

Abstract
The p53 tumor suppressor gene is the most frequently mutated gene in cancer. Significant progress has been made to discern the importance of p53 in coordinating cellular responses to DNA damage, oncogene activation, and other stresses. Noncoding RNAs are RNA molecules functioning without being translated into proteins. In this work, we discuss the dichotomy of p53 regulation by noncoding RNAs with four unconventional questions. First, is overexpression of microRNAs responsible for p53 inactivation in the absence of p53 mutation? Second, are there somatic mutations in the noncoding regions of the p53 gene? Third, is there a germline mutant in the noncoding regions of the p53 gene that predisposes carriers to cancer? Fourth, can p53 activation mediated by a noncoding RNA mutation cause cancer? This work highlights the prominence of noncoding RNAs in p53 dysregulation and tumorigenesis.
Keywords: noncoding RNA, p53, activation, mutation, 3′UTR, polymorphism, ribosomopathies
Introduction
The p53 tumor suppressor is known as the ‘guardian of the genome’. p53 is activated in response to a variety of stress signals, such as DNA damage and oncogene activation, allowing p53 to coordinate transcription programs (Vousden and Prives, 2009; Farnebo et al., 2010). As a transcription factor, p53 regulates the expression of a battery of genes that promote apoptotic cell death, cell cycle arrest, cellular senescence, metabolism, and other processes. Loss of p53 function is a common feature in the majority of human cancers, as somatic mutation in the coding sequence (CDS) of the p53 gene (also known as TP53 in human and Trp53 in mouse) is the most frequent genetic alteration. Almost 75% of the CDS mutations result in a mutant p53 protein that is defective in its tumor suppression functions; some may exert a dominant-negative regulation over remaining wild-type (WT) p53 and others acquire oncogenic functions that are independent of WT p53 (Muller and Vousden, 2013). For >30 years, p53 has been one of the most studied molecules in oncology, and significant progress has been made regarding its role in cancer and other pathophysiological conditions. Noncoding RNAs (also known as functional RNAs) are RNA molecules functioning without being translated into proteins. Although the fundamental role of noncoding RNAs like rRNA and tRNA in cellular functions and organismal evolution has been known for decades, only recently have other noncoding RNAs received a remarkable level of attention from biologists. In this review, we will discuss a new frontier in the study of p53 biology, that is, noncoding RNA-based regulatory mechanisms have a relevant role in p53 dysregulation and cancer development.
p53 inactivation in the absence of coding sequence mutations—implications of microRNA deregulation in multiple myeloma
p53 does not function properly in most human tumors, yet p53 is inactivated via CDS mutations in only about 50% of human tumors. In many tumors, p53 is inactivated indirectly through the amplification of the MDM2 gene encoding the murine double minute 2 (Mdm2) protein or its homolog MDMX, allelic deletion of CDKN2A encoding the p14ARF protein, viral proteins inhibiting p53, or mislocalization of p53 to the cytoplasm (Vogelstein et al., 2000). Multiple myeloma is a suitable model to investigate the role of microRNA overexpression as an alternative mechanism of p53 inactivation as p53 is rarely mutated in this blood cancer. Multiple myeloma is an incurable neoplasm characterized by an excessive clonal proliferation of abnormal plasma cells in the bone marrow and overproduction of monoclonal immunoglobulins. Various studies of p53 mutations in multiple myeloma cases with clinical heterogeneity and small sample sizes (n < 100) have demonstrated p53-mutation prevalence rates ranging from 0% to 20% (Fonseca et al., 2004). A comprehensive study with the largest number of newly diagnosed multiple myeloma patients reported that p53 mutations were rare, with only 9 of 268 samples (3%) testing positive (Chng et al., 2007). Moreover, the incidence of alternative mechanisms of p53 inactivation is relatively low in multiple myeloma as summarized (Kumar et al., 2011). Over the past 5 years, we and others have identified 11 microRNAs (miRNAs) that negatively regulate p53 expression by directly targeting the 3′ untranslated region (3′UTR): miR-25, miR-30d (Kumar et al., 2011), miR-125b (Le et al., 2009), miR-504 (Hu et al., 2010), miR-380-5p (Swarbrick et al., 2010), miR-92 (Neveu et al., 2010), miR-141 (Neveu et al., 2010), miR-1285 (Tian et al., 2010), miR-200a (Becker et al., 2012), miR-15a, and miR-16 (Fabbri et al., 2011). Strikingly, in at least one report, expression of 9 of these p53-targeting miRNAs was higher in multiple myeloma cells than in plasma cells obtained from healthy donors: miR-25 (Pichiorri et al., 2008; Kumar et al., 2011), miR-30d (Pichiorri et al., 2008; Corthals et al., 2011; Kumar et al., 2011), miR-125b (Pichiorri et al., 2008; Lionetti et al., 2009; Corthals et al., 2011), miR-92 (Pichiorri et al., 2008), miR-200a (Chi et al., 2011; Corthals et al., 2011), miR-504 (Corthals et al., 2011), miR-141 (Corthals et al., 2011), miR-15a (Pichiorri et al., 2008), and miR-16 (Pichiorri et al., 2008) (Figure 1). Interestingly, progression-free survival is improved in multiple myeloma patients with low expression of DICER, a gene essential for miRNA maturation (Corthals et al., 2011). These data support the hypothesis that p53 inactivation in multiple myeloma cells is mediated by miRNA overexpression. It should be noted that p53-targeting miRNAs are overexpressed in many cancers other than multiple myeloma. For instance, miR-25 was also found to be overexpressed in cancers of pancreas, prostate, and stomach (Volinia et al., 2006).
Figure 1.
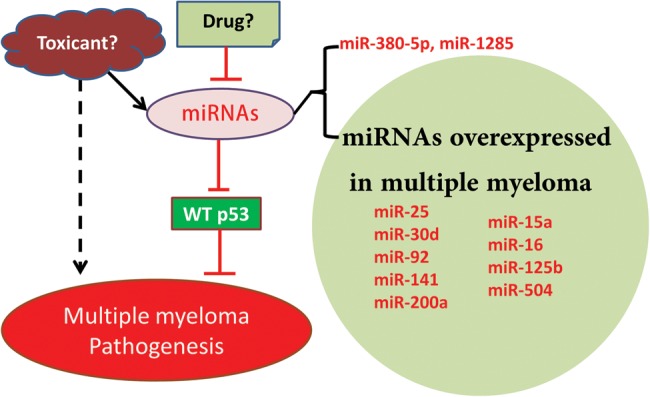
Schematic representation of the potential role of miRNA overexpression in the etiology of multiple myeloma.
Multiple myeloma is regarded as an environmental disease, yet its etiology remains largely unclear. The association of multiple myeloma with smoking has been investigated, with some studies suggesting a strong association (Williams and Horm, 1977; Mills et al., 1990), while others refuting this relationship (Fernberg et al., 2007; Castillo et al., 2012; Psaltopoulou et al., 2013). Multiple myeloma is preceded by monoclonal gammopathy of undetermined significance (MGUS), which is one of the most common pre-cancerous conditions afflicting 3% of the population over 50 years old. MGUS progresses to incurable multiple myeloma at a relentless rate of ∼1% per year (Kyle et al., 2006), yet little information is available about the molecular events that mediate this progression. It is notable that p53-targeting miRNAs, such as miR-25, are also overproduced in MGUS (Pichiorri et al., 2008). Continued progress in this area of investigation will open new venues for cancer prevention for millions of MGUS patients at risk of developing a deadly malignancy. On the other hand, identifying compounds that inhibit the expression of p53-targeting miRNAs will provide a novel therapeutic option for patients afflicted with MGUS or multiple myeloma (Figure 1).
Finding all p53 mutations—somatic passenger mutations in the 3′UTR of p53
The mRNA structure of most protein coding genes in mammals consists of four regions: the 5′UTR, the CDS, the 3′UTR, and a poly-A tail. However, the vast majority of p53 sequencing efforts focus on the CDS (from ATG start codon to the TGA stop codon), particularly exons 5–9 (Figure 2A). The exons covering the UTRs are unfortunately neglected. Recently, we completed a systematic analysis of the p53 UTR in diffuse large B-cell lymphoma (DLBCL) patients treated with R-CHOP regimen (Li et al., 2013). DLBCL is chosen because its p53 CDS mutation rate (∼20%) is moderate and our prior study reveals that p53 CDS mutation predicts poor survival. Hundreds of novel mutations were found in the p53 3′UTR, which occurred at a higher frequency than mutations observed within the CDS. The seed match binding sites of 8 out of 11 p53-targeting miRNAs are disrupted by mutations. Using miR-125b as a case of study, the importance of 3′UTR mutations in p53 regulation was examined to find that mutations disrupting the interaction between p53 3′UTR and miR-125b abolished p53 suppression mediated by miR-125b. From an evolutional perspective, 3′UTR mutations disrupt p53 suppression by miRNAs to produce more p53 tumor suppressors, which is opposite to coding mutations that produce oncogenic p53 proteins. 3′UTR mutations, presumably disfavor for tumor growth, could occur during B-cell clonal expansion as a self-defense mechanism to prevent malignant development. We did not find 3′UTR mutations that lead to the creation of new binding sites for the 11 validated p53-targeting miRNAs. Yet, there are many sites generated from these mutations for predicted p53-targeting miRNAs (Li et al., 2013). Next, patients were divided into two groups by p53 CDS status to analyze survival in relation to 3′UTR mutations. In the presence of WT CDS, patients without 3′UTR mutations seemed to have poorer survival than those with 3′UTR mutations, but the difference was not statistically significant. When 5-year survival rates were calculated, 3′UTR mutations conferred better overall survival in patients with WT CDS. In contrast, patients with both mutant CDS and 3′UTR mutations had poorer survival than patients with mutant CDS and no 3′UTR mutations (Figure 2B). Indeed, all patients with both mutant CDS and 3′UTR died within 76 months after diagnosis (Li et al., 2013). These findings identified the p53 3′UTR as a prognostic indicator of survival in DLBCL patients that is dependent on the status of the CDS, and further investigation is needed to ascertain the role of the 3′UTR alterations in p53 regulation and cancer prognosis.
Figure 2.

Differential impacts of mutations in the CDS or the 3′UTR of the p53 gene. (A) Schematic representation of the p53 reference mRNA. (B) 3′UTR mutations in DLBCL prognosis are dependent on the status of the CDS.
In the past decade, comprehensive deep sequencing has revealed the coding genomic landscapes of common forms of human cancer, including DLBCL. For most cancer types, this landscape consists of mutations in ∼50 genes. These mutations are classified into two groups: a driver mutation that directly or indirectly confers a selective growth advantage to the cell in which the mutation occurs, and a passenger mutation that has no effect on tumor cell growth. As such, driver mutations are believed to play major roles in tumorigenesis and cancer progression, while passenger mutations are assumed to be biologically neutral and pathologically irrelevant. In DLBCL, three exome sequencing efforts have reported that the overall load of tumor-acquired mutations range from 16 to 135 (Morin et al., 2011; Pasqualucci et al., 2011; Lohr et al., 2012). Mutations in the p53 CDS occur in ∼20% of DLBCL cases and those with p53 mutations had worse survival compared with those without mutation regardless of the treatment or disease subtype (Xu-Monette et al., 2012). These findings suggest that p53 CDS mutations are driver genetic alterations. In contrast, somatic mutations in the p53 3′UTR enhance neither tumor initiation nor progression, but render tumors less aggressive intrinsically and/or more susceptible to treatment. On the other hand, when patients have a mutant p53 CDS, 3′UTR mutations reduce the suppression mediated by miRNAs and subsequently increase the expression of oncogenic p53, causing poor survival (Figure 2B). On the basis of computer-aided simulation, it was proposed that passenger mutations could have deleterious effects on cancer progression (McFarland et al., 2013). The study of p53 CDS and 3′UTR mutations provides experimental evidence to support the idea that driver mutations and passenger mutations occur in the same gene (p53) and that passenger mutations can significantly impact cancer prognosis.
A unique noncoding germline variant of p53—rs78378222 as a model to study omnipresent low-penetrance cancer risk alleles
The p53 locus spans ∼20 kb in the human genome and germline mutations in the p53 CDS cause Li–Fraumeni syndrome (LFS). There are over 200 naturally occurring germline variants of p53 in human populations with few causing measurable perturbation of p53 function (Whibley et al., 2009). More than 90% of p53 variants occur in 10 introns alleged to have no cancer-related biological consequences. None of the 20 germline variants in the p53 CDS have been reproducibly linked to cancer predisposition (Whibley et al., 2009; Palmero et al., 2010). Genome-wide association studies (GWAS) reported no significant association between any germline p53 polymorphism and any cancer with a P-value ≤10−7 (a threshold for genome-wide significance) until a single nucleotide polymorphism (SNP) rs78378222 was found to be associated strongly with skin basal cell carcinoma with an odds ratio (OR) of 2.16 (P = 2.2 × 10−20) (Stacey et al., 2011). This SNP is located in the 3′UTR of p53 mRNA, changing the polyadenylation signal (PAS) AATAAA to AATACA, resulting in impaired 3′-end processing of p53 mRNA. Further investigation revealed its association with prostate cancer, glioma, and colorectal adenoma. Later, the association between this SNP and glioma was independently confirmed (Egan et al., 2012; Enciso-Mora et al., 2013), and its association with esophageal squamous cell carcinoma was also reported (Zhou et al., 2012; Li et al., 2013) (Table 1). However, this p53 noncoding variant is not related to breast cancer, a common LFS tumor; and at least in non-Hispanic Caucasians, this SNP does not increase the susceptibility to melanoma, squamous cell carcinoma of head and neck, and lung cancer (Guan et al., 2013). Stacey et al. (2011) showed that rs78378222[A/C] heterozygotes (n = 7) expressed ‘somewhat less’ p53 transcript than WT homozygotes in human blood specimens (P = 0.041). We tested the role of the alternative PAS in p53 regulation using a p53-null cell line H1299 with exogenous p53 constructs. The C allele, when compared with the A allele, dramatically lowered p53 mRNA levels and resulted in reduced p53 protein expression and cellular apoptosis. This finding provides direct evidence that rs78378222 negatively impacts p53 expression and function and is in agreement with previous reports that simian virus 40 mRNA with AATACA as PAS exhibits an 8-fold decrease in cleavage efficiency compared with the canonical site (Wickens and Stephenson, 1984). As rs78378222 occurs in the p53 PAS, none of the downstream p53 protein modifications are affected. Thus, this variant presents a unique instance of p53 alleles with unambiguous reduced-function, in contrast to most CDS mutants that are associated with a ‘loss-of-function’ and/or a ‘gain-of-function’ (Oren and Rotter, 2010).
Table 1.
Association between rs78378222[C] and cancers.
| Cancers | Frequency (%)a | OR |
|---|---|---|
| Colon adenoma | 1.85–1.92 | 1.39 |
| Prostate cancer | 0.28–1.92 | 1.44 |
| Glioma | 0.87–1.92 | 2.35 |
| 1.1 | 3.54 | |
| 1.3 | 3.74 | |
| Skin basal cell carcinoma | 0.46–1.92 | 2.16 |
| Esophageal squamous cell carcinoma | 1.0 | 3.22 |
aThe frequency of the C allele in general population (controls). Risk association to glioma is found in three studies.
The architecture of inherited cancer genetic susceptibility shows a montage of predisposition alleles with different levels of risk and prevalence in the population (Fletcher and Houlston, 2010). At one end of the spectrum are very-rare to rare frequency, high-penetrance, causative variants with risk allele frequencies typically ≤0.1% and cancer risks as high as 10%–20%. At the other end are common low-penetrance susceptibility alleles with risk allele frequencies >5% and OR of 1.1–1.6 (Figure 3). The p53 PAS variant is uniquely positioned among cancer susceptibility alleles (Hindorff et al., 2011) and characterized by: (i) low frequency (∼2% in the general populations), (ii) intermediate cancer risk (OR 1.39–3.54; Table 1), and (iii) predisposing carriers to multiple types of cancer. It is notable that cancer susceptibility of this p53 PAS variant does not strictly mirror that of p53 CDS germline mutations (LFS patients develop predominantly breast cancer, brain tumors, acute leukemia, sarcoma, and adrenal cortical carcinoma) (Lalloo et al., 2003; Gonzalez et al., 2009). Virtually all low-penetrance cancer susceptibility alleles implicated by GWAS with OR 1.1–1.6 are located in noncoding regions of the human genome (Hindorff et al., 2011). There are few mouse models generated for these variants to test the biological and functional relevance because it is difficult and often impossible to identify a mouse ortholog at the nucleotide level. Given the conservation of p53 PAS between human and mouse, generating a mouse model of this p53 germline variant affords an opportunity to test the causality of low-penetrance genetic alleles in cancer. Within this context, p53+/+, p53+/−, and p53−/− mice displayed different tumor spectra from each other (Donehower and Lozano, 2009) and from mice with LFS mutations (Olive et al., 2004). As the p53 PAS alleles have residual p53 expression and activity, it is likely that tumor profiles from mice carrying the PAS variant will be different from those of mice with other p53 alleles. Therefore, a mouse model of this p53 germline noncoding variant will offer novel insights into the impact of distinct p53 gene dosages and tumor spectra for potential cancer prevention and intervention research.
Figure 3.
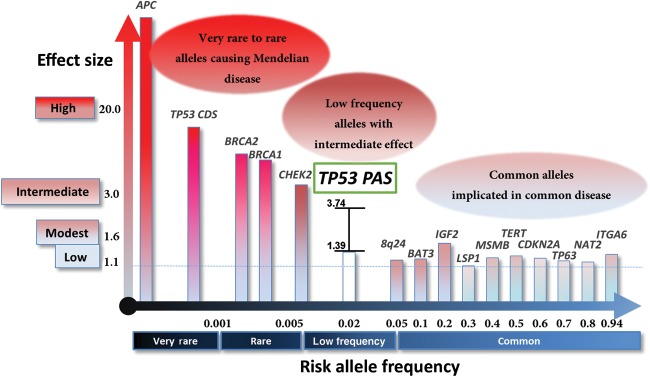
Risk allele frequency and effect sizes for genetic polymorphisms associated with cancer. Allele frequencies and odds ratios are taken from Hindorff et al. (2011) and are not portrayed to scale. Please be noted that alleles are associated with increased risk of different types of solid tumors.
p53 activation in ribosomopathies and cancer predisposition
In 1999, recurrent mutations in a ribosomal protein gene, RPS19, were reported in patients with Diamond–Blackfan anemia (DBA), a rare congenital bone marrow failure syndrome (BMFS) (Draptchinskaia et al., 1999). Since then, mutations in other ribosomal protein genes have been identified in up to 50% of patients with DBA. In addition, other congenital syndromes, Schwachman–Diamond syndrome, X-linked dyskeratosis congenita (DKC), cartilage–hair hypoplasia (CHH), and Treacher Collins syndrome (TCS), have been linked to defects in ribosome biogenesis. In the 5q− syndrome, a subtype of adult myelodysplastic syndrome, acquired haploinsufficiency for RPS14 was found to be causative (Ebert et al., 2008). Liu and Ellis (2006) summarized this group of disorders and pointed out their association with ribosome biogenesis with a hypothesis that defects in ribosomal biogenesis underlie bone marrow failure, and 4 years later Narla and Ebert (2010) coined the term ‘ribosomopathies’. All of the above mentioned diseases except TCS have manifested hematological defects (Table 2). Among ribosomopathies, CHH is unique as it is the only one with a mutant noncoding RNA gene called RMRP (Ridanpaa et al., 2001). In addition, CHH is associated with five other noncoding RNAs: the precursor rRNA, the mitochondrial RNA, the 5′UTR of the CLB2 mRNA, the endogenous siRNAs from double-stranded RMRP RNA, and the TERC RNA (Figure 4A). The Rmrp RNA has multiple functions associated with these noncoding RNAs. (i) It forms an endoribonuclease RNase mitochondrial RNA processing (RNase MRP) enzyme complex with multiple proteins in the nucleolus to process the precursor rRNA at the A3 site. This function has only been demonstrated unequivocally in yeast (Lygerou et al., 1996; Martin and Li, 2007). (ii) In mitochondria, the RNase MRP enzyme processes primer mitochondrial RNA for the leading-strand origin of mitochondrial DNA replication; and this is experimentally validated in both yeast (Saccharomyces cerevisiae) and mice (Chang and Clayton, 1989; Schmitt and Clayton, 1992). It has been recently found that yeast nucleolar RNase MRP and mitochondrial RNase MRP enzymes are distinct entities with differing activities and protein components, but sharing the same RMRP RNA (Lu et al., 2010). (iii) During mitosis, nucleolar RNase MRP localizes to the cytoplasm and forms a temporal asymmetric MRP body that processes the 5′UTR of the cyclin B2 mRNA to regulate cell cycle progression (Gill et al., 2004). (iv) The RMRP RNA forms a complex with the catalytic subunit of telomerase (TERT), which acts as an RNA-dependent RNA polymerase to synthesize the reverse strand of the RMRP RNA. The resultant double-stranded RNAs can be processed by DICER to generate endogenous siRNAs, causing downregulation of RMRP (Maida et al., 2009) and possibly other genes (Maida et al., 2013). The proliferative function of TERT is shown to be dependent on the RMRP–TERT complex (Mukherjee et al., 2011). It is notable that DKC1 (the causative gene for X-linked DKC) is a part of telomerase. Rosenbluh et al. (2011) have knocked out the RMRP allele in mice and found that heterozygous mice are phenotypically normal whereas homozygous RMRP-null mice are embryonic lethal. This is consistent with the finding that there are no homozygous RMRP-null alleles in CHH patients (Martin and Li, 2007) and that CHH is an autosomal recessive disease (McKusick et al., 1965). It would be intriguing to see whether mice with point mutations in the RMRP gene would phenocopy human counterparts including bone marrow failure and cancer predisposition.
Table 2.
Cancer predisposition and bone marrow failure in ribosomopathies.
| Disease | Gene | Gene function | p53 activation | Cancer predisposition | BMFS | Other clinical features |
|---|---|---|---|---|---|---|
| Cartilage–hair hypoplasia | RMRP | RNase MRP in processing the A3 site of pre-rRNA, a component of the RMRP-TERT complex | ? | Non-Hodgkin's lymphoma and basal cell carcinoma | Macrocytic anemia and defective proliferation of T- and/or B-cells | Dwarfism and hypoplastic hair |
| 5q− syndrome | RPS14 | pre-rRNA processing of the 18S rRNA, formation of the 40S ribosome subunit | Yes | Acute myeloid leukemia | Macrocytic anemia | Hypolobulated micromegakaryocytes |
| Diamond–Blackfan anemia | RPS19 and other ribosomal protein genes | pre-rRNA processing of the 18S rRNA, formation of the 40S or 60S ribosome subunit | ? | Acute myeloid leukemia and osteosarcoma | Macrocytic anemia and myelodysplastic syndrome | Growth retardation and craniofacial defects |
| Shwachman–Diamond syndrome | SBDS | 60S ribosome subunit joining/transportation | ? | Acute myeloid leukemia | Anemia and thrombocytopenia | Pancreatic insufficiency, growth defect, and neutropenia |
| X-linked dyskeratosis congenita | DKC1 | rRNA modification (pseudo-uridylation), a component of telomerase | ? | Acute myeloid leukemia and head and neck tumors | Cytopenias in the bone marrow | Oral leukoplakia, abnormal skin pigmentation, and nail dystrophy |
| Treacher Collins syndrome | TCOF1 | Transcription of ribosomal DNA and methylation of rRNA | Yes | None reported | No hematologic abnormalities | Craniofacial defects |
Figure 4.
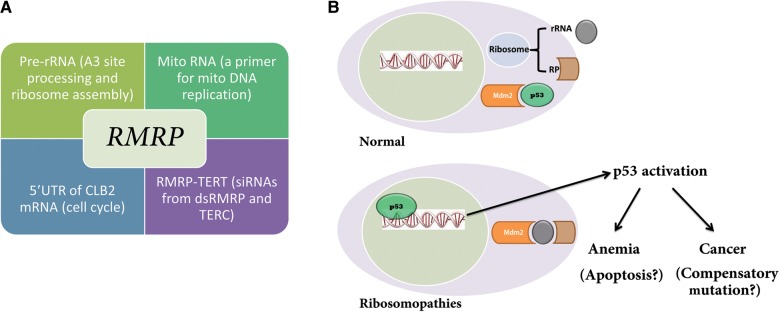
Ribosomopathies and p53 activation. (A) CHH is a genetic disease associated with multiple noncoding RNAs. (B) A proposed model of p53 activation in response to ribosome disassembly. In normal cells, ribosomes are assembled from rRNAs and ribosomal proteins and Mdm2 ubiquitinizes p53 for its degradation. When nascent ribosome biogenesis is disrupted, ribosomal proteins (such as RPL11) bind to MDM2 (with 5S rRNA), leading to p53 accumulation.
The direct link between ribosomopathies and p53 activation comes from the study of TCOF1 mutant mice. Mice haploinsufficient for TCOF1 exhibit diminished production of ribosomes and decreased proliferation of both neural ectoderm and neural crest cells (Dixon et al., 2006). Remarkably, chemical and genetic inhibition of p53 activity in these mice prevents craniofacial abnormalities (Jones et al., 2008). The idea that p53 activation underlies the pathology of ribosomopathies is further advanced with studies on 5q−. Mice with 5q− display macrocytic anemia, prominent erythroid dysplasia, and monolobulated megakaryocytes in the bone marrow (Barlow et al., 2010). Intercrossing with p53-deficient mice completely rescued bone marrow defects, providing strong evidence to suggest that a p53-dependent mechanism underlies the pathophysiology of the 5q− syndrome (Barlow et al., 2010). In cultured cell models, MDM2, along with 5S rRNA, binds multiple free ribosomal proteins, including RPL5, RPL23, RPL11, RPS7, and RPL26, and this binding inhibits Mdm2 function and increases the accumulation of p53 and the p53 transactivational activities in the nucleus (Figure 4B) (Zhang and Lu, 2009). This suggests that p53 activation could also underlie the pathology of DBA and other ribosomopathies beyond 5q–, yet animal models are needed to provide conclusive evidence.
In addition to anemia, a common clinical feature among patients with ribosomopathies is cancer predisposition (again, with the exception of TCS, Table 2). How does p53 activation lead to increased cancer risk in these patients? Amsterdam et al. (2004) screened a zebrafish mutant library and identified 12 lines with elevated cancer incidence, 11 of which were heterozygous for a mutation in a different ribosomal protein gene. Furthermore, tumor profiles of these zebrafish lines closely resembled that of p53-null zebrafish. In an ensuing study, it was found that p53 synthesis is impaired in tumors from zebrafish with ribosomal haploinsufficiency, yet there is neither mutation in the p53 CDS nor detectable gene amplifications of MDM2 or deletions of CDKN2A (MacInnes et al., 2008). ‘Super p53’ mice were generated with supernumerary fully functional copies of the p53 gene and these animals were protected from carcinogen-induced tumorigenesis and showed no increased cancer incidence (Garcia-Cao et al., 2002). Collectively, these data suggest unknown novel functions of ribosomal protein genes in addition to p53 activation induced by nucleolar stress. One potential mechanism of cancer predisposition in ribosomopathies is that cells accumulate somatic mutations to compensate unwanted p53-medicated apoptotic death (Figure 4B).
Conclusions and future directions
Despite intensive and fruitful investigations on p53 since its discovery >30 years ago, there are still many unresolved mysteries surrounding this master tumor suppressor. Intriguing paradoxical findings have been extensively reported in the history of p53 research. First, WT p53 is a tumor suppressor, whereas at least a portion of mutant p53 proteins are oncogenic. Second, p53 deficiency in mice leads to earlier onset of tumorigenesis and significantly shortens their life span (Donehower and Lozano, 2009), yet hyperactive mutant p53 and extra copies of p53 shorten life span in mice (Tyner et al., 2002; Maier et al., 2004). Third, patients with ribosomopathies have abnormal p53 activation, yet they are susceptible to cancer. In this review, we discuss the role of cancer-related noncoding RNAs in p53 inactivation and activation. Based on miRNA expression profiles, we postulate that in multiple myeloma with few p53 CDS mutations, partial p53 inactivation is mediated by overexpression of p53-targeting miRNAs. This alternative mechanism of p53 inactivation is critical for p53-dependent therapeutics and implicated in other cancers with WT p53 CDS. With information about 3′UTR mutation in lymphoma, a personalized prognosis and regimen based on the mutational status of both the UTR and CDS regions of p53 will be preferred. Therapeutics to increase p53 mRNA and protein levels could be applied to patients with WT CDS. In patients with p53 CDS mutation, reducing endogenous p53 expression or targeting the mutant p53 protein (Yu et al., 2012) might improve survival. As virtually every individual carries a low-penetrance cancer risk allele, a mouse model of the p53 noncoding variant (rs78378222) represents a unique opportunity to study the majority of cancers involving the interaction of genes with the environment, particularly in instances involving common low-penetrance variants that increase cancer risk moderately, and yet occur at higher frequencies in the population. As provocative hypotheses have been proposed to link p53 activation to cancer susceptibility in ribosomopathies including CHH that is caused by a noncoding RNA mutation, it is possible that other physiological functions are disrupted and/or compensatory genetic or non-genetic alterations are initiated due to ribosome biogenesis abnormality and p53 activation. Further dissection of noncoding RNAs in the p53 pathway at the molecular and cellular level and by using mouse models will gain insights into the underlying mechanisms of ribosomopathies and devise novel avenues in drug development against cancer and other p53-associated diseases.
Funding
Research in Li laboratory is supported by NIH grants CA138688 and GM103492.
Conflict of interest: none declared.
References
- Amsterdam A., Sadler K.C., Lai K., et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:e139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow J.L., Drynan L.F., Hewett D.R., et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q– syndrome. Nat. Med. 2010;16:59–66. doi: 10.1038/nm.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker L.E., Lu Z., Chen W., et al. A systematic screen reveals microRNA clusters that significantly regulate four major signaling pathways. PLoS One. 2012;7:e48474. doi: 10.1371/journal.pone.0048474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo J.J., Dhami P.K., Curry S., et al. No association between cigarette smoking and incidence of plasma cell myeloma: a meta-analysis of 17 observational studies. Am. J. Hematol. 2012;87:729–731. doi: 10.1002/ajh.23220. [DOI] [PubMed] [Google Scholar]
- Chang D.D., Clayton D.A. Mouse RNAase MRP RNA is encoded by a nuclear gene and contains a decamer sequence complementary to a conserved region of mitochondrial RNA substrate. Cell. 1989;56:131–139. doi: 10.1016/0092-8674(89)90991-4. [DOI] [PubMed] [Google Scholar]
- Chi J., Ballabio E., Chen X.H., et al. MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biol. Direct. 2011;6:23. doi: 10.1186/1745-6150-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng W.J., Price-Troska T., Gonzalez-Paz N., et al. Clinical significance of TP53 mutation in myeloma. Leukemia. 2007;21:582–584. doi: 10.1038/sj.leu.2404524. [DOI] [PubMed] [Google Scholar]
- Corthals S.L., Sun S.M., Kuiper R., et al. MicroRNA signatures characterize multiple myeloma patients. Leukemia. 2011;25:1784–1789. doi: 10.1038/leu.2011.147. [DOI] [PubMed] [Google Scholar]
- Dixon J., Jones N.C., Sandell L.L., et al. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl Acad. Sci. USA. 2006;103:13403–13408. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower L.A., Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer. 2009;9:831–841. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- Draptchinskaia N., Gustavsson P., Andersson B., et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- Ebert B.L., Pretz J., Bosco J., et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451:335–339. doi: 10.1038/nature06494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan K.M., Nabors L.B., Olson J.J., et al. Rare TP53 genetic variant associated with glioma risk and outcome. J. Med. Genet. 2012;49:420–421. doi: 10.1136/jmedgenet-2012-100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enciso-Mora V., Hosking F.J., Di Stefano A.L., et al. Low penetrance susceptibility to glioma is caused by the TP53 variant rs78378222. Br. J. Cancer. 2013;108:2178–2185. doi: 10.1038/bjc.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M., Bottoni A., Shimizu M., et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnebo M., Bykov V.J., Wiman K.G. The p53 tumor suppressor: a master regulator of diverse cellular processes and therapeutic target in cancer. Biochem. Biophys. Res. Commun. 2010;396:85–89. doi: 10.1016/j.bbrc.2010.02.152. [DOI] [PubMed] [Google Scholar]
- Fernberg P., Odenbro A., Bellocco R., et al. Tobacco use, body mass index, and the risk of leukemia and multiple myeloma: a nationwide cohort study in Sweden. Cancer Res. 2007;67:5983–5986. doi: 10.1158/0008-5472.CAN-07-0274. [DOI] [PubMed] [Google Scholar]
- Fletcher O., Houlston R.S. Architecture of inherited susceptibility to common cancer. Nat. Rev. Cancer. 2010;10:353–361. doi: 10.1038/nrc2840. [DOI] [PubMed] [Google Scholar]
- Fonseca R., Barlogie B., Bataille R., et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64:1546–1558. doi: 10.1158/0008-5472.can-03-2876. [DOI] [PubMed] [Google Scholar]
- Garcia-Cao I., Garcia-Cao M., Martin-Caballero J., et al. ‘Super p53’ mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21:6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill T., Cai T., Aulds J., et al. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: novel method of mRNA degradation. Mol. Cell. Biol. 2004;24:945–953. doi: 10.1128/MCB.24.3.945-953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez K.D., Noltner K.A., Buzin C.H., et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009;27:1250–1256. doi: 10.1200/JCO.2008.16.6959. [DOI] [PubMed] [Google Scholar]
- Guan X., Wang L.E., Liu Z., et al. Association between a rare novel TP53 variant (rs78378222) and melanoma, squamous cell carcinoma of head and neck and lung cancer susceptibility in non-Hispanic Whites. J. Cell. Mol. Med. 2013;17:873–878. doi: 10.1111/jcmm.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff L.A., Gillanders E.M., Manolio T.A. Genetic architecture of cancer and other complex diseases: lessons learned and future directions. Carcinogenesis. 2011;32:945–954. doi: 10.1093/carcin/bgr056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Chan C.S., Wu R., et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol. Cell. 2010;38:689–699. doi: 10.1016/j.molcel.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones N.C., Lynn M.L., Gaudenz K., et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M., Lu Z., Takwi A.A., et al. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene. 2011;30:843–853. doi: 10.1038/onc.2010.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle R.A., Therneau T.M., Rajkumar S.V., et al. Prevalence of monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2006;354:1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- Lalloo F., Varley J., Ellis D., et al. Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet. 2003;361:1101–1102. doi: 10.1016/S0140-6736(03)12856-5. [DOI] [PubMed] [Google Scholar]
- Le M.T., Teh C., Shyh-Chang N., et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Gordon M.W., Xu-Monette Z.Y., et al. Single nucleotide variation in the TP53 3′ untranslated region in diffuse large B-cell lymphoma treated with rituximab-CHOP: a report from the International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;121:4529–4540. doi: 10.1182/blood-2012-12-471722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionetti M., Biasiolo M., Agnelli L., et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood. 2009;114:e20–e26. doi: 10.1182/blood-2009-08-237495. [DOI] [PubMed] [Google Scholar]
- Liu J.M., Ellis S.R. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood. 2006;107:4583–4588. doi: 10.1182/blood-2005-12-4831. [DOI] [PubMed] [Google Scholar]
- Lohr J.G., Stojanov P., Lawrence M.S., et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl Acad. Sci. USA. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q., Wierzbicki S., Krasilnikov A.S., et al. Comparison of mitochondrial and nucleolar RNase MRP reveals identical RNA components with distinct enzymatic activities and protein components. RNA. 2010;16:529–537. doi: 10.1261/rna.1893710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygerou Z., Allmang C., Tollervey D., et al. Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science. 1996;272:268–270. doi: 10.1126/science.272.5259.268. [DOI] [PubMed] [Google Scholar]
- MacInnes A.W., Amsterdam A., Whittaker C.A., et al. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc. Natl Acad. Sci. USA. 2008;105:10408–10413. doi: 10.1073/pnas.0805036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maida Y., Yasukawa M., Furuuchi M., et al. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461:230–235. doi: 10.1038/nature08283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maida Y., Kyo S., Lassmann T., et al. Off-target effect of endogenous siRNA derived from RMRP in human cells. Int. J. Mol. Sci. 2013;14:9305–9318. doi: 10.3390/ijms14059305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B., Gluba W., Bernier B., et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A.N., Li Y. RNase MRP RNA and human genetic diseases. Cell Res. 2007;17:219–226. doi: 10.1038/sj.cr.7310120. [DOI] [PubMed] [Google Scholar]
- McFarland C.D., Korolev K.S., Kryukov G.V., et al. Impact of deleterious passenger mutations on cancer progression. Proc. Natl Acad. Sci. USA. 2013;110:2910–2915. doi: 10.1073/pnas.1213968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick V.A., Eldridge R., Hostetler J.A., et al. Dwarfism in the Amish. Ii. cartilage-hair hypoplasia. Bull. Johns Hopkins Hosp. 1965;116:285–326. [PubMed] [Google Scholar]
- Mills P.K., Newell G.R., Beeson W.L., et al. History of cigarette smoking and risk of leukemia and myeloma: results from the adventist health study. J. Natl Cancer Inst. 1990;82:1832–1836. doi: 10.1093/jnci/82.23.1832. [DOI] [PubMed] [Google Scholar]
- Morin R.D., Mendez-Lago M., Mungall A.J., et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Firpo E.J., Wang Y., et al. Separation of telomerase functions by reverse genetics. Proc. Natl Acad. Sci. USA. 2011;108:E1363–E1371. doi: 10.1073/pnas.1112414108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P.A., Vousden K.H. p53 mutations in cancer. Nat. Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- Narla A., Ebert B.L. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveu P., Kye M.J., Qi S., et al. MicroRNA profiling reveals two distinct p53-related human pluripotent stem cell states. Cell Stem Cell. 2010;7:671–681. doi: 10.1016/j.stem.2010.11.012. [DOI] [PubMed] [Google Scholar]
- Olive K.P., Tuveson D.A., Ruhe Z.C., et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Oren M., Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero E.I., Achatz M.I., Ashton-Prolla P., et al. Tumor protein 53 mutations and inherited cancer: beyond Li-Fraumeni syndrome. Curr. Opin. Oncol. 2010;22:64–69. doi: 10.1097/CCO.0b013e328333bf00. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L., Trifonov V., Fabbri G., et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichiorri F., Suh S.S., Ladetto M., et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl Acad. Sci. USA. 2008;105:12885–12890. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaltopoulou T., Sergentanis T.N., Kanellias N., et al. Tobacco smoking and risk of multiple myeloma: a meta-analysis of 40 observational studies. Int. J. Cancer. 2013;132:2413–2431. doi: 10.1002/ijc.27898. [DOI] [PubMed] [Google Scholar]
- Ridanpaa M., van Eenennaam H., Pelin K., et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage-hair hypoplasia. Cell. 2001;104:195–203. doi: 10.1016/s0092-8674(01)00205-7. [DOI] [PubMed] [Google Scholar]
- Rosenbluh J., Nijhawan D., Chen Z., et al. RMRP is a non-coding RNA essential for early murine development. PLoS One. 2011;6:e26270. doi: 10.1371/journal.pone.0026270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M.E., Clayton D.A. Yeast site-specific ribonucleoprotein endoribonuclease MRP contains an RNA component homologous to mammalian RNase MRP RNA and essential for cell viability. Genes Dev. 1992;6:1975–1985. doi: 10.1101/gad.6.10.1975. [DOI] [PubMed] [Google Scholar]
- Stacey S.N., Sulem P., Jonasdottir A., et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 2011;43:1098–1103. doi: 10.1038/ng.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarbrick A., Woods S.L., Shaw A., et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat. Med. 2010;16:1134–1140. doi: 10.1038/nm.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S., Huang S., Wu S., et al. MicroRNA-1285 inhibits the expression of p53 by directly targeting its 3′ untranslated region. Biochem. Biophy. Res. Commun. 2010;396:435–439. doi: 10.1016/j.bbrc.2010.04.112. [DOI] [PubMed] [Google Scholar]
- Tyner S.D., Venkatachalam S., Choi J., et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Vogelstein B., Lane D., Levine A.J. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Volinia S., Calin G.A., Liu C.G., et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl Acad. Sci. USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden K.H., Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Whibley C., Pharoah P.D., Hollstein M. p53 polymorphisms: cancer implications. Nat. Rev. Cancer. 2009;9:95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- Wickens M., Stephenson P. Role of the conserved AAUAAA sequence: four AAUAAA point mutants prevent messenger RNA 3′ end formation. Science. 1984;226:1045–1051. doi: 10.1126/science.6208611. [DOI] [PubMed] [Google Scholar]
- Williams R.R., Horm J.W. Association of cancer sites with tobacco and alcohol consumption and socioeconomic status of patients: interview study from the Third National Cancer Survey. J. Natl Cancer Inst. 1977;58:525–547. doi: 10.1093/jnci/58.3.525. [DOI] [PubMed] [Google Scholar]
- Xu-Monette Z.Y., Wu L., Visco C., et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with rituximab-CHOP: a report from an International DLBCL Rituximab-CHOP Consortium Program study. Blood. 2012;120:3986–3996. doi: 10.1182/blood-2012-05-433334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Vazquez A., Levine A.J., et al. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Yuan Q., Yang M. A functional germline variant in the P53 polyadenylation signal and risk of esophageal squamous cell carcinoma. Gene. 2012;506:295–297. doi: 10.1016/j.gene.2012.07.007. [DOI] [PubMed] [Google Scholar]