

Abstract
Purpose.
Mutations in the photoreceptor cell-specific gene encoding aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) lead to Leber congenital amaurosis (LCA4), retinitis pigmentosa, and cone–rod dystrophy. Gene therapy appears to be promising in the treatment for AIPL1-mediated vision loss in humans. Prior to initiating these treatments, however, it is crucial to understand how the retinal neurons remodel themselves in response to photoreceptor cell degeneration. In this study, using an animal model for AIPL1-LCA, Aipl1−/− mice, we investigate the changes in postreceptoral retinal neurons during the course of photoreceptor cell loss.
Methods.
Morphology of the Aipl1−/− retina from postnatal day 8 to 150 was compared to that of age-matched, wild-type C57Bl6/J retina (WT) by immunocytochemistry using cell-specific markers.
Results.
Expression of postsynaptic proteins in bipolar cells is reduced prior to photoreceptor cell degeneration at postnatal day 8. Bipolar and horizontal cells retract their dendrites. Cell bodies and axons of bipolar and horizontal cells are disorganized during the course of degeneration. Müller cell processes become hypertrophic and form a dense fibrotic layer outside the inner nuclear layer.
Conclusions.
An early defect in photoreceptor cells in the AIPL1-LCA mouse model affects the expression of postsynaptic markers, suggesting abnormal development of bipolar synapses. Once degeneration of photoreceptor cells is initiated, remodeling of retinal neurons in the Aipl1−/− animal is rapid.
Keywords: photoreceptor degeneration, remodeling, retina, LCA, childhood blindness
We show abnormal development of the photoreceptor–bipolar synapse in an animal model for Leber congenital amaurosis. As degeneration of rod and cone photoreceptor was complete, we observed rapid and severe alterations in inner retinal neurons.
Introduction
Light perception is primarily initiated in the retinal photoreceptor cells, rods and cones, lining the inner surface of the eye.1–3 The electrochemical signals generated by photoreceptor cells are processed by the circuitry containing a diverse population of second- and third-order neurons in the retina.1–3 The retinal circuit formed postnatally undergoes changes or remodels itself during physiological processes such as development, aging, injury, and disease.4–8 Alterations in the retinal circuitry are observed in both humans affected with retinal degenerative blinding diseases and animal models of the human diseases.7,9–11
A model for retinitis pigmentosa (RP), the rd1 mouse, shows that the primary loss of rod photoreceptor cells triggers dramatic changes in the morphology of second-order neurons.12,13 In rd1 mice, 97% of rod cell death occurs rapidly by postnatal day 17, whereas cone photoreceptor cells are lost at 2 to 4 months of age, with some cones surviving for the lifetime of the animal.14–16 As a result of the rapid rod degeneration, rod-driven bipolar and horizontal cell axon terminals retract their fine dendrites, and rod bipolar cell axon terminals assume immature synaptic structures.12,13 Defects extend to the cone circuit during the late phase of degeneration. In this case, both cones and cone horizontal cells sprout new neurites, whereas cone bipolar cells retract their dendrites.12,13,16,17 Similar alterations of the second-order neurons were observed in rd10, Stargardt (STGD3)-like mouse models, and crx−/− (cone–rod homeobox) mice in which degeneration of photoreceptor cells is slower.18–20
In humans, mutations in the photoreceptor cell-specific gene encoding aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) are associated with an early childhood blinding disease called Leber congenital amaurosis (LCA), RP, and cone–rod dystrophy.10,21–26 The AIPL1-LCA patients are clinically characterized by early loss of photoreceptor cells, low visual acuity at young ages, no measurable visual fields, and nondetectable electroretinograms (ERG). Degeneration of photoreceptor layer and signs of remodeling of inner retinal neurons were observed in AIPL1-LCA patients.10,27
A mouse model for LCA, Aipl1−/− animals, exhibited rapid degeneration of both rods and cones and did not produce any light-dependent electrical response at any age tested.22 Histological studies done on Aipl1−/− animals show loss of photoreceptor cells from postnatal day (P) 9, and by P30 photoreceptor degeneration is complete (Supplementary Fig. S1). Biochemical studies show that Aipl1 plays a crucial role in the stability of phosphodiesterase 6 (PDE6) in rods and cones.28 Recently, several studies have shown that the photoreceptor layer can be rescued in Aipl1−/− mice with early administration of an adeno-associated viral (AAV) vector encoding human AIPL1.29–31 However, rapid degeneration of photoreceptor cells poses a significant challenge for long-term efficacy of gene therapy. A better understanding of the cell-specific retinal remodeling is needed for improvement of current approaches to AAV-mediated gene therapy and for the development of alternative targeted therapies, such as cell transplantation or conversion of downstream neurons into light-sensitive cells, to treat AIPL1-LCA patients.7
Retinal remodeling has been investigated mostly in animal models such as the rd1 mouse in which the primary defect lies in rods and consequently leads to rod photoreceptor cell death followed by slower elimination of cones as a “bystander” effect. To understand retinal remodeling in a model where there is rapid rod and cone photoreceptor cell degeneration, we studied the retinal phenotype in a mouse in which a functional Aipl1 gene is absent.22 We examined the retina of Aipl1−/− and wild-type C57black 6/J (WT) mice at various ages using a panel of cell type–specific antibodies, providing a systematic description of retinal reorganization in this mouse model. We found differences in expression of synaptic proteins at an early age followed by rapid remodeling of second-order neurons and bipolar and horizontal cells.
Materials and Methods
Animals
All procedures used in this study conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Additionally, the procedures used in this work were approved by Institutional Animal Care and Use Committee at West Virginia University. Aipl1 knockout (Aipl1−/−) mice were backcrossed with C57black/6J (WT; Jackson Laboratories, Bar Harbor, ME, USA) for 10 generations and characterized as described in our earlier work.22 The Aipl1−/− mouse retinas were studied at P8, P10, P14, P18, P30, P60, P100, and P150 (n = 5). The control mice (C57Bl/6J) were examined at the same ages (n = 5). Animals were maintained on a 12-hour light/dark cycle and had access to standard mouse chow and water ad libitum.
Fixation, Sectioning, and Immunohistochemistry
Mouse eyes were enucleated, punctured with a fine needle, and incubated for 10 minutes in fixative, 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) in phosphate-buffered saline (1× PBS) at room temperature. To make eye cups, eyes were removed from the fixative, and the cornea and lens were dissected away. The eye cups were further fixed for 20 minutes at room temperature, then cryoprotected in PBS containing a sucrose gradient series starting at 10% and increasing up to 30% at 4°C followed by incubation in a 1:1 ratio of PBS containing 30% sucrose and optimal cutting temperature compound (OCT; Fisher Scientific, Pittsburgh, PA, USA) for 2 hours at 4°C. The eye cups were embedded in OCT and stored at −80°C. Retinal cross sections were cut (12 μm thick) parallel to the temporonasal axis through the optic nerve head using a Leica CM1850 (Leica Microsystems, Wetzlar, Germany) cryostat and were mounted on Superfrost Plus slides (Fisher Scientific).
For immunohistochemistry, retinal sections mounted on slides were washed (three times for 10 minutes) in 1× PBST (1× PBS with 0.1% Triton X-100) and incubated for 1 hour with blocking buffer (2% goat serum [Invitrogen, Grand Island, NY, USA], 0.1% Triton X-100, and 0.05% sodium azide in 1× PBS). After a brief wash, primary antibodies (Supplementary Table S1) were applied to retinal sections and incubated overnight at 4°C. Slides were washed with 1× PBST (three times for 10 minutes) and incubated with mouse, rabbit, or guinea pig secondary antibody (Alexa Fluor 488 or Alexa Fluor 568; Invitrogen) for 1 hour at room temperature. The nuclear marker 4′,6-diamidino-2-phenylindole (DAPI, 1:5000 dilution; Invitrogen) was added for 10 minutes, and after three washes with 1× PBST, sections were mounted with Prolong Gold antifade reagent (Invitrogen) and coverslipped.
Preparation of Whole-Mount Retina
Whole-mount tissue was processed as previously described.32 Briefly, whole eyes were enucleated, and the dorsal side was marked by puncturing the cornea with a 25-gauge 5/8 precisionglide needle for orientation purposes. The whole eye was fixed in 4% paraformaldehyde in 1× PBS for 30 minutes. The eye was then removed from fixative, and the cornea and lens were dissected away. The dorsal region of the retina was then marked by a linear cut to preserve the known orientation. Free-floating retinas were returned to 4% paraformaldehyde for 6 hours.
For immunohistochemistry, retina was washed with 1× PBS (three times for 30 minutes each) and then incubated with blocking buffer (2% goat serum [Invitrogen], 0.1% Triton X-100, and 0.05% sodium azide in 1× PBS) for 4 hours. After blocking, the tissue was incubated overnight with primary antibody (calbindin [CB]). After removal of primary antibody, retina were washed in 1× PBST (twice for 30 minutes each) and 1× PBS (once for 30 minutes) before being incubated overnight in secondary antibodies, goat anti-mouse Alexa 566 (diluted 1:1000; LI-COR Biosciences, Lincoln, NE, USA). Retina was further washed in 1× PBST (twice for 30 minutes each) and 1× PBS (once for 30 minutes). Before imaging, the retina was placed on a Superfrost Plus slide (Fisher Scientific) with the outer segments of the photoreceptors facing down. Radial cuts 2 to 3 mm in length were made to flatten the tissue. Finally, the whole retinal tissue was flat mounted, vitreal side up, onto the slide and was coverslipped for imaging.
Antibody Characterization
The primary antibody sources, the host in which the antibody was produced, and dilution used for immunohistochemistry are shown in Supplementary Table S1. Metabotropic glutamate receptor (mGluR6) antibody (gift from Shigetada Nakanishi, Kyoto University, Japan) provides punctate staining at the dendritic tips of rod bipolar cells and ON-type cone bipolar cells in the outer plexiform layer (OPL).19 C-terminal binding protein 2 (CtBP2) antibody produced a distinct horseshoe-shaped staining in the synaptic ribbons along OPL and dense punctate in the inner plexiform layer (IPL), which is identical to findings in previous reports.33,34 Transient receptor potential cation channel subfamily M member 1 (TRPM1) antibody (gift from T. Furukawa) provides staining at the dendritic tips in the OPL and rod bipolar cell somas.35 Pikachurin antibody showed a punctate staining pattern in the OPL.36 Antibodies against vesicular glutamate transporter (VGLUT1) and synaptophysin stain exclusively the IPL and OPL of the murine retina.37–40 Protein kinase C-α (PKCα) antibody labels rod bipolar cells and a subset of amacrine cells.19,41 Secretagogin (SCGN) antibody (gift from Ludwig Wagner, University of Vienna, Austria) labels cone bipolar cells.42 Calbindin mouse monoclonal antibody recognizes a single band of 28 kDa in Western blot of brain homogenate (Sigma-Aldrich, St. Louis, MO, USA). In the mouse retina, it stains horizontal cells and a subset of amacrine cells.12,41 Neurofilament 200 (NF-200) antibodies recognize the 200-kDa heavy chain neurofilament polypeptide in a Western blot (Sigma-Aldrich). In the murine retina, neurofilament labels axonal complexes of horizontal cells.12,41 Calretinin was raised against recombinant rat calretinin. Western blot studies conducted by the manufacturer (Chemicon International, Temecula, CA, USA) indicate that it is specific for calretinin and recognizes both calcium-bound and -unbound conformations of this protein. In retina, calretinin staining was confined to the starburst amacrine cells, tyrosine hydroxylase 2 amacrine cells, and ganglion cells.41 Glutamine synthetase (GS) specificity was confirmed with a Western blot using a rat cerebellum lysate where it reacted with a 45-kDa band that corresponds to the expected size of the GS protein, amino acids 1 to 373 (BD Transduction Laboratories, San Diego, CA, USA). In retina, GS antibody specifically stains the entire Müller cell.41,43 Glial fibrillary acidic protein (GFAP antibody specificity was observed in Western blots of mouse retina.19 In retina, GFAP antibody stains retinal astrocytes and Müller cells.19,43
Image Capture and Quantification of Images
Immunostained retinal sections were imaged using a Zeiss LSM 510 laser scanning confocal microscope (Carl Zeiss Meditec, Inc., Dublin, CA, USA) on an upright Zeiss AxioImager microscope using a Zeiss Plan-Apochromat 40×/1.4 and 63×/1.4 oil objective. Pinholes were set to 1 airy unit for each channel, line averaging of 4, 1024 × 1024 image format. Images were collected using 405-, 488-, and 561-nm laser lines for excitation and BP420-480, BP505-530, and LP560 emission filters for DAPI, Alexa 488, and Alexa 568 fluorophores, respectively. The WT and Aipl1−/− retinal sections were processed under identical conditions and were examined at room temperature. For each experiment, five independent samples were imaged using a single scan technique to ensure unbiased cell numbers; only the central one-third of the retinal region was considered for image analysis.
Counts of mGluR6 and CtBP2 puncta in the OPL were performed from an area of 66.8 × 66.8 μm. A region of interest that encompassed the OPL was drawn, and only puncta within this region were counted using ImageJ software (National Institutes of Health, Bethesda, MD, USA). For quantification of rod and cone bipolar cells, cell counting was done on previously taken images from an area of 133 × 133 μm. Immunopositive cells had to meet the criteria of distinct soma labeling above background level irrespective of the labeling intensity (weak or strong labeling). Data in graphs (see Figs. 1, 4, 5) represent average ± standard deviation. Statistical analysis (one-way ANOVA) was performed using an online program (http://www.vassarstats.net [in the public domain]).
Results
Abnormal Expression of Proteins at Photoreceptor–Bipolar Synapse
The timing of rapid degeneration of photoreceptor cells in Aipl1−/− animals coincides with synaptogenesis. Therefore, we wanted to determine if the photoreceptor–bipolar cell connection develops normally in Aipl1−/− animals. We examined the distribution of pre- and postsynaptic markers prior to and during photoreceptor loss in both Aipl1−/− and wild-type (WT) animals.
We used an antibody against CtBP2, a presynaptic marker that labels the synaptic ribbons in rod and cone terminals.34 In WT retina, at P8, punctate CtBP2 staining was evident, which then developed into a distinct horseshoe-shaped staining pattern along the synaptic ribbons in the OPL (Figs. 1A–E). A similar level and pattern of CtBP2 staining was observed in the retina from Aipl1−/− animals prior to photoreceptor degeneration at P8 (Fig. 1F, 1U). As photoreceptors degenerated at P10 to P30, CtBP2 labeling was reduced and appeared dispersed at OPL (Figs. 1G–I). At P30, when photoreceptor degeneration was complete, CtBP2 staining was undetectable (Fig. 1J). Similar to our observations with CtBP2, localization of pikachurin, a protein present in the synaptic cleft in the photoreceptor ribbon synapse, was unaffected prior to photoreceptor cell death at P8 (Supplementary Figs. S2E, S2G). These results suggest that presynaptic terminals of photoreceptor cells develop normally in animals lacking Aipl1 prior to significant photoreceptor degeneration.
Figure 1.

Reduced expression of mGluR6 prior to photoreceptor loss in Aipl1−/− animals. Immunostaining for CtBP2 (A–J) and mGluR6 (K–T) in WT and Aipl1−/− retina at specified ages. Arrow indicates abnormal staining of CtBP2 (H, I) and mGluR6 (R, S) in the outer plexiform layer of retina from Aipl1−/− animals. Scale bar: 10 μm. Images were obtained using confocal microscopy at 63× magnification with 2× zoom. Average number (±SD) of CtBP2 (U) and mGluR6 (V) puncta in a retinal cross window of 66.8 × 66.8 μm in Aipl1−/− and WT retina at P8. ns, nonsignificant; ****P < 0.0001.
To evaluate the integrity of dendritic tips of rods and cone ON bipolar cells, we tested the expression and localization of mGluR6, a postsynaptic marker. In WT retina, mGluR6 immunoreactivity was observed as a layer of punctate staining in the OPL, representing the dendritic tips of rod ON bipolar cells and a disc-like formation, which represents cone pedicles connected to the ON cone bipolar cell (Figs. 1K–O). Weak staining was also observed in the cell bodies of rod and cone bipolar cells (data not shown). In Aipl1−/− retina, there was reduced mGluR6 immunoreactivity in the OPL (Figs. 1P–1T). At an early stage (P8) of retinal development, an approximately 40% reduction in mGluR6 expression was observed in retina from Aipl1−/− animals (Figs. 1P, 1V). At P14 to P18, when photoreceptor degeneration was occurring, mGluR6 was distributed abnormally in the OPL (Figs. 1R, 1S). The majority of punctate mGluR6 receptors were lost from the OPL. Spots of mGluR6 immunoreactivity were found along cell bodies and some bipolar axons (Figs. 1R, 1S). When photoreceptor degeneration was complete, mGluR6 staining was undetectable in the OPL of Aipl1−/− retina (Fig. 1T). We also confirmed the early reduction in expression of postsynaptic proteins by investigating the localization of TRPM1, a channel protein expressed in ON bipolar cells (Supplementary Figs. S2C, S2D). Based on our results from immunolocalization, we conclude that expression of postsynaptic proteins in bipolar cells is defective, likely due to abnormal development of the photoreceptor–bipolar synapse in Aipl1−/− animals.
Early Changes in the Inner Plexiform Layer
Vesicular glutamate transporter is needed for replenishing synaptic vesicles with glutamate at the synaptic terminal.38 In WT retina, VGLUT1 expression was confined to the OPL and IPL where vesicular glutamate transmission occurs (Figs. 2A–E). More specifically, discrete and punctate immunostaining was observed across the full extent of the IPL. The larger rod puncta observed at the distal end of the IPL are typically identified as rod bipolar cell terminals, and the smaller cone bipolar termini stratify just above them (Figs. 2B–E; see also Supplementary Fig. S3).
Figure 2.

Reduction in VGLUT1 expression as photoreceptors degenerate. Labeling for VGLUT1 antibody at specified ages in WT (A–E) and Aipl1−/− (F–J) retina. Arrows in WT retina indicate cone terminals (C) and arrowheads indicate rod terminals (D, E). Cone and rod terminals are also marked in (E). Arrows in Aipl1−/− retina indicate reduction in VGLUT1 expression at P14 and P18 in the OPL (G, H). VGLUT1 expression is abolished at P30 in the OPL (I). Scale bar: 20 μm.
In the Aipl1−/− retina, at P8, VGLUT1 staining was similar to that in WT retina, confirming our earlier result indicating that photoreceptor presynaptic terminals develop normally. However, the number of VGLUT1-positive terminals in the OPL was dramatically reduced and was uneven at P14 (Fig. 2G). In P18 Aipl1−/− retina, discrete VGLUT1 labeling was observed in the OPL (Fig. 2H). This residual VGLUT1 labeling was absent in the OPL at P30 to P60 (Figs. 2I, 2J). Vesicular glutamate transporter expression in the IPL at earlier time points was not significantly different between Aipl1−/− and WT retina (compare Figs. 2A, 2B with Figs. 2F, 2G). However, at P30 to P60 in the Aipl1−/− retina, VGLUT1 reactivity in the IPL appeared disorganized (Figs. 2G–J). The plexus of large VGLUT1-positive rod terminals in the innermost portion of the IPL was reduced and difficult to distinguish from cone terminals (compare Figs. 2C–E with Figs. 2H–J; Supplementary Fig. S3).
Synaptophysin (SYP) is a synaptic vesicle protein expressed in all vesicular synaptic terminals in the OPL and IPL of the retina. In WT retina, SYP immunoreactivity was prominent and distributed throughout the synapse-rich OPL and IPL at all ages (Figs. 3A–E). In contrast, in the Aipl1−/− retina, SYP immunoreactivity was discontinuous in the OPL (Figs. 3F–J). When photoreceptors were completely lost, weak SYP labeling was found in the cell bodies near the collapsed pigment epithelium adjacent to OPL at P30 and P60 (Figs. 3I, 3J). Synaptophysin labeling in the IPL is similar between Aipl1−/− and WT retina at P14 (Figs. 3B, 3G). As photoreceptors degenerated from P18 to P60, SYP labeling in the rod and cone bipolar axon terminals was reduced and difficult to distinguish (compare Figs. 3C–E with Figs. H–J). In sum, our results show changes in synaptic terminals in the Aipl1−/− retina.
Figure 3.

Loss of synaptophysin (SYP) from photoreceptor synaptic terminals in Aipl1−/− retina. Synaptophysin labeling reveals changes in the synapse at indicated ages in WT (A–E) and Aipl1−/− (F–J) retina. Arrows indicate discrete labeling of SYP in OPL in the Aipl1−/− retina (G, H). Arrowhead (I, J) indicates SYP labeling in retinal pigment epithelium collapsed with OPL. Scale bar: 20 μm.
Morphologic Changes in the Rod Bipolar Cell Dendrites and Axon Terminals
Rod bipolar cells (RBC) are second-order retinal neurons that connect to rod spherules by a large dendritic arbor; their long axons project to the IPL with large club-shaped terminals.19 We used PKCα antibody to specifically label the RBC. In WT retina, PKCα staining was observed in the fine dendrites projecting to the rod terminals at all ages tested (Figs. 4A–F). Morphology of RBC was similar in Aipl1−/− and WT retinas in the early stage (P14) of photoreceptor degeneration (Fig. 4G). At P18, when photoreceptor cells are reduced to a single layer in the retina lacking Aipl1, PKCα staining was noticeably different than in control animals. The dendritic arborization was reduced and was retracted from the OPL in the Aipl1−/− retina (compare Figs. 4B, 4H). However, the number of axons remained similar in Aipl1−/− and WT retinas. At P30 and P60, when degeneration of photoreceptors was complete in retinas lacking Aipl1, axons were disorganized; cell bodies were poorly oriented and were reduced in numbers (Figs. 4I, 4J). At P100 and P150 the number of PKCα-immunopositive cells decreased, and the cell bodies were not aligned properly (Figs. 4K, 4L). A cell count done on vertical sections using a PKCα antibody showed significant decline in bipolar cell number during P30 to P150 (Fig. 4M).
Figure 4.

Loss of rod bipolar neurons in Aipl1−/− retina. Rod bipolar cell morphology was assessed in retina from WT (A–F; A’–F’) and Aipl1−/− (G–L; G’–L’) animals at indicated ages using an antibody against PKCα. Arrows at top indicate loss of dendrites (H), radial orientation of rod bipolar cell soma (J, K) in Aipl1−/− animals. Arrowhead at bottom indicates rod bipolar axonal endings that show stunted axonal arborization and appear as rudimentary dots in Aipl1−/− animals (J'–L'). Scale bar: 20 μm. Average number (±SD) of rod bipolar cells in a retinal cross window of 133 × 133 μm as a function of age (M). ***P < 0.001 and ****P < 0.0001.
Besides labeling RBC bodies, PKCα staining was restricted to large lobular axon terminals stratified in the IPL. In WT retina, PKCα-labeled RBC axonal terminals have a large varicose ending at the inner margin of the IPL (Figs. 4A'–F', bottom). The axonal bouton of RBC in the Aipl1−/− retina showed dramatic alterations in morphology (Figs. 4H'–J', bottom). Rod bipolar axonal terminals were small and appeared as rudimentary dots at P100 and P150 (Figs. 4K', 4L', bottom). Altogether, our results demonstrate early retraction of bipolar dendrites, reduction in the number of RBC, and reduced arborization of rod bipolar axonal terminals.
Changes in Cone Bipolar Cell Dendrites
Cone bipolar cells receive input from cone photoreceptors through dendritic arborization and redirect the signal to ganglion cell synapses in the IPL. Since Aipl1−/− animals undergo rapid rod and cone degeneration, we investigated the remodeling of cone bipolar cells. We used a cone bipolar–specific SCGN antibody that labels both ON-type and OFF-type cone bipolar cells.42 Secretagogin immunoreactivity was observed in dendrites extending into the OPL and axon terminals stratifying in the ON and OFF sublaminae of the IPL in the WT retina (Figs. 5A–F). In addition, the cytoplasm and entire axon of bipolar cells were labeled by SCGN.35
Figure 5.

Dendritic changes in cone bipolar cells. Cone bipolar cells labeled with an antibody against secretagogin in WT (A–F) and Aipl1−/− (G–L) retina. Dendrites are disorganized and retract in the Aipl1−/− retina from P60 (J, arrows). At P100 and P150, arrows indicate disorganized cell bodies in the Aipl1−/− retina (K, L). Scale bar: 20 μm. (M) Average number (±SD) of cone bipolar cells in a retinal cross window of 133 × 133 μm as a function of age. ***P < 0.001 and ****P < 0.0001.
Secretagogin expression was similar in WT controls and Aipl1−/− animals until P30, indicating normal development of cone bipolar cells (Figs. 5G–I). However, at P60, the uniform dendritic plexus formed in the OPL by dendritic arbors in the control retina was disrupted in the Aipl1−/− retina (Fig. 5J). At P100 and P150, cone bipolar cell dendrites were absent (Figs. 5K, 5L). Secretagogin-stained cell bodies were discontinuously arranged in the IPL (Figs. 5K, 5L). The axons of cone bipolar cells were shorter and their terminals appeared disordered. A significant reduction in cone bipolar cell number was observed at P100 to P150 (Fig. 5M).
Changes in Horizontal Cell Axons and Dendrites
Horizontal cells are interneurons that connect with cone pedicles through dendrites and with rod spherules through axonal arborization.44 We used CB antibody to stain horizontal cell bodies and dendrites in OPL. Calbindin immunoreactivity was seen in the OPL, dendrites, and cell bodies in WT retina (Figs. 6A–F). In comparison, retina from Aipl1−/− animals exhibited a different pattern of CB staining (Figs. 6G–L). Calbindin immunoreactivity was absent from puncta as early as P14 (Fig. 6G). The branches of dendrites became thinner and formed a loose meshwork in the OPL (Figs. 6H–L). At P60 to P150, horizontal cell bodies were oriented irregularly in the OPL (Fig. 6K). Flat-mount staining revealed thinning of horizontal cell processes and reduction in CB immunoreactivity in the retina from Aipl1−/− animals (Supplementary Figs. S4A, S4B).
Figure 6.
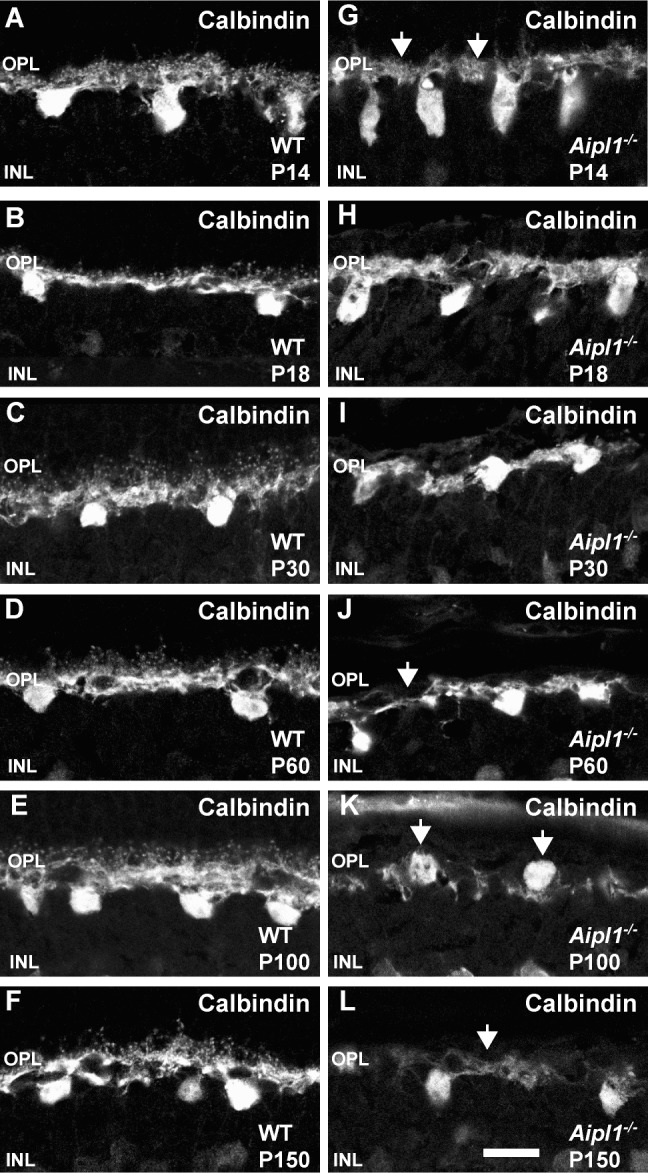
Altered horizontal cell morphology in the degenerating retina. Calbindin staining reveals changes in the horizontal cell morphology in WT (A–F) and Aipl1−/− (G–L) retina. Arrows point to the loss of puncta from OPL at P14 in retina from animals lacking Aipl1−/− compared to controls (G). (J–L) Arrows indicate thinning of horizontal cell processes and abnormal orientation of cell bodies as animal ages. Scale bar: 20 μm.
To stain horizontal cell axons, we used antibody against NF-200.
Neurofilament 200 labeling in the WT retina revealed axonal complexes that formed a tight mesh around the OPL (Figs. 7A–F). In Aipl1−/− retina, axonal complexes were poorly organized and became sparse as retinal degeneration proceeded. Sprouting of axonal branches of horizontal cells was observed at P14 (Fig. 7G). At P30, prolific extension of thin axonal branches toward the INL and to the border of the IPL was observed (Figs. 7I–K). At P100 and P150, axonal complexes were sparse with uneven, large holes formed by their loose processes in the OPL (Figs. 7K, 7L). To confirm that the sprouting of processes from horizontal cells were axonal complexes, we performed double labeling for CB and neurofilament in P30 Aipl1−/− retina. The sprouts stained with both antibodies, indicating that they originated from axons and not from dendrites (Supplementary Fig. S5). Interestingly, we also saw sprouting of optic nerve fiber from the ganglion cell layer toward the inner nuclear layer (INL) and IPL (Supplementary Fig. S5). Based on our immunostaining results, we conclude that a dramatic change occurs in horizontal cell axons and dendrites in the retina lacking Aipl1.
Figure 7.

Sprouting of horizontal cell axons in response to photoreceptor degeneration. Horizontal cell axon morphology was assessed at indicated ages using an antibody against neurofilament 200 in WT (A–F) and Aipl1−/− (G–L) retina. Arrow points to sprouting of horizontal cell axonal branches in the Aipl1−/− retina at P14 (G). Sprouting of horizontal cell axons was extensive at P30 to P60 ([I, J], arrow). At P100 to P150, arrowhead indicates axonal complexes that are sparse with uneven and large gaps in NF-200 staining pattern (K, L). Scale bar: 20 μm.
Reactive Changes in Müller Glial Cells and the Formation of the Glial Seal
Müller cells are radial glial cells that span the entire depth of the neural retina. Müller cell bodies are in the INL and project processes to the outer limiting membrane (OLM) and to the inner limiting membrane (ILM).43 Retinal Müller glial cells are known to undergo reactive changes, including gliosis in various retinal diseases. Müller cell reactivity was estimated from GS and GFAP expression in WT and Aipl1−/− animals.
In WT retina, GS antibodies label the entire Müller glial cell including the soma, processes, and end feet at the inner and outer limiting membranes (Figs. 8A–F). Glutamine synthetase staining revealed that Müller cell bodies were organized uniformly in the middle of the INL (Fig. 8A). In the Aipl1−/− retina, increased GS staining was observed in OLM starting from P14 (Fig. 8G). As photoreceptor degeneration progressed in the Aipl1−/− retina (P14–P30), the Müller glial cell processes in the outer retina started collapsing and forming a thick layer between the ONL and OPL called the fibrotic glial seal layer (Fig. 8I). The cell bodies were distributed unevenly and translocated to the outermost region of the INL in the Aipl1−/− retina after photoreceptor degeneration (Figs. 8J–L).
Figure 8.

Formation of Müller glial seal in the Aipl1−/− retina. Müller cells in the WT (A–F) and Aipl1−/− (G–L) retina were identified with an antibody against glutamine synthetase (GS). Arrows indicate glial seal formation in the Aipl1−/− retina at P30 (I). Uneven distribution of GS-stained cell bodies is indicated at P60 to P150 in the Aipl1−/− retina (J–L). Scale bar: 20 μm.
Glial fibrillary acidic protein, a marker for gliosis, was used to confirm the gliotic changes observed in the Aipl1−/− retina. In the WT retina, GFAP expression was found in astrocytes in the nerve fiber layer NFL; Figs. 9A–F). In contrast, in the Aipl1−/− retina, GFAP labeling progressed extensively toward the OPL at all ages tested (Figs. 9G–L). At P100 to P150, GFAP labeling regressed in the Aipl1−/− retina (Fig. 9L). Overall, our results demonstrate extensive gliotic changes in response to rapid photoreceptor degeneration.
Figure 9.

Activation of Müller glia in the degenerating retina. Müller glial cell morphology and activation were assessed at indicated ages using an antibody against glial fibrillary acidic protein (GFAP; green) in WT (A–F) and Aipl1−/− (G–L) retina. In Aipl1−/− retina, GFAP immunoreactivity is found from nerve fiber layer to the outer retina. DAPI nuclear staining is shown in blue. Scale bar: 20 μm.
Morphology of Amacrine and Ganglion Cells
Amacrine cells are interneurons present in the inner retina. Amacrine and ganglion cell morphology was estimated by staining WT and Aipl1−/− retina with calretinin antibody. In WT retina, antibody against calretinin stained amacrine cells, the synaptic layer of the IPL, displaced amacrine cells, and the ganglion cell layer (Figs. 10A–F). The general morphology and immunoreactive pattern of calretinin-labeled amacrine and ganglion cells in the retina from Aipl1−/− animals (at least up to the age of 5 months) were similar to observations in control WT retina (Figs. 10G–L). These results suggest that third-order neurons are unaffected by degeneration of photoreceptors.
Figure 10.

Morphology of amacrine cells in WT and Aipl1−/− retina. Amacrine cell morphology was assessed in WT (A–F) and Aipl1−/− (G–L) at indicated ages using an antibody against calretinin. Scale bar: 20 μm.
Discussion
A unique feature of vision among different sensory modalities is the extensive processing of stimuli that occurs in the retina prior to signal transmission to the visual cortex. This feature can be exploited to restore visual perception by imparting light sensitivity to downstream neurons when photoreceptor cells degenerate.7,45 However, the downstream neurons are dynamic and remodel in response to loss of photoreceptor cells, and thus may limit our ability to successfully restore vision. In order to address this limitation, we characterized the nature of remodeling in the retina of an animal model for AIPL1-LCA. We showed major alteration of inner retinal neurons in animals lacking Aipl1 as a consequence of both rod and cone photoreceptor loss. In particular, we observed improper development of synaptic connections, pruning of bipolar dendrites, and sprouting of horizontal cell processes during the course of remodeling. Müller glial cells underwent extensive gliosis with radial processes extending throughout the entire retina and formed a thick glial seal over the INL. The aberrant organization of neural retina observed in Aipl1−/− is likely due to a combination of defective development and remodeling in response to photoreceptor degeneration.
Remodeling of retinal neurons occurs in animal models exhibiting retinal degeneration and in humans afflicted with blinding diseases.5,7,9–11,18–20,46 Three distinct stages have been identified during the process of retinal remodeling.4 The initial phase is characterized by primary insult to photoreceptors leading to a stress in rod and/or cone photoreceptor cells. Phase II is a result of the earlier events ultimately leading to photoreceptor cell death. During these phases, the cells in the inner retina remain largely intact, although gliosis results as a consequence of photoreceptor cell death accompanied by retraction of bipolar cell dendrites. In phase III, extensive remodeling of the inner layer of retinal cells leads to morphological alteration of neural dendrites, rewiring of neural processes, and glial scars surrounding the retinal layers.4,7,46
Aipl1 protein expression can be detected in murine tissues starting at postnatal day 2 (data not shown). Early expression of AIPL1 was also noted in primate retina including that of humans.47 The role for Aipl1 in stability of PDE6, a protein crucial for maintenance and function of adult photoreceptors, is established.22,28 However, the need for Aipl1 during early development of photoreceptors is still not clear. Irrespective of the role for Aipl1, the insult to photoreceptors starts early in Aipl1−/− animals. Prior to photoreceptor degeneration at P8, we found that postsynaptic bipolar markers (mGluR6 and TRPM1) were not expressed at normal levels, implying that synapses did not develop normally in retina lacking Aipl1. These findings are in contrast to our observation in rd1 animals with similar rate of rod photoreceptor degeneration. In rd1 animals, at P8, the level of postsynaptic bipolar marker mGluR6 is similar to that in WT animals (Supplementary Fig. S6). In addition, previous studies show no changes in mGluR6 levels until P10 in rd1 animals.13 It appears that synaptogenesis is arrested at an early age, and synapses between rod and cone terminals with second-order neurons are not properly formed in Aipl1−/− animals. Further, ultrastructural studies are needed to confirm the observed synaptic abnormalities in our animal model.
As photoreceptor degeneration proceeded from P9 to P14, the synaptic machinery was severely affected. Metabotropic glutamate receptor was mislocalized to axons and the cell bodies of RBC (Fig. 11). Dramatic reduction in the expression of synaptic markers, such as VGLUT1 and SYP, was observed in the OPL at P18 (Fig. 11). Parallel to the synaptic changes in the OPL, RBC retracted their dendrites. In addition, atrophy of horizontal cell dendrites was observed as early as P14 to P18 (Fig. 11).
Figure 11.

A scheme summarizing changes observed in the Aipl1−/− retina. See text for details. PR, photoreceptors; CBC, cone bipolar cell; HC, horizontal cell.
In comparison to the OPL, no changes were found in the IPL at an early age. However, after photoreceptor degeneration is complete, P30 onward (phase III), we saw changes in VGLUT1 and SYP labeling at bipolar terminals in the IPL. Rod bipolar axonal endings in the IPL did not elaborate and appeared developmentally arrested (Fig. 4). This finding indicates that rod bipolar synaptogenesis is affected not only in the OPL of Aipl1−/− retina but also in the IPL. Interestingly, despite the rapid degeneration of rod and cone photoreceptors, the morphology of cone bipolar cells was well preserved. Dendritic trees connecting cone bipolar cell bodies were present until P30. However, after photoreceptor degeneration, discontinuity in the dendritic plexus was seen in the INL (Fig. 5). In addition, cone bipolar termini deteriorated progressively with time. The number of cone bipolar cells declined at a slower rate in comparison to their rod counterparts (Fig. 11). Our findings suggest an inherent difference in survival of bipolar cells in the absence of input from photoreceptor cells. The exact reason behind the longevity of cone bipolar cells is not clear. Another possibility is that variation in mechanisms behind rod (increased cyclic guanosine monophosphate [cGMP]) versus cone (reduced cGMP) cell death in Aipl1−/− animals may contribute to the difference in the survival of bipolar cells.48 Further studies are needed to decipher mechanisms that contribute to the difference in bipolar cell survival.49
We observed neurite extensions as early as P14 and extensive sprouting of axonal branches from the horizontal cell axons toward the INL and IPL at P30 (Fig. 7, Supplementary Fig. S5). Parallel experiments performed on rd1 animals and the RCS rat show similar results.13,50 Overexpression of neurofilaments and neurite extension are characteristic of axonal injury in several neurodegenerative disorders and occur in LCA and other retinal diseases.7,9,27 The significance of neurite extension from horizontal cells is unknown, but neurite outgrowth may alter existing synaptic connections and interfere with visual processing in the inner retina. In the Aipl1−/− retina, CB expression showed thinning of horizontal cell processes along with attenuation of CB staining at P100 (Fig. 6, Supplementary Fig. S4). The loss of dendrites and atrophy of axonal processes could be due to deprivation of neurotrophic factors released from photoreceptor cells or lack of interaction with photoreceptor synaptic termini.7
In agreement with previous observations on various retinal degenerative animal models, Müller glial cells in the Aipl1−/− retina showed extensive reactivity with GS and GFAP antibodies as early as P14 (Fig. 11). Glial seal formation was complete by P30 (Fig. 8). This might be due to structural compression of Müller cell processes in the outer retina.43 The Müller cell processes become hypertrophic in Aipl1−/− retinas compared to controls, suggesting an increase in new Müller cell processes (Fig. 9). We did not see major changes in the number and morphology of amacrine and ganglion cells, at least up to the age of 5 months. This observation is in line with previous studies on various animal models for photoreceptor degeneration.12,18–20 Lack of changes in the inner retinal circuit is likely due to their independent development without the need for input from photoreceptor cells.19
Similar to observations in the Aipl1−/− animal model, progressive but slower reorganization of bipolar and horizontal cells has been described in various models of photoreceptor degeneration, such as the P23H line 1 homozygous albino rat and the Royal College of Surgeons (RCS) rat.50,51 Changes in inner retinal neurons in response to photoreceptor cell degeneration have also been observed in AIPL1-LCA patients.10,27
Using virus-mediated gene therapy, we demonstrated an efficient vision rescue in the Aipl1−/− mouse model for LCA when treated at P10.29 While this strategy is suitable to restore vision prior to significant degeneration of photoreceptor cells, it will not be effective once the rapid degeneration of photoreceptor cells is in full swing. Alternate approaches such as stem cell transplantation, conversion of downstream cells to light-sensing neurons, or artificial retina implantation are likely to succeed in this scenario provided that strategies are devised to overcome the observed glial seal.7
Acknowledgments
We thank Ludwig Wagner, Shigetada Nakanishi, Takahisa Furukawa, and Albert Berrebi for their generous gifts of secretagogin, mGluR6, TRPM1, and calbindin antibodies. We also thank the members of the Ramamurthy laboratory and Peter Mathers for helpful discussion. All images were obtained at the West Virginia University Imaging Facility with the assistance of Karen Martin.
Supported by National Institutes of Health Grant RO1EY017035 (VR), a Knight Templar Eye Foundation grant (RKS), West Virginia Lions, Lions Club International Foundation, and an unrestricted challenge grant from Research to Prevent Blindness to West Virginia University.
Disclosure: R.K. Singh, None; S. Kolandaivelu, None; V. Ramamurthy, None
References
- 1. Masland RH. The fundamental plan of the retina. Nat Neurosci. 2001; 4: 877–886 [DOI] [PubMed] [Google Scholar]
- 2. Masland RH. Neuronal diversity in the retina. Curr Opin Neurobiol. 2001; 11: 431–436 [DOI] [PubMed] [Google Scholar]
- 3. Masland RH. The neuronal organization of the retina. Neuron. 2012; 76: 266–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jones BW, Kondo M, Terasaki H, Lin Y, McCall M, Marc RE. Retinal remodeling. Jpn J Ophthalmol. 2012; 56: 289–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones BW, Watt CB, Frederick JM, et al. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003; 464: 1–16 [DOI] [PubMed] [Google Scholar]
- 6. Marc RE, Jones BW, Anderson JR, et al. Neural reprogramming in retinal degeneration. Invest Ophthalmol Vis Sci. 2007; 48: 3364–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marc RE, Jones BW, Watt CB, Strettoi E. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003; 22: 607–655 [DOI] [PubMed] [Google Scholar]
- 8. Terzibasi E, Calamusa M, Novelli E, Domenici L, Strettoi E, Cellerino A. Age-dependent remodelling of retinal circuitry. Neurobiol Aging. 2009; 30: 819–828 [DOI] [PubMed] [Google Scholar]
- 9. Fariss RN, Li ZY, Milam AH. Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol. 2000; 129: 215–223 [DOI] [PubMed] [Google Scholar]
- 10. Jacobson SG, Cideciyan AV, Aleman TS, et al. Human retinal disease from AIPL1 gene mutations: foveal cone loss with minimal macular photoreceptors and rod function remaining. Invest Ophthalmol Vis Sci. 2011; 52: 70–79 [DOI] [PubMed] [Google Scholar]
- 11. Jacobson SG, Sumaroka A, Aleman TS, Cideciyan AV, Danciger M, Farber DB. Evidence for retinal remodelling in retinitis pigmentosa caused by PDE6B mutation. Br J Ophthalmol. 2007; 91: 699–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strettoi E, Pignatelli V, Rossi C, Porciatti V, Falsini B. Remodeling of second-order neurons in the retina of rd/rd mutant mice. Vision Res. 2003; 43: 867–877 [DOI] [PubMed] [Google Scholar]
- 13. Strettoi E, Porciatti V, Falsini B, Pignatelli V, Rossi C. Morphological and functional abnormalities in the inner retina of the rd/rd mouse. J Neurosci. 2002; 22: 5492–5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carter-Dawson LD, LaVail MM, Sidman RL. Differential effect of the rd mutation on rods and cones in the mouse retina. Invest Ophthalmol Vis Sci. 1978; 17: 489–498 [PubMed] [Google Scholar]
- 15. LaVail MM, Matthes MT, Yasumura D, Steinberg RH. Variability in rate of cone degeneration in the retinal degeneration (rd/rd) mouse. Exp Eye Res. 1997; 65: 45–50 [DOI] [PubMed] [Google Scholar]
- 16. Lin B, Masland RH, Strettoi E. Remodeling of cone photoreceptor cells after rod degeneration in rd mice. Exp Eye Res. 2009; 88: 589–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chua J, Fletcher EL, Kalloniatis M. Functional remodeling of glutamate receptors by inner retinal neurons occurs from an early stage of retinal degeneration. J Comp Neurol. 2009; 514: 473–491 [DOI] [PubMed] [Google Scholar]
- 18. Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J Comp Neurol. 2007; 500: 222–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuny S, Gaillard F, Mema SC, et al. Inner retina remodeling in a mouse model of stargardt-like macular dystrophy (STGD3). Invest Ophthalmol Vis Sci. 2010; 51: 2248–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pignatelli V, Cepko CL, Strettoi E. Inner retinal abnormalities in a mouse model of Leber's congenital amaurosis. J Comp Neurol. 2004; 469: 351–359 [DOI] [PubMed] [Google Scholar]
- 21. Pennesi ME, Stover NB, Stone EM, Chiang PW, Weleber RG. Residual electroretinograms in young Leber congenital amaurosis patients with mutations of AIPL1. Invest Ophthalmol Vis Sci. 2011; 52: 8166–8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramamurthy V, Niemi GA, Reh TA, Hurley JB. Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 2004; 101: 13897–13902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008; 27: 391–419 [DOI] [PubMed] [Google Scholar]
- 24. Dharmaraj S, Leroy BP, Sohocki MM, et al. The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch Ophthalmol. 2004; 122: 1029–1037 [DOI] [PubMed] [Google Scholar]
- 25. Sohocki MM, Perrault I, Leroy BP, et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol Genet Metab. 2000; 70: 142–150 [DOI] [PubMed] [Google Scholar]
- 26. Testa F, Surace EM, Rossi S, et al. Evaluation of Italian patients with leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest Ophthalmol Vis Sci. 2011; 52: 5618–5624 [DOI] [PubMed] [Google Scholar]
- 27. van der Spuy J, Munro PM, Luthert PJ, et al. Predominant rod photoreceptor degeneration in Leber congenital amaurosis. Mol Vis. 2005; 11: 542–553 [PubMed] [Google Scholar]
- 28. Kolandaivelu S, Huang J, Hurley JB, Ramamurthy V. AIPL1, a protein associated with childhood blindness, interacts with alpha-subunit of rod phosphodiesterase (PDE6) and is essential for its proper assembly. J Biol Chem. 2009; 284: 30853–30861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ku CA, Chiodo VA, Boye SL, et al. Gene therapy using self-complementary Y733F capsid mutant AAV2/8 restores vision in a model of early onset Leber congenital amaurosis. Hum Mol Genet. 2011; 20: 4569–4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun X, Pawlyk B, Xu X, et al. Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther. 2010; 17: 117–131 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Tan MH, Smith AJ, Pawlyk B, et al. Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum Mol Genet. 2009; 18: 2099–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kirschman LT, Kolandaivelu S, Frederick JM, et al. The Leber congenital amaurosis protein, AIPL1, is needed for the viability and functioning of cone photoreceptor cells. Hum Mol Genet. 2010; 19: 1076–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dumitrescu ON, Pucci FG, Wong KY, Berson DM. Ectopic retinal ON bipolar cell synapses in the OFF inner plexiform layer: contacts with dopaminergic amacrine cells and melanopsin ganglion cells. J Comp Neurol. 2009; 517: 226–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tom Dieck S, Altrock WD, Kessels MM, et al. Molecular dissection of the photoreceptor ribbon synapse: physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol. 2005; 168: 825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koike C, Obara T, Uriu Y, et al. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc Natl Acad Sci U S A. 2010; 107: 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Omori Y, Araki F, Chaya T, et al. Presynaptic dystroglycan-pikachurin complex regulates the proper synaptic connection between retinal photoreceptor and bipolar cells. J Neurosci. 2012; 32: 6126–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cordeiro S, Guseva D, Wulfsen I, Bauer CK. Expression pattern of Kv11 (Ether a-go-go-related gene; erg) K+ channels in the mouse retina. PLoS ONE. 2011; 6: e29490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson J, Tian N, Caywood MS, Reimer RJ, Edwards RH, Copenhagen DR. Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. J Neurosci. 2003; 23: 518–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Melone M, Burette A, Weinberg RJ. Light microscopic identification and immunocytochemical characterization of glutamatergic synapses in brain sections. J Comp Neurol. 2005; 492: 495–509 [DOI] [PubMed] [Google Scholar]
- 40. Phillips MJ, Otteson DC, Sherry DM. Progression of neuronal and synaptic remodeling in the rd10 mouse model of retinitis pigmentosa. J Comp Neurol. 2010; 518: 2071–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haverkamp S, Wassle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000; 424: 1–23 [PubMed] [Google Scholar]
- 42. Puthussery T, Gayet-Primo J, Taylor WR. Localization of the calcium-binding protein secretagogin in cone bipolar cells of the mammalian retina. J Comp Neurol. 2010; 518: 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chua J, Nivison-Smith L, Fletcher EL, Trenholm S, Awatramani GB, Kalloniatis M. Early remodeling of Muller cells in the rd/rd mouse model of retinal dystrophy. J Comp Neurol. 2013; 521: 2439–2453 [DOI] [PubMed] [Google Scholar]
- 44. Peichl L, Gonzalez-Soriano J. Morphological types of horizontal cell in rodent retinae: a comparison of rat, mouse, gerbil, and guinea pig. Vis Neurosci. 1994; 11: 501–517 [DOI] [PubMed] [Google Scholar]
- 45. Caporale N, Kolstad KD, Lee T, et al. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011; 19: 1212–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones BW, Marc RE. Retinal remodeling during retinal degeneration. Exp Eye Res. 2005; 81: 123–137 [DOI] [PubMed] [Google Scholar]
- 47. Hendrickson A, Bumsted-O'Brien K, Natoli R, Ramamurthy V, Possin D, Provis J. Rod photoreceptor differentiation in fetal and infant human retina. Exp Eye Res. 2008; 87: 415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kolandaivelu S, Singh RK, Ramamurthy V. AIPL1, a protein linked to blindness, is essential for the stability of enzymes mediating cGMP metabolism in cone photoreceptor cells. Hum Mol Genet. 2013; 23: 1002–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen M, Wang K, Lin B. Development and degeneration of cone bipolar cells are independent of cone photoreceptors in a mouse model of retinitis pigmentosa. PLoS ONE. 2012; 7: e44036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cuenca N, Pinilla I, Sauve Y, Lund R. Early changes in synaptic connectivity following progressive photoreceptor degeneration in RCS rats. Eur J Neurosci. 2005; 22: 1057–1072 [DOI] [PubMed] [Google Scholar]
- 51. Cuenca N, Pinilla I, Sauve Y, Lu B, Wang S, Lund RD. Regressive and reactive changes in the connectivity patterns of rod and cone pathways of P23H transgenic rat retina. Neuroscience. 2004; 127: 301–317 [DOI] [PubMed] [Google Scholar]