

Abstract
Psychosis occurs in 40–60% of Alzheimer's disease (AD) subjects, is heritable, and indicates amore rapidly progressive disease phenotype. Neuroimaging and postmortem evidence support an exaggerated prefrontal cortical synaptic deficit in AD with psychosis. Microtubule-associated protein tau is a key mediator of amyloid-β-induced synaptotoxicity in AD, and differential mechanisms of progressive intraneuronal phospho-tau accumulation and interneuronal spread of tau aggregates have recently been described. We hypothesized that psychosis in AD would be associated with greater intraneuronal concentration of phospho-tau and greater spread of tau aggregates in prefrontal cortex. We therefore evaluated prefrontal cortex phospho-tau in a cohort of 45 AD cases with and without psychosis. Intraneuronal phospho-tau concentration was higher in subjects with psychosis, while a measure of phospho-tau spread, volume fraction, was not. Across groups both measures were associated with lower scores on the Mini-Mental State Examination and Digit Span Backwards test. These novel findings indicate that tau phosphorylation may be accelerated in AD with psychosis, indicating a more dynamic, exaggerated pathology in AD with psychosis.
Keywords: Alzheimer's disease, Braak stage, Mini-Mental State Examination, psychosis, tau
Introduction
Alzheimer's disease (AD) is a common dementia characterized by insidious decline in cognition and function. The presence of psychotic symptoms in AD (AD+P) is associated with worse cognitive impairment, and portends a more severe trajectory of decline and greater likelihood of institutionalization [1, 2]. Psychotic symptoms are likely not just an epiphenomenon of more advanced AD, as they occur in only 40–60% of subjects with AD [3–6] and aggregate within families [7, 8]. The estimated heritability of any psychotic symptom in AD is 30%, and increases to 61% for multiple and/or recurrent psychotic symptoms [9]. These findings all suggest a distinct neurobiological basis for the development of AD+P.
Evidence is accumulating that cortical synapses in AD+P are more vulnerable to the pathological factors of AD, which contributes to the development of psychotic symptoms, or at least more pronounced cognitive impairments [10, 11]. This is congruent with findings that synapse loss is the strongest correlate of cognitive decline in AD [12–14]. Support for this hypothesis includes findings from neuroimaging studies, which have pointed to exaggerated neocortical dysfunction in AD+P (reviewed in [11]). SPECT studies comparing AD+P to AD without psychosis (AD−P) have described hypoperfusion in frontal, dorsolateral prefrontal cortex (DLPFC), and parietal regions in AD+P [15–17]. Positron emission tomography studies have found reduced frontal lobe metabolism in AD+P [18, 19], and Bruen et al. identified reduced frontal and parietal gray matter density in AD+P, but no difference in medial temporal atrophy compared to AD−P [20].
Support for this synaptic vulnerability hypothesis of AD+P also stems from postmortem studies, which have correspondingly described worse neocortical pathology in AD+P [21, 22]. Recently we determined that kalirin, a protein essential for cortical dendritic spine maintenance and growth [23], is reduced in AD+P DLPFC [22]. An earlier study in our laboratory found increased membrane breakdown products in several cortical regions, including DLPFC but not medial temporal lobe, in a comparison of AD+P and AD−P subjects [21]. Thus it appears that in AD+P at least the postsynaptic components of cortical synapses are less sustainable, and loss of cortical synapses is accelerated.
The classical view of AD pathology, which focused on the aggregation of amyloid-β (Aβ) into neuritic plaques and the aggregation of microtubule associated protein tau into neurofibrillary tangles (NFTs), has evolved into a more dynamic view of how Aβ and tau contribute to synaptic impairments and cognitive decline. Studies are highlighting that soluble oligomeric forms of Aβ are primary contributors to synaptic dysfunction in AD [24, 25]. In fact, neuritic plaques may serve as reservoirs of Aβ oligomers [26], existing in equilibrium with the surrounding soluble Aβ species. Tau may also be essential to the cascade of synaptic pathology induced by soluble Aβ, as tau knockout transgenic models fail to show Aβ-induced neurite degeneration [27], Aβ-induced impairment of long-term potentiation [28], or Aβ-induced cognitive deficits [29].
It has been known for some time that hyperphosphorylated tau (pTau) deposits in the AD brain in a characteristic regional progression associated with disease severity [30]. Tau is also phosphorylated in AD in a progressive manner, with different phosphorylation sites corresponding to different degrees of cytopathology, from intraneuronal tau inclusions to ghost tangles [31]. Recent work in animal models with localized high levels of expression of mutant tau found that regions monosynaptically connected to the site of expression subsequently demonstrated pathologic tau, indicating that misfolded and/or pTau may actually spread across synapses [32, 33]. This suggests that the hierarchical pattern of tau deposition may reflect direct interneuronal spread of tau aggregates, although such spreading has not been shown to occur in humans. These important findings underscore the possibility that the regional spread of pTau and its intraneuronal accumulation are distinct phenomena that could contribute in different ways to disease phenotype.
In keeping with the view of soluble Aβ as a key driver and pTau as a mediator of synaptic toxicity in AD, we found that the soluble Aβ1-42/Aβ1-40 ratio is increased in AD+P DLPFC [22]. Two studies of tau, that did not differentiate phosphorylation from regional spread, similarly identified differences. Farber and colleagues found a greater area density of NFTs in AD+P in multiple neocortical regions compared to AD−P [5]. Mukaetova-Ladinska and colleagues found increased paired helical filament tau protein (PHF tau) in cortical gray matter extracts of AD+P subjects [34]. These findings all suggest that the effects of soluble Aβ and pathological tau in the progression of AD may be accelerated in individuals who develop psychosis during AD.
In the present study, we examined two complementary measures of pTau in AD+P and AD−P subjects: mean immunoreactive fluorescence intensity (MI), a measure of local pTau concentration, and volume fraction (VV, the fraction of the gray matter consisting of pTau), a measure of the extent of local spread of pTau pathology. VV can be understood to result from increased spread of pTau within individual neurons (which is likely to be closely related to MI) and/or from development of pTau pathology in additional neurons in the region. We focused our investigations on DLPFC, a region implicated as affected in AD+P by neuroimaging studies [11], by evidence of greater impairments of executive function and working memory [35, 36], and in which we previously identified altered Aβ1-42/Aβ1-40 ratio and kalirin levels [22]. We hypothesized that both VV and MI would be associated with Braak stage, and that these measures would be higher in AD+P DLPFC. Greater VV and MI of pTau in the DLPFC could lead to the more severe cognitive dysfunction and rapid decline associated with the development of psychosis in AD.
Material and Methods
Subjects
Forty-five subjects (Table 1) underwent neurologic, neuropsychologic, and psychiatric diagnostic evaluations as part of their participation in the Clinical Core of the Alzheimer Disease Research Center (ADRC) at the University of Pittsburgh [3, 22, 37]. Psychiatrists with specialized training in geriatric psychiatry conducted semi-structured examinations, and the patient, primary caregiver, and all other available informants were interviewed. As part of the semi-structured examinations, the presence or absence of hallucinations and delusions was determined and rated on the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Behavioral Rating Scale [38]. Hallucinations were defined as sensory perceptions with no basis in reality. Delusions were defined as a false belief based on incorrect inference about reality and not attributable to membership in a social or cultural group. Psychosis was the presence of hallucinations or delusions at any visit [21, 22]. Subjects with psychosis occurring solely during an episode of delirium were not rated as AD+P [3]. No patient had a history of schizophrenia, schizoaffective disorder, or other idiopathic psychosis. Dates of death ranged from January 2000 to December 2003.
Table 1. Descriptive information of the subjects involved in the study.
| Variable | AD−P n = 19 |
AD+P; n = 26 |
p |
|---|---|---|---|
| Age, y | 80.4 ± 8.9 | 81.4 ± 8.1 | 0.691 |
| Age of onset, y | 69.7 ± 10.0 | 71.2 ± 9.4 | 0.629 |
| Duration of illness, y | 10.6 ± 4.5 | 10.2 ± 5.1 | 0.813 |
| Gender | 0.436 | ||
| Male | 11 (58) | 12 (46) | |
| Female | 8 (42) | 14 (54) | |
| Postmortem interval, h | 4.6 ± 1.3 | 6.1 ± 2.4 | 0.018 |
| Lewy body stage | 0.580 | ||
| Negative | 11 (58) | 11 (42) | |
| 0-6 | 4 (21) | 7 (27) | |
| 7-10 | 4 (21) | 8 (31) | |
| Braak | 0.276 | ||
| III | 4 (21) | 1 (4) | |
| IV | 3 (16) | 3 (12) | |
| V | 4 (21) | 6 (23) | |
| VI | 8 (42) | 16 (62) | |
| MMSE | 14.6 ± 7.2 | 9.9 ± 6.4 | 0.026 |
| DSB | 2.26 ± 2.1 | 1.58 ± 1.9 | 0.259 |
Mean values ± SD or number of subjects with percentage of total in parentheses. AD−P, Alzheimer's disease without psychosis; AD+P, Alzheimer's disease with psychosis; MMSE, Mini-Mental State Examination; DSB, Digit Span Backwards test.
A neuropathological diagnosis of AD was made at postmortem examination [39]. At the time of brain removal, the postmortem interval in hours was recorded, and the brain was removed intact, examined grossly, and divided in the midsagittal plane. The left hemibrain was immersion fixed in 10% buffered formalin for at least one week, sectioned into 1.0–2.0 cm coronal slabs, and sampled according to CERAD protocol for neuropathological diagnosis [39]. Samples from the midfrontal cortex, cingulate gyrus and caudate nucleus, hippocampus at the level of the lateral geniculate nucleus, inferior parietal lobule, superior and middle temporal cortex, primary visual cortex, amygdala, and transentorhinal cortex were stained using the Bielschowsky technique according to the Yamamoto and Hirano modification [40]. Samples from amygdala, midbrain, pons, and medulla were immunostained for ubiquitin and α-synuclein, and Lewy body pathology was rated [41, 42]. If any of these sections were positive, sections from cortex, hippocampus, and remaining brainstem were also stained for α-synuclein. Neuritic plaques were semiquantiatively scored according to CERAD criteria [39]; plaque density per 100x microscopy field was rated as “none,” “sparse,” “moderate,” or “frequent,” with most subjects scored as “frequent”. Braak staging was done on Bielschowsky stained sections of mesial temporal lobe (hippocampus with entorhinal, transentorhinal, and inferior temporal lobe cortex), mid-frontal, inferior parietal, superior temporal gyri, and occipital lobe containing the primary visual cortex and adjacent cortex [30].
A single coronal block, ∼1.5 cm thick, at the level of the genu of the corpus callosum, was cut through the left hemibrain (or, in 10 subjects, the right hemibrain) at the time of autopsy. This block was placed in 4% paraformaldehyde at 4°C for 48 hours, after which fixation was terminated by transfer through a series of progressive sucrose gradients. The block was then stored in cryoprotective solution at −30°C until tissue sectioning (Fig. 1). The middle frontal gyrus was dissected from the block and 50 μm thick coronal sections were cut using a cryostat and stored in cryoprotectant solution at −30°C [43].
Fig. 1.
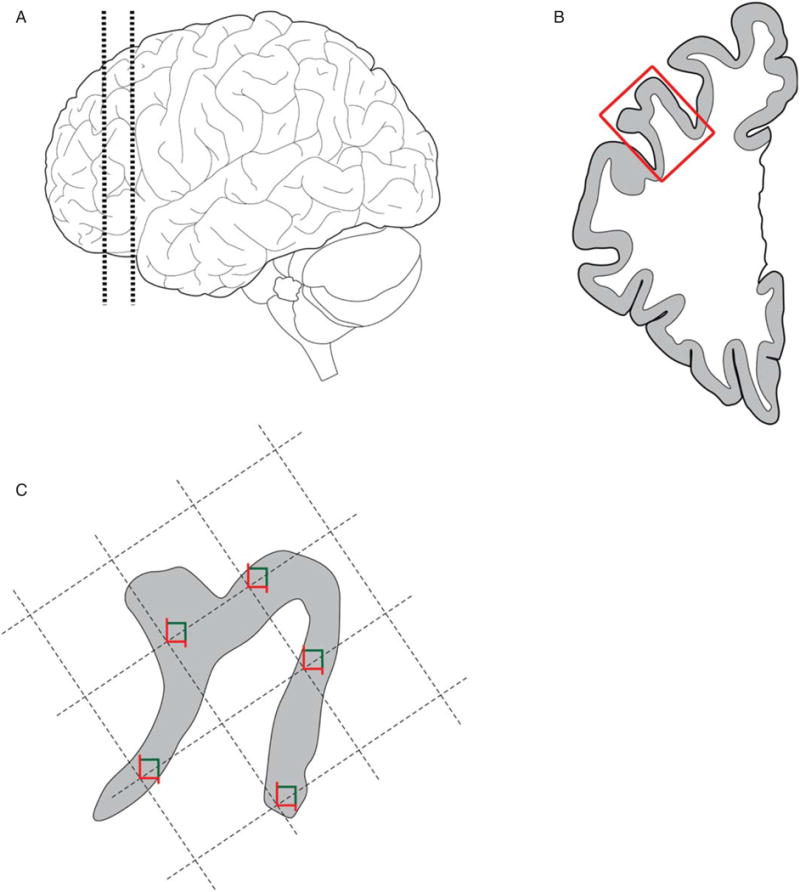
Tissue processing and sampling. A) 1.5 cm thick coronal block at the level of the genu of the corpus callosum was cut through the left hemibrain in most subjects. B) The middle frontal gyrus (box) was dissected from the block and cut into 50 μm thick sections. C) The gray matter mantle was outlined in StereoInvestigator and a random sampling grid was generated, with 5 sites per section.
Immunohistochemistry
For each subject, a section was randomly selected from the first 20 sections, and a second section was pulled with a separation of 3000 μm. To minimize effects of interassay variability, subjects were grouped into sets, stratified on psychosis presence. Subjects within sets were matched to the extent possible on age and Braak stage. Sections within sets were assayed together in the same wellplate for each run.
The AT8 antibody (Pierce, Rockford, IL, USA; product #MN1020) was used to label pTau; AT8 recognizes the pathological tau phosphorylation epitope of serine 199/serine 202/threonine 205 [44, 45]. Using a variation of a previously described methods [46], free-floating sections were rinsed in phosphate buffered saline (PBS) and incubated in 1% NaBH4 for 30 minutes to reduce autofluorescence. Following additional PBS rinses, sections were incubated in PBS containing 0.3% Triton X-100 for 30 minutes, followed by incubation in PBS containing 20% normal human serum, 20% normal goat serum, 1% bovine serum albumin (BSA), 0.1% lysine, and 0.1% glycine for 150 minutes. Sections were then incubated for 24 hours in PBS containing 5% normal human serum, 5% normal goat serum, 1% BSA, 0.1% lysine, and 0.1% glycine (incubation buffer), and a 1:1000 dilution of AT8 at 4°C. Sections were rinsed then incubated for 24 hours at 4°C in incubation buffer with a 1:500 dilution of goat anti-mouse secondary antibody conjugated to Alexa Fluor® 488 (Invitrogen, Carlsbad, CA, USA). Sections were rinsed, mounted on gelatincoated slides, dried, rehydrated, and coverslipped with Vectashield hard set mounting medium (Vector Laboratories, Burlingame, CA, USA) using #1.5 (0.17 mm) coverslips (Brain Research Laboratories, Newton, MA, USA).
Microscopy
Spinning disc confocal imaging was conducted using an Olympus BX51 WI upright microscope (Center Valley, PA, USA) equipped with an Olympus DSU, Olympus UPlanSApo 10X 0.40 N.A. air objective, and Olympus PlanAPO N 60X SC 1.40 N.A. oil immersion objective. Additional equipment included an ORCAR2 camera (Hamamatsu, Bridgewater, NJ, USA), MBF CX9000 front mounted digital camera (MicroBright-Field Inc., Natick, MA, USA), BioPrecision2 XYZ motorized stage with linear XYZ encoders (Ludl Electronic Products Ltd., Hawthorne, NY, USA), excitation and emission filter wheels (Ludl Electronic Products Ltd., Hawthorne, NY, USA), Sedat Quad 89000 filter set (Chroma Technology Corp., Bellows Falls, VT, USA), and a Lumen 220 metal halide lamp (Prior Scientific, Rockland, MA, USA). Software included Slidebook 5.27 (Intelligent Imaging Innovations Inc., Denver, CO, USA) and Stereo Investigator (Micro-BrightField Inc., Natick, MA, USA).
On each section, the entire mantle of the middle frontal gyrus grey matter was outlined using Stereo Investigator software. A sampling grid was generated, with grid size calculation based on the outlined grey matter area for the two sections from the individual, to target 10 sampling sites per individual (Fig. 1). The grid was randomly rotated over the contour, and sampling sites were identified at each intersection between the grid and the contour. At each site, Slidebook was used to collect 3-D image stacks consisting of a series of 2-D images of 512 × 512 pixels (3057.6 μm2) separated by 250 nm in the z-axis, and beginning 12 μm below the tissue surface closest to the coverglass.
Image acquisition was carried out with the following filters and exposure times: AT8 immunoreactivity was captured using 490/20 nm excitation, 525/36 nm emission with a 25% neutral density filter and exposure time of 300 ms; 645/30 nm excitation, 705/72 nm emission was used to record autofluorescence signal for removal from our analyses, with an exposure time of 1500 ms and no neutral density filter.
Image processing
Images were processed using Slidebook 5.27. Image stacks were deconvolved using the no neighbor (subtraction constant = 1 with no edge padding) and constrained iterative algorithms (maximum 20 iterations with 3-D frequency filtering). Object masks for AT8 immunoreactivity were generated on the no neighbor-deconvolved image using a variation of our iterative approach [47]. Briefly, the initial intensity threshold was set to 20 gray levels, and subsequent iterations were increased until a threshold value of 500 was reached. After each iteration, mask operations were used to combine object masks with minimum volumes of 20 voxels (Fig. 2). A mask was also generated for autofluorescence, and mask operations were used to subtract the autofluorescence mask from the AT8 mask (Fig. 3). This method of autofluorescence removal was piloted extensively for the current application. In a random subsample of imaging sites, the AT8 mask volume was reduced by only 1.5% after subtraction of the autofluorescense mask. The resultant mask was copied to the constrained iterative deconvolved image for quantification of AT8 fluorescence intensity levels.
Fig. 2.

Image processing and masking. A) Raw image capture in grayscale. B) Same image as a pseudocolor heatmap. C) Raw image was deconvolved using the no-neighbors deconvolution deblurring algorithm, which attenuates lower intensities in the image and preserves spatial fidelity. D) No-neighbors deconvolved image was masked using our iterative masking approach (white outlines). E) Raw image was deconvolved using the constrained iterative deconvolution algorithm, which accurately restores the signal source in the image using the point-spread function for our imaging system. F) Mask generated in D overlaid on the constrained iterative deconvolved image. Adjacent color bars reflect grayscale levels images captured at 60x magnification.
Fig. 3.
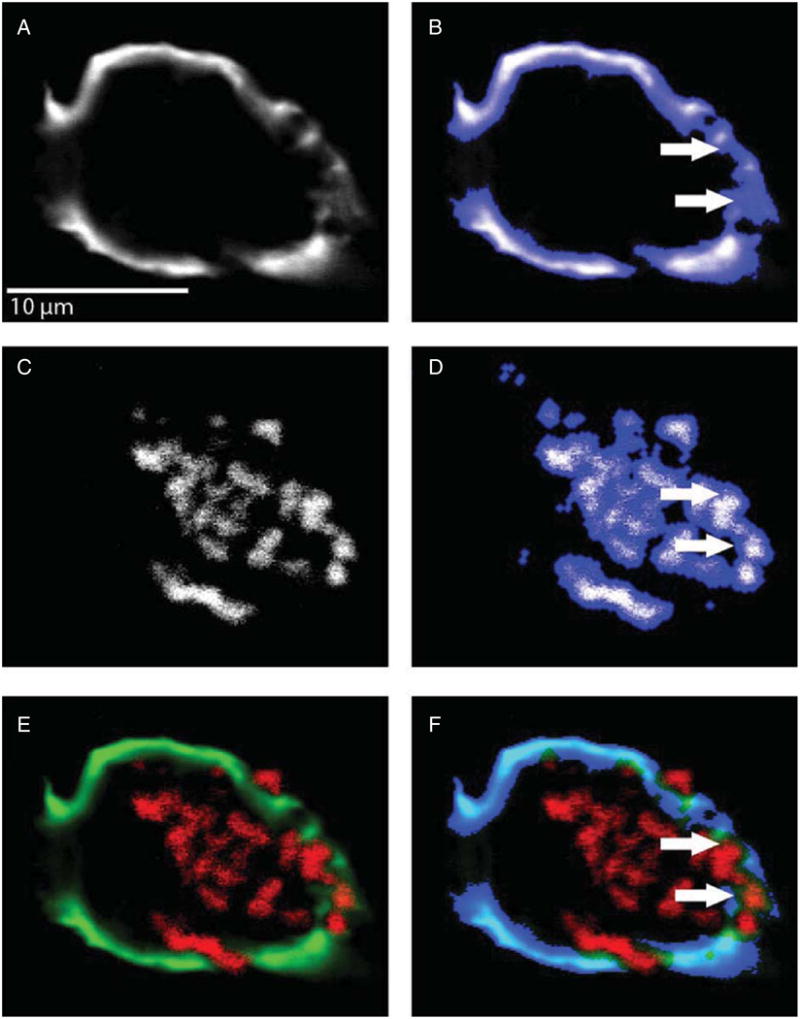
Removal of autofluorescence from the AT8 mask. A) A no-neighbors-deconvolved grayscale image of AT8 signal. B) The same image, shown overlaid by an interative intensity and morphologic mask. C) A no-neighbors-deconvolved grayscale image of intraneuronal lipofuscin accumulation. D) Lipofuscin, similarly overlaid by an interative intensity and morphologic mask. E) The combined image with AT8 in green and autofluorescence in red. F) The resulting mask of autofluorescence subtracted from the AT8 mask. Arrows highlight autofluorescence signal under the AT8 mask and show this signal has been eliminated from the final subtracted mask in F.
Image stacks were cropped to 492 × 492 pixels (2823.4 μm2) to provide a guard zone for edge artifacts, and a single plane was then selected 2.5 μm below the section surface in each of the deconvolved stacks. The VV was estimated using the mask objects of the no neighbor deconvolution within the selected plane. The ratio of the mask area to the selected plane area (AA, for a plane intersecting an object within a larger structure) is an estimator of VV, the fraction of the total structure consisting of the contained object [48], which in this case is the fraction of gray matter containing AT8 immunoreactive structures. The MI of AT8 immunoreactivity was extracted from the mask objects of the constrained iterative deconvolution within the selected plane.
Statistical analyses
Analysis of variance (ANOVA), t-tests, and χ2 tests were used to evaluate demographic, clinical, and pathologic variables between groups. The response variables for our main analyses were the AT8 VV and MI. Linear mixed models were employed, with psychosis, postmortem interval, and assay run as fixed effects, set assignment as a random effect, and sections within subject as repeated measures. To evaluate how DLPFC VV and MI related to different aspects of cognition, we examined the most recent scores of our subjects on the MMSE, a global measure of cognition that is consistently lower in AD+P relative to AD−P. We further evaluated whether DLPFC pathology may relate to Digit Span Backwards (DSB), a test of executive function that depends in part on DLPFC function. Analysis of covariance (ANCOVA) tests were used to examine the relationships between tertiles of Vv and MI and these two cognitive measures, with the time lag between most recent test and death as covariate. Post-hoc comparisons used the Tukey Honest Significant Difference test, for which only p values are reported.
All tests were 2-tailed with α = 0.05.
Results
We assessed how well-matched the groups (AD+P and AD−P) were on relevant clinical and pathologic variables. Groups did not differ with regard to gender or age. There was no difference with regard to Lewy body stage, neuritic plaque ratings, or Braak stage. Postmortem interval was different, but only slightly, with means of 4.58 hours for AD−P and 6.08 for AD+P. Overall use of antipsychotics was low (1 out of 19 in AD−P, 6 out of 26 in AD+P), and did not differ between the groups (Exact p = 0.111). Use of cognitive enhancers did not differ between groups (12 of 26 in AD+P, 9 of 19 in AD−P, χ21 = 0.007, p = 0.936).
Association with Braak stage
Our measures of VV and MI were significantly correlated with each other (r = 0.570, p < 0.001, Supplementary Fig. 1). VV was significantly correlated with Braak stage (rs = 0.758, p < 0.001), as was MI (rs = 0.491, p < 0.001) (Fig. 4). As expected, VV and MI did not differ due to α-synuclein pathology (F(1, 43) = 0.521, p = 0.597; F(1, 43) = 0.870, p = 0.427) because the groups were matched for Lewy body stage.
Fig. 4.

Association of VV and mean AT8 intensity with Braak stages. A) VV increased with Braak stage, but did not differ between AD+P and AD−P. B) Mean AT8 fluorescence intensity increased with Braak stages, and was consistently higher in AD+P.
AD+P
The VV was not significantly different between AD+P and AD−P (F(1, 31.984) = 3.225, p = 0.082). However, MI was 35.9% higher in AD+P (AD+P Est. Marginal Mean (SEM) 314.9 (22.2), AD−P 231.5 (24.9), F(1, 30.873) = 8.782, p = 0.006) (Figs. 4 and 5). These studies were conducted on two separate occasions, with one section per subject assayed, imaged, and processed in each experiment. In addition to our main analyses, where section was treated as a repeated measure, we also analyzed the results of each section assay independently to assess the reproducibility of our fluorescence imaging and analysis approach. The results were the same: VV was not significantly different between AD+P and AD−P in either experiment, but MI was significantly higher in AD+P in both experiments. Finally, because VV may in part depend on processes affecting MI, an interpretation supported by our finding that the two measures are correlated, we repeated our primary analysis of psychosis with VV, adding a fixed effect of MI. Once again, VV was not significantly different between AD+P and AD−P, and now without any evidence of a possible trend toward association (F(1, 40.580) = 0.190, p = 0.665).
Fig. 5.
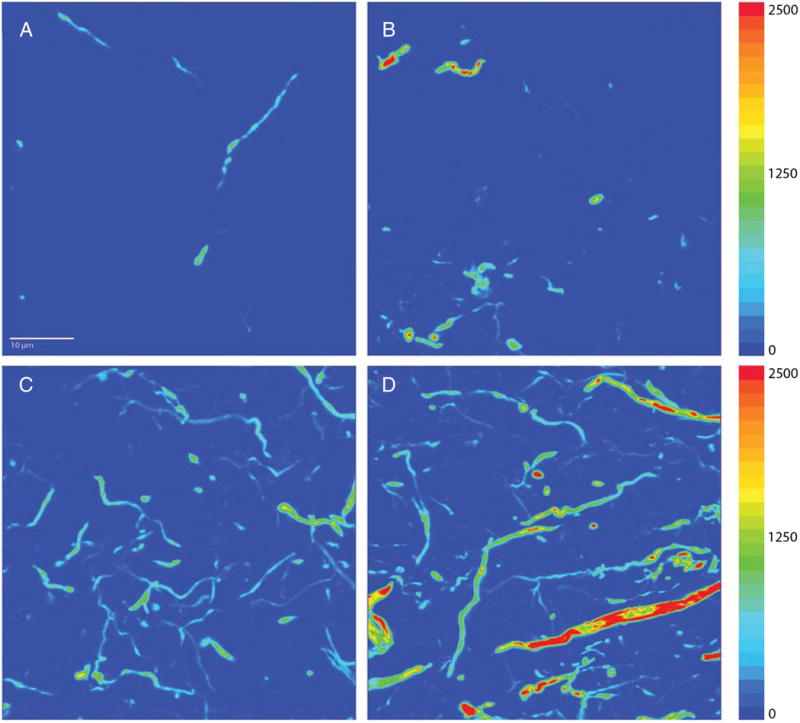
Representative images demonstrating the difference in mean AT8 intensity between AD−P (A, C) and AD+P (B, D) in subjects at Braak stages characterized by lower (stage IV; A, B) and higher (stage VI; C, D) degrees of pTau burden. These images were deconvolved using the no-neighbors algorithm. Adjacent color bars reflect grayscale levels.
We further evaluated the association of VV and MI with psychosis after accounting for other possible confounding variables. Entering Lewy Body Stage and the interaction of Lewy Body stage with Psychosis as fixed effects in our model did not alter the results. AD+P remained significantly associated with MI (F(1, 27.399) = 4.697, p = 0.039), but not with VV (F(1, 28.925) = 2.774, p = 0.107). Moreover, VV and MI were not significantly associated with Lewy Body Stage (MI: (F(2, 29.251) = 0.709, p = 0.5), VV: (F(2, 30.839) = 0.511, p = 0.605), nor with the interaction of Lewy Body Stage with Psychosis (MI: (F(2, 31.020) = 0.1.816, p = 0.180), VV: (F(2, 32.220) = 0.345, p = 0.711). Similarly, results were not changed after entering the most recent MMSE score and the time from MMSE testing to death as fixed effects in our model. AD+P remained significantly associated with MI (F(1, 27.853) = 8.362, p = 0.007), but not with VV (F(1, 28.317) = 2.241, p = 0.145).
Association with cognition
MMSE
The most recent available MMSE score was influenced by VV (F(2, 42) = 3.799, p = 0.030), with scores in subjects in the highest VV tertile 43.3% lower than scores in subjects in the lowest VV tertile (p = 0.024). MI also significantly influenced MMSE (F(2, 42) = 5.803, p = 0.006): compared to subjects in the lowest MI tertile, scores in subjects in the middle MI tertile were 41.7% lower (p = 0.015), and 42.1% lower in subjects in the highest MI tertile (p = 0.014) (Fig. 6). In both diagnostic groups, MMSE score was strongly correlated with Braak stage. In AD+P, rs = −0.389, p = 0.049. In AD−P, rs = −0.482, p = 0.037.
Fig. 6.

Measures of cognitive function by VV and mean AT8 intensity tertiles. A) Scores on the MMSE were significantly higher in the lowest VV tertile compared to the highest, and in the lowest mean intensity tertile compared to the middle and highest tertiles. B) Scores on the Digit Span Backwards test were significantly higher in the lowest VV tertile compared to the highest, and in the lowest mean intensity tertile compared to the middle and highest tertiles. *p < 0.05.
DSB
DSB test scores were influenced by VV (F(2, 42) = 5.507, p = 0.008), with scores in subjects in the highest VV tertile 75% lower than scores in subjects in the lowest VV tertile (p = 0.005). MI also influenced DSB score (F(2, 42) = 4.789, p = 0.013), with scores in subjects in the middle MI tertile 56.6% lower than scores in subjects in the lowest MI tertile (p = 0.036), and scores in subjects in the highest MI tertile were 60.8% lower than those in subjects in lowest tertile (p = 0.022) (Fig. 6).
Discussion
In this study we examined VV, as an index of the extent of local pTau spread, and MI, as an indicator of local intraneuronal concentration of pTau, in the DLPFC to determine how they relate to psychosis, Braak stage, and cognition. We found that MI of pTau, but not VV, was higher in AD+P. Both pTau VV and MI were strongly correlated with Braak stage, with VV more so than MI. Both VV and MI were associated with MMSE and DSB tests.
Tau phosphorylation is accelerated in AD+P DLPFC
The elevated DLPFC pTau MI we found in AD+P compared to AD−P subjects matched for duration of disease, neuritic plaque ratings, and Braak stage, points to an accelerated process of tau phosphorylation in AD+P that may be distinct from the propagation of pTau throughout the brain. The increased MI could reflect enhanced kinase and/or inhibited phosphatase activity, and as a result more of a phosphorylational drive, which occurs as an accompaniment to regional spread. While our findings are consistent with the possibility that greater intraneuronal pTau concentration may contribute to the risk of psychosis in AD, we also found that pTau concentrations in low Braak stage AD+P subjects were not above levels observed in AD−P subjects at later Braak stages. This latter observation suggests that either greater intraneuronal pTau concentration is not itself sufficient to cause psychosis in AD, or may be a correlate of the underlying process that leads to the more severe AD+P phenotype.
There have been prior findings of elevated numerical density and protein levels of tau aggregates reported in AD+P, although they are somewhat inconsistent. Farber et al. found that NFT density, measured as average number per square millimeter, is higher across the neocortex in AD+P including in DLPFC [5]. While providing evidence of increased tau pathology in AD+P, their approach, using a modified Bielschowsky method, evaluated fibrillar tau spread and not pTau directly. Although NFT area density measurements generate counts weighted by object volume [48], and therefore can be seen as a measure of tau spread by confounding NFT number and size, they do not account for the large contribution of tau aggregates confined to neurites. Mukaetova-Ladinska and colleagues presented similar findings with increased PHF-tau in AD+P using an enzyme-linked immunosorbent assay [34]. However, their use of total cortical extracts yields a measure that also confounds spread and PHF-tau concentration. In contrast, previous work in our lab using semiquantitative ratings of regional NFTs found no association with psychosis [3], a discrepancy that could be due to differences in methodology or subjects. It could also reflect the difficulty in detecting a small elevation in spread of pTau aggregates in AD+P. The present study clearly differentiates pTau spread and intraneuronal pTau concentration. In agreement with the majority of previous studies, we found evidence for increased pTau pathology in AD+P, but this was limited to greater cortical tau phosphorylation and not greater pTau spread.
Since the strongest correlate of cognitive impairment in AD is the loss of synapses [12–14], the more severe trajectory of cognitive decline in AD+P could reflect an exaggerated disruption of cortical synapses. There is neuroimaging and biochemical evidence to support this model [18, 20, 21]. Excess synaptic disruption in AD+P could stem from more severe pathological processes, whether by increased activity of Aβ and/or its downstream effectors, due to genetic factors or otherwise. Indeed the increased soluble Aβ1-42/Aβ1-40 ratio in AD+P cortex [22] may thereby promote greater synaptotoxicity [49]. However, while it is appreciated that Aβ drives missorting and phosphorylation of tau [50] and that reducing tau mitigates effects of Aβ [29], it is unclear how the ratio of Aβ isoforms may influence tau pathology.
A recent genome-wide association study identified serine/threonine kinase 11 (STK11), also known as liver kinase B1 (LKB1), as a possible candidate gene in AD+P [51]. LKB1 signaling triggers phosphorylation of tau [52], and AβPP overexpression promotes tau phosphorylation in an LKB1-dependent manner [53]. These findings may provide a mechanism downstream of Aβ that could lead to exaggerated tau phosphorylation in AD+P through LKB1. However, whether LKB1 expression is altered in AD+P is not currently known, and it is not known whether other pathways that phosphorylate tau downstream of Aβ [54] may be altered in AD+P. Certain tau phosphorylation sites that promote detachment from microtubules, such as Ser214, may actually inhibit the assembly of tau into paired helical filaments when phosphorylated [55]. Any slowed or disrupted progression of pTau to argyrophilic inclusions would likely increase pTau concentration. Aside from mechanisms of phosphorylation, the reduction of microtubules in AD independent of tau [56], along with detachment and missorting of tau [50], could influence the relative concentration of pTau.
DLPFC pTau is associated with Braak stage and related to cognitive decline
The strong correlations of Braak stage with our measures of DLPFC pTau, both AT8 MI and VV, support the validity of these measures as they relate to progression of disease. While DLPFC VV was more strongly associated with Braak stage, which shows that localized DLPFC pTau VV continues to increase as tau pathology spreads throughout the brain, intraneuronal pTau in DLPFC also increases in concentration as Braak stage progresses. We found DLPFC pTau in Braak stages III and IV cases (Fig. 5), stages which precede the appearance of neocortical NFTs [30]. The presence of these immunoreactive threads in the DLPFC, which are likely non-argyrophilic pTau [57], demonstrate pTau is spreading regionally even early in disease progression. We observed a clear distinction between Braak stages V and VI in the severity of cortical tau pathology, at least with regard to the DLPFC. Although these stages both represent neocortical infiltration, there may be regional differences in magnitude of tau load between these two stages. This is consistent with other data indicating substantial differences in degree of cognitive impairments between individuals at Braak stage V and VI [58]. Although neuropathologic diagnostic criteria emphasize combining these two stages [59, 60], for more subtle clinical pathologic correlations they are probably best analyzed separately.
Since MMSE score is strongly associated with the regional appearance of neurofibrillary pathology, both by discrete NFT counts [61] and by Braak stage stratification [58], we were interested in how our measures of DLPFC pTau might relate to global and regionally specific cognition. The MMSE score encapsulates a measure of overall cognitive function [62], and both VV and MI affected the scores. This finding is not surprising since it is well established that the MMSE score declines as the disease progresses [63]. Similarly, the DSB test, a measure of working memory or executive function, was also correlated with both pTau VV and MI. These findings provide a connection between tau pathology and cognitive decline, and show a relationship of DLPFC pTau with both global and executive domains of cognition.
Limitations
We chose to focus our observations on the DLPFC, based on multiple lines of evidence that it was likely to be affected in AD+P. However, our findings should not be interpreted as indicating that exaggerated tau pathology in AD+P is selective for DLPFC, as there is evidence for exaggerated tau pathology in AD+P across the neocortex [5, 34]. Similarly, psychotic symptoms are likely to reflect disturbances across a neocortical network, and not result solely from DLPFC pathology. Although postmortem interval was slightly longer in the AD+P group, the AT8 epitope undergoes a rapid initial dephosphorylation during the postmortem interval [64], thus increased MI could be even more pronounced in AD+P than what we report. While a direct, linear relationship specifically between MI and pTau concentration has not been firmly established, we have previously shown that detected fluorescence intensity is directly related to the intensity of fluorescent microspheres embedded in thick sections [47], and can be used to accurately measure protein concentration [47, 65, 66]. Since our findings were based on AT8 immunoreactivity, and pathological phosphorylation of tau obviously occurs at sites beyond those assayed by AT8, other tau phosphorylation states and conformations could be differentially associated with AD+P. Additionally, as time progresses and tau deposits transition to late extracellular ghost tangles, AT8 immunoreactivity diminishes [67–70], potentially affecting our measure of MI. Several factors may contribute to VV of pTau. For example, increased intraneuronal generation of pTau as described above for MI could lead to increased intraneuronal pTau volume and, consequently, increased VV. Alternatively, VV may increase due to development of pTau pathology in additional neurons in the region. This latter process could result from spread of an upstream source of pTau (e.g., more widespread Aβ pathology) or, as suggested by recent studies, by synaptic spread of pathologic tau itself. It must be noted, however, that as yet the evidence in support of the existence of synaptic spread of pathologic tau comes from animal models, and not from human studies. Although we failed to detect a difference in VV between diagnostic groups, the potential confounding of these independent contributions to VV could possibly differentially influence our VV findings. The range in days between last neuropsychological assessment and death was large, 1,358 ± 1161.4 days, but did not differ between groups (p = 0.114) and was taken into account as a covariate in our statistical tests.
Conclusions
The concentration of pTau is increased in AD+P DLPFC, potentially a sign of elevated kinase activity and consequent tau phosphorylation. This could reflect an enhanced sensitivity to factors that drive tau phosphorylation and highlights a network of more dynamic, exaggerated tau pathology in AD+P.
Supplementary Material
Acknowledgments
This work was supported by the Veterans Health Administration [BX000452 to R.A.S]; the National Institute of Mental Health [MH071533 to R.A.S.]; and the National Institute on Aging [AG005133 to O.L.L., AG027224 to R.A.S.]. P.S.M. is supported by the National Institute of Mental Health [MH019986] and seed grant funding from the National Institute on Aging [AG005133].
We wish to thank the staff members of the Alzheimer Disease Research Center and the Translational Neuroscience Program at the University of Pittsburgh. We also wish to thank Mrs. Mary Brady for assistance with the figures and Mr. Brad Rocco for critical review of image processing methodology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Veterans Affairs or the National Institutes of Health.
Footnotes
Authors' disclosures available online (www.jalz.com/disclosures/view.php?id=1987).
Supplementary Material: The supplementary figure is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-131166.
References
- 1.Scarmeas N, Brandt J, Albert M, Hadjigeorgiou G, Papadimitriou A, Dubois B, Sarazin M, Devanand D, Honig L, Marder K, Bell K, Wegesin D, Blacker D, Stern Y. Delusions and hallucinations are associated with worse outcome in Alzheimer disease. Arch Neurol. 2005;62:1601–1608. doi: 10.1001/archneur.62.10.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emanuel JE, Lopez OL, Houck PR, Becker JT, Weamer EA, DeMichele-Sweet MA, Kuller L, Sweet RA. Trajectory of cognitive decline as a predictor of psychosis in early Alzheimer disease in the cardiovascular health study. Am J Geriatr Psychiatry. 2011;19:160–168. doi: 10.1097/JGP.0b013e3181e446c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweet RA, Hamilton RL, Lopez OL, Klunk WE, Wisniewski SR, Kaufer DI, Healy MT, DeKosky ST. Psychotic symptoms in Alzheimer's disease are not associated with more severe neuropathologic features. Int Psychogeriatr. 2000;12:547–558. doi: 10.1017/s1041610200006657. [DOI] [PubMed] [Google Scholar]
- 4.Paulsen JS, Salmon DP, Thal L, Romero R, Weisstein-Jenkins C, Galasko D, Hofstetter CR, Thomas R, Grant I, Jeste DV. Incidence of and risk factors for hallucinations and delusions in patients with probable Alzheimer's disease. Neurology. 2000;54:1965–1971. doi: 10.1212/wnl.54.10.1965. [DOI] [PubMed] [Google Scholar]
- 5.Farber NB, Rubin EH, Newhouse PA, Kinscherf DA, Miller JP, Morris JC, Olney JW, McKeel DW. Increased neocortical neurofibrillary tangle density in subjects with Alzheimer's disease. Arch Gen Psychiatry. 2000;57:1165–1173. doi: 10.1001/archpsyc.57.12.1165. [DOI] [PubMed] [Google Scholar]
- 6.Forstl H, Burns A, Levy R, Cairns N. Neuropathological correlates of psychotic phenomena in confirmed Alzheimer's disease. Br J Psychiatry. 1994;165:53–59. doi: 10.1192/bjp.165.1.53. [DOI] [PubMed] [Google Scholar]
- 7.Sweet RA, Nimgaonkar VL, Devlin B, Lopez OL, DeKosky ST. Increased familial risk of the psychotic phenotype of Alzheimer disease. Neurology. 2002;58:907–911. doi: 10.1212/wnl.58.6.907. [DOI] [PubMed] [Google Scholar]
- 8.Sweet RA, Bennett DA, Graff-Radford NR, Mayeux R. Assessment and familial aggregation of psychosis in Alzheimer's disease from the National Institute on Aging Late Onset Alzheimer's Disease Family Study. Brain. 2010;133:1155–1162. doi: 10.1093/brain/awq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bacanu SA, Devlin B, Chowdari KV, DeKosky ST, Nimgaonkar VL, Sweet RA. Heritability of psychosis in Alzheimer disease. Am J Geriatr Psychiatry. 2005;13:624–627. doi: 10.1176/appi.ajgp.13.7.624. [DOI] [PubMed] [Google Scholar]
- 10.Geda YE, Schneider LS, Gitlin LN, Miller DS, Smith GS, Bell J, Evans J, Lee M, Porsteinsson A, Lanctot KL, Rosenberg PB, Sultzer DL, Francis PT, Brodaty H, Padala PP, Onyike CU, Ortiz LA, Ancoli-Israel S, Bliwise DL, Martin JL, Vitiello MV, Yaffe K, Zee PC, Herrmann N, Sweet RA, Ballard C, Khin NA, Alfaro C, Murray PS, Schultz S, Lyketsos CG. Neuropsychiatric symptoms in Alzheimer's disease: Past progress and anticipation of the future. Alzheimers Dement. 2013;9:602–608. doi: 10.1016/j.jalz.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray PS, Kumar S, DeMichele-Sweet MA, Sweet RA. Psychosis in Alzheimer's Disease. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 13.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: Correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 14.Scheff SW, Price DA. Alzheimer's disease-related alterations in synaptic density: Neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- 15.Mega MS, Lee L, Dinov ID, Mishkin F, Toga AW, Cummings JL. Cerebral correlates of psychotic symptoms in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2000;69:167–171. doi: 10.1136/jnnp.69.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staff RT, Shanks MF, Macintosh L, Pestell SJ, Gemmell HG, Venneri A. Delusions in Alzheimer's disease: Spet evidence of right hemispheric dysfunction. Cortex. 1999;35:549–560. doi: 10.1016/s0010-9452(08)70818-9. [DOI] [PubMed] [Google Scholar]
- 17.Kotrla KJ, Chacko RC, Harper RG, Jhingran S, Doody R. SPECT findings on psychosis in Alzheimer's disease. Am J Psychiatry. 1995;152:1470–1475. doi: 10.1176/ajp.152.10.1470. [DOI] [PubMed] [Google Scholar]
- 18.Sultzer DL, Mahler ME, Mandelkern MA, Cummings JL, Van Gorp WG, Hinkin CH, Berisford MA. The relationship between psychiatric symptoms and regional cortical metabolism in Alzheimer's disease. J Neuropsychiatry Clin Neurosci. 1995;7:476–484. doi: 10.1176/jnp.7.4.476. [DOI] [PubMed] [Google Scholar]
- 19.Koppel J, Sunday S, Goldberg TE, Davies P, Christen E, Greenwald BS. Psychosis in Alzheimer's disease is associated with frontal metabolic impairment and accelerated decline in working memory: Findings from the Alzheimer's Disease Neuroimaging Initiative. Am J Geriatr Psychiatry. 2013 doi: 10.1016/j.jagp.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 20.Bruen PD, McGeown WJ, Shanks MF, Venneri A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer's disease. Brain. 2008;131:2455–2463. doi: 10.1093/brain/awn151. [DOI] [PubMed] [Google Scholar]
- 21.Sweet RA, Panchalingam K, Pettegrew JW, McClure RJ, Hamilton RL, Lopez OL, Kaufer DI, DeKosky ST, Klunk WE. Psychosis in Alzheimer disease: Postmortem magnetic resonance spectroscopy evidence of excess neuronal and membrane phospholipid pathology. Neurobiol Aging. 2002;23:547–553. doi: 10.1016/s0197-4580(02)00009-x. [DOI] [PubMed] [Google Scholar]
- 22.Murray PS, Kirkwood CM, Gray MC, Ikonomovic MD, Paljug WR, Abrahamson EE, Henteleff RA, Hamilton RL, Kofler JK, Klunk WE, Lopez OL, Penzes P, Sweet RA. beta-Amyloid 42/40 ratio and kalirin expression in Alzheimer disease with psychosis. Neurobiol Aging. 2012;33:2807–2816. doi: 10.1016/j.neurobiolaging.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cahill ME, Xie Z, Day M, Photowala H, Barbolina MV, Miller CA, Weiss C, Radulovic J, Sweatt JD, Disterhoft JF, Surmeier DJ, Penzes P. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc Natl Acad Sci U S A. 2009;106:13058–13063. doi: 10.1073/pnas.0904636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 25.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 26.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shipton OA, Leitz JR, Dworzak J, Acton CE, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M. Tau protein is required for amyloid beta-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 30.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 31.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 32.deCalignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukaetova-Ladinska EB, Harrington CR, Xuereb J, Roth M, Wischik CM. Biochemical, neuropathological, and clinical correlations of neurofibrillary degeneration in Alzheimer's disease. In: Bergener M, Finkel SI, editors. Treating Alzheimer's and Other Dementias: Clinical Application of Recent Research Advances. Springer; New York: 1995. pp. 57–80. [Google Scholar]
- 35.Koppel J, Goldberg TE, Gordon ML, Huey E, Davies P, Keehlisen L, Huet S, Christen E, Greenwald BS. Relationships between behavioral syndromes and cognitive domains in Alzheimer disease: The impact of mood and psychosis. Am J Geriatr Psychiatry. 2012;20:994–1000. doi: 10.1097/JGP.0b013e3182358921. [DOI] [PubMed] [Google Scholar]
- 36.Paulsen JS, Ready RE, Stout JC, Salmon DP, Thal LJ, Grant I, Jeste DV. Neurobehaviors and psychotic symptoms in Alzheimer's disease. J Int Neuropsychol Soc. 2000;6:815–820. doi: 10.1017/s1355617700677081. [DOI] [PubMed] [Google Scholar]
- 37.Sweet RA, Hamilton RL, Healy MT, Wisniewski SR, Henteleff R, Pollock BG, Lewis DA, DeKosky ST. Alterations of striatal dopamine receptor binding in Alzheimer disease are associated with Lewy body pathology and antemortem psychosis. Arch Neurol. 2001;58:466–472. doi: 10.1001/archneur.58.3.466. [DOI] [PubMed] [Google Scholar]
- 38.Tariot PN, Mack JL, Patterson MB, Edland SD, Weiner MF, Fillenbaum G, Blazina L, Teri L, Rubin E, Mortimer JA, Stern Y Behavioral Pathology Committee of the Consortium to Establish a Registry for Alzheimer's Disease. The behavior rating scale for dementia of the Consortium to Establish a Registry for Alzheimer's Disease. Am J Psychiatry. 1995;152:1349–1357. doi: 10.1176/ajp.152.9.1349. [DOI] [PubMed] [Google Scholar]
- 39.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer's neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 41.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen ENH, Ballard C, de Vos RAI, Wilcock GK, Jellinger KA, Perry RH. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 42.McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, Dickson D, Dubois B, Duda JE, Feldman H, Gauthier S, Halliday G, Lawlor B, Lippa C, Lopez OL, Carlos MJ, O'Brien J, Playfer J, Reid W. Dementia with Lewy bodies. Lancet Neurol. 2004;3:19–28. doi: 10.1016/s1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- 43.Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- 44.Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 46.Moyer CE, Delevich KM, Fish KN, Asafu-Adjei JK, Sampson AR, Dorph-Petersen KA, Lewis DA, Sweet RA. Reduced glutamate decarboxylase 65 protein within primary auditory cortex inhibitory boutons in schizophrenia. Biol Psychiatry. 2012;72:734–743. doi: 10.1016/j.biopsych.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fish KN, Sweet RA, Deo AJ, Lewis DA. An automated segmentation methodology for quantifying immunoreactive puncta number and fluorescence intensity in tissue sections. Brain Res. 2008;1240:62–72. doi: 10.1016/j.brainres.2008.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howard CV, Reed MG. Estimation of component volume and volume fraction. In: Howard CV, Reed MG, editors. Unbiased Stereology: Three Dimensional Measurement in Microscopy. BIOS Scientific Publishers; Oxford, UK: 1998. pp. 55–65. [Google Scholar]
- 49.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der WK, Subramaniam V, Braeken D, Callewaert G, Bartic C, D'Hooge R, Martins IC, Rousseau F, Schymkowitz J, De SB. Neurotoxicity of Alzheimer's disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hollingworth P, Sweet R, Sims R, Harold D, Russo G, Abraham R, Stretton A, Jones N, Gerrish A, Chapman J, Ivanov D, Moskvina V, Lovestone S, Priotsi P, Lupton M, Brayne C, Gill M, Lawlor B, Lynch A, Craig D, McGuinness B, Johnston J, Holmes C, Livingston G, Bass NJ, Gurling H, McQuillin A, Holmans P, Jones L, Devlin B, Klei L, Barmada MM, Demirci FY, DeKosky ST, Lopez OL, Passmore P, Owen MJ, O'Donovan MC, Mayeux R, Kamboh MI, Williams J. Genome-wide association study of Alzheimer's disease with psychotic symptoms. Mol Psychiatry. 2012;17:1316–1327. doi: 10.1038/mp.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kojima Y, Miyoshi H, Clevers HC, Oshima M, Aoki M, Taketo MM. Suppression of tubulin polymerization by the LKB1-microtubule-associated protein/microtubule affinity-regulating kinase signaling. J Biol Chem. 2007;282:23532–23540. doi: 10.1074/jbc.M700590200. [DOI] [PubMed] [Google Scholar]
- 53.Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci. 2007;27:574–581. doi: 10.1523/JNEUROSCI.5094-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesne SE. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. 2012;32:16857–16871a. doi: 10.1523/JNEUROSCI.1858-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Schneider A, Biernat J, von BM, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–3558. doi: 10.1021/bi981874p. [DOI] [PubMed] [Google Scholar]
- 56.Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu X, Raina AK, Vinters HV, Tabaton M, Johnson AB, Paula-Barbosa M, Avila J, Jones PK, Castellani RJ, Smith MA, Perry G. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am J Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del TK. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: A practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 61.Nelson PT, Jicha GA, Schmitt FA, Liu H, Davis DG, Mendiondo MS, Abner EL, Markesbery WR. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: Neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–1146. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crum RM, Anthony JC, Bassett SS, Folstein MF. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA. 1993;269:2386–2391. [PubMed] [Google Scholar]
- 63.Teri L, McCurry SM, Edland SD, Kukull WA, Larson EB. Cognitive decline in Alzheimer's disease: A longitudinal investigation of risk factors for accelerated decline. J Gerontol A Biol Sci Med Sci. 1995;50A:M49–M55. doi: 10.1093/gerona/50a.1.m49. [DOI] [PubMed] [Google Scholar]
- 64.Gartner U, Janke C, Holzer M, Vanmechelen E, Arendt T. Postmortem changes in the phosphorylation state of tau-protein in the rat brain. Neurobiol Aging. 1998;19:535–543. doi: 10.1016/s0197-4580(98)00094-3. [DOI] [PubMed] [Google Scholar]
- 65.Glynn MW, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat Protoc. 2006;1:1287–1296. doi: 10.1038/nprot.2006.220. [DOI] [PubMed] [Google Scholar]
- 66.Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat Methods. 2005;2:677–684. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- 67.Bobinski M, Wegiel J, Tarnawski M, de Leon MJ, Reisberg B, Miller DC, Wisniewski HM. Duration of neurofibrillary changes in the hippocampal pyramidal neurons. Brain Res. 1998;799:156–158. doi: 10.1016/s0006-8993(98)00441-7. [DOI] [PubMed] [Google Scholar]
- 68.Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 70.Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.