

Abstract
Background
Verticillium dahliae is a soil-borne fungus that causes vascular wilt diseases in a wide range of plant hosts. V. dahliae produces multicelled, melanized resting bodies, also known as microsclerotia (MS) that can survive for years in the soil. The MS are the primary source of infection of the Verticillium disease cycle. Thus, MS formation marks an important event in the disease cycle of V. dahliae.
Results
In this study, next generation sequencing technology of RNA-Seq was employed to investigate the global transcriptomic dynamics of MS development to identify differential gene expression at several stages of MS formation in strain XS11 of V. dahliae, isolated from smoke tree. We observed large-scale changes in gene expression during MS formation, such as increased expression of genes involved in protein metabolism and carbohydrate metabolism. Genes involved in glycolytic pathway and melanin biosynthesis were dramatically up-regulated in MS. Cluster analyses revealed increased expression of genes encoding products involved in primary metabolism and stress responses throughout MS development. Differential expression of ubiquitin-dependent protein catabolism and cell death-associated genes during MS development were revealed. Homologs of genes located in the lineage-specific (LS) regions of V. dahliae strain VdLs.17, were either not expressed or showed low expression. Furthermore, alternative splicing (AS) events were analyzed, revealing that over 95.0% AS events involve retention of introns (RI).
Conclusions
These data reveal the dynamics of transcriptional regulation during MS formation and were used to construct a comprehensive high-resolution gene expression map. This map provides a key resource for understanding the biology and molecular basis of MS development of V. dahliae.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-324) contains supplementary material, which is available to authorized users.
Keywords: Verticillium dahliae, Microsclerotia development, RNA-Seq, Transcriptome, Gene expression, Alternative splicing
Background
Verticillium dahliae Kleb. (Eukaryota, Fungi, Ascomycota) is a ubiquitous soil-borne fungus that penetrates plant roots, enters the plant vascular system, and causes vascular wilt diseases collectively known as Verticillium wilts [1, 2]. V. dahliae can infect more than 200 plant species, including important crops, flowers, vegetables, trees, and shrubs, causing economically significant losses each year [1, 2].
Verticillium wilt is a threat to smoke tree (Cotinus coggygria Scop.) stands in China. Smoke trees are of primary importance as ornamentals, highly valued for the brilliant red leaf scenery that these trees provide in the Beijing region during autumn, especially in Fragrant Hills Park, a 160 hectare forest park in Beijing. Vascular wilt in smoke tree was reported in Fragrant Hills Park as early as 1990, and subsequently the disease spread to other smoke tree growing areas in Beijing [3]. Symptoms of vascular wilt in smoke trees include stunted growth of the stem, early senescence of leaves, and early mortality. Without adequate control, the famous “red leaf scenery” of Fragrant Hills and other areas is threatened by the detrimental effects of the Verticillium wilt. Currently available fungicides and other control measures are not effective in controlling the disease due in part to the ability of the fungus to survive for long periods in soil.
The survival of V. dahliae in soil depends on the production of melanized, multicellular structures known as microsclerotia (MS) [4]. Melanin deposition and a thickened cell wall enable the MS of the pathogen to resist UV irradiation, temperature extremes, enzymatic lysis, and fungicidal activities [5]. The MS can survive in soils in the absence of a host plant for as long as 10 years, and are the primary infectious propagules of the Verticillium wilt disease [6]. The MS germinate to form hyphae in the soil, and penetrate the plant roots, where the fungus colonizes the xylem tissue of the plant vascular system. As wilt symptoms progress, V. dahliae produces MS in dying plant tissues, which are returned to the soil to initiate new primary infections. Thus, the production of MS represents a significant developmental event in the life cycle of V. dahliae[7].
The morphological events of MS formation have been well studied by both light and electron microscopy [8–11]. In the early stages of MS development, hyphae become swollen, vacuolated, and form numerous septa. Subsequently, clusters of hyphal cells form in the swollen hyphae that resemble the microsclerotial initial [11, 12]. In the final phase of MS formation, melanin particles are extruded into the interhyphal spaces of the microsclerotium, and peripheral microsclerotial cells are killed by autolysis [8]. The genes involved in melanin biosynthesis have been identified and their functions have been characterized [10, 13–15]. The results indicate that melanin is necessary for the formation of fully functional MS [5]. However, little is known about the molecular pathways involved in MS formation.
Genome-wide identification of genes expressed during MS formation is a first step in elucidating the pathways and molecular mechanisms underlying MS formation in V. dahliae. Methods for genome-wide expression analyses include expressed sequence tag (EST) analysis [16, 17], suppression subtractive hybridization (SSH) [18, 19], serial analysis of gene expression (SAGE) [20], massive parallel signature sequencing (MPSS) [21–23] and RNA-Sequencing (RNA-Seq) [24–26]. Next-generation sequencing (NGS) technologies have provided new platforms for comprehensive transcriptional studies [27–31]. Transcriptome sequencing is an efficient means to generate transcriptomic data, and RNA-Seq is one approach transcriptome profiling that provides highly accurate measurements of gene expression by counting the number of sequencing reads, which map to a genome or annotated transcripts [28, 32], and further enable genome-wide identification of coding sequences, gene structure, alternative splicing [33]. Transcriptomic data produced by RNA-Seq methods have increased our understanding of gene expression involved in growth and development of pathogenic fungi [30, 34–39]. For example, transcriptional analysis of appressorium formation in the rice blast fungus Magnaporthe oryzae revealed the role of autophagy, lipid metabolism and melanin biosynthesis, and a Pmk1 MAPK kinase as a key global regulator in appressorium differentiation [39].
Resources available to facilitate transcriptome analyses of MS formation include transcript data for 10,535 genes and the 33.8 MB genome sequence for V. dahliae, strain VdLs.17 (Broad Institute Verticillium Group Database) [40]. Subsequent characterizations of this sequence resource also provide useful information to place transcriptomic studies in context. For example, comparison of the genome structure of V. dahliae strain VdLs.17 to that of the Verticillium alfalfae strain VaMs.102 revealed four lineage-specific (LS) regions of about 350 kb in length present in VdLs.17 but absent in the VaMs.102 strain. The VdLs.17 LS regions encoded 354 predicted genes, some of which had been associated with virulence and host range specificity. Only about 7.0% the LS genes encode predicted secreted proteins, and the LS regions were nearly devoid of “housekeeping” type genes [40]. Additional comparative genomics analyses of multiple V. dahliae strains indicated that the LS regions are diverse in length and gene content, enriched for in planta-expressed genes, and that chromosomal rearrangements associated with LS regions in V. dahliae are common [41]. An additional valuable resource includes expressed sequence tag (EST) libraries previously used to identify expressed genes in V. dahliae during pathogenic growth and MS development in V. dahliae[42]. Neumann and Dobinson [42] obtained about 1000 ESTs, many of which corresponded to melanin biosynthetic enzymes, exclusive to the developing MS culture type. The analysis of genes associated with specific ESTs has accelerated molecular characterization of MS formation. Recently, Duressa et al. performed a RNA-Seq analysis between MS producing cultures and those not producing MS in V. dahliae, revealing over 200 significantly expressed genes involved in melanin synthesis and other processes [43]. While this work focused on up- or down-regulation of gene expression between these two culture types, the focus was not on assessing gene expression associated with gradual developmental changes during MS formation.
In addition to those resources, several individual genes involved in MS formation have been characterized in V. dahliae[12, 44–47]. VDH1, encoding a class II hydrophobin, is one of the many genes significantly differentially expressed during MS formation, and VDH1 is required for MS formation of V. dahliae[12, 45]. VMK1, encoding a mitogen-activated protein kinase, modulates MS formation [46]; mutation of VdGARP1, encoding a glutamic acid-rich protein, significantly delays development of melanized MS [44]. The non-LS copy of VdHOG1, a homolog of the high osmolarity glycerol response protein kinase, positively regulates MS formation (Xiao, et al., unpublished data), and the G protein β subunit negatively controls MS formation [47]. Functional characterizations of these individual genes have provided valuable insight into the genetic control of MS formation, yet there remain major gaps in our understanding of the molecular determinants that trigger and regulate MS formation in V. dahliae.
The objectives of this work were to (1) examine developmental stage-specific gene expression during MS formation in V. dahliae and (2) to identify genes differentially expressed in this developmental process. To accomplish these aims, extensive microscopy analyses were carried out to initially characterize four different stages of MS development in a smoke tree strain of V. dahliae. RNA-Seq analyses were employed to analyze transcript profiles of 10,158 genes (96.4% of the total predicted reference genes of the VdLs.17 strain) and global patterns of gene expression in V. dahliae using four identified MS developmental stages as reference points. This enabled identification genes significantly differentially expressed during stages of MS development, revealing major metabolic processes and signal pathways associated with MS development in V. dahliae. Elucidation of the molecular mechanisms that govern MS formation in V. dahliae may be useful in designing novel strategies to control Verticillium wilt, not only for the smoke tree pathogen, but also related pathogenic strains and other Verticillium spp. that produce MS [48].
Results
Microscopic analyses of MS development
The dynamics of morphogenesis and pigment production were examined by light microscopy during MS development to chart cellular changes relative to gene expression during MS formation. Six stages were selected for these analyses based on phenotypic changes that were observed during MS formation. The stages of MS formation, as determined by light microscopy, were named MS1, MS2, MS3 and MS4 (Figure 1A). At 60 hpi (hours post incubation) or at MS1, cell clustering and swelling resembling the microsclerotial initial was observed at the margins of the cellulose membrane. This initial stage of MS development (60 hpi) gave rise to small masses of microsclerotial initial cells (Figure 1A) that were bright yellow. At 72 hpi (MS2; Figure 1A), developing MS were easily visible, and microsclerotial mass consisting of an accumulation of brown and lightly-pigmented cells appeared on the membrane (Figure 1A). At 96 hpi (MS3; Figure 1A), many pigmented MS were visible and the microsclerotial mass consisted of an accumulation of dark cells across the entire membrane. At 14 dpi (MS4; Figure 1A), mature and darkly pigmented MS were apparent. The individual cells of mature MS also possessed a thickened cell wall surrounded by melanin. In addition to these four stages of MS development, two additional morphological stages of the fungal conidia (CO) and germinating conidia (GC) were sampled for RNA-Seq and comparisons of differential gene expression.
Figure 1.
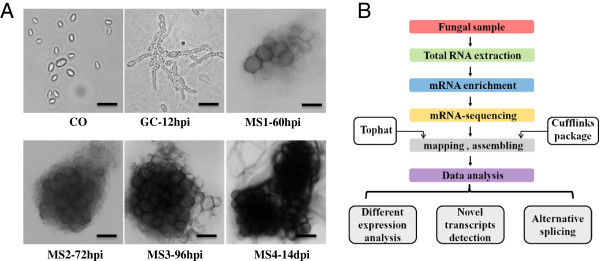
Microsclerotia developmental process of the smoke tree vascular wilt fungus, V. dahliae . and the transcriptome analysis pipeline. A. Micrographs showing microsclerotial development processes from conidia to microsclerotia in Verticillium dahliae. The panels represent different developmental stages of smoke tree wilt fungus cultured in CM broth and BM, respectively. Scale bar = 10 μm. MS1-MS4 represent four typical stages during the entire process of microsclerotia formation at 60 h, 72 h, 96 h and 14 d represent four stages of microsclerotia formation used for RNA-Seq. B. Flowcharts of the RNA-Seq method employed in this study. Tophat was used to align RNA-Seq reads to the genome of V. dahliae VdLs.17 (Broad Institute) and Cufflinks package was used to assemble and find differentially expressed genes and transcripts.
Overview of the V. dahliae transcriptome
The pipeline of RNA-Seq analysis is shown in Figure 1B, RNA libraries derived from the samples were pair-end sequenced using an Illumina high-throughput sequencing platform. After stringent quality checks and tag cleaning, we obtained a total of 158,501,901 reads from the six libraries with an average of 26,416,983 reads in each sample, ranging from 25,596,902 to 27,056,223 reads (Table 1, Additional file 1: Figure S1). The V. dahliae (strain VdLs.17) genome assembly and the 10,535 predicted genes of this strain were used as a reference sequence (Broad Institute, Verticillium group Database) [40]. In total, 85.8% of the total reads (135,989,758 reads) from all six samples were aligned to the genome of V. dahliae, strain VdLs.17 by Tophat with less than 2 bp mismatches [49]. The total mapped reads represented about 423-fold coverage of the genome and 955-fold coverage of the annotated V. dahliae transcriptome. Approximately 14.2% of total reads did not map to the genome, likely reflecting either sequence differences between strains (the smoke tree strain and the lettuce strain VdLs.17), gaps in the current assembly, sequencing errors, or alternative splicing that exists in the reference genome of the Broad Institute.
Table 1.
RNA-Seq statistics
| Sample | Insert size (bp)a | Total readsb | Mapped readsc | Perfect match readsd | Unmapped readse | Reads mapped to genesf |
|---|---|---|---|---|---|---|
| CO | 193.341 bp | 27056079 | 23660245 (87.45%g) | 19767391 (73.06%g) | 3395834 (12.55%g) | 15750739 (58.22%g/66.57%h) |
| GC | 195.521 bp | 27056223 | 23268435 (86.00%) | 19630309 (72.55%) | 3787788 (14.00%) | 15904017 (58.78%/68.35%) |
| MS1 | 188.009 bp | 26190923 | 22036154 (84.14%) | 18611864 (71.06%) | 4154769 (15.86%) | 14344871 (54.77%/65.10%) |
| MS2 | 194.213 bp | 25596902 | 21727696 (84.88%) | 18413763 (71.94%) | 3869206 (15.12%) | 14297493 (55.86%/65.80%) |
| MS3 | 195.993 bp | 26410805 | 22551758 (85.39%) | 19100267 (72.32%) | 3859047 (14.61%) | 14932675 (56.54%/66.22%) |
| MS4 | 190.443 bp | 26190969 | 22745470 (86.84%) | 19246089 (73.48%) | 3445499 (13.16%) | 14879393 (56.81%/65.42%) |
| Total | 158501901 | 135989758 (85.80%) | 114769683 (72.41%) | 22512143 (14.20%) | 90109188 (56.85%/66.26%) | |
aThe length of fragments used for sequencing.
bThe number of reads generated from sequencing after filtering low quality reads (Q ≤ 5).
cThe number of reads mapped to the reference genome within 2 bp mismatch.
dThe number of reads mapped to the reference genome with no mismatch.
eThe number of reads that could not be mapped to the reference genome within 2 bp mismatch
fThe number of reads mapped to the annotated genes within 2 bp mismatch.
gThe percentages of reads account for the total reads.
hThe percentages of reads account for the mapped reads.
Of the mapped reads, 114,769,683 (72.4% of total) reads were perfectly mapped to the V. dahliae genome without mismatch. Furthermore, 56.9% of the total reads could be mapped to the annotated genes with less than 2 bp mismatches, indicating that almost 30.0% of the total reads mapped to non-annotated regions, including the intergenic regions or other non-coding regions. The numbers of differentially expressed perfectly matched reads and reads mapped to the annotated genes in each stage were not significantly different (Table 1, Additional file 1: Figure S1). The unmapped percentages of reads were found to be lower (between 12.6% and 15.9%).
Gene expression levels were determined by Fragments Per Kilobase of gene per Million mapped fragments (FPKM). Mapping revealed that 10,158 (96.4% of 10,535 total annotated VdLs.17 genes) predicted genes were expressed, ranging from 9,412 (89.3%) to 9,939 (94.3%) among the six libraries (Table 2). Approximately 78.0% (8,222) –87.9% (9,267) of genes were expressed with FPKM >1. Expression patterns identified by RNA-Seq were validated by reverse transcription quantitative real-time PCR (qRT-PCR) analysis of selected genes from each group. Analysis of the RNA-Seq and qRT-PCR data sets revealed correlation between the two (Additional file 2: Figure S2). To examine the correlation of gene expression patterns among the six samples, the Pearson relationships of paired samples were calculated with FPKM of all expressed genes. Samples sharing similar gene expression patterns were clustered together according to the Pearson correlation. This correlation analysis revealed that stages MS1, MS2 and MS3 shared similar expression patterns as stage GC in the larger clade, while the expression patterns in stages MS4 and CO were distinct from each other. Furthermore, the pattern of gene expression in stages MS1-MS4 and GC clustered together, while the stage of CO was in a distinct clade (Figure 2A). Principal component analysis (PCA) was employed to examine the six samples according to their gene expression profiles. Principal components 1 and 2 explained 86.2% and 6.7% of the variance, respectively. Stage CO was distinct from the other five samples. MS1, MS2 and MS3 were clustered together and belonged to a cluster with GC. Stage MS4 was distinct from stages MS1-3 (Figure 2A, 2B). Therefore, the PCA result was consistent with the hierarchical clustering.
Table 2.
Distribution of gene expression values among developmental stages examined
| Sample | FPKMa > 0 | 0 < FPKM < =1 | 1 < FPKM < =10 | 10 < FPKM < =100 | FPKM > 100 |
|---|---|---|---|---|---|
| CO | 9412 (89.34%b) | 1190 (12.64%c) | 2879 (30.59%c) | 4319 (45.89%c) | 1024 (10.88%c) |
| GC | 9443 (89.63%) | 1188 (12.58%) | 2388 (25.29%) | 4805 (50.88%) | 1062 (11.25%) |
| MS1 | 9747 (92.52%) | 929 (9.53%) | 2335 (23.96%) | 5314 (54.52%) | 1169 (11.99%) |
| MS2 | 9858 (93.57%) | 757 (7.68%) | 2202 (22.34%) | 5696 (57.78%) | 1202 (12.19%) |
| MS3 | 9933 (94.29%) | 711 (7.16%) | 2199 (22.14%) | 5756 (57.95%) | 1267 (12.76%) |
| MS4 | 9939 (94.34%) | 672 (6.76%) | 2158 (21.71%) | 5743 (57.78%) | 1366 (13.74%) |
aFPKM, fragments per kilobase of transcript per million mapped fragments.
bThe percentage of genes accounting for all annotated genes in V. dahliae (10,535).
cThe percentage of genes accounting for all expressed genes in each library.
Figure 2.
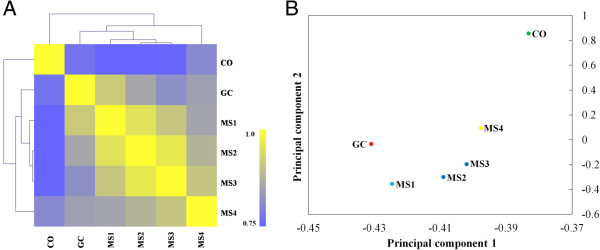
Correlation in expression patterns among the six RNA-sequenced libraries. A. Heatmap of the Pearson correlation of RNA-Seq samples according to gene expression level. Clustering was analyzed based on the expression data of 10,158 genes. B. Principal component analysis (PCA) of the transcriptomes during microsclerotia formation and germinating conidia. For the principal components 1 and 2, eigenvalues are 86.2% and 6.7%, respectively. The analysis was performed by the MultiExperiment Viewer using the expression data from 10,158 genes.
Clustering analysis reveals enrichment of particular gene categories expressed during MS formation
To elucidate dynamic changes in the V. dahliae transcriptome during MS formation, the clustering affinity search technique (CAST) was employed to generate clusters [50]. CAST analyses of the 10,158 expressed genes revealed 132 clusters, with gene numbers within clusters ranging from 1945 to 1. Most of the clusters had no more than 20 genes. However, each of the top 18 clusters contained more than 100 genes (a range from 1945 to 107), and contained about 77.1% of the total detected genes, illustrating the major gene expression clusters (Figure 3). Increased transcript levels were observed in clusters 1, 2, 5, 6, 11, 12, 13, 16 and 17 in stages MS1-MS4 as compared with stage CO, and comprised 47.0% of the total expressed genes (Figure 3B) whereas genes in clusters 3, 10 and 18 were down-regulated in stages MS1-MS4 compared to stage CO, and comprised 11.6% of the total expressed genes.
Figure 3.
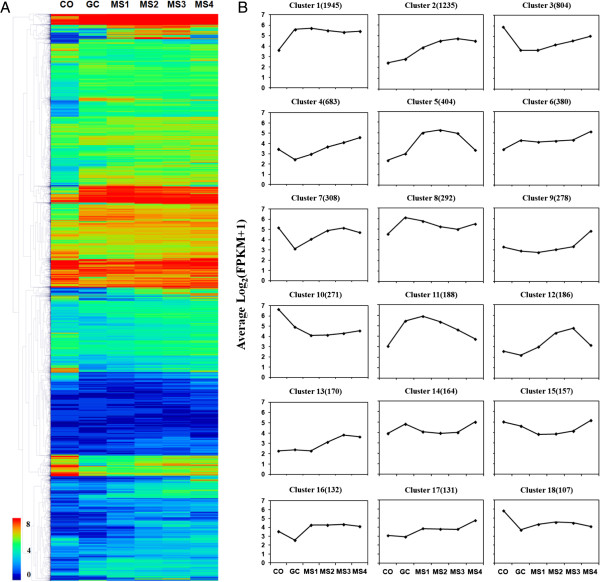
Clustering of gene expression. A. The log2 of FPKM for each gene was used for hierarchical analysis of the heat map at each of the six selected developmental stages (CO, GC, MS1, MS2, MS3 and MS4). The heat map illustrates that 10,158 expressed genes were classified into 132 gene-expression clusters generated by clustering. The colors correspond to the log2 of FPKM values, ranging from bright blue to red. B. Log2 average gene-expression levels of the top 18 clusters identified. The number of genes in each cluster is bracketed.
According to the gene expression pattern described earlier, and the cluster analysis results, a large variety of genes were differentially expressed during MS formation. To ascribe gene functions to those genes displaying differential expression patterns, gene ontology (GO) enrichment analysis was performed for genes from the top 18 clusters shown in Figure 3B by Blast2GO with Fisher’s Exact Test, and compared with the whole genome background filtered with false discovery rate (FDR) correction (≤0.01). The results of enriched GO terms revealed an overrepresentation of different gene functions in certain clusters. For example, genes encoding protein metabolic processes, such as ribonucleoprotein complex biogenesis, primary metabolic processes, and stress responses, were enriched in cluster 1, which was expressed at increased levels at each stage compared to the CO stage (Figure 3B). The genes in cluster 5 function mainly in signaling pathways, and increased in expression until MS3, at which point there were declining levels of expression (Figure 3B). Genes of cluster 7 were functionally enriched in transport. The functional enrichment of cluster 8 revealed cofactor metabolic processes, oxidation-reduction processes, and chromosome segregation. However, analyses did not reveal functional enrichment in other clusters, such as clusters 2, 4, 9, 10, 12, 13, 16 and 18. The full list of enriched GO terms and corresponding genes are provided in Additional file 3: Table S1.
Several gene families identified in other fungi, including in V. dahliae, have established functional roles of importance in growth, development, and pathogenesis. To further understand the expression patterns of these gene families, 16 different families or groups were selected and analyzed further (Figure 4). The results of this analysis illustrated diverse expression patterns, but relative to the GC stage, there was up-regulation of genes encoding products involved in fatty acid oxidation and the glyoxylate cycle (Figure 4A), calcium and GPCR-mediated signaling (Figure 4B), selected classes of transcription factors (Figure 4C), as well as secreted proteins and transporters (Figure 4D) during MS formation (MS1-MS4). As shown, selected branches of energy production pathways including glycolysis/gluconeogensis, TCA cycle, pentose phosphate pathway, fatty acid oxidation and glyoxylate cycle were up-regulated in MS formation compared to the CO stage (Figure 4A), while genes encoding proteins typical of signaling pathways, such as MAPK pathway members were slightly up-regulated during MS formation (Figure 4B, Additional file 4: Figure S3). The genome-wide expression analysis of transcription factors of V. dahliae suggested that certain transcription factors were also down-regulated in GC stages and up-regulated in stages MS1-MS4 (Figure 4C). Furthermore, more than 500 genes encoding secreted proteins showed an up-regulated pattern of expression in stages MS1-MS4 (Figure 4D); some of these genes were highly expressed (fold change >50) in MS formation, and 21 of these genes encoded small, cysteine-rich secreted proteins (<=300 aa) (Additional file 5: Figure S4, Additional file 6: Table S2).
Figure 4.
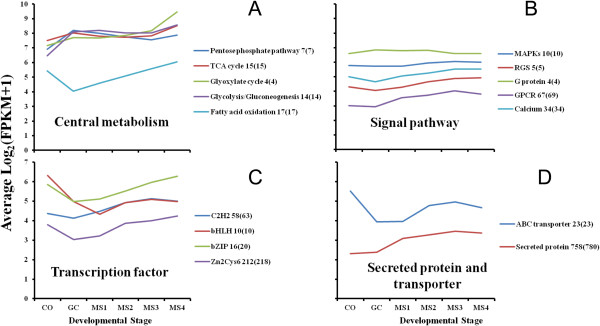
Global expression analysis shows distinct expression patterns of metabolic processes, signal pathways, transcription factors and secreted proteins during microsclerotia formation. A. to D. Expression trends of genes encoding proteins in four functional categories. A. The central metabolic pathways include the glyoxylate cycle, glycolysis/glucogeogenesis, TCA cycle, pentose phosphate pathway, fatty acid oxidation; B. Signal transduction and components, such as MAPK cascades, RGSs (regulators of G protein signaling), GPCR (G protein coupled receptors) and calcium signaling. C. Four main classes of transcription factors, i.e. C2H2, bHLH, bZIP and Zn2Cys6. D. Putative secreted proteins and ABC transporter genes. y axes are log2-transformed total intensities. The total numbers within a gene family are shown in numerals and the numbers of expressed genes within a family are shown in parentheses.
Gene expression profile during MS development
RNA-Seq provides digital readings of gene expression levels [51]. To determine which genes were expressed at each developmental stage, we examined the dynamics of gene expression throughout MS development in the genome-wide transcriptomic data. The distribution of gene expression values was different among the six stages studied. Nearly 12.0% of the expressed genes were detected with low expression values (0 < FPKM ≤ 1) in stages CO and GC, while only 8.0% (average) were detected in stages MS1-MS4, suggesting that many genes expressed with 0 < FPKM ≤1 in CO or GC stages were elevated transcript levels during the MS formation. However, the number of genes with moderate expression values (1 < FPKM ≤100) or high expression values (FPKM >100) in stages MS1-MS4 were increased relative to stages CO or GC, suggesting up-regulation of genes to meet the requirements of MS formation (Table 2). The number of expressed genes with FPKM >1 were gradually increased, from 8,818 (MS1) to 9,267 (MS4) (Table 2).
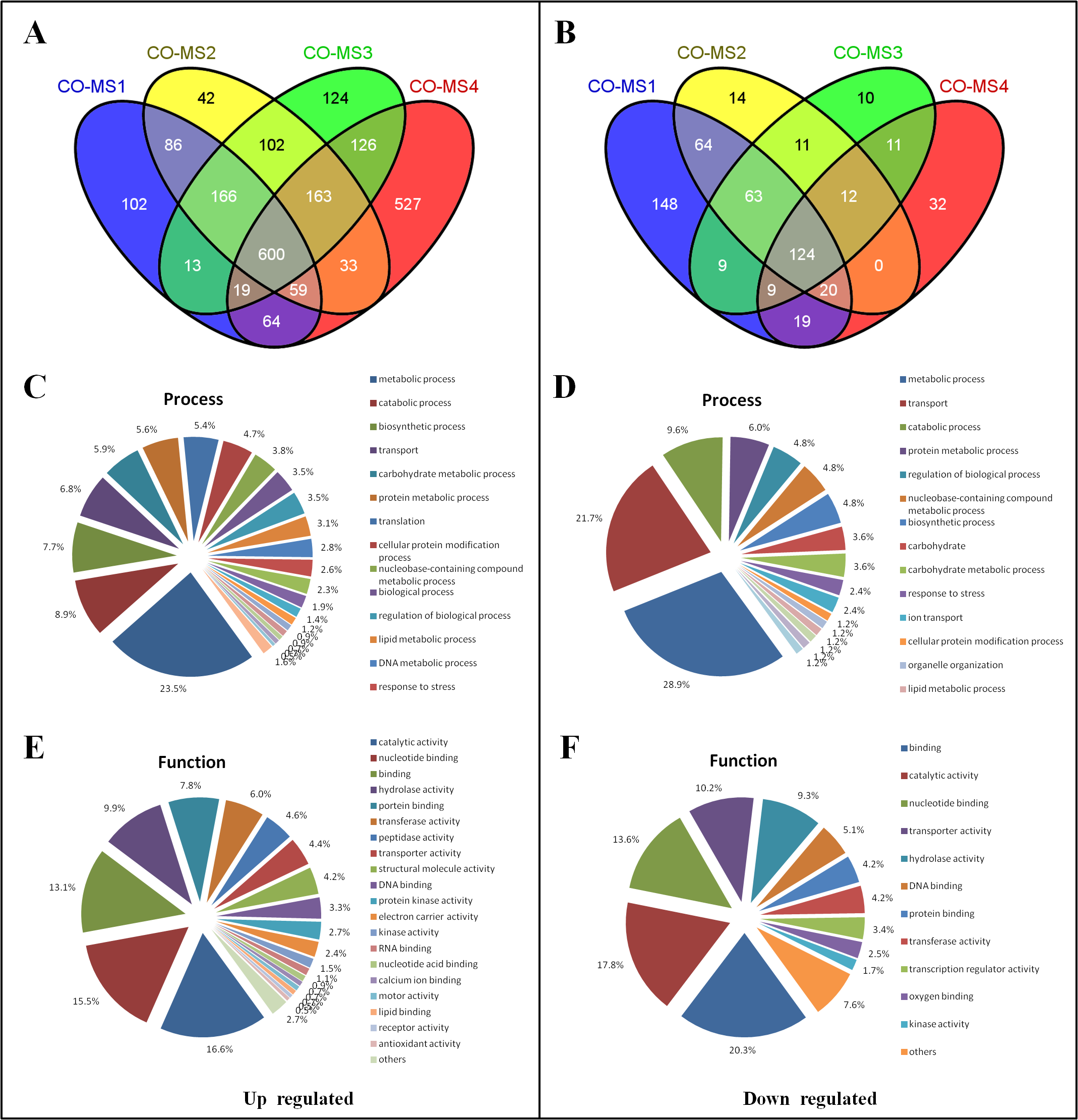
To determine which genes were differentially expressed during MS formation, pairwise comparisons were performed using counts of the significantly up- or down-regulated (adjusted p value < 0.05) genes. The number of significantly up-regulated genes was dramatically increased in stages MS1-MS4; however, the number of significantly down-regulated genes was reduced compared with the CO stage (Figure 5A). MS4 represented the stage with the largest number of significantly up-regulated genes and the least number of down-regulated genes compared with the CO stage, consistent with the distribution of gene expression at this stage. We further examined the expression level and log2 (fold change) distribution analysis of genes that were significantly differentially expressed at MS1-MS4 and GC stages compared with stage CO (Figure 5B). The number of genes with log2 (fold change) > 5 in each stage was consistent with that of significantly up-regulated genes. MS1-MS3 stages consisted of more genes with log2 (fold-change) >5 as compared with stage CO (Figure 5B). Using this approach, 600 significantly up-regulated genes and 124 significantly down-regulated genes (adjusted p value < 0.05) were identified (Additional file 7: Figure S5), and these were considered as candidate genes involved in MS development.
Figure 5.
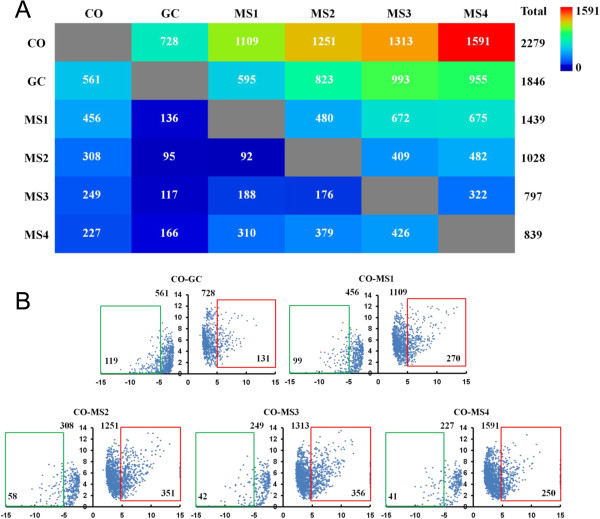
Significant changes in gene expression and the distribution of differentially expressed genes. A. The color-coded matrix shows number of significantly up-regulated genes in pairwise comparisons. Total represents the total number of genes significantly up-regulated compared to all other samples. Height, a complete gene significantly up-regulated genes (q value <0.05). B. The distribution of log2 fold-change and log2 expression value of the GC, and MS1-MS4 developmental stages compared to the CO stage. The y axes are log2 FPKM + 1of expressed genes in GC, and MS1-MS4 stages, and x axes are log2 fold change of the corresponding expression value compared with that of the CO stage. The numerical value on the top of each chart represents the number of differentially expressed genes, down- (left) or up-regulated (right). The numerical value in the green or red boxes indicates the number of up- or down- regulated genes with >5 fold-change.
GO analysis was performed for the 724 genes significantly differentially expressed in MS formation compared to the CO stage to examine potential molecular mechanisms regulating MS formation. Of the 724 genes, 365 up-regulated genes and 70 down-regulated genes were annotated with GO terms. GO analysis of the up-regulated genes identified processes related to metabolism, catabolism, transport, and the functions related to catalytic, transporter, hydrolase activities (Additional file 7: Figure S5, Additional file 8: Table S3). In addition, GO enrichment analysis was performed with the 600 up-regulated and 124 down-regulated genes compared with the whole genome with p value <0.05. Genes significantly up-regulated represented functional enrichment in structural molecules (GO:0005198), peptidase (GO:0008233), protein kinase (GO:0004672), hydrolase (GO:0016787) activities, and enrichment in processes including cellular carbohydrate metabolism (GO:0044262), cellular protein metabolism (GO:0044267), ribonucleoprotein complex biogenesis (GO:0022613), macromolecule biosynthesis (GO:0009059) and cellular component biogenesis (GO:0044085). On the other hand, the 124 significantly down-regulated genes represented functional enrichment in transporter (GO:0005215) and oxidoreductase (GO:0016491) activities, and in the transport processes (GO:0006810) (Table 3). In conclusion, the 600 up-regulated genes in the MS stages were mainly involved in protein metabolism, carbohydrate metabolism, and biosynthetic processes, including the macromolecule (ribosomal proteins) biosynthesis.
Table 3.
GO enrichment terms of significantly regulated genes during MS1-MS4 stages vs stage CO (p value <0.05)
| Terms | FDRa | P-value | Expression pattern | No. genesb |
|---|---|---|---|---|
| Ribonucleoprotein complex biogenesis (GO:0022613) | 0.089519378 | 0.002883868 | up | 23 |
| Cellular carbohydrate metabolic process (GO:0044262) | 0.132284495 | 0.006317295 | up | 25 |
| Cellular component biogenesis (GO:0044085) | 0.132284495 | 0.007286858 | up | 26 |
| Cellular protein metabolic process(GO:0044267) | 0.203232906 | 0.017429065 | up | 51 |
| Generation of precursor metabolites and energy (GO:0006091) | 0.203232906 | 0.017982239 | up | 14 |
| Gene expression (GO:0010467) | 0.203232906 | 0.01894544 | up | 24 |
| Structural molecule activity (GO:0005198) | 3.28E-04 | 1.39E-06 | up | 25 |
| Peptidase activity (GO:0008233) | 0.022419783 | 1.90E-04 | up | 27 |
| Motor activity (GO:0003774) | 0.233071632 | 0.022714608 | up | 4 |
| Oxidoreductase activity (GO:0016491) | 0.263043145 | 0.02675015 | up | 64 |
| Protein kinase activity (GO:0004672) | 0.332984226 | 0.038095653 | up | 15 |
| Hydrolase activity (GO:0016787) | 0.351323453 | 0.041682444 | up | 88 |
| Transport (GO:0006810) | 0.587336379 | 0.012443567 | down | 19 |
| Transporter activity (GO:0005215) | 0.418960736 | 0.001775257 | down | 13 |
| Oxidoreductase activity (GO:0016491) | 0.454356181 | 0.003850476 | down | 20 |
aFDR, False discovery rate, adjusted p-value of the test statistics.
bNumber of genes enrichment in each GO terms among the significantly regulated genes.
Differential expression of ubiquitin-dependent protein catabolism and cell death-associated genes during MS development
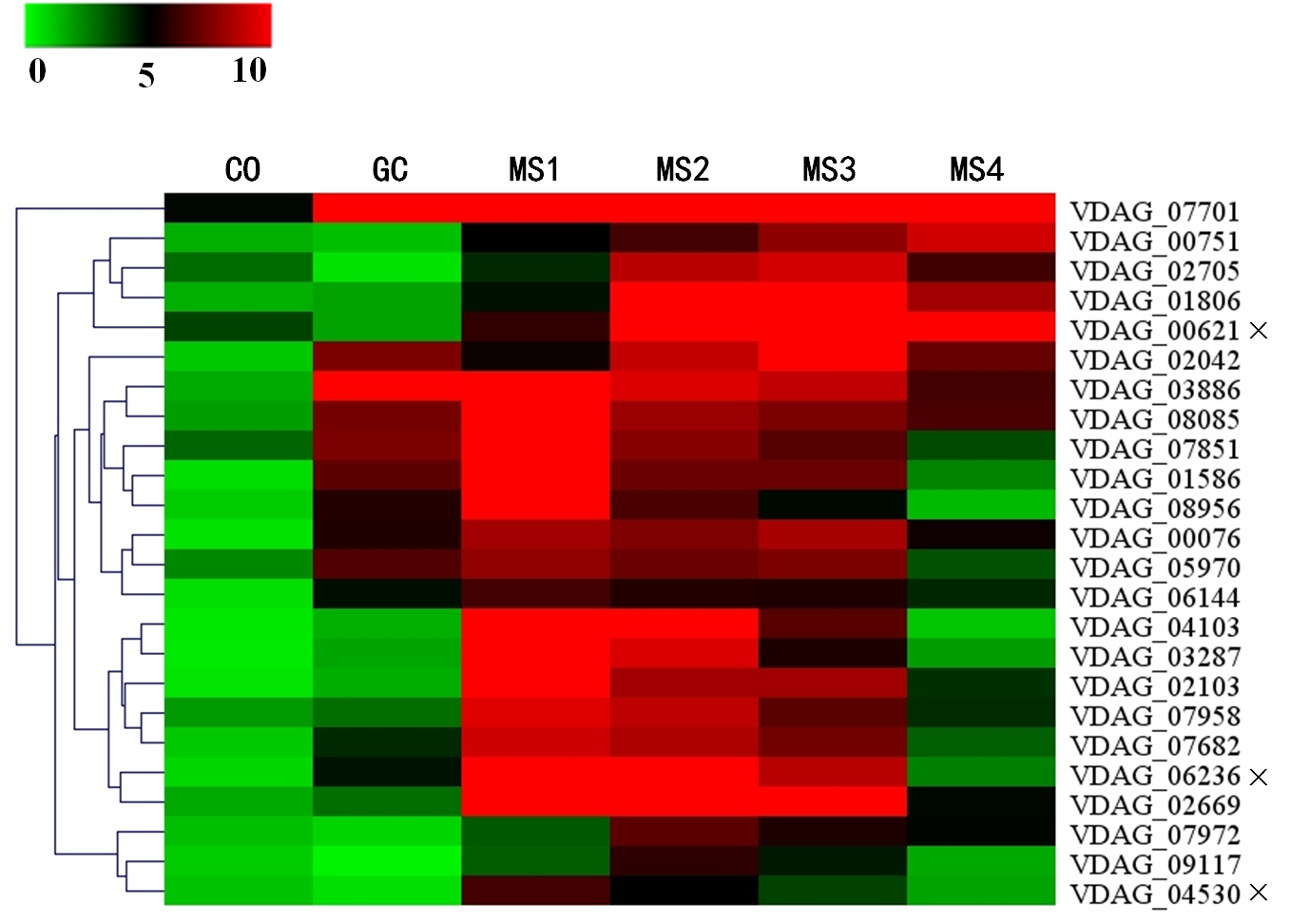
Some of the MS cells undergo autolysis or death during MS formation in V. dahliae[8]. According to the GO categories and enrichment analysis, there were more than twenty genes involved in protein metabolic processes, most of which participated in proteolysis. Among the genes involved in protein metabolic processes, five were involved in proteasome formation, and included VDAG_00111 (proteasome subunit alpha type 6), VDAG_08991 (proteasome component pup2), VDAG_02924 (proteasome component pre3 precursor), VDAG_03131 (proteasome component pup3), VDAG_04256 (proteasome subunit beta type 7 precursor) (Additional file 9: Figure S6A). Genes encoding products involved in protein modification processes were also enhanced during MS formation, such as ubiquitination, important for proteasome-mediated degradation of proteins [52].
We identified 51 genes involved in ubiquitin-dependent protein catabolic processes in V. dahliae using homology searches (Figure 6A). Expression profiles of the ubiquitin-dependent genes showed that almost 90.0% of these genes were up-regulated during MS formation (Figure 6A), suggesting that ubiquitination and subsequent protein degradation might play an important role in MS formation. The autophagy-related genes ATG3, ATG7, ATG8, ATG10 and ATG12 are involved in ubiquitin-like protein (UBL) degradation processes [52, 53]. Orthologous genes were identified in V. dahliae, such as VDAG_02434 (ATG3), VDAG_00045 (ATG7), VDAG_01225 (ATG8), VDAG_03183 (ATG10), VDAG_10057 (ATG12). Figure 6B shows the expression patterns in which the genes of the left branch of the UBL protein degradation process showed small changes in expression during MS formation, while those genes on the right branch showed relatively large expression changes, especially VDAG_03183. These results suggest that the proteolysis process was induced during MS formation, potentially contributing to protein degradation during MS formation.
Figure 6.
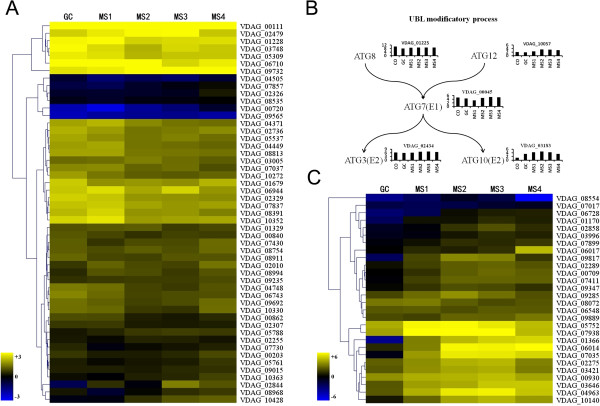
Ubiquitin-dependent protein catabolism and cell death. A. The heatmap shows levels of transcription abundance from genes involved in ubiquitin-dependent protein catabolism during microsclerotia formation (stages MS1-MS4). Levels of expression are represented as the log2 ratio of transcript abundance as compared to the CO stage. Genes showing similar expression patterns have been clustered. B. Two ubiquitin-like protein (UBL) modification pathways in V. dahliae and expression levels of genes encoding UBL proteins. For each protein, the bar graph shows abundance of transcripts encoding this protein in CO, GC and microsclerotia formation (MS1-MS4, black bars left to right). C. Heatmap of genes encoding a heterokaryon incompatibility protein (HET) domain.
Other genes involved in autophagy and cell death in V. dahliae were identified. In total, 23 autophagy genes were identified in V. dahliae, which were then divided into two groups, nonselective autophagy and selective autophagy [54]. Expression profiles of these genes showed stable expression patterns across developmental stages examined, except VDAG_03183 (nonselective) and VDAG_01208 (selective), which were up-regulated during MS formation (Additional file 9: Figure S6B). In addition, some genes encoding the Het domain were up-regulated during MS formation in V. dahliae, although over 90.0% of the Het-related genes were neither up- nor down-regulated during MS formation (Figure 6C). Vegetative incompatibility is an apoptotic-type heterokaryon incompatibility, and proteins containing a Het domain mediate the apoptotic-type pathway [55–59]. Potentially, cell death observed during MS formation in V. dahliae[11] is regulated through mechanisms that also mediate vegetative incompatibility.
Analyses of lineage-specific gene expression during MS development
There exist four lineage specific (LS) regions in V. dahliae strain VdLs.17, and these regions are of interest since they encode 354 predicted protein-encoding genes that play important roles such as secondary metabolism, transcription, and pathotype specificity [40]. To reveal changes in the expression of genes located in LS regions in the transcriptome during MS formation, the global gene expression profile in the V. dahliae genome in the six samples was visualized by Circos [60]. The genome-wide analyses of gene expression indicated that the majority of genes were expressed with FPKM values ranging from 10 to 100, and the expressed genes were uniformly distributed in the V. dahliae genome (Figure 7). However, the non-expressed genes were mainly distributed in the supercontig 4, supercontig 8 and supercontig 9 on Chr 3, and supercontig 23 on Chr 4 in all the six stages (Figure 7, red circles). Interestingly, the four LS regions in VdLs.17 are located within these particular supercontigs (Figure 7, red circles).
Figure 7.
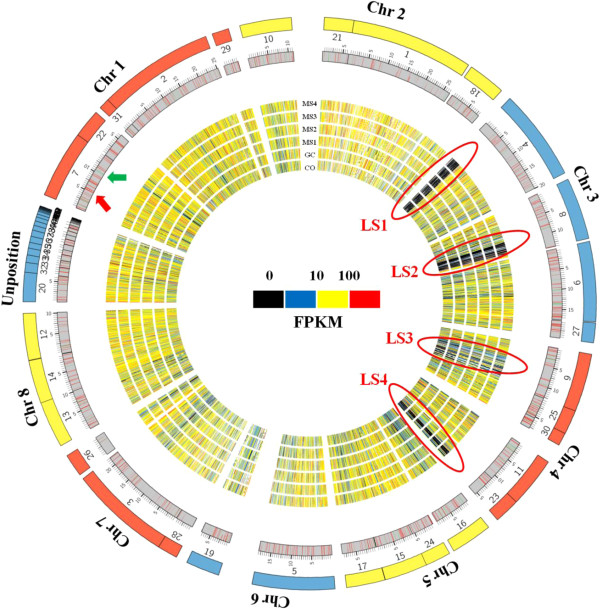
Global view of gene expression in hyphae, conidia, germinating conidia, and microsclerotial formation stages MS1-4. Seven concentric circles from inside to outside represent fungal stages CO, HY, GC, MS1, MS2, MS3 andMS4, respectively. Color representation (from black to red) illustrates the expression level (FPKM, fragments per kilobase of transcripts per million mapped fragments) of each transcript in each sample. The red and green colored bars in the gray circle represent genes significantly up- or down-regulated during microsclerotia development compared to the CO stage (q value <0.05), respectively. Each chromosome is shown by the number of the first outer circle, which represents the V. dahliae strain VdLs.17 scaffold number assigned by the Broad Institute. Red ovals represent the four LS regions in the V. dahliae, marked as LS1, 2, 3, 4. Numbers under the outer circle correspond to the respective supercontigs of the V. dahliae genome.
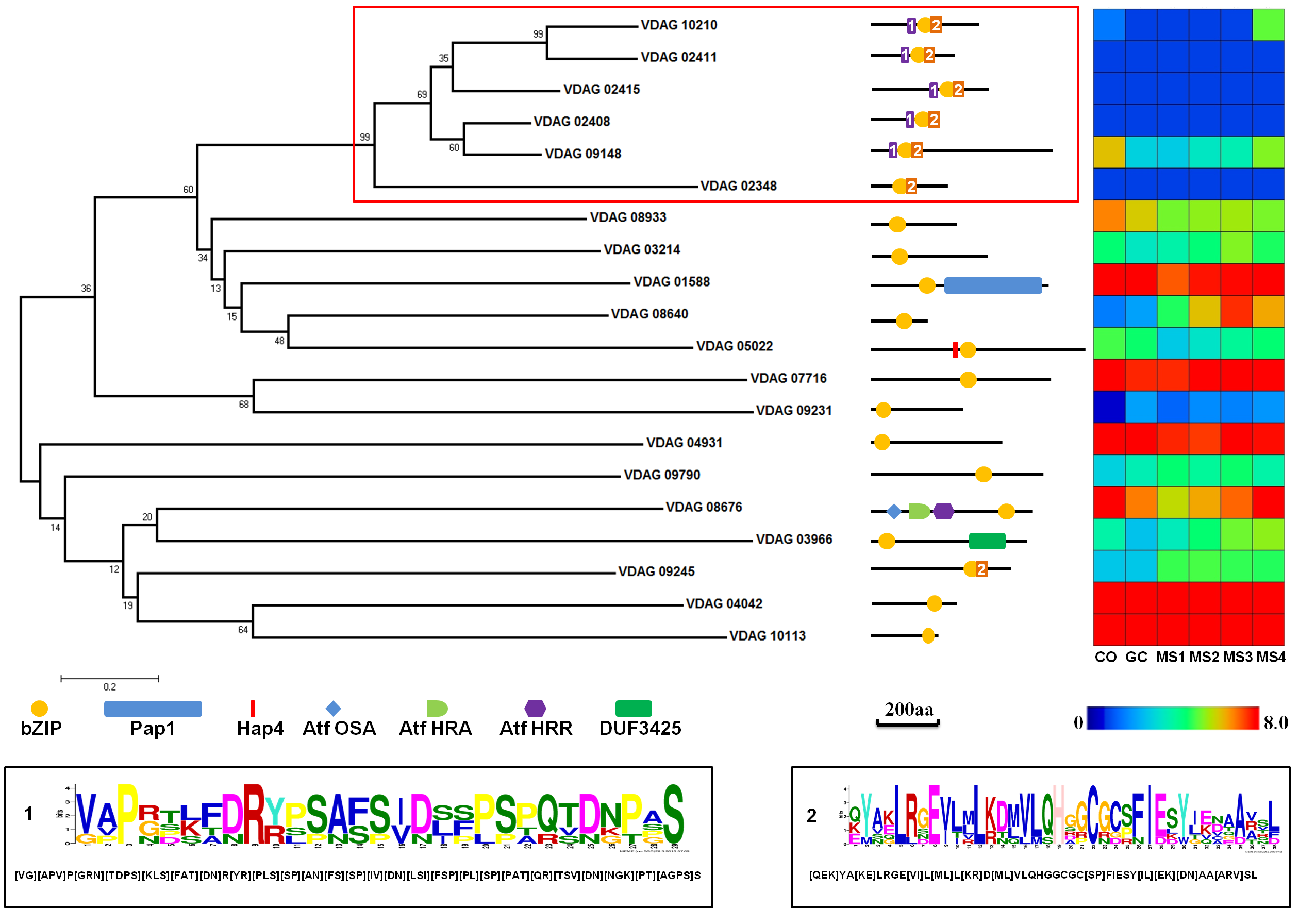
Transcription factors encoded within the LS regions potentially regulate gene expression of not only LS-associated genes, but also other core genes in the genome of V. dahliae that have roles in MS development. We followed the expression patterns of bZIP transcription factors to ascertain a potential role of these genes in regulating development in V. dahliae. Among 20 bZIP transcription factor-encoding genes of V. dahliae, strain VdLs.17, five genes (VDAG_02348, VDAG_02408, VDAG_02411, VDAG_02415 and VDAG_09148) are located within the LS regions and were separated in a distinct clade with other bZIP transcription factors in a phylogenetic analysis (Additional file 10: Figure S7), similar to those results previously reported [40] All five homologs of these LS-encoded genes, with the exception of VDAG_09148, were not differentially expressed during MS formation in the smoke tree strain of V. dahliae. Furthermore these genes are Verticillium-specific (Additional file 11: Figure S8).
Expression profiles of genes involved in carbohydrate metabolism and melanin biosynthesis during MS formation
Genes involved in carbohydrate metabolic process were significantly up-regulated during the MS formation based on GO annotation analysis. To further study the dynamics of hexose metabolism during MS formation, glycolysis/gluconeogenesis, tricarboxylic acid, pentose phosphate and glyoxylate pathways were examined (Figure 4A, Additional file 6: Table S2). Expression profiles showed that genes controlling hexose metabolism pathways were differentially expressed during MS formation (Additional file 12: Figure S9). For example, two key enzyme encoding genes VDAG_04087 (hexokinase) and VDAG_01206 (pyruvate kinase) in glycolysis were significantly up-regulated during MS1-MS4 stages, indicative of an up-regulation of glycolysis during MS formation. Conversely, two genes that are critical for gluconeogensis (VADG_07446 and VDAG_10101) were down-regulated.
Acetyl-CoA is an important intermediate product of metabolism and plays a very important role in the cell’s energy requirement, metabolic pathways, and appressorium formation in rice blast disease [39]. RNA-Seq analyses of genes expressed in MS formation revealed up-regulation of four genes in strain XS11 of V. dahliae (corresponding to VDAG_08164, VDAG_09433, VDAG_06356 and VDAG_01642) that are candidate genes involved in catalyzing the pyruvate to generate acetyl-CoA, suggesting an increase in the amount of acetyl-CoA during MS formation.
The polyketide pathway is one of the most important metabolic processes involved in the growth, development, and pathogenicity in filamentous fungi. Melanin is a prevalent polyketide in pathogenic fungi, and melanin biosynthesis is well studied in pathogenic fungi [61, 62]. Genes involved in melanin biosynthesis were identified in V. dahliae by homology searches, including polyketide synthase (VDAG_00190), scytalone dehydratase (VDAG_03393) and hydroxynaphthalene reductase (VDAG_03665). Expression profiles of these and other genes encoding enzymes of the melanin biosynthesis pathway are shown in Figure 8. VDAG_00190, VDAG_03393 and VDAG_00183 homologs in strain XS11 were all highly expressed and significantly up-regulated (over 10-fold change) during the MS formation as compared with stage CO (Figure 8). The expression profile was consistent with phenotypic observations of MS formation, during which the accumulation of melanin was dramatically increased at stages MS3 and MS4 (Figure 1A). Decreases in the expression levels of VDAG_03674, VDAG_00190, VDAG_03393 and VDAG_00183 were observed in MS4 as compared to MS3 in strain XS11, at the completion of melanin deposition in the MS (Figure 8).
Figure 8.
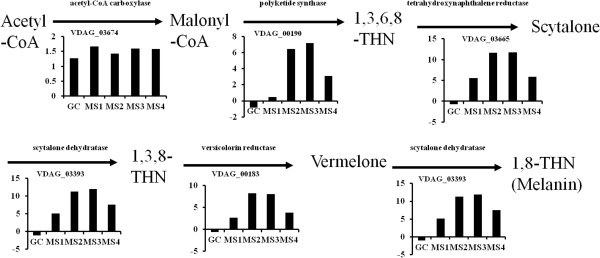
RNA-Seq reveals significantly high transcript abundance of genes involved in melanin biosynthesis. The diagram shows the process of fungal melanin biosynthesis as described by Soanes et al. [39]. For each enzyme, the histograms show the abundance of transcripts encoding enzymes in melanin biosynthesis, gene expression levels calculated by log2 FPKM of transcript abundances in stages GC, MS1, MS2, MS3 and MS4 compared with the CO stage.
Discovery of alternative splicing
Alternative splicing (AS) for the creation of new gene transcripts can increase regulatory complexity and control of developmental programs in higher organisms. A software SplicingViewer [63] was employed to identify putative AS events in the six different developmental stages of V. dahliae that were examined in this study. This analysis tool enabled detection of potential splicing junctions and identification of seven putative types of AS events, such as skipped exons (SE), mutually exclusive exons (MXE), alternative 5′ splicing sites (A5SS), alternative 3′ splicing sites (A3SS), alternative first exons (AFE), alternative last exons (ALE) and retained intron (RI) [64]. More than 50.0% (5,212) expressed genes were detected in the existing AS events representing 9,167 AS events in the six stages examined (Figure 9, Additional file 13: Table S4). Four primary types of AS events, namely, retained intron (RI), skipped exons (SE), alternative 5′ splicing sites (A5SS), alternative 3′ splicing sites (A3SS) were common in V. dahliae. The other three types of AS events were sporadic, and mutually exclusive exons (MXE) were rarely observed in V. dahliae. Furthermore, 44.8% (2,337) of the AS genes produced more than two isoforms (Additional file 14: Table S5). More than ten alternative transcripts were identified for a single gene, such as the VDAG_07349 homolog, encoding a cysteine-rich-protein that contains 14 exons. There were 31 different isoforms of VDAG_07349 that were observed, as concluded from all the six developmental stages examined. Over 95.0% of the AS events were RI in all stages, and the other AS events consisted of less than 1.0%, respectively (Figure 9), suggesting that the RI was the most common event observed in V. dahliae. We performed RT-PCR to validate the RI events of three genes, i.e. for homologs of VDAG_02669, VDAG_03183, VDAG_03665. All three genes exhibited alternatively spliced transcripts, consistent with the results of RNA-Seq (Additional file 15: Figure S10).
Figure 9.
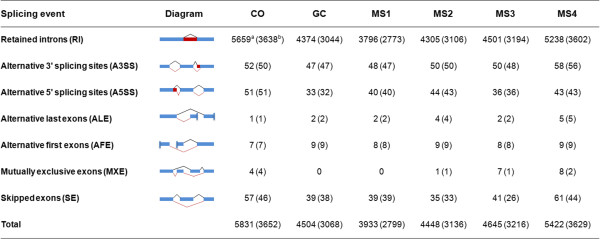
Statistics of alternative splicing events. a, represents number of alternative splicing events in each stage. b, represents number of genes that undergo alternative splicing events in each stage in parentheses.
Discussion
The formation of MS in V. dahliae is critically important for survival and propagation of this fungus, representing a significant developmental event in the disease cycle. The long term survival of the MS in soil poses a major obstacle for effective control of Verticillium wilts through cultural practices such as crop rotation. The structure of the MS, including a thickened cell wall and heavy melanin deposition, protects the pathogen from various environmental assaults and even fungicidal activity [5]. This study was undertaken to begin elucidating of the genetics and biochemical processes underpinning MS development, which may yield insight into control strategies to combat vascular wilt disease of smoke trees, and other plant hosts of economic or aesthetic importance.
In this work, RNA-Seq was performed on samples from six developmentally distinct stages, ranging from conidia (CO) and germinating conidia (GC) through four stages of MS formation and maturation (MS1 to MS4). Due to the actual difficulties of obtaining MS from real condition such as in plant or soil, especially during MS development, we obtained MS in vitro. The rationale for this approach was to define MS-associated gene expression during MS formation. Analyses of MS formation on an artificial surface ensured that gene expression data were exclusively from the fungus, rather than the smoke tree host. It is also technically difficult to sample different developmental stages of MS in plant tissues and soil. Likewise, MJ Neumann and KF Dobinson [42] employed a method to collect MS samples for studies of gene expression during MS development in V. dahliae using EST analyses from in vitro cultures. Given the limitations of EST-based analyses and advancements in NGS technologies, the dynamics of transcriptome expression during MS formation were studied using the relatively new NGS technology of RNA-Seq. This technology enabled identification of metabolic pathways of interest, as well as alternative splicing. Analyses of these transcriptome data revealed that this was an effective approach, providing an extensive catalog of expression values over the range of six distinct developmental stages. Moreover, of the 158.5 million sequenced reads, 114.8 million reads uniquely mapped to the genome without mismatch, and 90.1 million reads mapped perfectly to the annotated genes of the V. dahliae strain VdLs.17 sequence.
The increased expression of particular genes observed in this study was expected; including those involved in melanin biosynthesis, or selected genes that were previously identified as important in MS formation. Melanin biosynthetic genes in V. dahliae were expressed at relatively high levels during MS formation consistent with the culture phenotype (increased pigmentation) observed. In addition, VDH1 (VDAG_02273), a hydrophobin protein-encoding gene, is involved in MS formation, and VDH1 is specifically expressed in developing microsclerotia [12, 45]. In concurrence with the results of that study, the expression level of VDH1 was significantly increased in the stages of MS1-MS3, especially in MS1 and MS2 (Additional file 16: Table S6). The finding of the developmentally regulated expression of VDH1 in this study may also explain the lack of differential expression of VDH1 observed in RNA-Seq analyses of gene expression in which only 10 day MS-producing cultures and 10 day cultures not forming MS were compared [43]. In the study of Duressa et al. [43] stages equivalent to MS1 or MS2 were not analyzed.
Analyses of GO functional category enrichment indicated that carbohydrate and protein metabolic processes, and ribonucleoprotein biosynthetic processes were significantly enriched among up-regulated genes, while transport processes were significantly enriched among down-regulated genes. Functional classification revealed that peptidase, protein kinase, and hydrolase activities were significantly up-regulated during MS1-MS4 stages. Among the carbohydrate metabolic processes, several genes involved in energy production were enriched; such as VDAG_04087 (hexokinase), VDAG_01206 (pyruvate kinase), and VDAG_03029 (enolase). These results suggested that these differentially expressed genes are involved in a broad range of physiological functions, especially in proteolysis, protein modification, during MS formation. Enrichment of peptidase activity may also accompany processes associated with cell death or autolysis in MS formation [11]. On the other hand, most genes encoding plant cell wall-degrading enzymes, such as the polysaccharide lyase and carbohydrate esterase gene families, showed low expression levels during MS formation. The carbohydrate-binding module 1 gene family, which is significantly enriched in V. dahliae relative to most other fungi [40], also showed low expression at the MS developmental stages analyzed in this study.
Stage-specific differences were extensive in V. dahliae at the gene regulatory level during MS development. Among the genes annotated in V. dahliae, strain VdLs.17, dozens of genes that were differentially expressed during MS formation were identified (Additional file 7: Figure S5). This indicated that MS-stage-specific gene regulation occurs at the transcriptome level during MS formation. These data provided a basis for further assessing the roles of individual genes, especially those MS-specific genes potentially involved in cellular differentiation and MS formation.
The four major lineage-specific (LS) regions identified in the genome of strain VdLs.17 of V. dahliae are thought to contribute to adaptation to different host niches [40] and encode virulence factors [41]. While the total LS region contents differ between strains of V. dahliae[41], a total of 354 genes are located in the four LS regions of strain VdLs.17; and some of these genes encode proteins involved in lipid metabolism, plant-fungal interactions, and transcriptional regulation [40]. Interestingly, analyses of three EST libraries produced under low nutrient or complete medium revealed the LS genes of VdLs.17 were significantly up-regulated compared to genes in the genome core sequence. Furthermore, recent work indicates enrichment for in planta-expressed LS genes in the V. dahliae strain JR2 [41]. In this study, the majority of genes in LS regions of V. dahliae were either not expressed or showed low expression in MS1 to MS4, suggesting that genes of the LS regions are suppressed at the developmental stages examined relative to those of the genome core, and also may be influenced by the differing gene content of LS regions between different strains examined. Consistent with this hypothesis, expression of VDAG_02354, encoding a high osmolarity glycerol response (HOG1) protein, and located in LS region 1, was not detected in all six developmental stages examined in this study. The other copy of the HOG1 gene (VDAG_08982 was identified outside of the LS regions in the genome of VdLs.17 and expressed at a higher level during MS formation. The deletion of the HOG1 gene (VDAG_08982) from the smoke tree strain resulted in significantly reduced and delayed production and developmental progress of MS in vitro (Xiao et al., unpublished data). Additional analyses revealed that genes encoding LS region-associated bZIP transcription factors were not expressed, or showed low expression in the stages of MS formation, while other bZIP transcription factors located in non-LS regions were expressed during MS formation.
The RNA-Seq analysis provided useful information on alternatively spliced transcripts in V. dahliae. These analyses are critical for studies that aim to assess gene regulation and gene function in MS development and in other developmental processes. The number of genes undergoing AS were mainly estimated based on overlapping introns, which does not take intron retention and splice junctions into account [65]. Over 95.0% AS events were due to intron retention. As in other fungi, such as Aspergillus oryzae and Ustilago maydis, RI is the predominant form of AS [66, 67]. A total of 5,212 genes underwent AS events, while there was an average of 3,250 genes that underwent AS events in each stage. This suggests that stage-specific genes undergo AS at different times during MS development. Further screens of these genes are required to increase understanding the role of AS in the development and pathogenesis in the smoke tree wilt fungus. Assembly of transcription from short sequencing reads remains a computational challenge. Therefore, it is essential to validate the predicted AS events by laboratory and field experiments.
In summary, an RNA-Seq strategy was employed to gain insight on the biology and molecular basis of MS development of smoke tree vascular wilt fungus, V. dahliae. Functional categories of genes such as those involved in carbohydrate metabolism, proteolysis, and cell death were differentially regulated during MS formation. Comprehensive, high-resolution gene expression maps enabled detection of a large number of AS events that provide a key resource for further studies that aim to understand the molecular underpinnings of MS development and other developmental processes in this fungus. Further, de novo assembly of transcripts from RNA-Seq data represents a potential avenue for gene annotation [68–71]. The analysis of splicing events detected herein may shed light on alternate splicing in V. dahliae, and help to understand the roles of AS in MS formation and other developmental processes.
Conclusion
In this study, we have conducted a RNA-Seq analysis of the MS developmental process in smoke tree wilt fungus V. dahliae XS11. A global view of gene expression profiles and a large-scale stage-specific transcriptome alterations during MS development are revealed. Further analysis show that genes involved in glycolytic pathway, melanin biosynthesis and protein catabolism are dramatically up-regulated in MS stages. In addition, a large number of AS events are detected among CO, GC and MS stages. Our results provides a key resource for understanding the biological and molecular basis of MS development of V. dahliae.
Methods
Fungal strain and growth conditions
Verticillium dahliae strain XS11, which was single spore-isolated from a smoke tree in Fragrant Hills Park, Beijing, was used in these experiments. Cultures were initially grown on PDA (potato dextrose agar). Conidia were harvested from cultures grown in liquid CM, as previously described [72]. Conidia were collected by filtering through two layers of Miracloth (Calbiochem, USA), and the conidial suspension was sedimented by low speed (4000 rpm) centrifugation. The conidia were cultured at a concentration 105 conidia/ml for germination in the liquid basal medium (BM, 10 g/L glucose, 0.2 g/L sodium nitrate, 0.52 g/L KCl, 0.52 g/L MgSO4.7H2O, 1.52 g/L KH2PO4, 3 μM thiamine HCl, 0.1 μM biotin, 15 g/L agar, kindly provided by Dr. Katherine Dobinson, Agriculture and Agri-Food Canada, London, Canada) by shaking at 30 rpm for 12 hours at 24°C. Germinating conidia were harvested similarly.
Microsclerotial developmental stages
To observe the developmental process of MS formation of V. dahliae in stages MS1-MS4, a cellulose membrane (Ø =80 mm; pore size = 0.22 μm) was placed on BM agar and a suspension of 105 conidia/ml of V. dahliae strain XS11 was spread over the cellulose membrane, and incubated in the dark at 24°C. Developmental stages were observed under light microscopy (DM2500, Leica) at 12 hour intervals after incubation on the membrane until 7 days post incubation (dpi). After 7 dpi, the observations were conducted every 2 days for 1 week.
RNA extraction and validation of expression by RT-PCR and qRT-PCR
Total RNA was extracted from conidia, germinating conidia, and the MS1-MS4 stages of MS formation by using TRIzol Reagent (Invitrogen) and purified with the RNA Mini Kit (Ambion) according to the manufacturer’s instructions. All samples were ground to a fine powder with a mortar and pestle in liquid nitrogen. Total RNA was eluted in RNase-free water and stored at −80°C until further use. For each sample, two biological replicates were used for library preparations. The integrity and quantity of RNA was determined using a Qubit fluorometer (Invitrogen), agarose electrophoresis and the Agilent Bioanalyzer 2100 (Additional file 17: Table S7).
The RNA samples for RT-PCR were incubated at 37°C for 30 min with DNase I (RNase-free) (TaKaRa) to remove DNA contamination before reverse transcription. The mRNA were enriched using Oligo DT, then were transcribed to cDNA using SuperScript III Reverse Transcriptase (Invitrogen). The qRT-PCR was carried out using SYBR green (SuperReal Premix Plus; TIANGEN, China) methodology and the ABI 7500 real-time PCR system (Applied Biosystems, USA). The V. dahliae β-tublin gene was used as internal reference for all the qPCR analyses. Analyses of each gene were conducted in quadruplicate. Relative gene expression was calculated according to the ΔΔCT method. The primers used are described in Additional file 18: Table S8.
Library preparation for RNA-Seq
Libraries were prepared using RNA-Seq sample preparation kit from Illumina and poly(A) mRNA was enriched from total RNA using oligo (dT) beads. Fragmentation buffer was added for breaking mRNA to short fragments. Using these fragments as template, first-strand cDNA was synthesized by reverse transcription with a random hexamer primer. The second-strand cDNA was synthesized using buffer, dNTPs, RNase H and DNA polymerase I according to kit manufacturer instructions.. Short fragments were purified with QiaQuick PCR extraction kit (Qiagen) and resolved with EB buffer. Sequencing adaptors were added and amplified with PCR. Agarose gel electrophoresis was used to select the fragments with about 200 bp in size. Finally, the libraries were sequenced on Illumina HiSeq™ 2000 (Beijing Genomics Institute, Shenzhen) to produce 90 bp paired-end reads.
Mapping reads to the V. dahliae reference genome
Low quality (Q ≤5) reads containing adapters were removed from the raw reads, and Tophat software (version 2.0.0) [49] was used to align the filtered reads to the published reference genome of V. dahliae strain VdLs.17 (http://www.broadinstitute.org/annotation/genome/verticillium_dahliae/MultiHome.html) and to predict exon splice sites allowing less than two mismatches. The pipeline of RNA-Seq analysis is shown in Figure 1B. Total mapped reads were obtained with the parameters “-G (Vd) -r 20 --segment-length 30” provided by Tophat and perfect mapped reads were obtained using the parameters “-G (Vd) –r 20 --segment-length 30 –read-mismatches 0” provided by Tophat. In addition, reads only mapped to the annotated genes of V .dahliae were carried out with the parameters “-T -G (Vd) –r 20 --segment-length 30”.
Transcript assembly and genes expression analysis
Cufflinks software (version1.30) [73] was used to assemble the individual transcripts from RNA-Seq reads which had been aligned to the genome of V. dahliae, strain VdLs.17, with Tophat. Gene expression level was calculated using FPKM (fragments per kilobase of transcript per million mapped fragments) in Cufflinks. Due to their low reliability for assembly purposes, Cufflinks filtered low abundance transcripts using the default parameter. Cuffmerge, a component of Cufflinks, was used to merge the transcripts of several samples. Cuffdiff, a package of Cufflinks, was used to identify differentially expressed genes among samples (p value ≤0.05), and the CummeRbund R package [74] was used to visualize differentially expressed genes. Fold changes in gene expression were calculated with log2 FPKM compared with that of the control sample, CO. A MultiExperiment Viewer [50] was used to visualize changes in gene expression, cluster analyses, PCA analyses and CAST analyses with threshold affinity value 0.9 and other default parameters. The global views of gene expression patterns were visualized by Circos [60]. Pearson correlation coefficient was calculated among the six samples according to genes’ expression profiles. Venn diagram was drawn through the interactive tool of VENNY [75].
Gene ontology and functional annotation
Gene Ontology was identified in the GO database through Blast2GO [76] software using the in silico translated sequence and default parameters. In addition, functional annotation, classification, and enrichment analysis were performed by Blast2GO software with the default parameters.
Alternative splicing analysis
Alternative splicing events were identified by SplicingViewer [63]. First, reads were aligned to the reference genome by Bwa allowing less than two mismatches [77]. Second, Samtool was used to obtain unmapped reads from the first step [78], to detect the splice junctions, and obtain the splice junction sequences. Unmapped reads were mapped to the splice junction sequences by Bwa with less than two mismatches. Seven types of alternative splicing events were identified by AlternativeSplicing.jar (http://bioinformatics.zj.cn/splicingviewer/index.php).
Phylogenetic analysis
Amino acid sequences were aligned using Clustal X (version 1.83) without masking unreliable aligned positions [79]. Phylogenetic trees were constructed using Mega 5.0 by Maximum Likelihood method with at least 1000 bootstrap replications [80].
Availability of supporting data
RNA-Seq data were submitted to the NCBI SRA database (http://www.ncbi.nlm.nih.gov/Traces/sra/) with the accession number: SRR1232601, SRR1232602, SRR1232630, SRR1232631, SRR1232632, SRR1232674, respectively.
Electronic supplementary material
{kind=link}
Additional file 1: Figure S1: Overview of the RNA-Seq data. All of the developmental stages share similar sets of mapped reads. The number of reads produced from the Illumina platform (blue bar); the number of reads mapped to the reference genome within 2 bp mismatch (red bar); the number of reads perfectly mapped to the reference genome (green bar); the number of reads mapped to the annotated genes (purple bar). (PNG 62 KB)
{kind=link}
Additional file 2: Figure S2: Validation of RNA-Seq expression patterns. The RT-qPCR results of the selected genes show similar expression patterns to those detected by RNA-Seq. (PNG 68 KB)
Additional file 3: Table S1: GO enrichment analysis of genes in 18 clusters (p_value < 0.01). (XLSX 12 KB)
{kind=link}
Additional file 4: Figure S3: Expression profiles of MAPK cascades. Heatmap showed that four types of MAPK cascades during MS formation. Expression pattern of genes encoding high osmolarity (HOG1) pathway were clustered together. The non-LS HOG1-MAPK maintained the highest expression value compared to other MAPKs. (PNG 685 KB)
{kind=link}
Additional file 5: Figure S4: Expression profiles of small secreted proteins (length ≤300aa). Expression profiles of those genes encoding small secreted proteins with at least 50-fold up-regulated during MS formation compared with the CO stage. The levels of expression represent log2 FPKM Value +1. * represents genes with cysteine residues <4. (PNG 317 KB)
Additional file 6: Table S2: List of genes involved in metabolism, signal pathway and identified transcription factor. (XLSX 161 KB)
{kind=link}
Additional file 7: Figure S5: Functional categorization of genes differentially expressed (up- or down-regulated) during microsclerotia development. (A, C, E) Intersection of MS1-4 stages revealed 600 significantly up-regulated genes vs CO stage, and functional categorization of these genes. (B, D, F) Intersection of MS1-4 stages revealed 124 significantly down-regulated genes vs CO stage, and functional categorization of these genes. (PNG 584 KB)
Additional file 8: Table S3: Significantly regulated genes during microsclerotia formation vs CO stage (p_value < 0.05). (XLSX 46 KB)
{kind=link}
Additional file 9: Figure S6: Genes involved in protein metabolic processes and autophagy. A. Genes involved in protein metabolic processes by functional categorization; genes labeled with asterisks are the subunits associated with proteasome formation. B. Heatmap representation of genes involved in autophagy processes. Heatmap shows levels of transcripts abundance; relative levels of expression are presented by moderated log2 ratio of transcript abundance in MS developmental stages relative to the CO stage. (PNG 747 KB)
{kind=link}
Additional file 10: Figure S7: Phylogenetic analysis and expression profile of bZIP transcription factors of V. dahliae. The full-length amino acid sequences of bZIP transcription factors of V. dahliae strain VdLs.17 were aligned using Clustal X and the phylogenetic tree was constructed using Mega 5.0 using the maximum-likelihood method with 1000 replicates. Domain structures are drawn to represent their relative positions. The black solid line represents the corresponding protein and its length. The different-colored boxes represent different domains and their positions in each protein predicted by Sanger Pfam program (http://pfam.xfam.org/) and the yellow ovals represent bZIP domains. The red box indicates an independent cluster of bZIP transcription factors genes, which contain two specific motifs represented at the bottom. The right panel represents heat maps showing the expression pattern of bZIP transcription factors of V. dahliae. (PNG 522 KB)
Additional file 11: Figure S8: Phylogenetic analysis of bZIP transcription factors of V. dahliae and other fungi. The amino acid sequences of bZIP transcriptional factors of V. dahliae strain VdLs.17, and other fungi were aligned using Clustal X and the phylogenetic tree was constructed using Mega 5.0 using the maximum-likelihood method with 1000 replicates. Fungal species are Sc, Saccharomyces cerevisiae; Vd, Verticillium dahliae; Va, Verticillium alfalfae (formerly V. albo-atrum); Fv, Fusarium verticillioides; Fo, Fusarium oxysporum; Fg, Fusarium graminearum; Mg, Magnaporthe oryzae; Nc, Neurospora crassa. Pa, Podospora anserine; Hj, Hypocrea jecorina. (PDF 42 KB)
{kind=link}
Additional file 12: Figure S9: Expression patterns of genes involved in glycometabolism. Genes involved in carbohydrate metabolism including Glycolysis/Gluconeogensis, Glyoxylate cycle, TCA cycle, Pentose phosphate pathway. Heatmap shows levels of transcripts abundance; Level of expression are presented by moderated log2 ratio of transcript abundance vs CO stage. (PNG 556 KB)
Additional file 13: Table S4: List of genes undergoing AS events. (XLSX 575 KB)
Additional file 14: Table S5: Statistics of the AS events genes. (XLSX 9 KB)
{kind=link}
Additional file 15: Figure S10: RT-PCR validation of three genes undergo RI events. Primers (black arrows) were designed spanning intron region. gDNA represents genomic DNA. cDNA represents complementary DNA. (PNG 163 KB)
Additional file 16: Table S6: Gene expression value of all annotated genes. (XLSX 804 KB)
Additional file 17: Table S7: Quality of Total RNA by Agilent 2000. (XLSX 11 KB)
Additional file 18: Table S8: RT-PCR primers used in this study. (XLSX 11 KB)
Acknowledgement
We are grateful to the Center for Computational Biology, Beijing Forestry University for providing the Linux platform. The research was supported by National Natural Science Foundation of China (31370013 and 31000302), the Fundamental Research Funds for the Central Universities (NO. TD2011-06 and YX2013-10), and the Research Fund for the Doctoral Program of Higher Education (20100014120018) to YW.
Abbreviations
- NGS
Next generation sequencing
- CO
Conidia
- GC
Conidia germination
- MS1
Microsclerotia formation 60 h
- MS2
Microsclerotia formation 72 h
- MS3
Microsclerotia formation 96 h
- MS4
Microsclerotia formation 14d
- CM
Complete medium
- GO
Gene Ontology
- dpi
days post incubation
- FPKM
Fragments per kilobase of transcript per million mapped fragments
- UBL
Ubiquitin-like protein
- CWDEs
Cell wall degrading enzymes
- ESTs
Expressed sequence tags
- ORF
Open reading frame
- MAPK
Mitogen-activated protein kinase
- HOG1
High osmolarity glycerol
- GPCR
G protein–coupled receptor
- RGS
Regulator of G-protein signaling
- SE
Skipped exon
- MXE
Mutually exclusive exon
- A5SS
Alternative 5′ splicing site
- A3SS
Alternative 3′ splicing site
- AFE
Alternative first exon
- ALE
Alternative last exon
- RI
Retained intron.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: YW and CT. Performed the experiments: DX and YW. Analyzed the data: DX, YW, JM and SX. Contributed analysis: YW, DX, CT and SK. Wrote the paper: YW, DX, SK and CT. All authors read and approved the final manuscript.
Contributor Information
Dianguang Xiong, Email: xiongdianguang@126.com.
Yonglin Wang, Email: ylwang@bjfu.edu.cn.
Jie Ma, Email: majie@bjfu.edu.cn.
Steven J Klosterman, Email: Steve.Klosterman@ars.usda.gov.
Shuxiao Xiao, Email: jessiecain4ever@163.com.
Chengming Tian, Email: chengmt@bjfu.edu.cn.
References
- 1.Klosterman SJ, Atallah ZK, Vallad GE, Subbarao KV. Diversity, pathogenicity, and management of Verticillium Species. Annu Rev Phytopathol. 2009;47:39–62. doi: 10.1146/annurev-phyto-080508-081748. [DOI] [PubMed] [Google Scholar]
- 2.Fradin EF, Thomma BP. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol. 2006;7:71–86. doi: 10.1111/j.1364-3703.2006.00323.x. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Xiao S, Xiong D, Tian C. Genetic transformation, infection process and qPCR quantification of Verticillium dahliae on smoke-tree Cotinus coggygria. Australasian Plant Pathol. 2013;42:33–41. doi: 10.1007/s13313-012-0172-0. [DOI] [Google Scholar]
- 4.Hawke MA, Lazarovits G. Production and manipulation of individual microsclerotia of Verticillium dahliae for use in studies of survival. Phytopathology. 1994;84:883–890. doi: 10.1094/Phyto-84-883. [DOI] [Google Scholar]
- 5.Bell AA, Wheeler MH. Biosynthesis and functions of fungal melanins. Annu Rev Phytopathol. 1986;24:411–451. doi: 10.1146/annurev.py.24.090186.002211. [DOI] [Google Scholar]
- 6.Wilhelm S. Longevity of Verticillium wilt fungus in the laboratory and the field. Phytopathology. 1955;45:180–181. [Google Scholar]
- 7.Pegg G, Brady B. Verticillium Wilts. Wallingford, Oxfordshire: CABI Publishing; 2002. [Google Scholar]
- 8.Griffiths DA. The fine structure of developing microsclerotia of Verticillium dahliae Kleb. Arch Mikrobiol. 1970;74:207–212. doi: 10.1007/BF00408881. [DOI] [Google Scholar]
- 9.Perry JW, Evert RF. Structure of microsclerotia of Verticillium dahliae in roots of ‘Russett Burbank’ potatoes. Can J Bot. 1982;62:396–401. doi: 10.1139/b84-060. [DOI] [Google Scholar]
- 10.Wheeler MH, Tolmsoff WJ, Meola S. Ultrastructure of melanin formation in Verticillium dahliae with (+)-scytalone as a biosynthetic intermediate. Can J Microbiol. 1976;22:702–711. doi: 10.1139/m76-103. [DOI] [PubMed] [Google Scholar]
- 11.Griffiths DA. The fine structure of Verticillium dahliae Kleb. colonizing cellophane. Can J Microbiol. 1971;17:79–81. doi: 10.1139/m71-013. [DOI] [PubMed] [Google Scholar]
- 12.Klimes A, Amyotte SG, Grant S, Kang S, Dobinson KF. Microsclerotia development in Verticillium dahliae: Regulation and differential expression of the hydrophobin gene VDH1. Fungal Genet Biol. 2008;45:1525–1532. doi: 10.1016/j.fgb.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Bell AA, Puhalla JE, Tolmsoff WJ, Stipanovic RD. Use of mutants to establish (+)-scytalone as an intermediate in melanin biosynthesis by Verticillium dahliae. Can J Microbiol. 1976;22:787–799. doi: 10.1139/m76-115. [DOI] [PubMed] [Google Scholar]
- 14.Wheeler MH, Tolmsoff WJ, Bell AA, Mollenhauer HH. Ultrastructural and chemical distinction of melanins formed by Verticillium dahliae from (+)-scytalone, 1,8-dihydroxynaphthalene, catechol, and L-3,4-dihydroxyphenylalanine. Can J Microbiol. 1978;24:289–297. doi: 10.1139/m78-049. [DOI] [PubMed] [Google Scholar]
- 15.Wheeler MH. Comparisons of fungal melanin biosynthesis in ascomycetous, imperfect and basidiomycetous fungi. Trans Br Mycol Soc. 1983;81:29–36. doi: 10.1016/S0007-1536(83)80200-9. [DOI] [Google Scholar]
- 16.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 17.Adams MD, Kerlavage AR, Fleischmann RD, Fuldner RA, Bult CJ, Lee NH, Kirkness EF, Weinstock KG, Gocayne JD, White O. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995;377:3–174. [PubMed] [Google Scholar]
- 18.Diatchenko L, Lukyanov S, Lau YF, Siebert PD. Suppression subtractive hybridization: a versatile method for identifying differentially expressed genes. Methods Enzymol. 1999;303:349–380. doi: 10.1016/S0076-6879(99)03022-0. [DOI] [PubMed] [Google Scholar]
- 19.Diatchenko L, Lau YF, Campbell AP, Chenchik A, Moqadam F, Huang B, Lukyanov S, Lukyanov K, Gurskaya N, Sverdlov ED, Siebert PD. Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Acad Sci U S A. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 21.Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, Roth R, George D, Eletr S, Albrecht G, Vermaas E, Williams SR, Moon K, Burcham T, Pallas M, DuBridge RB, Kirchner J, Fearon K, Mao J, Corcoran K. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- 22.Torres TT, Metta M, Ottenwalder B, Schlotterer C. Gene expression profiling by massively parallel sequencing. Genome Res. 2008;18:172–177. doi: 10.1101/gr.6984908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinartz J, Bruyns E, Lin JZ, Burcham T, Brenner S, Bowen B, Kramer M, Woychik R. Massively parallel signature sequencing (MPSS) as a tool for in-depth quantitative gene expression profiling in all organisms. Brief Funct Genomic Proteomic. 2002;1:95–104. doi: 10.1093/bfgp/1.1.95. [DOI] [PubMed] [Google Scholar]
- 24.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O'Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 26.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacLean D, Jones JDG, Studholme DJ. Application of ‘next-generation’ sequencing technologies to microbial genetics. Nat Rev Microbiol. 2009;7:287–296. doi: 10.1038/nrmicro2122. [DOI] [PubMed] [Google Scholar]
- 30.Bruno VM, Wang Z, Marjani SL, Euskirchen GM, Martin J, Sherlock G, Snyder M. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010;20:1451–1458. doi: 10.1101/gr.109553.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aanes H, Winata CL, Lin CH, Chen JP, Srinivasan KG, Lee SG, Lim AY, Hajan HS, Collas P, Bourque G, Gong Z, Korzh V, Aleström P, Mathavan S. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 2011;21:1328–1338. doi: 10.1101/gr.116012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr Protoc Mol Biol. 2010;89:4.11.1–4.11.13. doi: 10.1002/0471142727.mb0411s89. [DOI] [PubMed] [Google Scholar]
- 33.Metzker ML. Sequencing technologies- the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 34.Guida A, Lindstadt C, Maguire SL, Ding C, Higgins DG, Corton NJ, Berriman M, Butler G. Using RNA-seq to determine the transcriptional landscape and the hypoxic response of the pathogenic yeast Candida parapsilosis. BMC Genomics. 2011;12:628. doi: 10.1186/1471-2164-12-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riccombeni A, Butler G. Role of Genomics and RNA-seq in Studies of Fungal Virulence. Curr Fungal Infect Rep. 2012;6:267–274. doi: 10.1007/s12281-012-0104-z. [DOI] [Google Scholar]
- 36.Zhao C, Waalwijk C, de Wit PJ, Tang D, van der Lee T. RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genomics. 2013;14:21. doi: 10.1186/1471-2164-14-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teichert I, Wolff G, Kuck U, Nowrousian M. Combining laser microdissection and RNA-seq toChart the transcriptional landscape of fungal development. BMC Genomics. 2012;13:511. doi: 10.1186/1471-2164-13-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibbons JG, Beauvais A, Beau R, McGary KL, Latge JP, Rokas A. Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot Cell. 2012;11:68–78. doi: 10.1128/EC.05102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soanes DM, Chakrabarti A, Paszkiewicz KH, Dawe AL, Talbot NJ. Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 2012;8:e1002514. doi: 10.1371/journal.ppat.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klosterman SJ, Subbarao KV, Kang S, Veronese P, Gold SE, Thomma BP, Chen Z, Henrissat B, Lee YH, Park J, Garcia-Pedrajas MD, Barbara DJ, Anchieta A, de Jonge R, Santhanam P, Maruthachalam K, Atallah Z, Amyotte SG, Paz Z, Inderbitzin P, Hayes RJ, Heiman DI, Young S, Zeng Q, Engels R, Galagan J, Cuomo CA, Dobinson KF, Ma LJ. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 2011;7:e1002137. doi: 10.1371/journal.ppat.1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Jonge R, Bolton MD, Kombrink A, van den Berg GC, Yadeta KA, Thomma BP. Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 2013;23:1271–1282. doi: 10.1101/gr.152660.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neumann MJ, Dobinson KF. Sequence tag analysis of gene expression during pathogenic growth and microsclerotia development in the vascular wilt pathogen Verticillium dahliae. Fungal Genet Biol. 2003;38:54–62. doi: 10.1016/S1087-1845(02)00507-8. [DOI] [PubMed] [Google Scholar]
- 43.Duressa D, Anchieta A, Chen D, Klimes A, Garcia-Pedrajas MD, Dobinson KF, Klosterman SJ. RNA-seq analyses of gene expression in the microsclerotia of Verticillium dahliae. BMC Genomics. 2013;14:607. doi: 10.1186/1471-2164-14-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao F, Zhou B, Li G, Jia P, Li H, Zhao Y, Zhao P, Xia G, Guo H. A glutamic acid-rich protein identified in Verticillium dahliae from an insertional mutagenesis affects microsclerotial formation and pathogenicity. PLoS One. 2010;5:e15319. doi: 10.1371/journal.pone.0015319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klimes A, Dobinson KF. A hydrophobin gene, VDH1, is involved in microsclerotial development and spore viability in the plant pathogen Verticillium dahliae. Fungal Genet Biol. 2006;43:283–294. doi: 10.1016/j.fgb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Rauyaree P, Ospina-Giraldo MD, Kang S, Bhat RG, Subbarao KV, Grant SJ, Dobinson KF. Mutations in VMK1, a mitogen-activated protein kinase gene, affect microsclerotia formation and pathogenicity in Verticillium dahliae. Curr Genet. 2005;48:109–116. doi: 10.1007/s00294-005-0586-0. [DOI] [PubMed] [Google Scholar]
- 47.Tzima AK, Paplomatas EJ, Tsitsigiannis DI, Kang S. The G protein beta subunit controls virulence and multiple growth- and development-related traits in Verticillium dahliae. Fungal Genet Biol. 2012;49:271–283. doi: 10.1016/j.fgb.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Inderbitzin P, Bostock RM, Davis RM, Usami T, Platt HW, Subbarao KV. Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS One. 2011;6:e28341. doi: 10.1371/journal.pone.0028341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 51.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 52.Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 54.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 55.Glass NL, Dementhon K. Non-self recognition and programmed cell death in filamentous fungi. Curr Opin Microbiol. 2006;9:553–558. doi: 10.1016/j.mib.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 56.Paoletti M, Clave C. The fungus-specific HET domain mediates programmed cell death in Podospora anserina. Eukaryot Cell. 2007;6:2001–2008. doi: 10.1128/EC.00129-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saupe SJ. Molecular genetics of heterokaryon incompatibility in filamentous ascomycetes. Microbiol Mol Biol Rev. 2000;64:489–502. doi: 10.1128/MMBR.64.3.489-502.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saupe SJ, Clave C, Begueret J. Vegetative incompatibility in filamentous fungi: Podospora and Neurospora provide some clues. Curr Opin Microbiol. 2000;3:608–612. doi: 10.1016/S1369-5274(00)00148-X. [DOI] [PubMed] [Google Scholar]
- 59.Begueret J, Turcq B, Clave C. Vegetative incompatibility in filamentous fungi: het genes begin to talk. Trends Genet. 1994;10:441–446. doi: 10.1016/0168-9525(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 60.Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stipanovic RD, Bell AA. Pentaketide metabolites of Verticillium dahliae. 3. identification of (−)-3,4-dihydro-3,8-dihydroxy-1(2 h)-naphtalenone((−)-vermelone) as a precursor to melanin. J Org Chem. 1976;41:2468–2469. doi: 10.1021/jo00876a026. [DOI] [PubMed] [Google Scholar]
- 62.Chumley FG, Valent B. Genetic analysis of melanin-deficient, nonpathogenic mutants of Magnaporthe grisea. Mol Plant Microbe Interact. 1990;3:135–143. doi: 10.1094/MPMI-3-135. [DOI] [Google Scholar]
- 63.Liu Q, Chen C, Shen E, Zhao F, Sun Z, Wu J. Detection, annotation and visualization of alternative splicing from RNA-Seq data with SplicingViewer. Genomics. 2012;99:178–182. doi: 10.1016/j.ygeno.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 64.Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, Davuluri RV. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 2011;21:1260–1272. doi: 10.1101/gr.120535.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marquez Y, Brown JW, Simpson C, Barta A, Kalyna M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012;22:1184–1195. doi: 10.1101/gr.134106.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang B, Guo G, Wang C, Lin Y, Wang X, Zhao M, Guo Y, He M, Zhang Y, Pan L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010;38:5075–5087. doi: 10.1093/nar/gkq256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ho EC, Cahill MJ, Saville BJ. Gene discovery and transcript analyses in the corn smut pathogen Ustilago maydis: expressed sequence tag and genome sequence comparison. BMC Genomics. 2007;8:334. doi: 10.1186/1471-2164-8-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yassour M, Kaplan T, Fraser HB, Levin JZ, Pfiffner J, Adiconis X, Schroth G, Luo S, Khrebtukova I, Gnirke A, Nusbaum C, Thompson DA, Friedman N, Regev A. Ab initioConstruction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci U S A. 2009;106:3264–3269. doi: 10.1073/pnas.0812841106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gan P, Ikeda K, Irieda H, Narusaka M, O’Connell RJ, Narusaka Y, Takano Y, Kubo Y, Shirasu K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013;197:1236–1249. doi: 10.1111/nph.12085. [DOI] [PubMed] [Google Scholar]
- 70.Mayer KF, Waugh R, Brown JW, Schulman A, Langridge P, Platzer M, Fincher GB, Muehlbauer GJ, Sato K, Close TJ, Wise RP, Stein N, International Barley Genome Sequencing C A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–716. doi: 10.1038/nature11543. [DOI] [PubMed] [Google Scholar]
- 71.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, Brown JB, Cherbas L, Davis CA, Dobin A, Li R, Lin W, Malone JH, Mattiuzzo NR, Miller D, Sturgill D, Tuch BB, Zaleski C, Zhang D, Blanchette M, Dudoit S, Eads B, Green RE, Hammonds A, Jiang L, Kapranov P, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dobinson KF, Lecomte N, Lazarovits G. Production of an extracellular trypsin-like protease by the fungal plant pathogen Verticillium dahliae. Can J Microbiol. 1997;43:227–233. doi: 10.1139/m97-031. [DOI] [PubMed] [Google Scholar]
- 73.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 75.Oliveros JC. VENNY. An interactive tool for comparing lists with Venn Diagrams. 2007. [Google Scholar]
- 76.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 77.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 80.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Overview of the RNA-Seq data. All of the developmental stages share similar sets of mapped reads. The number of reads produced from the Illumina platform (blue bar); the number of reads mapped to the reference genome within 2 bp mismatch (red bar); the number of reads perfectly mapped to the reference genome (green bar); the number of reads mapped to the annotated genes (purple bar). (PNG 62 KB)
Additional file 2: Figure S2: Validation of RNA-Seq expression patterns. The RT-qPCR results of the selected genes show similar expression patterns to those detected by RNA-Seq. (PNG 68 KB)
Additional file 3: Table S1: GO enrichment analysis of genes in 18 clusters (p_value < 0.01). (XLSX 12 KB)
Additional file 4: Figure S3: Expression profiles of MAPK cascades. Heatmap showed that four types of MAPK cascades during MS formation. Expression pattern of genes encoding high osmolarity (HOG1) pathway were clustered together. The non-LS HOG1-MAPK maintained the highest expression value compared to other MAPKs. (PNG 685 KB)
Additional file 5: Figure S4: Expression profiles of small secreted proteins (length ≤300aa). Expression profiles of those genes encoding small secreted proteins with at least 50-fold up-regulated during MS formation compared with the CO stage. The levels of expression represent log2 FPKM Value +1. * represents genes with cysteine residues <4. (PNG 317 KB)
Additional file 6: Table S2: List of genes involved in metabolism, signal pathway and identified transcription factor. (XLSX 161 KB)
Additional file 7: Figure S5: Functional categorization of genes differentially expressed (up- or down-regulated) during microsclerotia development. (A, C, E) Intersection of MS1-4 stages revealed 600 significantly up-regulated genes vs CO stage, and functional categorization of these genes. (B, D, F) Intersection of MS1-4 stages revealed 124 significantly down-regulated genes vs CO stage, and functional categorization of these genes. (PNG 584 KB)
Additional file 8: Table S3: Significantly regulated genes during microsclerotia formation vs CO stage (p_value < 0.05). (XLSX 46 KB)
Additional file 9: Figure S6: Genes involved in protein metabolic processes and autophagy. A. Genes involved in protein metabolic processes by functional categorization; genes labeled with asterisks are the subunits associated with proteasome formation. B. Heatmap representation of genes involved in autophagy processes. Heatmap shows levels of transcripts abundance; relative levels of expression are presented by moderated log2 ratio of transcript abundance in MS developmental stages relative to the CO stage. (PNG 747 KB)
Additional file 10: Figure S7: Phylogenetic analysis and expression profile of bZIP transcription factors of V. dahliae. The full-length amino acid sequences of bZIP transcription factors of V. dahliae strain VdLs.17 were aligned using Clustal X and the phylogenetic tree was constructed using Mega 5.0 using the maximum-likelihood method with 1000 replicates. Domain structures are drawn to represent their relative positions. The black solid line represents the corresponding protein and its length. The different-colored boxes represent different domains and their positions in each protein predicted by Sanger Pfam program (http://pfam.xfam.org/) and the yellow ovals represent bZIP domains. The red box indicates an independent cluster of bZIP transcription factors genes, which contain two specific motifs represented at the bottom. The right panel represents heat maps showing the expression pattern of bZIP transcription factors of V. dahliae. (PNG 522 KB)
Additional file 11: Figure S8: Phylogenetic analysis of bZIP transcription factors of V. dahliae and other fungi. The amino acid sequences of bZIP transcriptional factors of V. dahliae strain VdLs.17, and other fungi were aligned using Clustal X and the phylogenetic tree was constructed using Mega 5.0 using the maximum-likelihood method with 1000 replicates. Fungal species are Sc, Saccharomyces cerevisiae; Vd, Verticillium dahliae; Va, Verticillium alfalfae (formerly V. albo-atrum); Fv, Fusarium verticillioides; Fo, Fusarium oxysporum; Fg, Fusarium graminearum; Mg, Magnaporthe oryzae; Nc, Neurospora crassa. Pa, Podospora anserine; Hj, Hypocrea jecorina. (PDF 42 KB)
Additional file 12: Figure S9: Expression patterns of genes involved in glycometabolism. Genes involved in carbohydrate metabolism including Glycolysis/Gluconeogensis, Glyoxylate cycle, TCA cycle, Pentose phosphate pathway. Heatmap shows levels of transcripts abundance; Level of expression are presented by moderated log2 ratio of transcript abundance vs CO stage. (PNG 556 KB)
Additional file 13: Table S4: List of genes undergoing AS events. (XLSX 575 KB)
Additional file 14: Table S5: Statistics of the AS events genes. (XLSX 9 KB)
Additional file 15: Figure S10: RT-PCR validation of three genes undergo RI events. Primers (black arrows) were designed spanning intron region. gDNA represents genomic DNA. cDNA represents complementary DNA. (PNG 163 KB)
Additional file 16: Table S6: Gene expression value of all annotated genes. (XLSX 804 KB)
Additional file 17: Table S7: Quality of Total RNA by Agilent 2000. (XLSX 11 KB)
Additional file 18: Table S8: RT-PCR primers used in this study. (XLSX 11 KB)