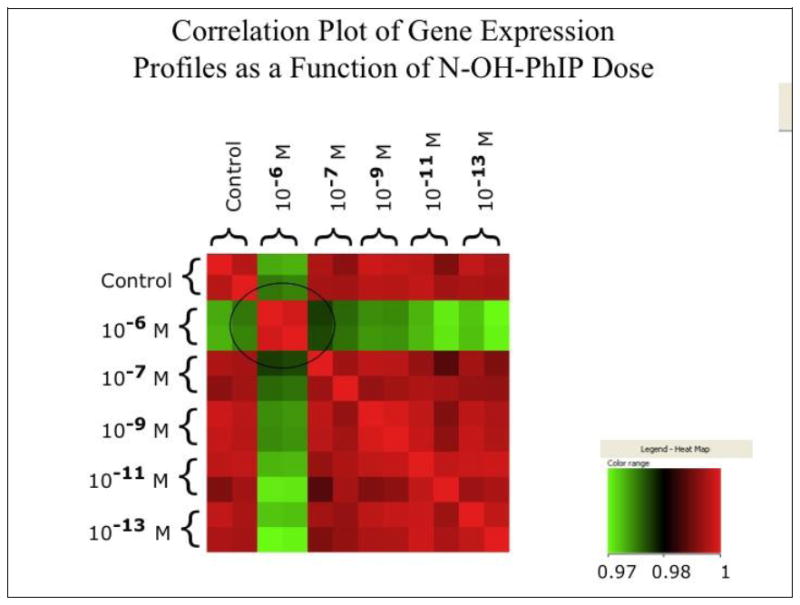
Gene expression analyses were performed in duplicate at the indicated doses of N-hydroxy-PhIP using Affymetrix GeneChip Human Genome U133 Plus™ arrays. After normalization and background subtraction, the similarity in gene expression patterns between all samples was evaluated pair-wise using the Array Assist software package. The heat map in the legend indicates the relationship between color and the correlation coefficient across all genes of a given pair of arrays. Circle highlights a region which illustrates that a dose of 10-6 yields a pattern of gene expression that is substantially different from controls and lower doses.
While these results were highly suggestive of a NOTEL, it was important to perform detailed bioinformatic analysis of the gene expression profiles at the various doses to examine the responses at the level of biochemical pathways (data not shown). Given the small number of microarray replicates relative to genes tested in most microarray studies, the frequentist approach to statistical analysis is prone to significant error due to the multiple testing problem, which requires correction of P values for the number of tests performed on each sample [31]. As an alternative, we used Bayesian Robust Inference for Differential Gene Expression or BRIDGE [32] to test for differentially expressed genes on microarray data. The Bayesian hierarchical model is a technique to combine information from different replicate measurements to produce a summary statistic over all of the replicate measurements. BRIDGE allows each gene to have a different variance and allows for the detection of differentially expressed genes under multiple experimental conditions. We used BRIDGE to compare gene expression patterns at each dose to the controls and extracted sets of genes that were significantly different. These sets of genes were individually subjected to pathway analysis using Ingenuity Pathway Analysis™ software. The preliminary analysis of gene expression data indicated that at doses above the NOTEL, the affected pathways were related to carcinogenesis and apoptosis. At the lower doses closer to the NOTEL, the affected pathways were related to an oxidative stress response. While these results are still preliminary, similar findings were also reported from in vitro and in vivo studies performed by other groups [33-35]. Taken together, available data suggest that NOTEL may be a much more sensitive indicator of biologically relevant exposure than the classical toxicology endpoints (NOEL, NOAEL or benchmark dose) that rely on adverse physiological or overt toxic responses. Moreover, it is possible to detect a NOTEL for genotoxic compounds, hormones [28] and chemical mixtures [36].
One interpretation of the NOTEL data is that the response of cells to a toxicant is dosedependent, shifting from what might be called a physiological or adaptive response, to a toxic response depending on the level of cellular damage. If correct this interpretation suggests that there is a threshold for the response of cells to genotoxic damage. If damage is minimal, cellular repair pathways are activated to maintain homeostasis without significant change in cellular physiology. Once the level of damage exceeds a threshold, cells induce repair mechanisms, alter their growth and in extreme cases, induce apoptosis. In some instances these self-preserving repair mechanisms, such as DNA repair by-pass polymerases [37], can in fact be the drivers of mutagenesis and hence carcinogenesis. Thus, the pattern of gene expression induced as a function of dose and time could therefore be indicative of increased risk. In addition, the pattern of gene expression induced as a function of dose and time may suggest a mode of action that leads to adverse outcomes. Gene expression profiling may also predict risk in situations where mixtures of xenobiotics, but not their individual components, can induce an adverse effect [36]. Genomic approaches may help identify patterns of gene expression associated with a mechanism or mode of action that lead to adverse outcomes, and allow the identification of a no observable adverse transcription effect level (NOATEL). Combining genomic data with risk assessment methodologies such as benchmark dose modeling, dose extrapolation, and the mode of action framework may therefore provide important new opportunities in risk assessment and risk management [33-35].
High-throughput genotyping approaches are also being used to provide a more accurate assessment of population variability, particularly with respect to gene environment interactions [23, 27]. Although not discussed here, responses to xenobiotics like benzene [38], and by inference their toxicological thresholds, are subject to modulation by polymorphisms in genes associated with the mechanism of toxicity [23, 27]. A more accurate assessment of population variability can better inform the use of genetic safety factors, which are currently set arbitrarily, usually to a value of 10. In addition, NOTEL or NOAEL values for individuals would also be modulated by and therefore integrate the effects other host, epigenetic, and environmental factors. Variations in thresholds could therefore provide another useful measure of variable susceptibility within a population.
Toxicogenomics of Benzene Exposure: Applications to the Study of Bone Marrow Toxicity
The leukemogenic activity of benzene was first noted more than a hundred years ago by Le Noire, who reported multiple cases of leukemia among cobblers [cited in 39]. The mechanism by which benzene induces leukemia is as yet unresolved and remains an area of intensive research [39- 41]. The metabolism of benzene is complex [42], generating numerous DNA reactive intermediates, phenols and quinones that can react with DNA directly, or generate DNA reactive oxygen species [43]. Although the metabolism of benzene to DNA-reactive intermediates is required for leukemogenesis, most of the products produced are weak mutagens per se. Epidemiological evidence indicates that prolonged occupational exposure to high levels of benzene is associated with increased risk of aplastic anemia, acute myeloid leukemia, and chronic lymphocytic leukemia [44-46]. Paradoxically, benzene is also a prototypical hematotoxicant that induces progressive depression of bone marrow function as a function of exposure [47]. As a result there is a narrow dose range for leukemogenicity, since higher doses result in bone marrow aplasia. This enigma is partially explained by studies showing rapid recovery of bone marrow stem cell growth shortly after cessation of exposure [39]. As a result, the stem cell compartment has been shown to decrease during the typical weekend lapse in occupation exposures and the resulting oscillations in hematopoiesis have been linked to leukemogenesis. Consistent with such a model, benzene has been shown to decrease the number of hematopoietic stem cells via cell cycle regulation and apoptosis [48]. Chronic exposure was also associated with an increase in persistent DNA damage in this cell compartment.
Given the complexity of benzene-induced hematotoxicity and leukemogenesis, researchers have used genomic technologies to investigate the effects of benzene exposure on the pattern of gene expression in animal models and in occupationally-exposed workers [48] By analyzing the set of differentially expressed genes in the target organ or cell compartment in the context of biochemical pathways, these studies have 1) implicated benzene-induced effects on cell cycle checkpoints, apotosis, and DNA repair systems on leukemogenesis [41]; 2) identified subsets of genes that serve as biomarkers of exposures [49, 50]; 3) demonstrated the importance of looking at the effects of benzene on gene expression in the cell compartment of interest, namely hematopoietic stem cells [48]; 4) the importance of examining the effects in the context of cyclical exposures [39]; and 5) the confounding effects of co-exposures that synergize or antagonize the effects of benzene and actually can result in the induction of genes not induced by either compound alone [36]. However, none of these studies have systematically evaluated the benzene-induced changes in gene expression patterns as a function of dose. The study by Hendriksen [36], which examined the effects of co-exposures, did examine the response at the Lowest Observed Adverse Effect level (LOAEL) and at doses that were two- to three-fold lower (sub-LOAEL). Significantly, these investigators not only found synergy at the level of benzene-induced gene expression, but observed these changes at doses well below those that are detectable by classical toxicological endpoints. These latter findings support our contention that using genomic approaches to identify the NOTEL or NOATEL of benzene and other compounds may be a valuable new approach to incorporating mechanistic or mode of action data into risk assessment.
Conclusion
In the final analysis, genomics may not be able to provide a definitive response to the vanishing zero debate. Those who consider the vanishing zero as myth will simply argue that the observed thresholds are due to a lack of technical sensitivity or reproducibility, or due to the possibility that at low doses, only a fraction of the cells are impacted by the exposure, albeit adversely. Perhaps the most important contribution of genomics to risk assessment will be in more accurately defining the doses that adversely affect cellular function. In either case, our data and those from other studies suggest that exposures to genotoxic/carcinogenic and non-genotoxic xenobiotics can induce transcriptional responses indicative of incipient disease at doses that are well below those required for induction of more classical endpoints such as change in organ weight, histopathology, or carcinogenicity [23, 30], or time-points [24] well before detection of overt tumors. Toxicogenomic-based benchmark doses, NOTELs, and NOATELs are also more intimately linked to the mechanism of toxicity or the mode of action. Importantly, in vivo studies with acetaminophen have shown that the transcriptional response of white blood cells to can be 97% correlated with gene expression profile indicative of incipient histopathological changes in hepatocytes [23]. These findings suggest that white blood cells may serve as a suitable substitute for target organ cell in the determination of genomic endpoints such as the NOTEL.
Evaluating the potential benefits of incorporating genomic endpoints such as NOTEL, NOATEL, or benchmark doses estimated from array data (Figure 1) in risk assessment is therefore an important area for research. Given its wealth of available mode of action data, the defined thresholds for multiple endpoints, the abundance of human exposure data, and the availability of blood samples collected from occupationally exposed workers, benzene may be the ideal compound for a case study in genomics-based risk assessment. To be successful, this research must be designed and carried out as a collaborative effort between assessors and researchers, as is presently done for the Mode of Action Framework. Any change or amendment to accepted risk assessment methodology and the regulatory process will require significant experimental testing and validation of the NOTEL concept for a wide variety of compounds. Given the limitations of existing approaches in establishing mechanism-based exposure guidelines, the vast potential of genomic approaches to inform mode of action and thresholds of toxicity indicate that the effort required is warranted. Once validated, the combined use of these genomic approaches could improve risk assessment by facilitating the development of exposure guidelines that are more biologically relevant, even in the face of the vanishing zero.
Acknowledgments
This work was supported by Public Health Services Grants (NIH):
RO1CA112231 (P. Vouros, P.I.), U19ES011387 (H. Zarbl, P.I.), and P30ES005022 (H. Zarbl, P.I.)
Footnotes
Conflict of Interest Statement
The authors declare that are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zweig G, editor. The Vanishing Zero: The Evolution of Pesticide Analysis. Academic Press, Inc.; New York and London: 1970. [Google Scholar]
- 2.Flarakos J, Xiong W, Glick J, Vouros P. A deoxynucleotide derivatization methodology for improving LC-ESI-MS detection. Anal Chem. 2005;77(8):2373–2380. doi: 10.1021/ac0483724. [DOI] [PubMed] [Google Scholar]
- 3.Dingley KH, Roberts ML, Velsko CA, Turteltaub KW. Attomole detection of 3H in biological samples using accelerator mass spectrometry: application in low-dose, dual-isotope tracer studies in conjunction with 14C accelerator mass spectrometry. Chem Res Toxicol. 1998;11(10):1217–1222. doi: 10.1021/tx9801458. [DOI] [PubMed] [Google Scholar]
- 4.Turteltaub KW, Dingley KH. Application of accelerated mass spectrometry (AMS) in DNA adduct quantification and identification. Toxicol Lett. 1998;102-103:435–439. doi: 10.1016/s0378-4274(98)00344-0. [DOI] [PubMed] [Google Scholar]
- 5.Randall KL, Argoti D, Paonessa JD, Ding Y, Oaks Z, Zhang Y, Vouros P. An improved liquid chromatography-tandem mass spectrometry method for the quantification of 4-aminobiphenyl DNA adducts in urinary bladder cells and tissues. J Chromatogr A. 2009 doi: 10.1016/j.chroma.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saltelli A, Funtowicz S. The Precautionary Principle: implications for risk management strategies. Int J Occup Med Environ Health. 2004;17(1):47–57. [PubMed] [Google Scholar]
- 7.Goldstein BD, Carruth RS. Implications of the Precautionary Principle: is it a threat to science? Int J Occup Med Environ Health. 2004;17(1):153–161. [PubMed] [Google Scholar]
- 8.Science and Decisions. National Research Council; Washington, D.C.: 2009. [Google Scholar]
- 9.Omenn GS. Assessing the risk assessment paradigm. Toxicology. 1995;102(1-2):23–28. doi: 10.1016/0300-483x(95)03034-d. [DOI] [PubMed] [Google Scholar]
- 10.Lee KM, Han S, Park WY, Kang D. Identification and application of biomarkers in molecular and genomic epidemiologic research. J Prev Med Public Health. 2009;42(6):349–355. doi: 10.3961/jpmph.2009.42.6.349. [DOI] [PubMed] [Google Scholar]
- 11.La DK, Swenberg JA. DNA adducts: biological markers of exposure and potential applications to risk assessment. Mutat Res. 1996;365(1-3):129–146. doi: 10.1016/s0165-1110(96)90017-2. [DOI] [PubMed] [Google Scholar]
- 12.Swenberg JA, Fryar-Tita E, Jeong YC, Boysen G, Starr T, Walker VE, Albertini RJ. Biomarkers in toxicology and risk assessment: informing critical dose-response relationships. Chem Res Toxicol. 2008;21(1):253–265. doi: 10.1021/tx700408t. [DOI] [PubMed] [Google Scholar]
- 13.Swenberg JA, Ham A, Koc H, Morinello E, Ranasinghe A, Tretyakova N, Upton PB, Wu K. DNA adducts: effects of low exposure to ethylene oxide, vinyl chloride and butadiene. Mutat Res. 2000;464(1):77–86. doi: 10.1016/s1383-5718(99)00168-0. [DOI] [PubMed] [Google Scholar]
- 14.Marsden DA, Jones DJ, Britton RG, Ognibene T, Ubick E, Johnson GE, Farmer PB, Brown K. Dose-response relationships for N7-(2-hydroxyethyl)guanine induced by low-dose [14C]ethylene oxide: evidence for a novel mechanism of endogenous adduct formation. Cancer Res. 2009;69(7):3052–3059. doi: 10.1158/0008-5472.CAN-08-4233. [DOI] [PubMed] [Google Scholar]
- 15.Preston RJ, Fennell TR, Leber AP, Sielken RL, Jr, Swenberg JA. Reconsideration of the genetic risk assessment for ethylene oxide exposures. Environ Mol Mutagen. 1995;26(3):189–202. doi: 10.1002/em.2850260303. [DOI] [PubMed] [Google Scholar]
- 16.Doak SH, Jenkins GJ, Johnson GE, Quick E, Parry EM, Parry JM. Mechanistic influences for mutation induction curves after exposure to DNA-reactive carcinogens. Cancer Res. 2007;67(8):3904–3911. doi: 10.1158/0008-5472.CAN-06-4061. [DOI] [PubMed] [Google Scholar]
- 17.Bailey GS, Reddy AP, Pereira CB, Harttig U, Baird W, Spitsbergen JM, Hendricks JD, Orner GA, Williams DE, Swenberg JA. Nonlinear cancer response at ultralow dose: a 40800-animal ED(001) tumor and biomarker study. Chem Res Toxicol. 2009;22(7):1264–1276. doi: 10.1021/tx9000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenkins GJ, Doak SH, Johnson GE, Quick E, Waters EM, Parry JM. Do dose response thresholds exist for genotoxic alkylating agents? Mutagenesis. 2005;20(6):389–398. doi: 10.1093/mutage/gei054. [DOI] [PubMed] [Google Scholar]
- 19.Johnson GE, Doak SH, Griffiths SM, Quick EL, Skibinski DO, Zair ZM, Jenkins GJ. Non-linear dose-response of DNA-reactive genotoxins: Recommendations for data analysis. Mutat Res. 2009 doi: 10.1016/j.mrgentox.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Gocke E, Muller L. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutat Res. 2009;678(2):101–107. doi: 10.1016/j.mrgentox.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Muller L, Gocke E, Lave T, Pfister T. Ethyl methanesulfonate toxicity in Viracept--a comprehensive human risk assessment based on threshold data for genotoxicity. Toxicol Lett. 2009;190(3):317–329. doi: 10.1016/j.toxlet.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Pottenger LH, Carmichael N, Banton MI, Boogaard PJ, Kim J, Kirkland D, Phillips RD, van Benthem J, Williams GM, Castrovinci A. ECETOC workshop on the biological significance of DNA adducts: Summary of follow-up from an Expert Panel Meeting. Mutat Res. 2009;678(2):152–157. doi: 10.1016/j.mrgentox.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Bushel PR, Heinloth AN, Li J, Huang L, Chou JW, Boorman GA, Malarkey DE, Houle CD, Ward SM, Wilson RE, Fannin RD, Russo MW, Watkins PB, Tennant RW, Paules RS. Blood gene expression signatures predict exposure levels. Proc Natl Acad Sci U S A. 2007;104(46):18211–18216. doi: 10.1073/pnas.0706987104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas RS, Bao W, Chu TM, Bessarabova M, Nikolskaya T, Nikolsky Y, Andersen ME, Wolfinger RD. Use of short-term transcriptional profiles to assess the long-term cancer-related safety of environmental and industrial chemicals. Toxicol Sci. 2009;112(2):311–321. doi: 10.1093/toxsci/kfp233. [DOI] [PubMed] [Google Scholar]
- 25.Toxicity Testing in the 21st Century. A Vision and a Strategy. National Research Council; Washington. D.C.: 2007. [Google Scholar]
- 26.Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science. 2008;319(5865):906–907. doi: 10.1126/science.1154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Applications of Toxicogenomic Technologies to Predictive Toxicology and Risk Assessment. National Research Council; Washington, D.C.: 2007. [PubMed] [Google Scholar]
- 28.Lobenhofer EK, Cui X, Bennett L, Cable PL, Merrick BA, Churchill GA, Afshari CA. Exploration of low-dose estrogen effects: identification of No Observed Transcriptional Effect Level (NOTEL) Toxicol Pathol. 2004;32(4):482–492. doi: 10.1080/01926230490483324. [DOI] [PubMed] [Google Scholar]
- 29.Naciff JM, Hess KA, Overmann GJ, Torontali SM, Carr GJ, Tiesman JP, Foertsch LM, Richardson BD, Martinez JE, Daston GP. Gene expression changes induced in the testis by transplacental exposure to high and low doses of 17{alpha}- ethynyl estradiol, genistein, or bisphenol A. Toxicol Sci. 2005;86(2):396–416. doi: 10.1093/toxsci/kfi198. [DOI] [PubMed] [Google Scholar]
- 30.Poynton HC, Loguinov AV, Varshavsky JR, Chan S, Perkins EJ, Vulpe CD. Gene expression profiling in Daphnia magna part I: concentration-dependent profiles provide support for the No Observed Transcriptional Effect Level. Environ Sci Technol. 2008;42(16):6250–6256. doi: 10.1021/es8010783. [DOI] [PubMed] [Google Scholar]
- 31.Benjamini Y, Heller R. Screening for partial conjunction hypotheses. Biometrics. 2008;64(4):1215–1222. doi: 10.1111/j.1541-0420.2007.00984.x. [DOI] [PubMed] [Google Scholar]
- 32.Gottardo R, Raftery AE, Yeung KY, Bumgarner RE. Bayesian robust inference for differential gene expression in microarrays with multiple samples. Biometrics. 2006;62(1):10–18. doi: 10.1111/j.1541-0420.2005.00397.x. [DOI] [PubMed] [Google Scholar]
- 33.Thomas RS, Allen BC, Nong A, Yang L, Bermudez E, Clewell HJ, 3rd, Andersen ME. A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol Sci. 2007;98(1):240–248. doi: 10.1093/toxsci/kfm092. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Allen BC, Thomas RS. BMDExpress: a software tool for the benchmark dose analyses of genomic data. BMC Genomics. 2007;8:387. doi: 10.1186/1471-2164-8-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pottenger LH, Gollapudi BB. A case for a new paradigm in genetic toxicology testing. Mutat Res. 2009 doi: 10.1016/j.mrgentox.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Hendriksen PJ, Freidig AP, Jonker D, Thissen U, Bogaards JJ, Mumtaz MM, Groten JP, Stierum RH. Transcriptomics analysis of interactive effects of benzene, trichloroethylene and methyl mercury within binary and ternary mixtures on the liver and kidney following subchronic exposure in the rat. Toxicol Appl Pharmacol. 2007;225(2):171–188. doi: 10.1016/j.taap.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Friedberg EC, Wagner R, Radman M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science. 2002;296(5573):1627–1630. doi: 10.1126/science.1070236. [DOI] [PubMed] [Google Scholar]
- 38.Lv L, Kerzic P, Lin G, Schnatter AR, Bao L, Yang Y, Zou H, Fu H, Ye X, Gross SA, Armstrong TW, Irons RD. The TNF-alpha 238A polymorphism is associated with susceptibility to persistent bone marrow dysplasia following chronic exposure to benzene. Leuk Res. 2007;31(11):1479–1485. doi: 10.1016/j.leukres.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 39.Hirabayashi Y, Yoon BI, Li GX, Kanno J, Inoue T. Mechanism of benzene-induced hematotoxicity and leukemogenicity: current review with implication of microarray analyses. Toxicol Pathol. 2004;32(Suppl 2):12–16. doi: 10.1080/01926230490451725. [DOI] [PubMed] [Google Scholar]
- 40.Snyder R, Witz G, Goldstein BD. The toxicology of benzene. Environ Health Perspect. 1993;100:293–306. doi: 10.1289/ehp.93100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon BI, Li GX, Kitada K, Kawasaki Y, Igarashi K, Kodama Y, Inoue T, Kobayashi K, Kanno J, Kim DY, Hirabayashi Y. Mechanisms of benzene-induced hematotoxicity and leukemogenicity: cDNA microarray analyses using mouse bone marrow tissue. Environ Health Perspect. 2003;111(11):1411–1420. doi: 10.1289/ehp.6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snyder R, Hedli CC. An overview of benzene metabolism. Environ Health Perspect. 1996;104(Suppl 6):1165–1171. doi: 10.1289/ehp.961041165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laskin JD, Rao NR, Punjabi CJ, Laskin DL, Synder R. Distinct actions of benzene and its metabolites on nitric oxide production by bone marrow leukocytes. J Leukoc Biol. 1995;57(3):422–426. doi: 10.1002/jlb.57.3.422. [DOI] [PubMed] [Google Scholar]
- 44.Bird MG, Greim H, Snyder R, Rice JM. International symposium: Recent advances in benzene toxicity. Chem Biol Interact. 2005;153-154:1–5. doi: 10.1016/j.cbi.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Irons RD, Lv L, Gross SA, Ye X, Bao L, Wang XQ, Ryder J, Armstrong TW, Zhou Y, Miao L, Le AT, Kerzic PJ, Ni W, Fu H. Chronic exposure to benzene results in a unique form of dysplasia. Leuk Res. 2005;29(12):1371–1380. doi: 10.1016/j.leukres.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 46.Liu H, Liang Y, Bowes S, Xu H, Zhou Y, Armstrong TW, Wong O, Schnatter AR, Fang J, Wang L, Nie L, Fu H, Irons R. Benzene exposure in industries using or manufacturing paint in China--a literature review, 1956-2005. J Occup Environ Hyg. 2009;6(11):659–670. doi: 10.1080/15459620903249646. [DOI] [PubMed] [Google Scholar]
- 47.Synder CA, Goldstein BD, Sellakumar A, Wolman SR, Bromberg I, Erlichman MN, Laskin S. Hematotoxicity of inhaled benzene to Sprague-Dawley rats and AKR mice at 300 ppm. J Toxicol Environ Health. 1978;4(4):605–618. doi: 10.1080/15287397809529683. [DOI] [PubMed] [Google Scholar]
- 48.Faiola B, Fuller ES, Wong VA, Recio L. Gene expression profile in bone marrow and hematopoietic stem cells in mice exposed to inhaled benzene. Mutat Res. 2004;549(1-2):195–212. doi: 10.1016/j.mrfmmm.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 49.Smith MT, Vermeulen R, Li G, Zhang L, Lan Q, Hubbard AE, Forrest MS, McHale C, Zhao X, Gunn L, Shen M, Rappaport SM, Yin S, Chanock S, Rothman N. Use of ‘Omic’ technologies to study humans exposed to benzene. Chem Biol Interact. 2005;153-154:123–127. doi: 10.1016/j.cbi.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 50.Forrest MS, Lan Q, Hubbard AE, Zhang L, Vermeulen R, Zhao X, Li G, Wu YY, Shen M, Yin S, Chanock SJ, Rothman N, Smith MT. Discovery of novel biomarkers by microarray analysis of peripheral blood mononuclear cell gene expression in benzene-exposed workers. Environ Health Perspect. 2005;113(6):801–807. doi: 10.1289/ehp.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]