

Abstract
Trauma patients who suffer cardiac arrest (CA) from exsanguination rarely survive. Emergency preservation and resuscitation using hypothermia was developed to buy time for resuscitative surgery and delayed resuscitation with cardiopulmonary bypass (CPB), but intact survival is limited by neuronal death associated with microglial proliferation and activation. Pharmacological modulation of microglia may improve outcome following CA. Systemic injection of liposome-encapsulated clodronate (LEC) depletes macrophages. To test the hypothesis that intrahippocampal injection of LEC would attenuate local microglial proliferation after CA in rats, we administered LEC or PBS into the right or left hippocampus, respectively. After rapid exsanguination and 6 min no-flow, hypothermia was induced by ice-cold (IC) or room-temperature (RT) flush. Total duration of CA was 20 min. Pre-treatment (IC, RTpre) and post-treatment (RTpost) groups were studied, along with shams (cannulation only) and CPB controls. On day 7, shams and CPB groups showed neither neuronal death nor microglial activation. In contrast, the number of microglia in hippocampus in each individual group (IC, RTpre, RTpost) was decreased with LEC vs. PBS by ~34–46% (P < 0.05). Microglial proliferation was attenuated in the IC vs. RT groups (P < 0.05). Neuronal death did not differ between hemispheres or IC vs. RT groups. Thus, intrahippocampal injection of LEC attenuated microglial proliferation by ~40%, but did not alter neuronal death. This suggests that microglia may not play a pivotal role in mediating neuronal death in prolonged hypothermic CA. This novel strategy provides us with a tool to study the specific effects of microglia in hypothermic CA.
Keywords: Inflammation, Iba-1, Cardiac arrest, Brain ischemia, Resuscitation, Emergency preservation, Hypothermia
1. Introduction
Patients who suffer a cardiac arrest (CA) from trauma, often from exsanguination, have <10% chance of survival, even with aggressive fluid resuscitation and an emergency department thoracotomy. Emergency preservation and resuscitation (EPR) is a novel approach for resuscitation of exsanguination CA victims.1 EPR uses cold aortic flush to induce deep hypothermic preservation for prolonged CA to buy time for transport, damage control surgery, and delayed resuscitation with cardiopulmonary bypass (CPB).
Prolonged CA results in neuronal death and a reactive glial response. Specifically, microglial activation and proliferation has been linked to delayed neuronal death, presumably via releasing neurotoxic substances, including reactive oxygen radicals, nitric oxide (NO), and pro-inflammatory cytokines.2 Microglial activation could contribute to neuronal death or microglial-mediated synaptic injury and/or neuronal dysfunction – which could mediate cognitive deficits even in the absence of overt neuronal death.
Microglia could also have beneficial effects, contributing to delayed repair after injury via elaboration of growth factors,3 or their presence could represent an epiphenomenon. The effect of microglia could also depend on the severity of the primary insult, resulting in neurotoxicity vs. neuroprotection. Thus, there may be a specific time window for benefit from inhibition of the microglial contribution to damage, as well as specific scenario in which inhibiting microglia could be helpful. Therapeutical modulation of the microglial response for insults even less than the threshold for neuronal death may help to improve outcome following global brain ischemia.4
Pharmacological modulation of microglial proliferation may help to improve outcome following CA. Recently, studies in several CNS insults have shown benefit from treatment with minocycline, an agent that attenuated microglial activation and proliferation.5,6
Liposome-encapsulated clodronate (LEC) is an agent that –when used systemically – depletes macrophages7 and has been shown to deplete microglia in vitro, including brain slices.8 In brain ischemia, however, the local inflammatory response is predominated by microglial rather than macrophage accumulation.9 Since LEC does not cross the blood–brain barrier (BBB), we hypothesized that intraparenchymal injection of LEC into the brain would selectively deplete microglia and attenuate hippocampal neuronal degeneration.
2. Methods
We used the rat EPR model described in detail previously (Fig. 1).10 All rats received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” (www.nap.edu/catalog/5140.html). The study protocol has been approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Fig. 1.

Experimental protocol including rapid hemorrhagic shock and 6 min normothermic cardiac arrest (CA), followed by emergency preservation and resuscitation (EPR) using ice-cold (IC) or room-temperature (RT) aortic flush and resuscitation with cardiopulmonary bypass (CPB). Bold solid line, mean arterial pressure (MAP) in IC and RT groups; dotted line, temperature in IC group; thin solid line, temperature in RT groups; dashed line; MAP and temperature in sham and CPB control groups.
Five groups were studied: (1) rats pre-treated 24 h prior to CA and subjected to deep hypothermia during CA using ice-cold (IC) flush (IC, n = 6); (2) rats pre-treated 24 h prior to CA and subjected to moderate hypothermia during CA using room-temperature (RT) flush (RTpre, n = 3); (3) rats injected 24 h after CA and subjected to moderate hypothermia during CA using RT flush (RTpost, n = 3); (4) shams (n = 4), subjected to the same cannulations and duration of anesthesia; (5) CPB controls (n = 3), subjected to the same cannulations, anesthesia and 60 min of normothermic CPB.
2.1. Intrahippocampal injections
Adult male Sprague-Dawley rats (350–375 g) were obtained from Hilltop Lab Animals (Scottdale, PA) and housed for at least three days before the experiment under 12-h light/dark cycle with unrestricted access to food and water. Rats were anesthetized with 4% isoflurane in a transparent acrylic jar. After tracheal intubation with a 14 gauge (G) intravenous catheter (Becton Dickinson; Sandy, UT), rats were mechanically ventilated using a piston ventilator (Harvard Ventilator Model 683, Harvard Rodent Apparatus; South Natick, MA) with a tidal volume of 0.8 ml/100 g and a frequency 36–42/min to maintain normocapnia, and a positive end-expiratory pressure (PEEP) of 4 cm H2O. Anesthesia was maintained with 1.5–2% isoflurane in FiO2 0.5.
Using a stereotaxic frame, burr holes (diameter 0.45 mm) were created bilaterally (−4.3 mm dorsoventral, −2.0 mm lateral from bregma). A 27G needle was then inserted 3.5 mm deep into the hippocampus. Each rat received simultaneous intrahippocampal injections of either 5 μL of liposome-encapsulated phosphate-buffered saline (PBS) (left hemisphere) or 5 μL of LEC (right hemisphere) over 10 min via a 27G needle connected by a polyethenylene tubing to a 10 μL Hamilton syringe (Hamilton, 701N) and an infusion pump (Harvard Apparatus; South Natick, MA). Clodronate was a gift of Roche Diagnostics GmbH (Mannheim, Germany). It was encapsulated in liposomes as described previously.11 Using a different treatment in each hemisphere, each rat served as its own control. After a 3 min additional period with the needle in place to allow distribution of the compound, the needle was withdrawn at the rate of 1 mm/min to prevent leakage through the burr hole.
In addition, we tested if intrahippocampal injections of 10 μl would cause an increase in intracranial pressure (ICP), and thus potentially alter our model. ICP was monitored in selected rats (n = 4) via a 1 French intraparenchymal ICP probe (SPR-1000; Millar Instruments, Houston, TX) inserted from a separate burr hole in the frontal lobe. The ICP monitoring was discontinued and the probe was withdrawn after the completion of the injections.
After completion of the injections and ICP monitoring, the burr holes were sealed with a bone wax and skin was closed by layers using 2.0 silk. Anesthesia was discontinued, rats were extubated and allowed to recover in the cage.
2.2. Cardiac arrest
Rats were anesthetized, intubated and mechanically ventilated as mentioned above. After shaving and prepping with povidone iodine, bilateral femoral and right jugular cutdowns were performed. The left femoral artery and vein were cannulated for blood pressure monitoring and blood sampling. EKG, respiration, arterial and central venous pressure were continuously monitored and recorded (Polygraph; Grass Instruments, Quincy, MA). The right femoral artery was cannulated with a 20G catheter (Becton Dickinson; Sandy, UT) that served as an arterial CPB cannula. The right jugular vein was cannulated with a modified five-hole 14G intravenous cannula advanced to the right atrium to be used for venous drainage during the hemorrhage phase and later as a venous CPB cannula. Rectal and tympanic probes were used to monitor the temperature. Baseline blood samples were obtained, and hemodynamic values were recorded. Removed blood volume was replaced with an electrolyte-balanced crystalloid (Plasma-Lyte A Injection pH 7.4; Baxter; Deerfield, IL) in a ratio 1:3 (blood:crystalloid). Heparin sodium was administered to achieve activated clotting time (ACT) > 400 s (Haemochron Jr. Signature, ITC; Edison, NJ) to prevent clotting of the CPB circuit; the effects of heparin were not reversed.
After instrumentation, intubated rats were weaned to spontaneous ventilation of isoflurane 2% at FiO2 0.25% via a nose cone mask. After 5 min equilibration period, rapid exsanguination (12.5 ml of blood over 5 min) was performed via the internal jugular catheter. The shed blood was collected. After the rapid exsanguination phase, asystole was induced with intravenous administration of 9 mg (0.9 ml) of esmolol (Baxter; Deerfield, IL) and 0.2 mEq (0.1 ml) of potassium chloride (Hospira; Lake Forest, IL). After 5 min of CA, 270 ml of either an IC or RT flush solution (Plasma-Lyte A Injection pH 7.4) was instilled via the right femoral artery catheter at 50 ml/min. The flush was drained from the jugular vein catheter.
After 20 min of CA, resuscitation was started with CPB. Heating and cooling were achieved with a circulating water bath around the oxygenator. Blood samples for biochemistry and hematology were obtained at 5, 15, 30, 45, and 60 min CPB time and processed immediately using a point-of-care blood analyzer (Stat Profile, Nova Biomedical; Waltham, MA).
Arterial blood gas management followed alpha-stat principles. pH and electrolyte values outside of the normal range were corrected during CPB and ICU phases by adjustments in ventilation and/or administration of sodium bicarbonate, calcium chloride, or potassium chloride. Neuromuscular blockade was induced with cisatracurium (Nimbex; Abbott, North Chicago, IL) to prevent gasping during CPB. Additional blood obtained from an isoflurane-anesthetized donor rat was used to maintain hematocrit > 25%. CPB support was gradually weaned after 60 min. Mechanical ventilation with a FiO2 of 1.0 was continued while maintaining normocapnia for additional 2 h.
Using a midline laparotomy incision, a Mini-Mitter telemetric probe (Mini-Mitter Co; Sunriver, OR) was introduced into the peritoneal cavity to allow post-operative temperature control and continuous monitoring of heart rate and movement. Surviving rats were extubated 2 h later after removal of catheters, and placed separately in a temperature-controlled cage (34.5 °C for 6 h) with supplemental oxygen for 18 h, and free access to food and water. Neurological outcome was assessed daily by determining overall performance categories (OPC; 1 = normal, 5 = death) and neurologic deficit score (NDS; 0–10% = normal, 100% = maximum deficit).12 At 7 days after resuscitation, rats were euthanized with an isoflurane overdose and perfused via left ventricle with normal saline followed by 10% neutral-buffered formalin.
2.3. Histology
The tissue samples were processed for embedding in paraffin. The resulting paraffin blocks were sequentially sectioned at 5 μm. All sections were stained with Fluoro-Jade C (FJC) to indicate neuronal degeneration13 and with anti-Iba-1 staining visualizing microglia. Iba-1 is a calcium-binding protein expressed specifically in activated microglia,14 with its peak occurring at 4–7 days in experimental stroke.15 For the Iba-1 staining, sections were washed in TBST, incubated in 0.3% H2O2 in TBST for 30 min to inhibit endogenase peroxidase activity, washed in TBST, and blocked in TBST containing 3% normal goat serum for 30 min. The sections were incubated with a rabbit anti-Iba1 polyclonal antibody (1:250, Wako, Richmond, VA) overnight at 4 °C. The sections were then washed in TBST and incubated with a FITC-conjugated goat anti-rabbit IgG antibody (Invitrogen, Carlsbad, CA) for 1 h at room temperature. After the reactions, sections were washed and coverslipped with Vectashield Mounting media containing 4′,6-diamidino-2-phenylindole (DAPI) counterstain.
In addition, colorimetric visualization of Iba-1 immunostaining using diaminobenzamide (DAB) (Vector, CA) was used as a secondary confirmatory method to visualize microglia. In short, sections were processed the same as for fluorescent labeling on day 1, using a 1:250 dilution of anti-rabbit Iba-1 overnight at 4 °C. Sections were washed with TBST, incubated at RT for 1 h with a biotinylated anti-rabbit IgG, followed by 1 h of avidin-biotin complex binding using an ABC kit (Vector, CA). Sections were washed and incubated for 10 min with DAB followed by hematoxylin counterstaining. Tissue was dehydrated, cleared and coverslipped for microscopic analysis. For control staining, normal rabbit IgG was used as the primary antibody.
Adjacent sections obtained at approximately 4.3 mm from bregma were used to assess neuronal degeneration and microglial proliferation within the CA1 region of the hippocampus (Fig. 2). A photograph of the representative section of the CA1 region was taken under 10× magnification. FJC positive neurons and Iba-1 positive activated microglia (characterized by ameboid cell body and retracted processes without thin ramifications)16 were then quantitated morphometrically by two independent researchers (KJ, CDW) in a CA1 region of the hippocampus marked in Fig. 2 using the National Institutes of Health Image-J software. No automated features of the software were used. Image-J was used solely to track the cell counts and provide a controlled feedback between the independent evaluators.
Fig. 2.
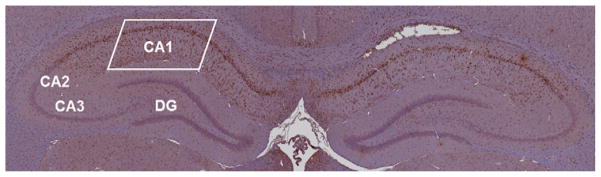
A microscopic pan-scan of both left and right hippocampi with identification of its main regions. A rectangle in the CA1 area shows the area of interest where cell counts were performed. The DAB staining used in this slide shows activated microglia. CA, cornu ammonis; DG, dentate gyrus.
2.4. Statistical analysis
Repeated measures ANOVA was performed, followed by Tukey post hoc tests, to identify differences in hemodynamic parameters and temperature between groups. For the aforementioned comparisons, data from RTpre and RTpost groups were pooled since they did not differ. One-way ANOVA was used to compare histologic damage, biochemical and hematologic data between groups. Kruskal–Wallis H test was used to compare NDS among groups. Mann–Whitney U test was used to compare two groups if Kruskal–Wallis H test indicated differences between groups existed. A P value < 0.05 was considered statistically significant.
3. Results
There were no differences in baseline characteristics between individual study groups. Induction of hypothermia IC flush resulted in lower intra-arrest temperature compared to the RT groups (P < 0.05 IC vs. RT). Physiologic parameters (heart rate, blood pressure) and temperature profiles during resuscitation are shown in Figs. 3–6. Prolonged CA in IC and RT flush groups resulted in marked physiologic derangements in the acid–base status with extremely low pH, BE up to −20, and increased lactate up to 6 mmol/L. These changes were gradually improved during resuscitation and were largely ameliorated by the end of the ICU phase. Minimal changes between groups were observed after 7 d (Table 1).
Fig. 3.
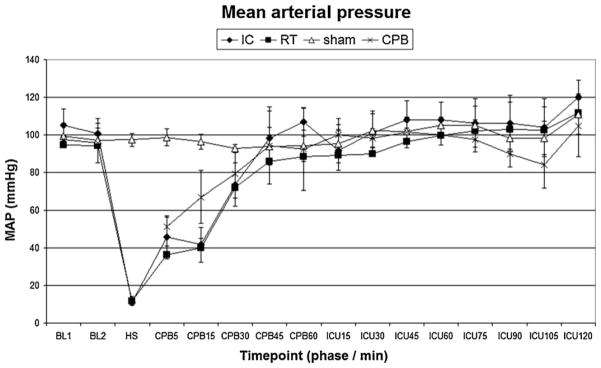
Mean arterial pressure. IC, ice-cold flush; RT, room-temperature flush; CPB, cardiopulmonary bypass control group. P < 0.05 RT vs. sham.
Fig. 6.
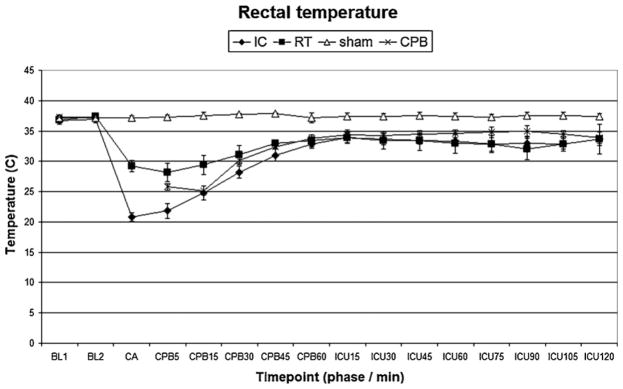
Rectal temperature. IC, ice-cold flush; RT, room-temperature flush; CPB, cardiopulmonary bypass control group. P < 0.05 sham vs. IC, RT or CPB groups.
Table 1.
Selected biochemical and hematological values after 20 min hypothermic cardiac arrest.
| BL | CPB5 | CPB60 | ICU120 | Final | |
|---|---|---|---|---|---|
| pHa | |||||
| IC | 7.36 ± 0.03 | 7.09 ± 0.07c,d | 7.41 ± 0.08 | 7.45 ± 0.01c,d | 7.45 ± 0.03 |
| RT | 7.35 ± 0.02 | 7.02 ± 0.04c,d | 7.40 ± 0.04 | 7.41 ± 0.03c,d | 7.48 ± 0.06 |
| sham | 7.42 ± 0.04 | 7.39 ± 0.06a,b,d | 7.37 ± 0.04 | 7.41 ± 0.02a,b | 7.44 ± 0.08 |
| CPB | 7.38 ± 0.03 | 7.22 ± 0.06 | 7.28 ± 0.04 | 7.42 ± 0.07a,b | 7.49 ± 0.06 |
| paO2 | |||||
| IC | 231 ± 32 | 600 ± 46b,c,d | 417 ± 34b,c,d | 233 ± 65 | 412 ± 194 |
| RT | 214 ± 24 | 351 ± 109a | 305 ± 75a | 194 ± 57 | 491 ± 164 |
| sham | 224 ± 27 | 200 ± 20a | 213 ± 19a | 163 ± 54 | 384 ± 73 |
| CPB | 233 ± 47 | 387 ± 182a | 246 ± 100a | 163 ± 114 | 452 ± 79 |
| paCO2 | |||||
| IC | 42 ± 4 | 32 ± 12 | 41 ± 9 | 42 ± 2 | 34 ± 4 |
| RT | 39 ± 5 | 30 ± 3 | 44 ± 7 | 40 ± 2 | 26 ± 5 |
| sham | 36 ± 3 | 37 ± 2 | 38 ± 4 | 34 ± 3 | 38 ± 7 |
| CPB | 39 ± 2 | 36 ± 8 | 42 ± 10 | 33 ± 3 | 37 ± 10 |
| BE | |||||
| IC | −1.0 ± 1.7 | −17.5 ± 6.9c,d | 1.8 ± 2.3c | 5.1 ± 1.4b,c,d | −0.3 ± 2.0 |
| RT | −2.7 ± 0.5 | −20.7 ± 1.0c,d | 2.0 ± 2.1c | 1.5 ± 1.7a,c,d | −2.1 ± 0.6d |
| sham | −1.0 ± 1.0 | −1.7 ± 2.6a,b | −3.1 ± 1.0a,b | −2.4 ± 1.0a,b | 1.4 ± 2.8 |
| CPB | −2.0 ± 1.1 | −4.7 ± 0.3a,b | −2.0 ± 2.2 | −3.9 ± 3.3a,b | 3.8 ± 2.9 |
| Lactate | |||||
| IC | 1.5 ± 0.5 | 4.2 ± 1.2b | 4.3 ± 1.2b | 2.7 ± 1.1 | 1.4 ± 0.4b,c |
| RT | 1.4 ± 1.2 | 5.9 ± 0.8a,c,d | 6.4 ± 1.0a,c,d | 3.3 ± 1.6 | 3.1 ± 1.4a |
| sham | 1.9 ± 0.4 | 2.5 ± 0.4b | 3.2 ± 0.9b | 3.4 ± 1.2 | 3.2 ± 0.8a |
| CPB | 1.4 ± 0.4 | 2.1 ± 0.6a,b | 3.5 ± 0.6b | 4.9 ± 1.9 | 2.0 ± 0.6 |
| Hct | |||||
| IC | 36 ± 2 | 24 ± 2 | 29 ± 2 | 30 ± 2 | 35 ± 4 |
| RT | 33 ± 2 | 25 ± 2 | 29 ± 2 | 32 ± 1 | 35 ± 1 |
| sham | 40 ± 3 | 41 ± 4 | 39 ± 1 | 37 ± 4 | 42 ± 2 |
| CPB | 40 ± 2 | 25 ± 2 | 25 ± 2 | 31 ± 1 | 35 ± 2 |
| Glucose | |||||
| IC | 228 ± 46 | 179 ± 45 | 229 ± 44 | 163 ± 16 | 224 ± 56 |
| RT | 196 ± 68 | 183 ± 13 | 206 ± 16 | 130 ± 30 | 251 ± 113 |
| sham | 322 ± 15 | 333 ± 25 | 316 ± 81 | 198 ± 89 | 232 ± 17 |
| CPB | 227 ± 6 | 247 ± 10 | 325 ± 102 | 152 ± 71 | 241 ± 26 |
IC, ice-cold flush; RT, room-temperature flush; CPB; cardiopulmonary bypass control group; BE, base excess; Hct, hematocrit.
P < 0.05 vs. IC.
P < 0.05 vs. RT.
P < 0.05 vs. sham.
P < 0.05 vs. CPB.
All rats improved in neurologic status over time but only rats from both RTpre and RTpost groups exhibited persistent neurologic deficits (P < 0.01 vs. IC, shams or CPB group, respectively; OPC, Table 2; NDS, Fig. 7).
Table 2.
Overall performance categories (OPC 1–5) at 7 d after prolonged hypothermic cardiac arrest and in control groups.
| IC | RTpre | RTpost | sham | CPB | |
|---|---|---|---|---|---|
| OPC 5 | |||||
| Death/brain death | |||||
| OPC 4 | |||||
| Severe disability | |||||
| OPC 3 | •• | •• | |||
| Moderate disability | |||||
| OPC 2 | • | ||||
| Mild disability | |||||
| OPC 1 | •••••• | • | •••• | ••• | |
| Normal |
Each dot represents one rat. IC, ice-cold flush; RTpre, pre-treatment, room-temperature flush; RTpost, post-treatment, room-temperature flush; CPB, cardiopulmonary bypass control group.
Fig. 7.

Neurologic deficit score between groups. Boxes represent interquartile ranges. The line across each box indicates the median, and the whiskers are the highest and lowest values. The round marker represents an outlier. IC, ice-cold flush; RTpre, pre-treatment, room-temperature flush; RTpost, post-treatment, room-temperature flush; CPB, cardiopulmonary bypass control group. *P < 0.05 vs. IC, sham or CPB group, respectively.
At 7 d after the insult a robust microglial response was seen in the hippocampus in groups subjected to CA. Minimal microglial proliferation was observed in the CPB group. No activated microglia were visualized in shams. The number of microglia in hippocampus was decreased in the hemisphere injected with LEC vs. PBS in all individual groups and when pooled together (P < 0.05). The number of FJC-positive neurons, however, did not differ between hemispheres in individual groups (P = 1.0) (Figs. 8 and 9, and Table 3). ICP did not significantly differ before (6 ± 1 mmHg) and after (5 ± 2 mmHg) LEC injection into hippocampus. Continuous ICP monitoring for 20 min during the LEC injection is shown (Fig. 10).
Fig. 8.

Histological outcome after prolonged hypothermic cardiac arrest. Representative samples of five animals are displayed, each row showing one animal from each individual group (A–E): A, shams; B, CPB controls; C, IC group; D, RTpre group; E, RTpost group. The two left columns (1–2) show degenerating neurons as visualized by the Fluoro-Jade C staining, in the left and right CA1 regions of hippocampus, respectively. The four right columns show microglial proliferation and activation detected by anti-Iba-1 FITC staining and visualized by fluorescence (columns 3 and 4) or with DAB and hematoxylin counterstaining (columns 5 and 6). Both shams and CPB controls showed no neuronal death and minimal microglial staining. All groups subjected to cardiac arrest showed extensive neuronal degeneration with no differences between groups and between left and right hemispheres in the individual groups. The microglial proliferation was significantly attenuated in all groups subjected to cardiac arrest in the right hippocampus, the site of LEC injection, vs. the corresponding left hippocampus, where PBS was injected. Blue staining in columns 1–4 is DAPI, identifying nuclei of intact neurons. Scale bar in panel A1 = 200 μm.
Fig. 9.
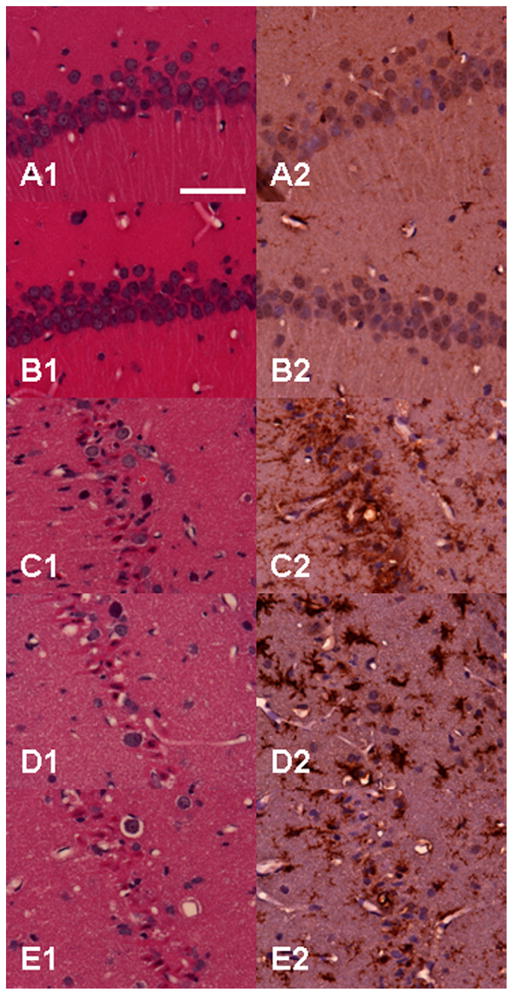
Histological outcome after prolonged hypothermic cardiac arrest. Same animals and groups as in Fig. 8 are shown here under high-power magnification (40×). No neuronal degeneration is seen in shams (A1) and CPB controls (B1), while large number of injured neurons with shrunken, acidophilic cytoplasm and pyknotic nuclei are present in the groups subjected to cardiac arrest (C1–E1). None or only a few scattered quiescent microglia are seen in shams (A2) or CPB controls (B2). In contrast, proliferated microglia with bushy appearance surround the degenerating neurons in the CA1 layer of hippocampus. Panels A1–E1, hematoxylin and eosin; panels A2–E2, DAB and hematoxylin. Scale bar in panel A1 = 50 μm.
Table 3.
Cell counts showing neuronal degeneration and microglial proliferation after prolonged cardiac arrest and in control groups. Control groups did not receive intrahippocampal injection. The numbers represent the sum of all counted cells from all animals in individual groups.
| Staining | Neuronal death FJC |
Microglial proliferation FITC |
DAB
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment/ratio | PBS | LEC | Ratio | PBS | LEC | Ratio | PBS | LEC | Ratio |
| Treated groups | |||||||||
| IC, n = 6 | 892 | 921 | 1.03 | 1520 | 1011 | 0.67 | 1731 | 993 | 0.57 |
| RTpre, n = 3 | 453 | 405 | 0.89 | 677 | 406 | 0.6 | 1474 | 838 | 0.57 |
| RTpost, n = 3 | 312 | 327 | 1.04 | 346 | 187 | 0.54 | 560 | 362 | 0.65 |
| Sum of all treated groups | 1657 | 1653 | 1 | 2543 | 1604 | 0.63* | 3765 | 2193 | 0.58* |
| 0% difference in FJC | 37% attenuation per FITC | 42% attenuation per DAB | |||||||
| Control groups | – | – | – | – | – | – | |||
| sham, n = 4 | 0 | 0 | 22 | 26 | 43 | 35 | |||
| CPB, n = 3 | 0 | 0 | 67 | 54 | 186 | 193 | |||
IC, ice-cold flush; RTpre, pre-treatment, room-temperature flush; RTpost, post-treatment, room-temperature flush; CPB, cardiopulmonary bypass control group.
P < 0.05 PBS vs. LEC.
Fig. 10.

Intracranial pressure during intrahippocampal injection. The tracing shows a representative sample from one rat during the simultaneous bilateral intrahippocampal injection of clodronate and PBS. No change in the intracranial pressure was observed. The tracing fluctuation seen at timepoints 0:00–1:00 and 22:00–25:00 min represent interference during the needle insertion and withdrawal, respectively.
4. Discussion
Using our established, clinically relevant model of EPR to study prolonged hypothermic CA in trauma resuscitation we found that both pre- and post-treatment with direct injection of LEC into the brain attenuates local microglial proliferation in hippocampus. This effect was not associated with a decrease in neuronal loss or a change in ICP.
The role of microglia in neuroinflammation in prolonged CA remains poorly defined. There is a large body of evidence documenting that microglia are a source of multiple potentially cytotoxic substances including NO, free radicals and pro-inflammatory cytokines, especially tumor necrosis factor α and interleukin-1β.17 Attenuation of microglial activation has shown benefit in multiple CNS injuries and neuroinflammatory diseases.18 In contrast, ablation of microglia in transgenic mice in stroke models showed detrimental effects,3 and administration of exogenous microglia had neuroprotective effects after ischemia,19,20 possibly linked to the production of neurotrophic factors like insulin-growth factor-1 or brain-derived neurotrophic factor.21 It has been postulated that the microglial reaction is dependent on the severity of the insult, and their role could be either “toxic” or “protective”.22,23 The tetracycline derivative minocycline has been used traditionally to attenuate microglial activity in multiple studies and showed potential as a neuroprotective agent.5 However, minocycline is non-specific.24 We specifically targeted microglia with local LEC administration.
Clodronate (dichloromethylene bisphosphonate – Cl2MDP) has been developed to eliminate macrophages in order to permit “in vivo” studies of their function. Clinically, it has been used in the treatment of osteoporosis,25 including prevention of skeletal events in patients with breast cancer.26 Prolonged administration of clodronate (oral, intravenous or intramuscular) seems to be safe.25
The exact mechanism of effects of clodronate is not yet fully elucidated. Systemically injected liposomes including encapsulated clodronate are ingested by macrophages which are then destroyed following phospholipase-mediated disruption of the liposomal bilayers and release of clodronate via a so-called macrophage “suicide” technique.27 Clodronate released from the liposomes has a short half-life that allows prompt removal from the circulation.28 Depletion of macrophages occurs rapidly (within 24 h after intravenous administration) and lymphocytes are not depleted. The effect persists for as long as one month after a single injection.29 A second dose was used to deplete bone-marrow residing macrophages and optimize the depletion.30 Despite profound effects, an increase in infectious complications has not been reported in long-term outcome models. Microglia remain unaffected because liposomes do not cross the BBB.31 We previously showed that our model of EPR is not associated with BBB disruption.32 Intraventricular injection of LEC resulted in a selective depletion only of perivascular and meningeal macrophages in the CNS. The macrophages started to repopulate in the given areas 14 d after the LEC depletion.33 Thus, we chose to target microglia in hippocampus with a direct intrahippocampal injection of LEC.
On a subcellular level, after its phagocytosis by macrophages, clodronate causes collapse of the mitochondrial membrane potential via inhibition of the ADP/ATP translocase by its metabolite AppCCl2p, resulting in delayed apoptosis.34
The effects of direct injection of LEC into tissues have not yet been fully explored. An intraparenchymal injection of LEC was used to induce macrophage depletion in rat testes. After local injection, the number of residing macrophages in testes were reduced at least by 90–97% at 14 d after injection, with repopulation observed at 60 d.35 These results suggest that the local spread of the liposomes and the depletion of tissue macrophages could be slower compared to rather rapid uptake of liposomes by circulating macrophages.
Stereotactic administration of LEC into hippocampus allowed us to selectively deplete resident microglia either before or after the insult. However, although successful, microglial depletion did not attenuate neuronal degeneration in CA1 region of hippocampus – a site of selective vulnerability that is accompanied by robust microglial activation and proliferation in our model.10 While we achieved significant microglial depletion at the area of injection, the exact distribution of LEC-induced microglia depletion could not be determined in our model.
It is plausible that microglia could mediate bi-phasic effects –initially contributing to secondary injury, while later being neuroprotective. In an in vitro study, LEC depleted microglia and inhibited microglial secretion of pro-inflammatory cytokines and NO in excitotoxically injured organotypic hippocampal slice cultures.36 Our newly developed method will enable us to study in the future the effects of microglia in temporal sequence.
Other methods of microglial depletion have been tested previously. Ganciclovir-treated transgenic mice that express a mutant form of herpes simplex virus type I thymidine kinase driven by a myeloid-specific CD11b promoter show 75% reduction in proliferating microglia after nerve injury.37 Using this technique in a brain ischemia model, a ~40% reduction of Iba-1 immunoreactivity was noted at 72 h, which is similar to the ~40% reduction observed with our technique. Mac-2, an alternative marker of activated/proliferating microglia, showed even higher reduction rate, up to 65%.3 However, technical limitations of currently available experimental CPB techniques do not allow CPB-assisted resuscitation, an integral part of our paradigm, in a mouse model. Rats are currently the smallest animal that can be resuscitated with CPB. Thus, it was necessary for us to explore alternative methods of microglial depletion to study the effects of microglia after CA in our rat model.
Minocycline has been widely used to deplete microglia in models of neuroinflammatory diseases and brain ischemia in rats.5 While the effects of minocycline in neuroinflammation have been generally positive, the outcomes of studies in brain ischemia models have been controversial. We and others showed minocycline-induced neuroprotection in both experimental6,38,39 and clinical settings.40 In contrast, others reported that selective ablation of proliferating microglial cells exacerbates ischemic brain injury.3 Minocycline effects are also non-specific. In a spinal cord injury model, minocycline reduced delayed oligodendrocyte death and attenuated axonal dieback, thus improving functional outcome.41 However, the effect of minocycline on oligodendrocytes could affect the neurological outcome by mechanisms other than microglial depletion.42,43 We have previously reported that our model of hypothermic CA is associated with persistent neurological and motor deficits.10,44 Minocycline was only marginally beneficial.10 These controversies underscore the need for a treatment paradigm that would enable us to study selective microglial depletion in a rat model.
Brain ischemia can also result in an early post-insult of the ischemic lesion by polymorphonuclear neutrophils (PMN) that could aggravate the injury. Microglia were previously shown to be neuroprotective against invading PMNs via their engulfment in an in vitro model.45 It is not clear whether LEC had any effect on PMNs in our model, but it seems unlikely given the small dose of LEC used in our study. Studies aimed at systemic depletion of PMN used repeated doses of LEC to achieve complete depletion of PMN.
We explored both pre-treatment and post-treatment with LEC in a brain region that previously showed an extensive damage in our model, using a proof-of-concept approach in which each animal served as its own control (only one hemisphere was treated with LEC). Only the intra-arrest temperature, but not the timing of the microglial depletion, had an effect on outcome. There may exist a certain window of opportunity for post-insult treatment given the fact that even delayed treatment with minocycline improved outcome after focal brain ischemia.43,46,47
Consistent with the lack of a role for microglia in affecting neuronal death in hypothermic CA, we reported that attenuation of microglial proliferation with deep hypothermia during CA in our model vs. moderate hypothermia resulted in improved neurological outcome, despite not preventing neuronal loss.10 We were not able to demonstrate this effect in the current study. It is possible that the intrahippocampal injections, with either clodronate or PBS containing liposomes, affected the microglial activation and proliferation in all groups, and the net effect of different levels of hypothermia were not as marked as they were in intact tissues. We did not use intrahippocampal injections in our control groups. We cannot exclude the possibility that intrahippocampal injections alone would produce a certain degree of gliosis and/or neuronal degeneration.
We did not explore the possibility of neurogenesis in our model. It has been postulated that delayed neuronal death is complete within 5–7 days. Several studies in both global and focal cerebral ischemia reported neurogenesis and/or synaptogenesis after an ischemic insult.48,49 However, neurogenesis starts at the subgranular zone of the dentate gyrus, and becomes maximal at two weeks after the insult.50 Based on these studies, we chose a 7 day outcome as a timepoint when neuronal loss is complete but repopulation of CA1 is unlikely. However, the role of neurogenesis in our model and the impact of hypothermia and microglia on this process remain to be determined in future studies, using bilateral LEC-induced depletion and complex neurobehavioral assessment with Morris water maze tasks.
In this feasibility trial, we showed that either pre- or post-treatment with direct injection of LEC into the hippocampus (1) attenuated local microglial proliferation in hippocampus by ~40%, and (2) did not acutely increase ICP. However, depletion did not alter neuronal degeneration in the hippocampus in our model of hypothermic CA. This suggests that microglia do not play a pivotal role in mediating neuronal death. However, LEC at the dose and treatment time chosen did not cause total ablation of microglial activity. An optimized dose and a timing of pretreatment may be needed to achieve higher depletion rate. Also, our insult produces marked loss of neurons and thus we cannot rule out the possibility that depletion of microglia could further exacerbate neuronal loss. It is also possible that the insult was too severe for LEC to have a robust impact. A detailed topographical map of microglial depletion needs to be characterized in future experiments. This novel strategy provides us with a tool to study the effects of microglia comprehensively in hypothermic CA, and in other models of neuroinflammation.
Fig. 4.
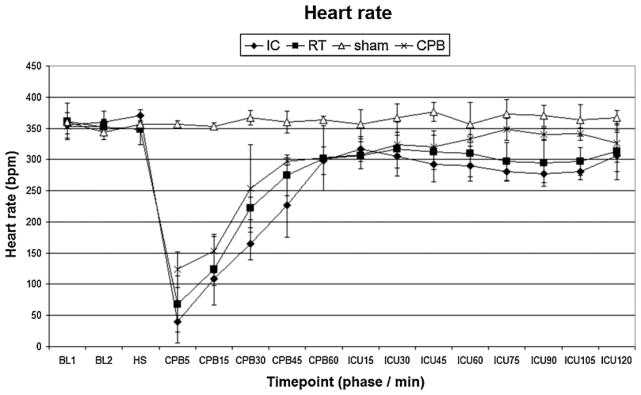
Heart rate. IC, ice-cold flush; RT, room-temperature flush; CPB, cardiopulmonary bypass control group. P < 0.05 IC vs. RT, sham or CPB; RT vs. IC or sham; sham vs. IC, RT or CPB.
Fig. 5.
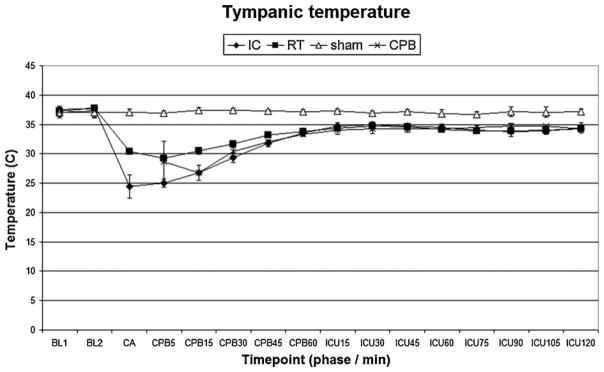
Tympanic temperature. IC, ice-cold flush; RT, room-temperature flush; CPB, cardiopulmonary bypass control group. P < 0.05 IC vs. RT or sham; RT vs. IC, sham or CPB; sham vs. IC, RT or CPB; CPB vs. RT or sham.
Acknowledgments
This work was supported by the AHA Beginning Grant-in-Aid # 09BGIA2310196 (TD), Society of Cardiovascular Anesthesiologists Starter Grant (TD), a Seed Grant from The Department of Anesthesiology, University of Pittsburgh (TD), by the Laerdal Foundation for Acute Medicine (TD), and by NS070003, NS30318 and NS38087 (PMK).
Samuel A. Tisherman and Patrick M. Kochanek are co-patent holders with the University of Pittsburgh on “Method of Inducing Suspended Animation Following Cardiopulmonary Arrest”. Patent pending, #60/692,722.
Cl2MDP (or clodronate) was a gift of Roche Diagnostics, GmbH, Mannheim, Germany.
Footnotes
A Spanish translated version of the abstract of this article appears as Appendix in the final online version at doi:10.1016/j.resuscitation.2011.09.016.
Conflict of interest statement
The authors declare that there is no conflict of interests.
URL: http://www.safar.pitt.edu/ (T. Drabek).
References
- 1.Safar P, Tisherman SA, Behringer W, et al. Suspended animation for delayed resuscitation from prolonged cardiac arrest that is unresuscitable by standard cardiopulmonary-cerebral resuscitation. Crit Care Med. 2000;28:N214–8. doi: 10.1097/00003246-200011001-00012. [DOI] [PubMed] [Google Scholar]
- 2.Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW. Reactive microglia in cerebral ischaemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol. 1995;21:277–89. doi: 10.1111/j.1365-2990.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 3.Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J, Bartels M, Lu A, Sharp FR. Microglia/macrophages proliferate in striatum and neocortex but not in hippocampus after brief global ischemia that produces ischemic tolerance in gerbil brain. J Cereb Blood Flow Metab. 2001;21:361–73. doi: 10.1097/00004647-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. Neuroscientist. 2005;11:308–22. doi: 10.1177/1073858405275175. [DOI] [PubMed] [Google Scholar]
- 6.Tang M, Alexander H, Clark RS, Kochanek PM, Kagan VE, Bayir H. Minocycline reduces neuronal death and attenuates microglial response after pediatric asphyxial cardiac arrest. J Cereb Blood Flow Metab. 2010;30:119–29. doi: 10.1038/jcbfm.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Rooijen N. The liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods. 1989;124:1–6. doi: 10.1016/0022-1759(89)90178-6. [DOI] [PubMed] [Google Scholar]
- 8.Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41:535–47. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- 9.Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2005;196:290–7. doi: 10.1016/j.expneurol.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Drabek T, Tisherman SA, Beuke L, et al. Deep hypothermia attenuates microglial proliferation independent of neuronal death after prolonged cardiac arrest in rats. Anesth Analg. 2009;109:914–23. doi: 10.1213/ane.0b013e3181b0511e. [DOI] [PubMed] [Google Scholar]
- 11.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 12.Neumar RW, Bircher NG, Sim KM, et al. Epinephrine and sodium bicarbonate during CPR following asphyxial cardiac arrest in rats. Resuscitation. 1995;29:249–63. doi: 10.1016/0300-9572(94)00827-3. [DOI] [PubMed] [Google Scholar]
- 13.Schmued LC, Hopkins KJ, Fluoro-Jade B. A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–30. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- 14.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 15.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–15. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 16.Koshinaga M, Suma T, Fukushima M, Tsuboi I, Aizawa S, Katayama Y. Rapid microglial activation induced by traumatic brain injury is independent of blood brain barrier disruption. Histol Histopathol. 2007;22:129–35. doi: 10.14670/HH-22.129. [DOI] [PubMed] [Google Scholar]
- 17.Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. doi: 10.1139/Y05-143. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematol Disord. 2004;4:65–84. doi: 10.2174/1568006043481284. [DOI] [PubMed] [Google Scholar]
- 19.Imai F, Suzuki H, Oda J, et al. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- 20.Kitamura Y, Takata K, Inden M, et al. Intracerebroventricular injection of microglia protects against focal brain ischemia. J Pharmacol Sci. 2004;94:203–6. doi: 10.1254/jphs.94.203. [DOI] [PubMed] [Google Scholar]
- 21.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 22.Lai AY, Todd KG. Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia. 2008;56:259–70. doi: 10.1002/glia.20610. [DOI] [PubMed] [Google Scholar]
- 23.Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56:149–71. doi: 10.1016/s0301-0082(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline upregulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279:19948–54. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 25.Frediani B, Cavalieri L, Cremonesi G. Clodronic acid formulations available in Europe and their use in osteoporosis: a review. Clin Drug Investig. 2009;29:359–79. doi: 10.2165/00044011-200929060-00001. [DOI] [PubMed] [Google Scholar]
- 26.Pavlakis N, Schmidt R, Stockler M. Bisphosphonates for breast cancer. Cochrane Database Syst Rev. 2005:CD003474. doi: 10.1002/14651858.CD003474.pub2. [DOI] [PubMed] [Google Scholar]
- 27.van Rooijen N, Sanders A, van den Berg TK. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. 1996;193:93–9. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 28.Fleisch H. Bisphosphonates: a new class of drugs in diseases of bone and calcium metabolism. Recent Results Cancer Res. 1989;116:1–28. doi: 10.1007/978-3-642-83668-8_1. [DOI] [PubMed] [Google Scholar]
- 29.Van Rooijen N, Kors N, vd Ende M, Dijkstra CD. Depletion and repopulation of macrophages in spleen and liver of rat after intravenous treatment with liposome-encapsulated dichloromethylene diphosphonate. Cell Tissue Res. 1990;260:215–22. doi: 10.1007/BF00318625. [DOI] [PubMed] [Google Scholar]
- 30.Barbe E, Huitinga I, Dopp EA, Bauer J, Dijkstra CD. A novel bone marrow frozen section assay for studying hematopoietic interactions in situ: the role of stromal bone marrow macrophages in erythroblast binding. J Cell Sci. 1996;109:2937–45. doi: 10.1242/jcs.109.12.2937. [DOI] [PubMed] [Google Scholar]
- 31.Micklus MJ, Greig NH, Tung J, Rapoport SI. Organ distribution of liposomal formulations following intracarotid infusion in rats. Biochim Biophys Acta. 1992;1124:7–12. doi: 10.1016/0005-2760(92)90118-f. [DOI] [PubMed] [Google Scholar]
- 32.Lahoud-Rahme MS, Stezoski J, Kochanek PM, Melick J, Tisherman SA, Drabek T. Blood–brain barrier integrity in a rat model of emergency preservation and resuscitation. Resuscitation. 2009;80:484–8. doi: 10.1016/j.resuscitation.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Polfliet MM, Goede PH, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmunol. 2001;116:188–95. doi: 10.1016/s0165-5728(01)00282-x. [DOI] [PubMed] [Google Scholar]
- 34.Lehenkari PP, Kellinsalmi M, Napankangas JP, et al. Further insight into mechanism of action of clodronate: inhibition of mitochondrial ADP/ATP translocase by a nonhydrolyzable, adenine-containing metabolite. Mol Pharmacol. 2002;61:1255–62. doi: 10.1124/mol.61.5.1255. [DOI] [PubMed] [Google Scholar]
- 35.Bergh A, Damber JE, van Rooijen N. Liposome-mediated macrophage depletion: an experimental approach to study the role of testicular macrophages in the rat. J Endocrinol. 1993;136:407–13. doi: 10.1677/joe.0.1360407. [DOI] [PubMed] [Google Scholar]
- 36.Dehghani F, Conrad A, Kohl A, Korf HW, Hailer NP. Clodronate inhibits the secretion of proinflammatory cytokines and NO by isolated microglial cells and reduces the number of proliferating glial cells in excitotoxically injured organotypic hippocampal slice cultures. Exp Neurol. 2004;189:241–51. doi: 10.1016/j.expneurol.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 37.Gowing G, Vallieres L, Julien JP. Mouse model for ablation of proliferating microglia in acute CNS injuries. Glia. 2006;53:331–7. doi: 10.1002/glia.20288. [DOI] [PubMed] [Google Scholar]
- 38.Fan LW, Lin S, Pang Y, Rhodes PG, Cai Z. Minocycline attenuates hypoxia–ischemia-induced neurological dysfunction and brain injury in the juvenile rat. Eur J Neurosci. 2006;24:341–50. doi: 10.1111/j.1460-9568.2006.04918.x. [DOI] [PubMed] [Google Scholar]
- 39.Xu L, Fagan SC, Waller JL, et al. Low dose intravenous minocycline is neuroprotective after middle cerebral artery occlusion-reperfusion in rats. BMC Neurol. 2004;4:7. doi: 10.1186/1471-2377-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lampl Y, Boaz M, Gilad R, et al. Minocycline treatment in acute stroke: an open-label, evaluator-blinded study. Neurology. 2007;69:1404–10. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- 41.Stirling DP, Khodarahmi K, Liu J, et al. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24:2182–90. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox C, Dingman A, Derugin N, et al. Minocycline confers early but transient protection in the immature brain following focal cerebral ischemia–reperfusion. J Cereb Blood Flow Metab. 2005;25:1138–49. doi: 10.1038/sj.jcbfm.9600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hewlett KA, Corbett D. Delayed minocycline treatment reduces long-term functional deficits and histological injury in a rodent model of focal ischemia. Neuroscience. 2006;141:27–33. doi: 10.1016/j.neuroscience.2006.03.071. [DOI] [PubMed] [Google Scholar]
- 44.Drabek T, Fisk JA, Dixon CE, et al. Prolonged deep hypothermic circulatory arrest in rats can be achieved without cognitive deficits. Life Sci. 2007;81:543–52. doi: 10.1016/j.lfs.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 45.Neumann J, Sauerzweig S, Ronicke R, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. 2008;28:5965–75. doi: 10.1523/JNEUROSCI.0060-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–9. doi: 10.1097/00006123-200106000-00051. [discussion 1399–401] [DOI] [PubMed] [Google Scholar]
- 47.Liu Z, Fan Y, Won SJ, et al. Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke. 2007;38:146–52. doi: 10.1161/01.STR.0000251791.64910.cd. [DOI] [PubMed] [Google Scholar]
- 48.Bendel O, Bueters T, von Euler M, Ove Ogren S, Sandin J, von Euler G. Reappearance of hippocampal CA1 neurons after ischemia is associated with recovery of learning and memory. J Cereb Blood Flow Metab. 2005;25:1586–95. doi: 10.1038/sj.jcbfm.9600153. [DOI] [PubMed] [Google Scholar]
- 49.Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–32. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- 50.Yamashima T, Tonchev AB, Borlongan CV. Differential response to ischemia in adjacent hippocampalsectors: neuronal death in CA1 versus neurogenesis in dentate gyrus. Biotechnol J. 2007;2:596–607. doi: 10.1002/biot.200600219. [DOI] [PubMed] [Google Scholar]