

Abstract
BACKGROUND
Hereditary hemochromatosis (HH) is a very rare disease in Iran and reported cases are all negative for HFE mutation. We report a family affected by severe juvenile hemochromatosis (JH) with a detailed molecular study of the family members.
METHODS
We studied a pedigree with siblings affected by juvenile HH and followed them for 3 years. Microsatellite and gene sequencing analysis was performed for all family members.
RESULTS
Two siblings (the proband and his sister, aged 26 and 30 years, respectively) were found to have clinical findings of JH. The proband’s brother, who presented with hyperpigmentation, died of probable JH at the age of 24 years. Gene sequencing analysis showed that the proband has a homozygote c.265T>C (p.C89R) HJV mutation + a heterozygote c.884T>C (p.V295A) mutation of HFE. The affected proband’s sister presented with the same HJV c.265T>C (p.C89R) homozygote mutation. In addition, we found the HJV c.98-6C>G polymorphic variant in both the sister and proband (homozygote). Sequencing of hepcidin (HAMP), TfR2, and FPN revealed no mutation.
CONCLUSION
We have shown that molecular analysis of the HH related gene is a powerful tool for reliable diagnosis of JH and, in conjunction with magnetic resonance imaging (MRI) and noninvasive liver stiffness measurement by elastography, is adequate tool for management and follow up of HH.
Keywords: Juvenile Hemochromatosis, Hemojuvelin Mutation, Genetic Study, Iran
INTRODUCTION
Hereditary hemochromatosis (HH) is a common autosomal recessive disease leading to iron over-load and end-organ damage. The most prevalent cause of HH is a missense mutation of HFE on chromosome 6 made by substitution of tyrosine for cysteine at amino acid 282 (C282Y).1 Despite the high prevalence of HFE mutation among European ancestry, studies of African and Asian ethnicities have found very low prevalence of the HFE mutation.2-6 A genetic study performed in 224 healthy individuals in Tehran showed that only 2.2% were heterozygous for the C282Y HFE mutation and there were no homozygous cases.2 Hemojuvelin mutation (HJV) located on chromosome 1 has been found to cause juvenile hemochromatosis (JH) with clinical symptoms in the second decade of life. This clinical consequences of this mutation is more severe than HFE related HH.7,8 Contrary to HFE related HH, in HJV hemochromatosis a range of different types of mutations has been reported. Papanikolaou et al. showed that 80% of HJV mutations are p.G320V, however until now more than 25 different mutations in the HJV gene have been described.7,9,10
In this report we aim to present clinical, laboratory, genetic and follow up data of a family with an HJV mutation in 3 affected members who are residents of Western Iran. This is the first report of an HJV mutation from Iran.
MATERIALS AND METHODS
Subjects
We studied a family pedigree from Western Iran that originated from Kurd and Lur ethnicity (I-1, I-4: of Lur and I-2, I-3: Kurd ethnicity) and has 43 members. Study approval was obtained by the Digestive Disease Research Institute (DDRI) Ethical Committee and all 9 subjects who gave blood samples signed informed consents to participate in this study.
History and physical examination
A thorough history was taken and complete physical examination was performed for the proband (III-9) and his family.
Laboratory and imaging investigations
A primary laboratory examination (complete blood count, iron indices, liver function test, and hepatic viral marker) was performed for all family members. In addition, serial laboratory examinations (liver elastography, abdominal MRI) were performed for the proband and his affected sister both before and after therapy.
Genetic studies
For analysis of the HFE, HAMP, TfR2, and HJV coding regions (exons plus intron–exon boundaries), PCR-amplified fragments were purified with the QIAquick PCR Purification Kit (Qiagen, Valencia, CA) and sequenced by using the Dye-terminato Cycle-sequencing Kit (Beckman Coulter Inc.). Fragments were then electrophoretically separated and analyzed with a CEQ 8000 XL Beckman Coulter DNA sequencer.
RESULTS
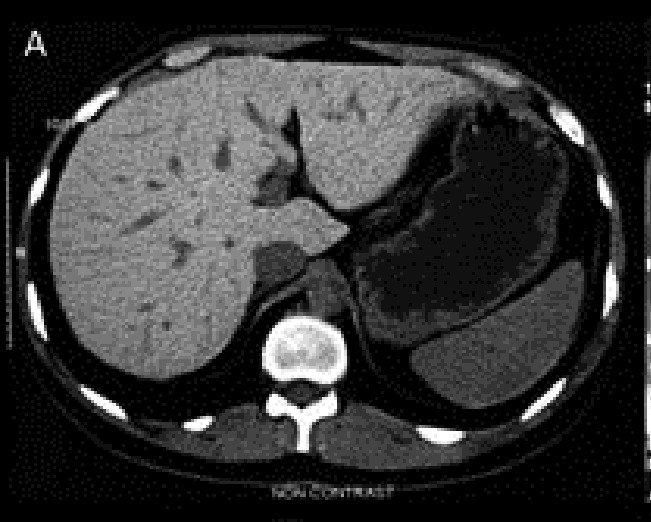
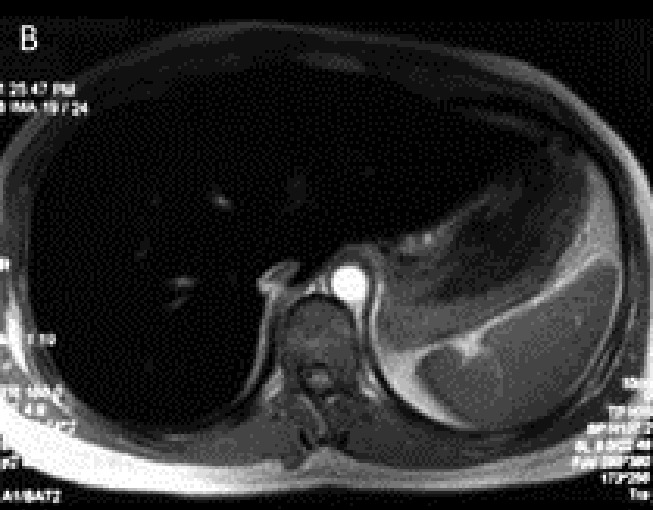
The proband was visited in our clinic in Tehran for the first time in 2009. He was a 26-year-old man who referred to our center because of weakness, skin hyperpigmentation and occasional dyspnea from three months before admission. He also complained of decreased libido. The only positive finding in physical examination was hyperpigmentation in the forehead area. His older brother who also had skin hyperpigmentation died at age 24 years of heart disease in the year 2005. The etiology of his heart disease was not known. With a clinical suspicion of JH, an iron profile and further studies were performed (Table 1). Liver transaminases and alkaline phosphatase were reported to be two times normal and liver elastography revealed moderate increase in liver stiffness [Fibroscan score=7.8 kilo pascal (KP)]. Abdominal CT scan revealed a relatively hyperdense liver in comparison with the spleen. Likewise, there was significant decrease in hepatic signal intensity without signal alteration of the spleen in magnetic resonance imaging (MRI) which was compatible with hereditary hepatic iron overload (Figures 1 A, B). Echocardiography was reported to be normal with a normal ejection fraction. All other work ups for underlying liver disease were negative. A clinical diagnosis of JH was made and a family screening for iron profile and genetic study was planned. Therapeutic biweekly phlebotomy was started after the second visit.
Table 1. Proband’s laboratory data.
| Visits |
First
visit |
After 6 mo | After 12 mo | After 18 mo | After 24 mo | After 30 mo | After 36 mo | |
| Blood count | Hb (g/dl) | 14.2 | 14.3 | 14.9 | 13 | 16.3 | 15.3 | 14.3 |
| Plat (mm3 ) | 135000 | 140000 | 145000 | 150000 | 183000 | 144000 | 192000 | |
| Iron indices | Iron (µg/dl) | 265 | 220 | 240 | 255 | 313 | 162 | 95 |
| TIBC (µg/dl) | 312 | 300 | 320 | 340 | 384 | 408 | 280 | |
| Tsat (%) | 85 | 73 | 75 | 75 | 81 | 40 | 34 | |
| Ferritin (ng/ml) | >2000 | 2990 | 2247 | 917 | 253 | 193 | 21 | |
| Liver function studies | AST (U/l) | 65 | 103 | 80 | 30 | 25 | 16 | 20 |
| ALT (U/l) | 69 | 90 | 80 | 32 | 29 | 26 | 21 | |
| ALK phos (U/l) | 570 | 551 | 456 | 440 | 389 | 363 | 250 | |
|
Liver Stiffness |
Score (KP) | 7.8 | 5.8 |
Hb: Hemoglobin, Plat: Platelets, TIBC: Transferrin iron binding capacity, Tsat: Transferrin phosphatase, GGT: Gamma glutamate transferase, PT: Prothrombin time, KP: Kilo pascal
Fig 1.
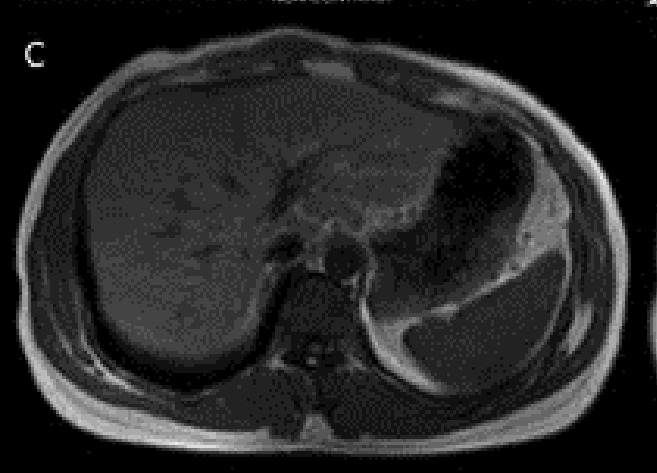
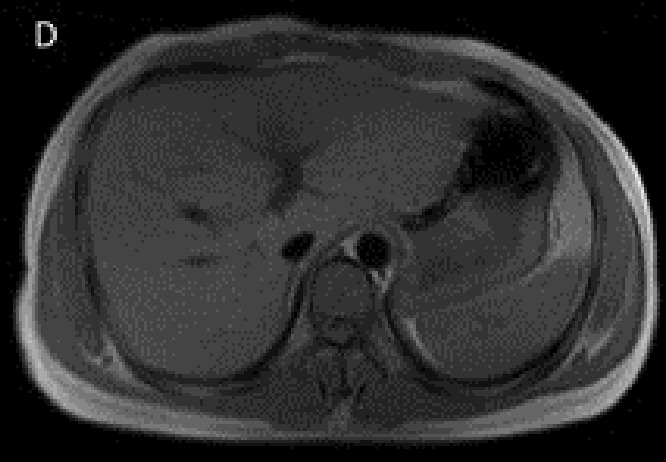
Proband’s pre- (A, B) and post-(C, D) treatment liver imaging studies. Increased density of the liver visualized on the CT scan (A) compared to the spleen with considerable hypointensity of the liver in axial T1-weighted MR image (B). Normal appearing hepatic signal intensity in axial T1-weighted MR image after treatment (C), confirmed by axial T2*-weighted MR image (D) as the most sensitive sequence for iron detection which revealed a normal mean T2* value of 22.4 msec.
A .
B .
C .
D .
Further investigation showed that the proband’s brother (III-10) (Figure 2) died of cardiac disease in year 2005 at the age of 24. Unfortunately no medical work up was made to search for hemochromatosis in this case. Other family members had no clinical symptoms or signs of hemochromatosis.
Fig .2 .
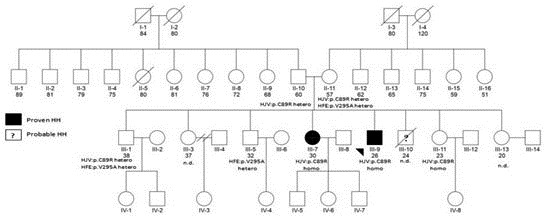
Pedigree of the family: Men and women family members are represented by squares and circles, respectively. Filled symbols indicate individuals with phenotypically expressed hemochromatosis. Below the symbols of the third generation and their parents are the genetic study results. The pedigree shows that cases III-9 and III-7 were homozygotes for HJV (p.C89R) and III-1, III-5, III-9 were heterozygotes for the HFE mutation (p.V295A). Both parents (II-10 and II-11) were heterozygotes for the HJV mutation (p.C89R). Additionally, the mother was a heterozygote for the HFE mutation. Case III-10 died before study due to probable hemochromatosis. HH: Hemochromatosis, homo: Homozygote, hetero: Heterozygote, nd: No data, HFE: HFE mutation, HJV: Hemojuvelin mutation.
In family member screening of iron indices in 2010, the proband’s 30 year old sister (III-7) although asymptomatic, was found to have a high serum ferritin level with increased liver stiffness on elastography (liver fibrosis score: 27 KP). She began biweekly phlebotomy treatments. Table 2 shows her laboratory data for sequential visits. With the exception of the proband’s father who was determined to be seropositive for HBSAg with a normal liver function test and negative serum HBV-DNA (inactive HBV carrier state) all other family members had normal laboratory studies (Table 3).
Table 2. Laboratory data for proband's sister.
| Complete blood count | Hb (g/dl) | 14.5 | 13.6 | 13 |
| Plat (mm3 ) | 154000 | 193000 | 189000 | |
| Iron indices | Iron (µg/dl) | 235 | 248 | 151 |
| TIBC (µg/dl) | 320 | 309 | 300 | |
| Tsat (%) | 73 | 80 | 50 | |
| Ferritin (ng/ml) | 4350 | 1305 | 192 | |
| Liver function studies | AST (U/l) | 58 | 36 | 29 |
| ALT (U/l) | 85 | 31 | 28 | |
| ALK phos (U/l) | 353 | 268 | 254 | |
| Fibroscan | Score (KP) | 27 | 6.8 | |
| Visits | First visit | After 1 year | After 3 years |
Hb: Hemoglobin, Plat: Platelets, TIBC: Transferrin iron binding capacity, Tsat: Transferrin saturation, AST:
Aspartate aminotransferase, ALT: Alanine aminotransferase, ALK phos: Alkaline phosphatase, GGT: Gamma glutamate transferase, KP: Kilo pascal
Table 3. Results of proband family member’s laboratory investigations .
|
Hb
(g/dl) |
PLT
(mm3 ) |
ALT
(U/lit) |
AST
(U/lit) |
ALP
(U/lit) |
Fe
(µg/dl) |
TIBC
(µg/dl) |
TS
(%) |
Ferritin
(ng/dl) |
HBsAg | |
| Father (61 y) | 14.9 | 211000 | 19 | 24 | 190 | 91 | 303 | 30 | 112.7 | Pos |
| Mother (58 y) | 14.4 | 254000 | 14 | 15 | 289 | 68 | 369 | 18 | 188.4 | Neg |
| Sibling 1 (34 y) | 12.7 | 282000 | 13 | 18 | 195 | 75 | 394 | 19 | 26.76 | Neg |
| Sibling 2 (31 y) | 15.2 | 224000 | - | - | - | 79 | 347 | 22 | 128.8 | Neg |
| Sibling 3 (24 y) | 13.9 | 224000 | 22 | 22 | 263 | 97 | 391 | 25 | 82.91 | Neg |
| Sibling 4 (21 y) | 13.8 | 202000 | 10 | 14 | 230 | 56 | 420 | 13 | 10.22 | Neg |
| Grandson (15 y) | 14.6 | 197000 | 13 | 15 | 220 | 50 | 400 | 12.5 | 20 | Neg |
| Granddaughter (10 y) | 12.2 | 289000 | 16 | 20 | 200 | 60 | 370 | 16 | 20 | Neg |
Hb: Hemoglobin, TIBC: Transferrin iron binding capacity, Tsat: Transferrin saturation, AST: Aspartate aminotransferase, ALT: Alanine aminotransferase, ALP: Alkaline phosphatase, HBsAg: Hepatitis B surface antigen, y: Year-old, Pos: Positive, Neg: Negative
Genetic studies of the third generation and their parents showed that the proband, case III-9 was a homozygote c.265T>C (p.C89R) HJV mutation + a heterozygote c.884T>C (p.V295A) mutation for HFE. The latter mutation has been described previously.11,12 The proband’s sister, case III-7, had evidence of the same homozygote HJV mutation found in the proband, namely c.265T>C (p.C89R). Also HJV c.98-6C>G (rs56025621) polymorphic variant was found in cis in the sister (homozygote) and the proband (homozygote). Cases III-1 and III- 5 were also heterozygous for the HFE c.884T>C (p.V295A) mutation. Both parents (II-10 and II-11) were heterozygous for the HJV c.265T>C (p.C89R) mutation and the mother was also heterozygous for the HFE c.884T>C (p.V295A) mutation (Figure 2).
Our case of JH presented with deceased libido, skin hyperpigmentation, and dyspnea. His symptoms all subsided following 3 years of phlebotomy. Currently he is alive and healthy. His post-treatment MRI image was almost normal. The pretreat ment MRI showed a very hypointense liver image, however on the post-treatment MRI the intensity of the liver became equal to that of the spleen. (Figures 2 C,D). The proband’s brother died of hemochromatosis early in life and his asymptomatic sister was homozygous for HJV with increased stiffness of liver as measured by liver elastography, which was suggestive of an underlying iron-induced liver disease.
DISCUSSION
JH occurs equally in both sexes. Patients usually present with hypogonadism and cardiac symptoms as early as the second decade of life. Delay in diagnosis and treatment of JH patients may lead to death from cardiac involvement as was likely the case in the proband’s brother.8 The etiology of JH is a genetic mutation in either the HJV or HAMP genes. It has been reported that about 80% of JH patients from central Europe have at least one copy of the G320V mutation.7 However, until now, studies showed numerous other mutations in the HJV gene.9,10,13
The proband was homozygous for HJV and heterozygous for HFE in comparison to his sister who was only homozygous for HJV (Figure 2). HFE heterozygosity might be a possible reason for the symptoms seen in the proband. Some studies have shown that HFE homozygotes with heterozygote mutations of the HJV or HAMP genes exhibited increased severity of symptoms,14,15 although others disagree.16,17 However, considering that subjects with heterozygote mutation of the HFE gene have lower degrees of iron-overload (if any) and no clinical symptoms,18 it seems unlikely that the reported HFE heterozygosis might have had a modifier effect on the proband’s clinical symptoms. Of note, when the proband’s brother died, the physician who treated his brother did not consider HH and told the father that his son died because of weakness and that the other sons should consume more meat. Therefore, the proband was placed on a high protein diet by the father and was forced to eat kebab even in the morning. His affected sister who was married and had two children, had regular menstruation. For this reason she was symptomless despite the presence of iron overload.
The HJV c.265T>C (p.C89R) mutation is a novel mutation and there is no information about its functional effect on the hemojuvelin protein. However, all of the five mutation function predicting algorithms (SIFT, PolyPhen2, Mutation Taster, Mutation Assessor, and FATHMM) of the dbNSFP database19 predicted this mutation to be pathogenic.
HH is reported to be very rare in Iran and the reported rate of HFE mutation is very low in the normal Iranian population.2,5 Of the few cases of HH that have been reported in Iran, all were non-HFE mutations. However these cases did not undergo additional genetic analyses to search for mutations in other HH genes.4,6 To the best of our knowledge this is the first report of an HJV mutation from Iran which is reported as a novel HJV mutation (p.C89R).
We were able to diagnose both patients with HH without performing a liver biopsy and the iron measurement in the biopsy samples as classically recommended. Recently, elastography has been shown to be an effective method for diagnosis of liver fibrosis and portal hypertension.20,21 One study has shown the effectiveness of elastography in measurement of liver fibrosis in patients with hemochromatosis.22 Therefore using noninvasive measures such as MRI imaging and elastography along with the availability of genetic information seems to be more convenient and safe. This may replace liver biopsy and iron measurements in biopsy specimens.
It is very important to know that HH due to a mutation in HJV and probably other genes when sought for can be found and prevent liver mortality from HH. Therefore clinicians in Iran and other regional countries should be more vigilant, consider HH when a compatible clinical presentation is evident, and have a higher index of suspicion for this treatable disease.
ACKNOWLEGMENTS
We express our appreciation to the proband (M.K.) and his family for their cooperation.
FUNDING
This study was funded by the Digestive Disease Research Institute, Tehran University of Medical Sciences, Tehran, Iran. Genetic studies were performed by the Division of Internal Medicine and Center for Hemochromatosis, University Hospital of Modena, Italy.
CONFLICT OF INTEREST
The authors declare no conflict of interest related to this work.
Please cite this paper as:
Malekzadeh MM, Radmard AR, Nouroozi AR, Akbari MR, Amini M, Navabakhsh B, Caleffi A, Pietrangelo A, Malekzadeh R. Juvenile Hemochromatosis, Genetic Study and Long-term Follow up after Therapy. Middle East J Dig Dis 2014;6:87-92.
References
- 1.Pietrangelo A. Hereditary hemochromatosis--a new look at an old disease. N Engl J Med. 2004;350:2383–97. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 2.Bakayev V, Ignatiev I, Jazayeri M, Mohaghegh H, Zborovsky S, Zali MR. Duplex polymerase chain reaction-restriction fragment length polymorphism assay for rapid detection of HFE mutations-C282Y occurs with a low frequency in Tehran’s population. J Hepatol. 2004;40:559–60. doi: 10.1016/j.jhep.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 3.Roth M, Giraldo P, Hariti G, Poloni ES, Sanchez-Mazas A, Stefano GF. et al. Absence of the hemochromatosis gene Cys282Tyr mutation in three ethnic groups from Algeria (Mzab), Ethiopia, and Senegal. Immunogenetics. 1997;46:222–5. doi: 10.1007/s002510050265. [DOI] [PubMed] [Google Scholar]
- 4.Nobakht H, Merat S, Malekzadeh R. Hereditary hemochromatosis: a rare disease in Iran. Arch Iran Med. 2006;9:78–80. [PubMed] [Google Scholar]
- 5.Karimi M, Yavarian M, Delbini P, Harteveld CL, Farjadian S, Fiorelli G. et al. Spectrum and haplotypes of the HFE hemochromatosis gene in Iran: H63D in beta-thalassemia major and the first E277K homozygous. Hematol J. 2004;5:524–7. doi: 10.1038/sj.thj.6200553. [DOI] [PubMed] [Google Scholar]
- 6.Mohamad Alizadeh AH, Masood M, Aghazadeh R, Ehsani Ardakani MJ, Forootan M, Zali MR. Non HFE-Related Hemochromatosis; A Case Report from Iran. Govaresh. 2003;9:66–9. [Google Scholar]
- 7.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP. et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 8.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393–408. doi: 10.1053/j.gastro.2010.06.013. .e1-2. [DOI] [PubMed] [Google Scholar]
- 9.Robson K, Merryweather-Clar A, Cadet E, Viprakasit V, Zaahl M, Pointon J. et al. Recent advances in understanding haemochromatosis: a transition state. J Med Genet. 2004;41:721–30. doi: 10.1136/jmg.2004.020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Gac G, Ferec C. The molecular genetics of haemochromatosis. Eur J Hum Genet. 2005;13:1172–85. doi: 10.1038/sj.ejhg.5201490. [DOI] [PubMed] [Google Scholar]
- 11.Jones DC, Young NT, Pigott C, Fuggle SV, Barnardo MC, Marshall SE. et al. Comprehensive hereditary hemochromatosis genotyping. Tissue antigens. 2002;60:481–8. doi: 10.1034/j.1399-0039.2002.600603.x. [DOI] [PubMed] [Google Scholar]
- 12.Bento MC, Ribeiro ML, Relvas L. Gene symbol: HFEDisease: Haemochromatosis. Hum Genet. 2004;114:405. [PubMed] [Google Scholar]
- 13.Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, Simard H. et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood. 2004;103:4317–21. doi: 10.1182/blood-2004-01-0192. [DOI] [PubMed] [Google Scholar]
- 14.Le Gac G, Scotet V, Ka C, Gourlaouen I, Bryckaert L, Jacolot S. et al. The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Hum Mol Genet. 2004;13:1913–8. doi: 10.1093/hmg/ddh206. [DOI] [PubMed] [Google Scholar]
- 15.Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A. et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet. 2003;12:2241–7. doi: 10.1093/hmg/ddg225. [DOI] [PubMed] [Google Scholar]
- 16.Barton JC, Rivers CA, Niyongere S, Bohannon SB, Acton RT. Allele frequencies of hemojuvelin gene (HJV) I222N and G320V missense mutations in white and African American subjects from the general Alabama population. BMC Medical Genetics. 2004;5:29. doi: 10.1186/1471-2350-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace DF, Dixon JL, Ramm GA, Anderson GJ, Powell LW, Subramaniam N. Hemojuvelin (HJV)-associated hemochromatosis: analysis of HJV and HFE mutations and iron overload in three families. Haematologica. 2005;90:254–5. [PubMed] [Google Scholar]
- 18.Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 19.Liu X, Jian X, Boerwinkle E. dbNSFP v20: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berzigotti A, Seijo S, Arena U, Abraldes JG, Vizzutti F, Garcia-Pagan JC. et al. Elastography, spleen size, and platelet count identify portal hypertension in patients with compensated cirrhosis. Gastroenterology. 2013;144:102–11. doi: 10.1053/j.gastro.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Castera L, Vergniol J, Foucher J, Le Bail B, Chanteloup E, Haaser M. et al. Prospective comparison of transient elastography, Fibrotest, APRI, and liver biopsy for the assessment of fibrosis in chronic hepatitis C. Gastroenterology. 2005;128:343–50. doi: 10.1053/j.gastro.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Adhoute X, Foucher J, Laharie D, Terrebonne E, Vergniol J, Castera L. et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: a prospective study. Gastroenterol Clin Biol. 2008;32:180–7. doi: 10.1016/j.gcb.2007.12.021. [DOI] [PubMed] [Google Scholar]