

Abstract
We constructed a deletion mutant of the pyrE gene in Bifidobacterium longum 105-A. A pyrE knockout cassette was cloned into pKKT427, a Bifidobacterium-Escherichia coli shuttle vector, and then introduced into B. longum 105-A by electroporation. The transformants were propagated and spread onto MRS plates containing 5-fluoroorotic acid (5-FOA) and uracil. 5-FOA-resistant mutants were obtained at a frequency of 4.7 × 10−5 integrations per cell. To perform pyrE gene complementation, the pyrE gene was amplified by PCR and used to construct a complementation plasmid (pKKT427-pyrE+). B. longum 105-A ∆pyrE harboring this plasmid could not grow on MRS plates containing 5-FOA, uracil and spectinomycin. We also developed a chemically defined medium (bifidobacterial minimal medium; BMM) containing inorganic salts, glucose, vitamins, isoleucine and tyrosine for positive selection of pyrE transformants. B. longum 105-A ∆pyrE could not grow on BMM agar, but the same strain harboring pKKT427-pyrE+ could. Thus, pyrE can be used as a counterselection marker in B. longum 105-A and potentially other Bifidobacterium species as well. We demonstrated the effectiveness of this system by constructing a knockout mutant of the xynF gene in B. longum 105-A by using the pyrE gene as a counterselection marker. This pyrE-based selection system will contribute to genetic studies of bifidobacteria.
Keywords: pyrimidine metabolism, gene knockout, homologous recombination, 5-FOA, B. longum 105-A, bifidobacterial minimal medium (BMM), gene inactivation
INTRODUCTION
Bifidobacteria are high GC-content, Gram-positive anaerobes that belong to the Actinobacteria phylum [1]. They inhabit the intestinal tracts of many animals, including humans. The genome sequences of bifidobacteria, such as B. longum and B. adolescentis, are available [2, 3]. However, only a few genetic investigations have been reported for bifidobacteria, because sufficient gene manipulation techniques had not yet been developed for this genus. Recently, some useful gene manipulation techniques have been developed [4,5,6,7].
Generally, the genetic approaches for anaerobes are difficult and very limited, because of their sensitivity to O2 and their low transformation efficiency. The primary reason for low transformation efficiency is attributed to restriction-modification systems, which defend the cells from genetic invasion. To improve transformation efficiency, we developed the plasmid artificial modification (PAM) method [4, 5]. Briefly, a shuttle vector was prepared from E. coli, in which the genes encoding modification enzymes are cloned and expressed. The plasmid became resistant to digestion by the restriction enzyme during the transformation.
We have also constructed temperature-sensitive plasmid (pKO403) that replicates at 37°C but not at 42°C [6]. A gene knockout experiment was successfully performed using pKO403 in B. longum 105-A.
In order to perform gene inactivation by homologous recombination and selection of transformants, antibiotic selection markers have been generally used in some microorganisms. Some antibiotic selection markers have been reported, including genes for spectinomycin (Sp) [8], tetracycline [9], erythromycin and chloramphenicol (Cm) resistance [10], for the selection of transformants in Bifidobacterium. However, only Sp (75 µg/ml) gave reproducible, low-background results in our observation. Due to the limited number of available selection markers, multiple gene knockout experiments are hard to perform. To overcome the limited availability of selection markers, we focused on a counterselection marker, which allowed us to conduct a sequential multigene knockout technique by marker recycling.
In some microorganisms, several counterselection markers are available [11]. Genes related to the pyrimidine metabolic pathway have been found in most microorganisms (Fig. 1). In the case of Thermococcus kodakaraensis, the pyrE and pyrF genes encode orotate phosphoribosyl transferase (OPRTase) and orotidine 5′-monophosphate decarboxylase (OMPase), respectively [12]. The pyrE or pyrF genes and their orthologs (URA5 and URA3, respectively) are also widely used as counterselection markers for gene manipulation in T. kodakaraensis, Rhodobacter capsulatus and yeasts [12,13,14,15,16,17].
Fig. 1.

Pyrimidine metabolism in B. longum.
(A) Orotic acid and 5-FOA are metabolized by the same enzymes. pyrE encodes orotate phosphoribosyl transferase (OPRTase, EC:2.4.2.10), and pyrF encodes orotidine 5′-monophosphate decarboxylase (OMPase, EC:4.1.1.23). 5-FOA, an analog of orotic acid, is sequentially converted to 5-fluoroorotidine monophosphate (5-FOMP) by pyrE and then 5-fluorouridine monophosphate (5-FUMP) by pyrF. 5-FUMP is converted to pyrimidine-containing nucleotides in further steps and is toxic to cells. The following abbreviations of genes were used: carA, carbamoyl phosphate synthase small subunit (EC:6.3.5.5); pyrB, aspartate carbamoyltransferase catalytic subunit (EC:2.1.3.2); pyrC, dihydroorotase (EC:3.5.2.3); pyrD, dihydroorotate dehydrogenase; pyrG, CTP synthetase; pyrH, uridylate kinase (EC:2.7.4.22); pyrI, aspartate carbamoyltransferase regulatory subunit (EC:2.1.3.2); pyrK, dihydroorotate dehydrogenase electron transfer subunit; upp, uracil phosphoribosyl transferase (EC:2.4.2.9). (B) Genomic organization of pyrimidine metabolic genes. pyrB-F and pyrI are clustered in the B. longum NCC2705 genome. pyrG (BL0874) and pyrH (BL1505) are located upstream and downstream of these genes, respectively.
Orotic acid, an intermediate in pyrimidine metabolism, is converted to orotidine 5′-monophosphate (OMP) by OPRTase, which is encoded by pyrE. 5-Fluoroorotic acid (5-FOA), an analog of orotic acid, is metabolized by the same enzyme. OPRTase (PyrE) can convert 5-FOA into 5-fluoroorotidine monophosphate (5-FOMP) instead of OMP. Then, 5-FOMP is converted to 5-fluorouridine monophosphate (5-FUMP), instead of UMP, by OMPase (PyrF). Accumulation of 5-FUMP is toxic and leads to cell death. In contrast, pyrE– or pyrF– cells can grow in the presence of 5-FOA, since pyrimidine metabolism is blocked at the corresponding steps so that 5-FUMP cannot be synthesized and accumulated in the cells (Fig. 1).
In addition, pyrE or pyrF mutants show uracil auxotrophy in a chemically defined medium without pyrimidine compounds, such as uracil, thymine, cytosine, or their nucleotide derivatives. In these mutant strains, the pyrE or pyrF genes are useful as positive selection markers. Mutant strains harboring the plasmids carrying these marker genes are able to grow in the same medium.
In molecular genetic experiments, if the pyrE (or pyrF) mutant strains are available, these genes can be applied as positive selection markers by auxotrophy in Ura– minimal medium and negative selection (= counterselection) markers in 5-FOA-supplemented medium. Thus, these types of markers are called bidirectional selection markers and can be applied in multigene knockout experiments. After constructing a knockout mutant using a pyrE (or pyrF) marker, the marker can be cured by addition of 5-FOA. The same marker can be reused in knocking out the next target gene. Such marker recycling is already widely used in some microorganisms for sequential gene knockout [15, 16].
In our recent study, the pyrE gene was knocked out using a temperature-sensitive plasmid for evaluating the frequency of gene knockout [6]. In this study, we aimed to generate pyrE knockout mutants and investigate their properties. Furthermore, we developed a new chemically defined medium (bifidobacterial minimal medium; BMM) for pyrE-based positive selection and also for investigation of metabolism of Bifidobacterium.
This selection system should be a powerful genetic tool in studies of Bifidobacterium requiring efficient gene knockout construction. In this paper, we demonstrated the utility of this system by constructing a knockout mutant of the xynF gene in B. longum 105-A by using the pyrE gene as a counterselection marker.
MATERIALS AND METHODS
Bacterial strains and media
The bacterial strains and plasmids used in this study are listed in Table 1. B. longum 105-A was used as the host strain. Escherichia coli TOP10 (Invitrogen, Carlsbad, CA, USA) was used as the cloning host for plasmid construction. B. longum was grown anaerobically in MRS medium (BD, Franklin Lakes, NJ, USA) at 37°C. E. coli was grown on LB medium (10 g tryptone, 5 g yeast extract and 5 g NaCl per l) at 37°C. For plate culture, 1.5% agar was added to the medium before autoclaving. Sp (75 µg/ml), Cm (30 µg/ml), 5-FOA (500 µg/ml) and uracil (200 µg/ml) were added as needed. A minimal medium was prepared based on the minimal medium for Bacteroides [18, 19]. Yeast extract (BD, Franklin Lakes, NJ, USA), vitamins and amino acids were added as required.
Table 1. Bacterial strains and plasmids used.
| Strain or plasmid | Descriptiona | Source |
| Bacterial strains | ||
| Escherichia coli TOP10 | F−, mcrA(mrr, hsdRMS-mcrBC), φ80(lacZ)∆M15 ∆lacX74, recA1, araD139, (ara-leu)7697, galU, galK, rpsL(Strr), endA1, nupG | Invitrogen |
| Bifidobacterium longum 105-A | Wildtype | 24 |
| B. longum 105-A ∆pyrE | 5-FOAr Ura–; ∆pyrE | This study |
| B. longum 105-A ∆pyrE/pKKT427-pyrE+ | 5-FOAs Ura+; pyrE+ | This study |
| B. longum 105-A ∆pyrE/pKEC58-∆xynF | 5-FOAs Ura+ Spr Cmr; ∆pyrE | This study |
| B. longum 105-A ∆pyrE ∆xynF | 5-FOAr Ura– Spr Cms; ∆pyrE ∆xynF | This study |
| Plasmids | ||
| pKKT427 | Spr; 3.9 kb shuttle vector between Bifidobacterium and E. coli | 4, 5, 22 |
| pKKT427-∆pyrE | Spr; pKKT427 carrying the up- and downstream flanking regions of pyrE | This study |
| pKKT427-pyrE+ | Spr; pKKT427 carrying the putative pyrE gene, promoter, and terminator | This study |
| pBAD28 | Cmr; 5.8 kb plasmid for E. coli | 23 |
| pKEC58 | Cmr; same as pKKT427-pyrE+ but Spr was replaced by the Cmr | This study |
| pKEC58-∆xynF | Spr and Cmr; pKEC58 carrying the xynF deletion DNA fragments. Spr was sandwiched by up- and downstream regions of the xynF gene | This study |
a 5-FOAr and Ura–, resistant to 5-fluoroorotic acid and uracil auxotrophy, respectively.
Molecular techniques
Genomic DNA of B. longum 105-A and its derivatives were extracted and purified as previously described [20]. Plasmid extractions from E. coli strains were performed using a QIAprep Spin Miniprep Kit (Qiagen, Hilden, Germany). DNA digestion with restriction enzymes was performed according to the manufacturer’s protocol (Takara Bio, Japan). DNA sequencing of plasmid and genomic DNA was performed on an ABI 3100 DNA sequencer using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA).
PCR conditions
The PCR conditions and primer sequences used in this study for generating each plasmid or confirming the deletion of target genes are described in Table 2. KOD-plus DNA polymerase (Toyobo, Osaka, Japan) was used in all experiments as described by the manufacturer’s protocol. The PCR products were analyzed by 1% (w/v) agarose gel electrophoresis. For cloning, the target fragments were extracted from the gel using a NucleoSpin kit (Nippon Genetics, Tokyo, Japan).
Table 2. Summary of PCR experiments.
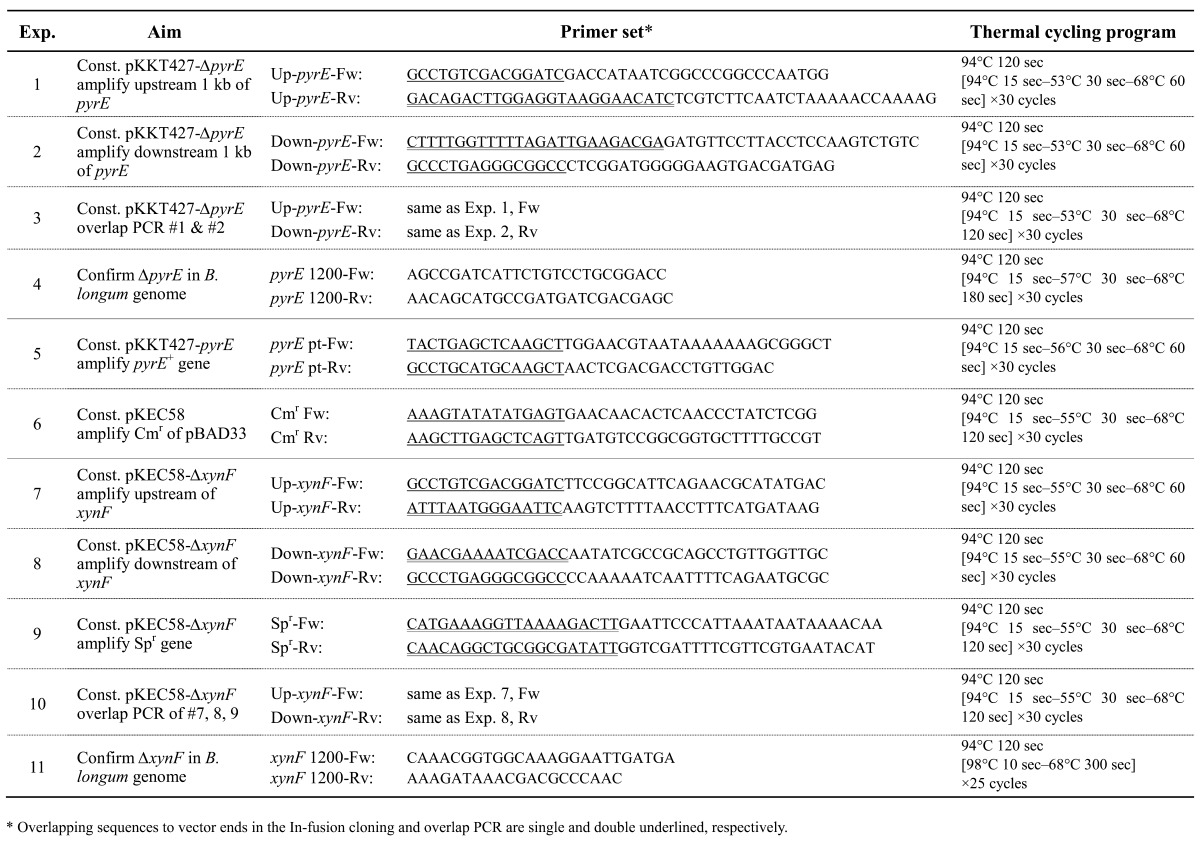
Plasmid construction
The pKKT427-∆pyrE plasmid was constructed as follows. The PCR primers were designed according to the putative pyrE gene (BL0788) and the flanking genomic regions of B. longum NCC2705 (GenBank Accession no. AE014295). To obtain the pyrE deletion DNA fragments, upstream (1.0 kb) and downstream regions (1.0 kb) of the putative pyrE gene of B. longum 105-A was amplified by PCR as summarized in Table 2 (Exp. 1 and 2). The produced DNA fragments were connected by an overlap PCR (Table 2, Exp. 3) [21]. The connected PCR product was cloned into a BamHI/NotI-digested pKKT427, a Bifidobacterium-E. coli shuttle vector [4, 5, 22], using an In-Fusion Dry-Down PCR Cloning Kit (Clontech, Mountain View, CA, USA).
The plasmid pKKT427-pyrE+ was constructed as follows. The 835 bp DNA fragment, including the putative promoter (79 bp), open reading frame (ORF) (696 bp) and putative terminator (60 bp) of the pyrE gene in B. longum 105-A, was PCR amplified (Table 2, Exp. 5) and then cloned into HindIII-digested pKKT427 using the In-Fusion method as described above.
pKKT427-pyrE+ was modified to change the selection marker from the Spr gene to the Cm-resistance (Cmr) gene. The Cmr gene was PCR amplified from pBAD28 [23] (Table 2, Exp. 6). The Cmr gene was cloned into ScaI-digested pKKT427-pyrE+ using the In-Fusion PCR method. The obtained plasmid was designated pKEC58.
The plasmid pKEC58-∆xynF was constructed as follows. PCR primers were designed according to the putative xynF gene (BL1544) and the flanking genomic regions of B. longum NCC2705. To obtain the xynF deletion DNA fragments, upstream (1.0 kb) and downstream regions (1.0 kb) of the putative xynF gene in B. longum 105-A, were amplified by PCR (Table 2, Exp.7 and 8, respectively). The Spr gene was PCR amplified from pKKT427 (Table 2, Exp. 9). The three produced DNA fragments (upstream, Spr gene and downstream) were connected by an overlap PCR [21] using the primers Up-xynF-Fw and Down-xynF-Rv (Table 2, Exp. 10). Approximately 3.1 kb DNA fragments were cloned into BamHI/NotI-digested pKEC58 using the In-Fusion method.
Gene knockout of pyrE
B. longum 105-A cells were transformed with pKKT427-∆pyrE by electroporation [24]. After transformation, cells were spread and cultured on MRS plates containing Sp. Transformants were selected and cultured in MRS liquid medium containing Sp. Cells were serially diluted and spread on MRS plates containing 5-FOA and uracil. After incubation at 37°C for 2 days, 5-FOAr colonies were selected and used for further analysis. Genomic DNA was extracted from 5-FOAr colonies and wild-type strains. The deletion of pyrE was confirmed by PCR using the pyrE 1200-Fw and pyrE 1200-Rv primers (Table 2, Exp. 4) from the genomic DNA template.
Gene knockout of xynF
B. longum 105-A ∆pyrE was transformed with pKEC58-∆xynF by electroporation. After transformation, cells were cultivated on MRS plates containing Sp at 37°C for 2 days. Spr transformants were inoculated in MRS liquid medium containing Sp. Propagated cells were diluted and spread on MRS plates containing 5-FOA, uracil and Sp. After incubation at 37°C for 2 days, cells resistant to 5-FOA and Sp were selected as the target of xynF knockout. Genomic DNA was extracted from candidates and ∆pyrE strains. The deletion of xynF was confirmed by PCR (Table 2, Exp. 11) from the genomic DNA template.
Acquisition frequencies of ΔxynF mutants were estimated at the same time. The ratios of the number of 5-FOAr and Spr cells to the number of total cells were calculated.
Frequency evaluation of homologous recombination
The frequency of homologous recombination events was examined. Spr cells harboring pKKT427-∆pyrE were prepared and spread on MRS plates containing 5-FOA and uracil for selection of pyrE knockout mutants generated by DCO homologous recombination and nonselective MRS plates for viable cell count. These plates were cultured at 37°C for 2 days, and then the number of colonies was counted. The frequency of DCO homologous recombination (integrations per cell, ipc) was calculated as the ratio of the number of 5-FOAr cells to the number of total cells. The number of total cells was corrected with the plasmid loss rate, which was estimated as the ratio of the number of Spr cells to the number of total cells.
To confirm the phenotypic observation, 5-FOAr colonies (B. longum 105-A ∆pyrE1 and B. longum 105-A ∆pyrE2) and wild-type strains were streaked on MRS plates containing uracil or 5-FOA and BMM plates with or without uracil. These plates were incubated at 37°C for 2 days.
HPLC conditions
Reverse-phase column chromatography was performed using an ODS column (Inertsil ODS-3V, 5 µm, 4.6 mm i.d. × 250 mm, GL Sciences, Tokyo, Japan). The HPLC system used was an LC-10ATVP with an SPD-M10A Diode Array Detector (Shimadzu, Kyoto, Japan). The mobile phase was composed of methanol (solvent A) and 0.05% trifluoroacetic acid (solvent B) with the following gradient elution: 20% A and 80% B initially, changing to 80% A and 20% B in 30 min. The column flow rate was 1 ml/min at 25°C with detection at 230 and 256 nm.
Growth assay using a 96-well microplate
The collected fractions were freeze-dried, redissolved in methanol at 5 mg/ml, individually added to a 96-well plate and air-dried, and then minimal medium was added. B. longum 105-A was cultured in these media at 37°C for 24 hr. The OD490 of growth was measured using a plate reader (Immunomini NJ-2300, Cosmo Bio, Tokyo, Japan).
1H NMR analysis
The growth-promoting fraction was used for 1H NMR. 1H NMR spectra were recorded using an ECA500 (JEOL, Tokyo, Japan).
RESULTS AND DISCUSSION
Generation of a B. longum 105-A pyrE knockout mutant
In the B. longum NCC2705 genome, BL0788 and BL0791 CDS are annotated as pyrE and pyrF, respectively. To obtain a pyrE knockout mutant, we constructed a plasmid, pKKT427-∆pyrE (Fig. 2-A). It was introduced into B. longum 105-A. For gene knockout experiments, the transformants were cultured in MRS liquid medium containing Sp, and propagated cells (100 µL culture ≈ 1 × 108 cells) were spread onto MRS plates containing 5-FOA and uracil. After incubation, 5-FOAr colonies were obtained at about 200 colonies per plate. We selected two candidates (B. longum 105-A ∆pyrE1 and B. longum 105-A ∆pyrE2) for further analysis. Genomic DNA was extracted from these candidates to confirm the pyrE deletion by PCR (Fig. 2-B). The resulting PCR for both mutants gave fragments of 2.4 kb, whereas the DNA amplified from the wild-type B. longum 105-A resulted in a 3.1 kb fragment. The DNA sequences of the 2.4 kb fragments were analyzed to confirm correct deletion. A 696 bp region of the pyrE ORF was successfully knocked out in the candidates.
Fig. 2.

Construction of B. longum 105-A pyrE knockout mutants. (A) Schematic diagram of the construction of pyrE knockout mutants. The plasmid pKKT427-∆pyrE was introduced into B. longum 105-A. Transformants were cultured on MRS plates containing 5-FOA and uracil at 37°C. The cells in which DCO homologous recombination had occurred between the plasmid and chromosome showed a 5-FOAr and Sps phenotype. The arrows indicate primers, pyrE 1200-Fw, pyrE 1200-Rv and theoretical products of PCR confirmation for the pyrE deletion in Panel B. Ori indicates the origin of replication of the plasmid pTB6. (B) PCR analysis of B. longum 105-A wild-type and B. longum 105-A pyrE knockout mutants. The region flanking pyrD, BL0787, was amplified by PCR (Table 2, No.4). Lane M indicates the DNA marker, λ-EcoT14I, with the following size order from top to bottom: 19,329 bp, 7,743 bp, 6,223 bp, 4,254 bp, 3,472 bp, 2,690 bp, 1,882 bp, 1,489 bp and 925 bp. Lane 1, B. longum 105-A wild-type; lane 2, B. longum 105-A ∆pyrE1; and lane 3, B. longum 105-A ∆pyrE2.
It was also possible that some spontaneous pyrE mutants (such as point mutations) were also selected during the 5-FOA selection. To estimate the frequency of spontaneous mutation, we tested 100 of the 5-FOAr colonies by colony PCR. However, all colonies showed correct deletion. These results indicated that most 5-FOAr colonies (>99%) were double crossover (DCO) recombination mutants. The calculated frequency of DCO homologous recombination was 4.7 × 10−5 ipc.
Development of chemically defined medium
We examined the growth characteristics of the ∆pyrE mutants (B. longum 105-A ∆pyrE1 and B. longum 105-A ∆pyrE2) on MRS plates with or without 5-FOA and uracil (Fig. 3-A). The resistance of ∆pyrE mutants to 5-FOA was clearly confirmed. However, uracil auxotrophy was not observed in MRS medium because MRS medium contains an adequate concentration of uracil or other pyrimidine compounds. Thus, we defined a new medium, BMM, based on a minimal medium for Bacteroides [18, 19]. Yeast extract, which is a component of the minimal medium, was analyzed to construct a chemically defined medium. Different volumes of yeast extract (5,000 mg, 500 mg and 50 mg/L) were added to the basal media. These media were used for cultivation of B. longum 105-A. As a result, normally growing colonies were detected in media containing more than 50 mg/L (data not shown).
Fig. 3.

Growth of transformants on the selection media.
(A) Growth of pyrE knockout mutants, B. longum 105-A ∆pyrE1 and B. longum 105-A ∆pyrE2, as compared with B. longum 105-A wild type. Each strain was spread on an MRS plate in the presence or absence of uracil (200 µg/ml) or with uracil and 5-FOA (500 µg/ml). (B) The pyrE deletion was complemented by pKKT427-pyrE+. B. longum 105-A ∆pyrE1 and B. longum 105-A ∆pyrE2 were transformed with pKKT427-pyrE+. Spr colonies were selected and streaked on an MRS plate containing Sp (75 µg/ml) or uracil and 5-FOA and on a BMM plate containing Sp. Sp is the selection marker for pKKT427-pyrE+.
To clarify additional factors to assist B. longum 105-A, the growth-promoting components were purified from yeast extract. Twenty-five grams of yeast extract powder was sequentially extracted with 500 mL each of ethyl acetate, acetone and methanol. The methanol fraction showed the strongest growth stimulation activity. Thus, we separated the fraction by reverse-phase column chromatography. As a result, nine assayed peaks were observed (Fig. 4-A). We examined the growth promoting activity for B. longum 105-A by adding each peak fraction to the basal media. The level of the third peak was the same as that for addition of yeast extract (Fig. 4-B). By the analysis using 1H NMR, it was found that the fraction consisted of isoleucine and tyrosine. The growth of B. longum 105-A was confirmed by adding these compounds to the basal medium. Addition of isoleucine and tyrosine resulted in the cells showing successful growth on the agar plate. In contrast, the cells showed very weak growth using liquid medium. These data revealed that some unknown factor(s) was required for the growth of B. longum 105-A (Fig. 4 and Table 3). The unknown factor should be some compound present in trace amounts in agar and also pure agarose. We are investigating this as a possible new growth factor for bifidobacteria.
Fig. 4.

Purification of growth-promoting compounds from yeast extract.
(A) Yeast extract powder was sequentially extracted with ethyl acetate, acetone and methanol. The methanol extract was separated by reverse-phase (RP) column chromatography using an ODS column as described in Materials and Methods. (B) Growth assays were performed in the basal medium supplemented with reverse-phase column chromatography fractions (panel A). The turbidity was measured using a 96-well plate reader at 490 nm. YE, yeast extract.
Table 3. Composition of bifidobacterial minimal medium (BMM).
| Components | Conc. (per liter) |
| Salts | |
| NaCl | 9.0 g |
| CH3COONa | 5.0 g |
| KH2PO4 | 4.5 g |
| Na2CO3* | 400 mg |
| (NH4)2SO4 | 400 mg |
| MgSO4 | 200 mg |
| CaCl2·2H2O | 250 mg |
| MnCl2·4H2O | 100 mg |
|
Carbon source | |
| D-Glucose* | 20 g |
|
Vitamins* | |
| L-ascorbate (Na) | 340 mg |
| Inositol | 1400 μg |
| 4-aminobenzoic acd | 800 μg |
| Nicotinic acid | 600 μg |
| Biotin | 300 μg |
| Choline | 300 μg |
| Pantothenic acid | 300 μg |
| Riboflavin | 120 μg |
| Pyridoxine | 40 μg |
| Folic acid | 1.5 μg |
|
Amino acids | |
| Cysteine·HCl* | 500 mg |
| Isoleucine | 64.5 mg |
| Tyrosine | 87.0 mg |
*Glucose, Na2CO3, vitamins and cysteine·HCl were sterilized with 0.33 µm filters (Sterivex, Millipore, Billerica, MA, USA) and added after the medium was autoclaved and cooled.
Uracil auxotrophy of the ∆pyrE mutants was examined on BMM plates with or without uracil. Consequently, these mutants were clearly confirmed to be uracil auxotrophic (Ura–) mutants that could grow only in the presence of uracil (Fig. 3-A).
We also found that the acetone fraction of the yeast extract assisted the growth of B. longum NCC2705. For other strains, it would need to be optimized with supplemental amino acids or vitamins for each strain [25, 26].
Complementation of pKKT427-pyrE+
We performed complementation experiments using the pyrE gene as a selection marker. The knockout mutants (above) harboring pKKT427-pyrE+ were streaked on MRS plates with or without 5-FOA, uracil and Sp. The uracil auxotrophy of B. longum 105-A ∆pyrE was successfully complemented by the plasmid pKKT427-pyrE+. The ∆pyrE strains harboring pKKT427-pyrE+ could not grow on MRS plates containing 5-FOA, uracil and Sp (Fig. 3-B, left), but the strain harboring pKKT427 grew normally. The complementation of the pyrE gene was observed on MRS plates. The pyrE+ strains (B. longum 105-A ∆pyrE1 and ∆pyrE2 harboring pKKT427-pyrE+) grew normally on BMM plates, but the pyrE– strain (B. longum 105-A ∆pyrE1/pKKT427) did not (Fig. 3-B, right). Thus, recombinant pyrE expressed a functional OPRTase, and BMM agar plates can be used for positive selection of pyrE transformants.
Gene knockout of the xynF gene
To evaluate the usefulness of a pyrE-based selection system, we demonstrated a gene knockout of xynF, which putatively encodes an extracellular exo-xylanase of B. longum 105-A, is conserved between B. longum strains and is annotated as BL1544 in B. longum NCC2705. We chose this as a target gene for the knockout study. β-1,4-Xylosidase is thought to be an essential enzyme in xylo-oligosaccharide metabolism, and B. longum consists of two possible β-1,4-xylosidase genes, xynF and BL0523 (classified in Family 31, possible α-glucosidase or β-xylosidase). The xynF gene encodes the ordinary β−xylosidase catalytic domain but also includes a putative signal peptide for secretion that is predicted to be an extracellular enzyme. The physiological roles of these genes are still unclear, and the knockout gene technique is essential to clarify such complex paralogue systems.
The gene knockout plasmid, pKEC58-∆xynF (Fig. 5-A), was constructed and introduced into B. longum 105-A ∆pyrE. To obtain gene knockout mutants, the transformants were inoculated on MRS plates containing 5-FOA, uracil and Sp. Spr and 5-FOAr colonies were obtained at a frequency of 8.5 × 10−4 ipc. To investigate the deletion of the xynF gene, 20 clones were analyzed by PCR. We confirmed the deletion of 5.3 kb of the xynF ORF by Spr gene integration (Fig. 5-B). These results indicate that gene knockout mutants might be easily constructed using this pyrE-based selection system. We also checked the deletion of the xynF gene by using sequence analysis. As a result, deletion of this gene was confirmed (data not shown).
Fig. 5.

Construction of B. longum 105-A xynF knockout mutants.
(A) Schematic diagram of the construction of xynF knockout mutants. The plasmid pKEC58-∆xynF was introduced into B. longum 105-A ∆pyrE. Transformants were cultured on MRS plates containing 5-FOA, uracil and Sp at 37°C. The cells in which DCO homologous recombination had occurred between the plasmid and chromosome showed a 5-FOAr and Spr phenotype. The arrows indicate primers, xynF 1200-Fw, xynF 1200-Rv and theoretical products of PCR. Ori indicates the origin of replication of the plasmid pTB6. (B) PCR analysis of B. longum 105-A ∆pyrE and B. longum 105-A xynF knockout mutants (Table 2, No. 10). Lane M: DNA size marker as described in Fig. 2. Lane 1, B. longum 105-A ∆pyrE; lane 2, B. longum 105-A ∆xynF1; and lane 3, B. longum 105-A ∆xynF2.
The phenotypes of the ∆xynF mutants were examined by means of BMM plates containing 5-FOA, uracil, Sp and xylo-oligosaccharides. Contrary to our expectation, both strains of B. longum 105-A ∆pyrE and B. longum 105-A ∆pyrE ∆xynF grew in this medium (data not shown). This result suggests that other β-1,4-xylosidase isozymes shows enough activity to utilize xylo-oligosaccharides in B. longum 105-A. Here, we demonstrated a xynF knockout experiment to prove the usefulness of pyrE as a counterselection marker. Further studies, including BL0523 knockout, are required to understand the functions of these genes and also xylan and xylo-oligosaccharide metabolism.
We have tried to construct genetic tools for bifidobacteria, such as high efficiency transformation methods [4, 5], and to improve gene expression by tuning promoter and ribosomal binding sequences [27, 28]. In this study, we reported a new selection marker in Bifidobacterium. Now we can use two gene knockout methods, Ts-plasmid [6] and pyrE bidirectional selection marker.
In the case of xynF gene knockout as described above, only 2 weeks are needed for construction, from primer design to PCR confirmation. In each experimental step, only 4–5 plates were needed, and around 100 colonies were obtained on the selection plate, in transformation and counterselection. Because of the high efficiency, around only 10 colonies were enough to screen for true knockout mutants by colony direct PCR confirmation.
In addition, a transformation efficiency of around 103 cfu/µg DNA is sufficient for the initial transformation experiment in this system because a few, such as 5–10, clones are enough to perform the subsequent procedure. We have achieved this efficiency with some strains, including B. longum and B. adolescentis strains, using the PAM method [4, 5]. We have already applied this method in some other strains and species of bifidobacteria, including B. longum and B. breve, and successfully obtained ΔpyrE mutants (data not shown). Using these quick and convenient tools, it becomes very easy to design reverse genetic experiments and perform strain engineering [29]. We hope these improvements accelerate genetic studies of bifidobacteria.
Acknowledgments
This research was supported by a Grant-in Aid for Scientific Research (C), 20510189, from the Ministry of Education, Culture, Sports, Science and Technology of Japan and a C19 Kiyomi Yoshizaki Grant for the Promotion of Scientific Research (2011).
REFERENCES
- 1.Stackebrandt E, Rainey FA, WardRainey NL. 1997. Proposal for a new hierarchic classification system, Actinobacteria classis nov. Int J Syst Bacteriol 47: 479–491 [Google Scholar]
- 2.Lee JH, Karamychev VN, Kozyavkin SA, Mills D, Pavlov AR, Pavlova NV, Polouchine NN, Richardson PM, Shakhova VV, Slesarev AI, Weimer B, O’Sullivan DJ. 2008. Comparative genomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genomics 9: 247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yasui K, Tabata M, Yamada S, Abe T, Ikemura T, Osawa R, Suzuki T. 2009. Intra-species diversity between seven Bifidobacterium adolescentis strains identified by genome-wide tiling array analysis. Biosci Biotechnol Biochem 73: 1422–1424 [DOI] [PubMed] [Google Scholar]
- 4.Yasui K, Kano Y, Tanaka K, Watanabe K, Shimizu-Kadota M, Yoshikawa H, Suzuki T. 2009. Improvement of bacterial transformation efficiency using plasmid artificial modification. Nucleic Acids Res 37: e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki T, Yasui K. 2011. Strain engineering: Methods and protocols, methods in molecular biology, In Williams, J. A. (ed.), Chapter 18, pp. 309–326, Humana Press, New York. [Google Scholar]
- 6.Sakaguchi K, He J, Tani S, Kano Y, Suzuki T. 2012. A targeted gene knockout method using a newly constructed temperature-sensitive plasmid mediated homologous recombination in Bifidobacterium longum. Appl Microbiol Biotechnol 95: 499–509 [DOI] [PubMed] [Google Scholar]
- 7.Hirayama Y, Sakanaka M, Fukuma H, Murayama H, Kano Y, Fukiya S, Yokota A. 2012. Development of a double-crossover markerless gene deletion system in Bifidobacterium longum: functional analysis of the α-galactosidase gene for raffinose assimilation. Appl Environ Microbiol 78: 4984–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Missich R, Sgorbati B, Leblanc DJ. 1994. Transformation of Bifidobacterium longum with pRM2, a constructed Escherichia-coli B. longum shuttle vector. Plasmid 32: 208–211 [DOI] [PubMed] [Google Scholar]
- 9.Flórez AB, Ammor MS, Alvarez-Martin P, Margolles A, Mayo B. 2006. Molecular analysis of tet (W) gene-mediated tetracycline resistance in dominant intestinal Bifidobacterium species from healthy humans. Appl Environ Microbiol 72: 7377–7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi M, Brigidi P, Rodriguez A, Matteuzzi D. 1996. Characterization of the plasmid pMB1 from Bifidobacterium longum and its use for shuttle vector construction. Res Microbiol 147: 133–143 [DOI] [PubMed] [Google Scholar]
- 11.Reyrat JM, Pelicic V, Gicquel B, Rappuoli R. 1998. Counterselectable markers: Untapped tools for bacterial genetics and pathogenesis. Infect Immun 66: 4011–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato T, Fukui T, Atomi H, Imanaka T. 2003. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J Bacteriol 185: 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yano T, Sanders C, Catalano J, Daldal F. 2005. sacB-5-Fluoroorotic acid-pyrE-based bidirectional selection for integration of unmarked alleles into the chromosome of Rhodobacter capsulatus. Appl Environ Microbiol 71: 3014–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boeke JD, LaCroute F, Fink GR. 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Molecular & general genetics MGG 197: 345–346. [DOI] [PubMed] [Google Scholar]
- 15.Hirashima K, Iwaki T, Takegawa K, Giga-Hama Y, Tohda H. 2006. A simple and effective chromosome modification method for large-scale deletion of genome sequences and identification of essential genes in fission yeast. Nucleic Acids Res 34: e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaneko S, Tanaka T, Noda H, Fukuda H, Akada R, Kondo A. 2009. Marker-disruptive gene integration and URA3 recycling for multiple gene manipulation in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 83: 783–789 [DOI] [PubMed] [Google Scholar]
- 17.Kitamoto K, Oda K, Gomi K, Takahashi K. 1990. Construction of uracil and tryptophan auxotrophic mutants from sake yeasts by disruption of URA3 and TRP1 genes. Agric Biol Chem 54: 2979–2987 [Google Scholar]
- 18.Seddon SV, Shah HN, Hardie JM, Robinson JP. 1988. Chemically defined and minimal media for Bacteroides gingivalis. Curr Microbiol 17: 147–149 [Google Scholar]
- 19.Varel VH, Bryant MP. 1974. Nutritional features of Bacteroides fragilis subsp. fragilis. Appl Microbiol 28: 251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: A Laboratory Manual. Cold Spring Harbour Laboratory Press, Woodbury, NY. [Google Scholar]
- 21.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61–68 [DOI] [PubMed] [Google Scholar]
- 22.Tanaka K, Samura K, Kano Y. 2005. Structural and functional analysis of pTB6 from Bifidobacterium longum. Biosci Biotechnol Biochem 69: 422–425 [DOI] [PubMed] [Google Scholar]
- 23.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose P-BAD promoter. J Bacteriol 177: 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsumura H, Takeuchi A, Kano Y. 1997. Construction of Escherichia coli Bifidobacterium longum shuttle vector transforming B. longum 105-A and 108-A. Biosci Biotechnol Biochem 61: 1211–1212 [DOI] [PubMed] [Google Scholar]
- 25.Tashiro Y. 1994. Bifidus Kin no Kenkyu (Study of Bifidobacteia). pp. 68–73. In Mitsuoka, T. (ed.), Japan Bifidus Foundation, Tokyo, Japan. [Google Scholar]
- 26.Tanaka R, Mutai M. 1980. Improved medium for selective isolation and enumeration of Bifidobacterium. Appl Environ Microbiol 40: 866–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He J, Sakaguchi K, Suzuki T. 2012. Determination of the ribosome-binding sequence and spacer length between binding site and initiation codon for efficient protein expression in Bifidobacterium longum 105-A. J Biosci Bioeng 113: 442–444 [DOI] [PubMed] [Google Scholar]
- 28.He J, Sakaguchi K, Suzuki T. 2012. Acquired Tolerance to oxidative stress in Bifidobacterium longum 105-A via expression of a catalase gene. Appl Environ Microbiol 78: 2988–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukiya S, Suzuki T, Kano Y, Yokota A. Lactic Acid Bacteria and Bifidobacteria: Current Progress in Advanced Research, In Sonomoto K, Yokota A. (eds), Chapter 2, pp. 33–51. [Google Scholar]