

Abstract
Background
Intrachromosomal segmental duplications provide the substrate for non-allelic homologous recombination, facilitating extensive copy number variation in the human genome. Many multi-copy gene families are embedded within genomic regions with high levels of sequence identity (>95%) and therefore pose considerable analytical challenges. In some cases, the complexity involved in analyzing such regions is largely underestimated. Rapid, cost effective analysis of multi-copy gene regions have typically implemented quantitative approaches, however quantitative data are not an absolute means of certainty. Therefore any technique prone to degrees of measurement error can produce ambiguous results that may lead to spurious associations with complex disease.
Results
In this study we have focused on testing the accuracy and reproducibility of quantitative analysis techniques. With reference to the C-C Chemokine Ligand-3-like-1 (CCL3L1) gene, we performed analysis using real-time Quantitative PCR (QPCR), Multiplex Ligation-dependent Probe Amplification (MLPA) and Paralogue Ratio Test (PRT). After controlling for potential outside variables on assay performance, including DNA concentration, quality, preparation and storage conditions, we find that real-time QPCR produces data that does not cluster tightly around copy number integer values, with variation substantially greater than that of the MLPA or PRT systems. We find that the method of rounding real-time QPCR measurements can potentially lead to mis-scoring of copy number genotypes and suggest caution should be exercised in interpreting QPCR data.
Conclusions
We conclude that real-time QPCR is inherently prone to measurement error, even under conditions that would seem favorable for association studies. Our results indicate that potential variability in the physicochemical properties of the DNA samples cannot solely explain the poor performance exhibited by the real-time QPCR systems. We recommend that more robust approaches such as PRT or MLPA should be used to genotype multi-allelic copy number variation in disease association studies and suggest several approaches which can be implemented to ensure the quality of the copy number typing using quantitative methods.
Keywords: Copy number variation, Multi-allelic, CCL3L1, Real-time quantitative PCR, Multiplex ligation-dependent probe amplification, Paralogue ratio test
Background
Structurally complex regions of the human genome contain a wealth of information that is yet to be fully understood within the context of evolution and disease. Here we use the term “complex” to describe genomic regions containing highly duplicated sequence of >95% sequence identity (eg. segmental duplications) demonstrating a range of diploid copies (>3 copy number classes). The C-C Chemokine Ligand-3-like-1 (CCL3L1) gene is one such example [1].
The quantitative methods commonly used to genotype multi-allelic copy number polymorphisms (CNPs) are not absolute, therefore accuracy and reproducibly are required to generate robust disease associations [2]. Single or multi locus genotyping methods including Multiplex Ligation-dependent Probe Amplification (MLPA), Paralogue Ratio Test (PRT) and real-time Quantitative PCR (QPCR) are flexible, and cost effective ways to analyze multi-allelic CNPs [3]. Although studies have shown that in some cases these techniques can provide an accurate assessment across a range of diploid copy numbers, their reproducibility can be influenced by a number of factors including assay design, sample quality and operator error.
A challenging aspect of multi-allelic copy number analysis is that the relative difference between copy number classes becomes smaller and more difficult to discriminate as copy number increases [4]. Despite this challenge, CNP analysis should produce data that clusters tightly around integer values [5], unless there is evidence for tissue mosaicism in which different cell types contain different copy numbers [6]. As such, tissue mosaicism can occur over a continuous spectrum (1-99%), and can result in extensive overlap between integer classes resulting in data that seemingly indicates a continuous copy number distribution. However, similar data can also be produced when measurements of non-mosaic tissues are imprecise and unable to discriminate CNVs with a high level of accuracy, potentially leading to erroneous conclusions.
Indeed, differences in these observations made in a number of studies support the conclusion that such measurement error can complicate data interpretation [7] and these effects are clearly evident in studies of the CCL3L1 gene and disease susceptibility [1, 8, 9]. In some cases, there could be misconceptions surrounding assay validation and/or data interpretation, and if this is coupled with a lack of transparency regarding the distribution of raw unrounded copy number measurements, it may lead to spurious results that are difficult to explain. The CCL3L1 gene represents a particularly intriguing locus for study, most notably for its implication in HIV AIDS susceptibility [1], but also because this region demonstrates several layers of complexity. In addition to being copy number variable, this locus is highly population stratified and the presence of extreme levels of sequence duplication can complicate assay design. As such due to the presence of the CC3L3 pseudogene, the analysis of certain CCL3L1 sequences differs with respect to copy number content and should be considered when attempting to compare methods and studies.
In this study we aim to demonstrate some simple approaches for assessing the quality of genotyping data when using methods such as real-time QPCR, PRT and MLPA for the analysis of multi-allelic copy number. These methods are evaluated by examining the copy number distribution of the CCL3L1 gene in DNA samples of European ethnicity.
Methods
DNA samples
We selected 150 DNA samples from patients of European origin. Genomic DNA was isolated from venous blood leukocytes using a phenol/chloroform extraction procedure as previously described [10]. The concentration and purity of the DNA samples was determined using the NanoDrop Specrophotometer (Thermo Scientific). We considered the genomic DNA samples to be free of contaminants or solvents if the 260/280 ratio resulted in a range between 1.7-2.1 and the 260/230 ratio was >1.5. The samples were collected as part of ongoing eye studies at the Royal Victorian Eye and Ear Hospital (RVEEH), Melbourne. Subjects were given a standard risk factor questionnaire and following clinical examination are considered healthy control individuals. The study was conducted in accordance with the Declaration of Helsinki and according to the National Health and Medical Research Council of Australia’s statement on ethical conduct in research involving humans, revised in 2000. Written informed consent was obtained from all individuals, and ethics approval for the project was provided by the Human Research and Ethics Committee of the Royal Victorian Eye and Ear Hospital (RVEEH), Melbourne.
Multiplex Ligation-dependent Probe Amplification (MLPA)
Two colour MLPA was conducted as previously described [11]. We designed probes targeting exon 1 and exon 3 of the CCL3L1 gene listed in Table 1 and these were was normalized against two reference probes known to have a diploid copy number state of two unless there is an obvious phenotype. The reference probes targeted the CREBBP and EP300 genes located on chromosome 16 and 22 respectively (Table 1). The reagents for the MLPA reaction were purchased from Fisher Biotec (Australia). Briefly, 150–250 ng of DNA in a final volume of 5 μl was denatured at 98°C for 5 minutes. After cooling to room temperature, 1.5 μl of probe mix containing CCL3L1 MLPA probes and two additional reference loci, was combined with 1.5 μl of Hybridization buffer and added to the sample, heat denatured at 95°C for 1 minute followed by hybridization at 60°C for 16 hours. Ligation was performed at 54°C by adding 32 μl of ligation reaction, and after 15 minutes the enzyme was inactivated by heating at 95°C for 5 minutes. Polymerase Chain Reaction (PCR) amplification was carried out under the following conditions: 1 cycle of 98°C for 1 minute; 35 cycles of 95°C 30 seconds, 57°C 30 seconds, 72°C 30 seconds; and 1 cycle of 72°C 20 minutes. The PCR reaction was performed in 25 μl. From each PCR reaction, 1 μl was mixed with 8.9 μl of HiDi formamide and 0.1 μl of ROX500 size standard (Applied Biosystems). Product separation was performed on the ABI 3130 Electrophoresis 16 capillary sequencer (Applied Biosystems). Data Analysis was performed as previously described [12] using the GeneMapper Software (Applied Biosystems) and the peak heights were exported to Microsoft Excel. Each peak height was normalized against the sum of two control loci to generate a ratio, and the normalized ratio was then divided by the median of all ratios for the corresponding peak. These values were ordered from low to high and subgroups corresponding to different DNA copy numbers were calculated to determine the relative differences between the subgroups.
Table 1.
MLPA probe sequences used for targeting the CCL3L1 gene and two reference loci EP300 and CREBBP
| Probe | Left probe | Right probe |
|---|---|---|
| CCL3L1 | GACTCTTGGCTCTGCTGACACTCGAGCCCACATTCCATCACCTGCTCCC | AATCATGCAGGTCTCCACTGCTGCCCTTGCC |
| exon 1 | ||
| CCL3L1 | CCTCCACCTTCCCTCACAGTGT | GTCTGGTGACAACCGAGTGGCT |
| exon 3 | ||
| CREBBP | CCAGCTAGTGGAATTCAAAACACAATTGGTTCTGTTGGCACA | GGGCAACAGAATGCCACTTCTTTAAGTAACCC |
| EP300 | CCAACCTAAGCACTGTTAGTCAGATTGATCCCAGCTCCAT | AGAAAGAGCCTATGCAGCTCTTGGACTACCCTATCA |
Real-time quantitative PCR (QPCR)
Real-time Quantitative PCR was performed according to the TaqMan® Copy Number Assays protocol (Applied Biosystems). We used 20 ng/μL of genomic DNA in a 10 μL reaction, consisting of 5 μL of 2× TaqMan® Genotyping Master Mix, 0.5 μL of RNaseP TaqMan assay 20× working stock, 0.625 μL of CCL3L1 Forward 10 μM, 0.625 μL of CCL3L1 Reverse 10 μM, 0.3 μL of CCL3L1 MGB probe 10 μM and 1 μL DH2O. The CCL3L1 MGB probe sequence is as follows MGB-6FA: TTCGAGGCCCAGCGACCTCA. The PCR primer sequences are as follows CCL3L1 Forward: GGGTCCAGAAATACGTCAGT and CCL3L1 Reverse: CATGTTCCCAAGGCTCAG as previously described [13]. Reactions were performed in duplicate, the cycling times were 1 cycle of 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. To assess the PCR efficiencies of the test and reference locus, we performed a series of 10-fold serial dilutions for both the CCL3L1 and RNaseP PCR reactions. We found that the PCR efficiencies were approximately equal, differing by less than 1%. We exported the CT values for both and analyzed them using CopyCaller™ software from Applied Biosystems. CopyCaller™ performs a comparative CT (ΔΔCT) relative quantitation analysis whereby the difference between the threshold cycles of the target and reference gene is calculated, the ΔCT of the test sample is compared to a calibrator of known copy number. We chose the calibrator sample which represented two copies of the CCL3L1 gene as tested by MLPA.
Paralogue Ratio Test (PRT)
We performed Single Plex Paralogue Ratio Test (PRT) using the method described previously [5]. We used 20 ng/μL of genomic DNA in a 25 μL reaction, comprising of 5 μL of 5× Bioline My Taq Buffer, 2 μL of 10 pmol/μL CCL3C Forward: GGC TAA GAC CCC TTC TAG AG and CCL3C Reverse: AAT CAT GCA GGT CTC CAC T Primer, 1 μL Dimethyl sulfoxide (DMSO), 0.25 μL Bioline My Taq Polymerase and 14.75 μL DH2O. The cycling times were 1 cycle of 95°C for 1 minute, 24 cycles of 95°C for 30 seconds, 55°C for 30 seconds and 70°C for 1 minute, followed by 1 cycle of 70°C for 40 minutes. The PCR primers were FAM labeled and for each PCR reaction, 1 μl was mixed with 8.9 μl of HiDi Formamide and 0.1 μl of ROX500 size standard (Applied Biosystems). PCR product separation was performed on the ABI 3130 Electrophoresis 16 capillary sequencer (Applied Biosystems). Data Analysis was performed using the GeneMapper Software (Applied Biosystems) and the peak heights were exported to Microsoft Excel. Copy number was calculated as the ratio of the CCL3L1 peak height against the reference peak height of the diploid CCL3 gene.
Results
Assay design
The CCL3L1 gene is approximately 1.9 kb in size and resides in a 90 kb segmental duplication on chromosome 17q12 (Figure 1). CCL3L1 contains 3 exons and shares >95% sequence identity with the CCL3 gene. As exons 2 and 3 are duplicated in the CCL3L pseudogene, a quantitative assay targeting exons 2 or 3 will in some instances, produce 1–2 copies more in comparison to an assay targeting exon 1. The PRT assay previously described by Carpenter et al. was specifically designed to avoid targeting the CCL3L pseudogene, an approach to only amplify the active gene in disease association studies [5]. In contrast, many studies (reviewed in [7]) using real-time QPCR, have targeted exon 3 also co-amplifying the CCL3L pseudogene. The real-time QPCR primers used in this study, have been described previously and target exon 3 [13]. In order to allow a direct comparison between these systems, we leveraged the multiplexing capability of MLPA, and designed two probes targeting exons 1 and 3 of the CCL3L1 gene (Figure 1). As the real-time QPCR assay targets exon 3 and the PRT assay targets exon 1, we could then compare both techniques to the two individual probe results generated by the MLPA assay.
Figure 1.
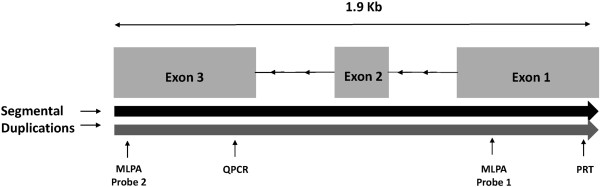
Schematic representation of the CCL3L1 gene located on chromosome 17q12. Overlapping segmental duplications are indicated by the black and grey arrows and the probe and primer positions for the PRT, real-time QPCR and MLPA assays are indicated by labels.
Validation of CNV genotyping methods
In order to validate each of the three genotyping methodologies, we compared 12 samples previously identified as having 1, 2, 3 or 4 CCL3L1 copies (three samples of each copy number class) using the MLPA assay, and compared these with results generated by both the PRT and real-time QPCR systems. We found that the genotype calls between all three methodologies were highly concordant, demonstrating that each assay represented data specific to each exon.
Assessment of assay performance and separation of integer copy number
We genotyped 150 genomic DNA samples using three quantitative assays MLPA, PRT and real-time QPCR. We plotted the raw unrounded copy number estimates on a histogram for all samples to provide an overall assessment on the quality of the data (Figures 2 and 3). Both the PRT and MLPA exon 1 systems demonstrated clear grouping of measures, consistent with integer clustering (Figure 2A and B). Clearly distinguishable from the histogram are 4 distinct clusters, each surrounding a normalized ratio of 0, 0.5, 1.5, and 2 consistent with 0, 1, 2, 3 and 4 diploid CCL3L1 copies. A direct comparison of copy number genotypes demonstrated 97% (145/150) concordance of copy number calls between the two assays with the exception of five samples within the three to five copy number range differing by only single copy (Figure 4A). Using a concordance plot to examine the degree of correlation between the MLPA and PRT systems, we demonstrated an R2 value of 0.91. The high degree of clustering exhibited by the histogram plots of raw unrounded copy number and the high degree of correlation and clustering shown in the concordance plot leaves little doubt as to the assignment of integer copy number (Figure 4A). We next examined the performance of real-time QPCR, with respect to cluster quality and assignment of integer copy number by comparison to data obtained by MLPA in the same 150 DNA samples. As the real-time QPCR and MLPA exon 3 assays amplify the CCL3L pseudogene, the range and frequency of integer copy number extends from 0–6 copies, with a higher frequency of 4 and 5 copy genotypes than would be expected if only exon 1 was amplified. Contrary to the data we observed with the MLPA system, the real-time QPCR assay performed quite poorly over a large sample range (Figure 3A). We found that overall, real-time QPCR produced a continuous spread of copy number values rather than discrete clusters (Figure 3A). Interestingly, real-time QPCR appeared to demonstrate a far more consistent clustering around 1 and 2 copy number classes, compared to reliably clustering the difference between 2, 3, 4 and higher copy number classes. As we could not adequately infer copy number genotype based on the clustering of unrounded measures, the 2-ΔΔCT values for the real-time QPCR assay were rounded to the nearest copy number integer. We achieved 81% (121/150) concordance with genotypes obtained with MLPA, with the highest discrepancies present in the 3 from 4 copy range (Table 2). Interestingly, when we performed the concordance plot between the two assays, the R2 between the two assays was higher than expected (R2 0.87), yet the overall cluster quality was quite poor (Figure 4B), suggesting that real-time QPCR has a reasonable level of accuracy but exhibits poor precision.
Figure 2.
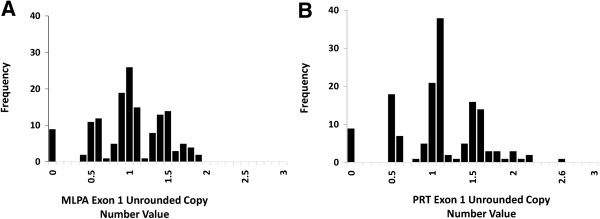
The distribution of unrounded CCL3L1 copy number measurements plotted on a histogram from the PRT and MLPA exon 1 assays. A. The MLPA assay demonstrates clear clustering, with peaks centered on integer values and gaps between each of the clusters ranging from 0–4 copies. B. The PRT assay demonstrates a distribution of 0–5 copies, with peaks clearly centered on integer values.
Figure 3.
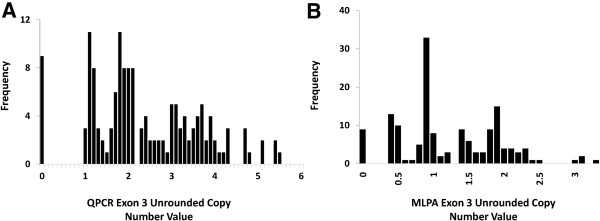
The distributions of unrounded CCL3L1 copy number measurements plotted on a histogram from real-time QPCR and MLPA exon 3 assays. A. The real-time QPCR assay demonstrates substantial variation around integer values. Without defined gaps between each of the clusters, assignment of whole copy number is performed by rounding the data to the nearest integer. B. The MLPA assay demonstrates a distribution of 0–6 copies with peaks centered on integer values. Copy number assignment can be performed by the groups of samples to a particular cluster.
Figure 4.
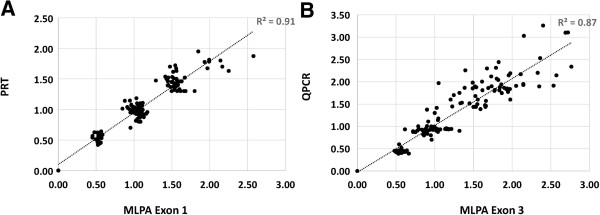
Comparison of the raw ratio data between MLPA, PRT and real-time QPCR using concordance plots. A. MLPA and PRT based measurements clearly demonstrates grouping around integer values and excellent correlation, as indicated by the R2 value of 0.91. Five samples represent a difference between PRT and MLPA of a single copy, however clustering both PRT and MLPA measurements allows copy number to be unambiguously assigned. B. A comparison between MLPA and real-time QPCR demonstrates a greater degree of variation between the two measurements. An almost vertical spread of measurements on the concordance plot demonstrates a poor degree of clustering in the 3–6 copy range between the two assays. The correlation between real-time QPCR and MLPA is high (R2 0.87), suggesting a low level of precision with respect to QPCR.
Table 2.
CNV genotyping calls from three independent techniques, QPCR, PRT and MLPA, when analyzing the CCL3L1 gene
| CCL3L1 exon 1 | CCL3L1 exon 3 | |||
|---|---|---|---|---|
| C# | PRT | MLPA | MLPA | QPCR |
| 0 copies | 9 | 9 | 9 | 9 |
| 1 copy | 25 | 25 | 24 | 26 |
| 2 copies | 67 | 67 | 50 | 53 |
| 3 copies | 39 | 43 | 22 | 28 |
| 4 copies | 9 | 6 | 35 | 25 |
| 5 copies | 1 | 0 | 6 | 7 |
| 6 copies | 4 | 2 | ||
Discussion
Measurement error has the power to drastically influence disease association studies, and this is particularly evident when performing analysis of multi-allelic CNPs. Studies have shown that even a small degree of differential bias, applied systematically to large case/control data sets, is likely to result in spurious association between multi-allelic CNPs and disease [2, 5] Our results demonstrate quite conclusively that real-time QPCR is prone to measurement error and performs poorly when applied to the CCL3L1 multi-allelic CNP region, when compared to alternative techniques such as MLPA or PRT.
The performance of real-time QPCR has previously been assessed for genotyping multi-allelic CNP regions such as the CCL3L1 gene [14] and the Beta-defensin gene cluster [2, 15, 16]. In each case, two main conclusions concerning the results have been reported 1. There is a lack of clear integer clustering 2. There are discrepancies between some copy number calls when compared to alternative approaches. Both of these findings are supported by the data presented here. Previous studies have attempted to compare the performance of CCL3L1 genotyping using the real-time QPCR and PRT systems [14]. However as the assays targeted different exons, the presence of a pseudogene excluded a more direct comparison of the techniques. Additionally, the cases and controls used in the study by Field et al. were extracted using different techniques potentially introducing variation in template quality as a contributor to the poor performance of real-time QPCR.
Several studies have suggested that the biological properties of the DNA including protein content, shearing, methods of extraction and storage conditions are potential contributors to the poor performance of real-time QPCR for analyzing multi-allelic CNPs [2, 14, 15, 17]. Additionally, it has been suggested that real-time QPCR will perform comparably to PRT if the concentration of DNA samples is normalized to a uniform concentration, and the efficiency between the test and reference loci differ by less than 5% [18]. With knowledge of these potential variables on assay performance, we felt that it would be unfair to perform a method comparison on DNA samples that were subject to such limitations. Therefore, we reasoned that if DNA quality is crucial to the success of real-time QPCR, then using high quality input DNA samples of similar concentration, generated using a single technique, and stored under uniform conditions, should produce high quality data. However, the results we present here do not support the conclusion that sample quality or PCR efficiency are the only reason for the variability observed with the real-time QPCR system, and thus we present an alternative hypothesis.
A question that has not been expressly raised when conducting disease association studies using real-time QPCR has been; what is the required number of technical replicates to generate reproducible data using an appropriate standard deviation? We contend that, due to the dynamic nature of multi-allelic CNPs a standardized number of technical replicates (for example 2–3), is inappropriate when the difference between copy number integers is not static. Evidence to support this was reported by Weaver et al. 2010 where using a standard deviation of 0.16, and false positive and false negative rate of 2.5%, would theoretically require a minimum of 4 replicates to distinguish 1 copy from 2 copies, 6 replicates to distinguish 2 copies from 3 copies, 11 replicates to distinguish 3 copies from 4 copies and 17 replicates to distinguish 4 copies from 5 copies [19]. We summarized some of the larger studies which have utilized real-time QPCR to analyze the CCL3L1 gene and the Beta defensins for disease association, and find that in all but one case, two technical replicates have been used to conduct the association study (Table 3). In the study presented here we also performed a duplicate testing approach to real-time QPCR, so that our data could be compared to previous work. However, under this model, it would appear that the use of real-time QPCR is questionable for analyzing the CCL3L1 gene which commonly varies between 0–4 copies per diploid genome [5] and totally inappropriate for beta defensins given the copy number range is much higher, commonly between 2–7 copies [20].
Table 3.
Reported number of technical replicates used in real-time QPCR studies for disease association
| Studies | Gene | Disease association | Technical replicates performed |
|---|---|---|---|
| Gonzalez et al.[1] | CCL3L1 | HIV related Outcomes | 2 |
| Urban et al.[9] | CCL3L1 | HIV related Outcomes | 2 |
| Bhattacharya et al.[8] | CCL3L1 | HIV related Outcomes | 3 |
| Field et al.[14] | CCL3L1 | T1 Diabetes | 2 |
| Mamtani et al.[21] | CCL3L1 | Systemic Lupus Erythematosus | 2 |
| Fellerman et al.[22] | Beta-defensin | Crohn’s Disease | 3 |
| Bentley et al.[23] | Beta-defensin | Crohn’s Disease | 2 |
| Mehlotra et al.[24] | Beta-defensin | HIV related Outcomes | 2 |
Previous studies have highlighted the critical importance of implementing copy number reference samples, plotting the raw unrounded copy number measurement data and observing the clustering around integer values in disease association studies [2, 5, 25]. We also demonstrate that assay validation should not consist solely in genotyping positive controls as real-time QPCR performed comparably to PRT and MLPA when a small number of positive controls were genotyped. Our data shows that because the variance for real-time QPCR measurements surrounding integer values is substantially greater than that of the PRT or MLPA systems, only by plotting and ordering the raw unrounded measurements from a larger sample size on a histogram can the short-comings of QPCR be identified. It is when a smaller sample size is utilized that the results can potentially be misleading. We would like to point out that this can be done by genotyping as few as 40 test samples, well before an entire cohort is genotyped. Therefore, we believe the use of positive controls should be strictly limited to assessing the ability of an assay to detect copy number variation, and then calibrating inter-run variation.
In some cases, studies using real-time QPCR have systematically removed uncertain data that fails to cluster around integer values in an attempt to reduce the impact of measurement error [26, 27]. However, calling samples with only a high degree of certainty may result in bias, as the failure to cluster is unlikely to be independent of genotype [28]. A more robust approach using real-time QPCR would be to firstly; perform a sufficient number of replicates as dictated by the copy number content of the given region; secondly repeat samples in which the inference of the most likely copy number has extended error bars, utilize the z-scores to highlight data with low confidence [29]; and thirdly, implement a statistical model such as CNVtools which specifically controls for differential bias in the case/control analysis while also allowing one to input data as probability weighted CNV scores as well as integers [28].
One of the more troubling aspects regarding the use of real-time QPCR in our study is the failure to form integer clusters on a sample set of high quality DNA of similar concentration. This suggests that real-time QPCR is inherently prone to measurement error. Moreover this error may be exacerbated by varying sample quality in large case/control cohorts potentially leading to false positive associations. However, it is also noteworthy that real-time QPCR is not substantially incorrect in its assessment of copy number genotype, as demonstrated in Table 2. Our data shows that even though under optimal conditions the variance of real-time QPCR is considerable across large sample numbers, however it still may have a place as purely a confirmatory assay for sequencing projects or MLPA and PRT data sets.
The consistent level of clustering observed by both the PRT and MLPA systems performed without replicates or repeat testing is unequivocal evidence of their accuracy and reliability on a large scale. Additionally, we have not fully utilized the multiplexing capacity of MLPA [11] or Triplex PRT [5] which would further improve the copy number typing of these systems. One important aspect to consider is that combining measurements from two independent techniques, such as PRT and MLPA, is very powerful with respect to accuracy and reproducibility. We found that MLPA and PRT calls differed by a single copy in five samples, notably in the 4–5 copy range. However, by combining the two techniques using a concordance plot we were able to unambiguously assign copy number to these samples (Figure 4A).
We argue that many multi-allelic CNP regions have been largely underestimated with regard to the complexity of analysis, as evidenced by the large number of contradictory evidence concerning disease association studies (reviewed in [7]). There also appears to be a fundamental need to understand how these techniques work, along with which variables can impact the reproducibility of the assay. We also highlight that real-time QPCR appears to be satisfactory for detecting differences between 1 and 2 copies, thus may be appropriate for the detection of di-allelic CNPs where the differences between the integers are at least 2-fold. Recently, water in oil digital droplet PCR (ddPCR) has been described for genotyping multi-allelic loci with an impressive level of reproducibility and accuracy [30, 31]. While this technology has not yet been widely implemented for association studies, it is likely that ddPCR will resolve a number of issues regarding measurement error in large cohorts. The development of a low cost genotyping assay that can provide accurate copy number content, and also possesses the ability to discriminate sequence content, including smaller insertion/deletions and paralogue specific variation, is required to fully understand the complex nature of multi-copy genomic regions. Recent work combining molecular inversion probes (MIPs) together with massively parallel DNA sequencing [32] appears perfectly suited for this type of analysis and will bridge the gap between locus specific quantitative measures and whole genome sequencing technologies. Although MIPs have immense promise for accurately genotyping multi-allelic CNPs such as CCL3L1, it is noteworthy that MIP technology is heavily reliant on high quality reference sequences and availability of singly unique nucleotides (SUNs) to accurately distinguish gene family paralogs. Therefore at present MIP- based genotyping cannot be universally applied to all multi-allelic CNP regions.
Conclusions
In conclusion the data presented here clearly demonstrates that real-time QPCR is prone to measurement error when applied to genotyping the CCL3L1 multi-allelic CNP region. We recommend that real-time QPCR data be treated with a high degree of caution and suggest that if it is used for the analysis of multi-allelic CNPs, that it is accompanied by independent genotyping measures such as MLPA or PRT. With reference to quantitative measures, ddPCR technology, MIPs or MLPA and PRT, represent far more reliable and robust approaches for accurately resolving multi-allelic CNPs and should be used in preference if possible.
Availability of supporting data
The data supporting the results of this article are included within the article.
Authors’ informations
Paul N Baird and Stefan J White are recognized as joint senior authors.
Acknowledgements
This work is supported by funding from Monash University awarded to PW and SW, and the National Health and Medical Research Council Centre for Clinical Research Excellence Grant Number #529923. Translational Clinical Research in Major Eye Diseases. NHMRC CJ Martin Biomedical Fellowship awarded to SC; Grant Number #1073726. NHMRC Senior Research Fellowship awarded to PNB Grant Number #1028444. MIMR-PHI Institute and The Centre for Eye Research Australia (CERA) receive operational infrastructure support from the Victorian Government.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SC, PW, PNB and SJW designed the study. SC performed the experimental work, data analysis and wrote the first draft of the manuscript. SC and SW performed the data analysis. SC, PW, PNB and SJW contributed to the final manuscript. All authors read and approved the final manuscript.
Contributor Information
Stuart Cantsilieris, Email: stuart.cantsilieris@monash.edu.
Patrick S Western, Email: patrick.western@monash.edu.
Paul N Baird, Email: pnb@unimelb.edu.au.
Stefan J White, Email: stefan.white@monash.edu.
References
- 1.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307(5714):1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 2.Aldhous M, Abu Bakar S, Prescott N, Palla R, Soo K, Mansfield J, Mathew C, Satsangi J, Armour J. Measurement methods and accuracy in copy number variation: failure to replicate associations of beta-defensin copy number with Crohn’s disease. Hum Mol Genet. 2010;19(24):4930–4938. doi: 10.1093/hmg/ddq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantsilieris S, Baird PN, White SJ. Molecular methods for genotyping complex copy number polymorphisms. Genomics. 2012;101(2):86–93. doi: 10.1016/j.ygeno.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Hollox EJ. The challenges of studying complex and dynamic regions of the human genome. Methods Mol Biol. 2012;838:187–207. doi: 10.1007/978-1-61779-507-7_9. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter D, Walker S, Prescott N, Schalkwijk J, Armour J. Accuracy and differential bias in copy number measurement of CCL3L1 in association studies with three auto-immune disorders. BMC Genomics. 2011;12(1):418. doi: 10.1186/1471-2164-12-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Notini AJ, Craig JM, White SJ. Copy number variation and mosaicism. Cytogenet Genome Res. 2009;123(1–4):270–277. doi: 10.1159/000184717. [DOI] [PubMed] [Google Scholar]
- 7.Cantsilieris S, White SJ. Correlating multi-allelic copy number polymorphisms with disease susceptibility. Hum Mutat. 2013;34(1):1–13. doi: 10.1002/humu.22172. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharya T, Stanton J, Kim EY, Kunstman KJ, Phair JP, Jacobson LP, Wolinsky SM. CCL3L1 and HIV/AIDS susceptibility. Nat Med. 2009;15(10):1112–1115. doi: 10.1038/nm1009-1112. [DOI] [PubMed] [Google Scholar]
- 9.Urban TJ, Weintrob AC, Fellay J, Colombo S, Shianna KV, Gumbs C, Rotger M, Pelak K, Dang KK, Detels R, Martinson JJ. CCL3L1 and HIV/AIDS susceptibility. Nat Med. 2009;15(10):1110–1112. doi: 10.1038/nm1009-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a Laboratory manual. Cold Spring Harbor Laboratory Press. 1989;1:E3–E7. [Google Scholar]
- 11.White SJ, Vink GR, Kriek M, Wuyts W, Schouten J, Bakker B, Breuning MH, den Dunnen JT. Two-color multiplex ligation-dependent probe amplification: detecting genomic rearrangements in hereditary multiple exostoses. Hum Mutat. 2004;24(1):86–92. doi: 10.1002/humu.20054. [DOI] [PubMed] [Google Scholar]
- 12.White SJ, Breuning MH, den Dunnen JT. Detecting copy number changes in genomic DNA: MAPH and MLPA. Methods Mol Biol. 2004;75:751–768. doi: 10.1016/s0091-679x(04)75032-3. [DOI] [PubMed] [Google Scholar]
- 13.Sudmant P, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. 1000 Genomes Project. Diversity of human copy number variation and multicopy genes. Science. 2010;330(6004):641–646. doi: 10.1126/science.1197005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Field SF, Howson JMM, Maier LM, Walker S, Walker NM, Smyth DJ, Armour JAL, Clayton DG, Todd JA. Experimental aspects of copy number variant assays at CCL3L1. Nat Med. 2009;15(10):1115–1117. doi: 10.1038/nm1009-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fode P, Jespersgaard C, Hardwick R, Bogle H, Theisen M, Dodoo D, Lenicek M, Vitek L, Vieira A, Freitas J, Andersen PS, Hollox EJ. Determination of beta-defensin genomic copy number in different populations: a comparison of three methods. PLoS One. 2011;6(2):e16768. doi: 10.1371/journal.pone.0016768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perne A, Zhang X, Lehmann LE, Groth M, Stuber F, Book M. Comparison of Multiplex Ligation-dependant Probe Amplification and real time PCR accuracy for gene copy number quantification using the Beta Defensin Locus. Biotechniques. 2009;47(6):1023–1026. doi: 10.2144/000113300. [DOI] [PubMed] [Google Scholar]
- 17.Cukier HN, Pericak-Vance MA, Gilbert JR, Hedges DJ. Sample degradation leads to false-positive copy number variation calls in multiplex real-time polymerase chain reaction assays. Anal Biochem. 2009;386(2):288–290. doi: 10.1016/j.ab.2008.11.040. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L, Gutierrez G, Irastorza I, Castan L, Vitoria J, Bilbao J. Accuracy in copy number calling by qPCR and PRT: a matter of DNA. PLoS One. 2011;6(12):e28910. doi: 10.1371/journal.pone.0028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weaver S, Dube S, Mir A, Qin J, Gang S, Ramakrishnan R, Jones RC, Livak K. Taking qPCR to a higher level: analysis of CNV reveals the power of high throughpout qPCR to enhance quantitative resolution. Methods. 2010;50:271–276. doi: 10.1016/j.ymeth.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Hollox EJ, Armour JA, Barber JCK. Extensive normal copy number variation of a β-defensin antimicrobial-gene cluster. A J Hum Genet. 2003;73(3):591–600. doi: 10.1086/378157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamtani M, Rovin B, Brey R, Camargo JF, Kulkarni H, Herrera M, Correa P, Holliday S, Anaya JM, Ahuja SK. CCL3L1 gene-containing segmental duplications and polymorphisms in CCR5 affect risk of systemic lupus erythaematosus. Ann Rheum Dis. 2008;67(8):1076–1083. doi: 10.1136/ard.2007.078048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, Reinisch W, Teml A, Schwab M, Lichter P, Radlwimmer B, Stange EF. A chromosome 8 gene-cluster polymorphism with Low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79(3):439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bentley RW, Pearson J, Gearry RB, Barclay ML, McKinney C, Merriman TR, Roberts RL. Association of higher DEFB4 genomic copy number with Crohn’s disease. Am J Gastroenterol. 2010;105(2):354–359. doi: 10.1038/ajg.2009.582. [DOI] [PubMed] [Google Scholar]
- 24.Mehlotra R, Dazard J, John B, Zimmerman P, Weinberg A, Jurevic R. Copy number variation within human β-defensin gene cluster influences progression to AIDS in the multicenter AIDS cohort study. J Aids Clin Res. 2012;3(10):1000184. doi: 10.4172/2155-6113.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollox EJ. Beta-Defensins and Crohn’s disease: confusion from counting copies. Am J Gastroenterol. 2010;105(2):360–362. doi: 10.1038/ajg.2009.573. [DOI] [PubMed] [Google Scholar]
- 26.McKinney C, Broen JCA, Vonk MC, Beretta L, Hesselstrand R, Hunzelmann N, Riemekasten G, Scorza R, Simeon CP, Fonollosa V, Carreira PE, Ortego-Centeno N, Gonzalez-Gay MA, Airo P, Coenen M, Martin J, Radstake TRDJ, Merriman TR. Evidence that deletion at FCGR3B is a risk factor for systemic sclerosis. Genes Immun. 2012;13(6):458–460. doi: 10.1038/gene.2012.15. [DOI] [PubMed] [Google Scholar]
- 27.McKinney C, Merriman ME, Chapman PT, Gow PJ, Harrison AA, Highton J, Jones PBB, McLean L, O’Donnell JL, Pokorny V, Spellerberg M, Stamp LK, Willis J, Steer S, Merriman TR. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2008;67(3):409–413. doi: 10.1136/ard.2007.075028. [DOI] [PubMed] [Google Scholar]
- 28.Barnes C, Plagnol V, Fitzgerald T, Redon R, Marchini J, Clayton D, Hurles ME. A robust statistical method for case–control association testing with copy number variation. Nat Genet. 2008;40(10):1245–1252. doi: 10.1038/ng.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010;50:262–270. doi: 10.1016/j.ymeth.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Boettger L, Handsaker R, Zody M, McCarroll S. Structural haplotypes and recent evolution of the human 17q21.31 region. Nat Genet. 2012;44(8):881–886. doi: 10.1038/ng.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP, Hodges SP, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nuttle X, Huddleston J, O’Roak BJ, Antonacci F, Fichera M, Romano C, Shendure J, Eichler EE. Rapid and accurate large-scale genotyping of duplicated genes and discovery of interlocus gene conversions. Nat Meth. 2013;10(9):903–909. doi: 10.1038/nmeth.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]