

Abstract
The importance of the post-translational lipid modifications farnesylation and geranylgeranylation in protein localization and function coupled with the critical role of prenylated proteins in malignant transformation has prompted interest in their biology and the development of farnesyl transferase and geranylgeranyl transferase inhibitors (FTIs and GGTIs) as chemical probes and anticancer agents. The ability to measure protein prenylation before and after FTI and GGTI treatment is important to understanding and interpreting the effects of these agents on signal transduction pathways and cellular phenotypes, as well as to the use of prenylation as a biomarker. Here we describe protocols to measure the degree of protein prenylation by farnesyl transferase or geranylgeranyl transferase in vitro, in cultured cells and in tumors from animals and humans. The assays use [3H]farnesyl diphosphate and [3H]geranylgeranyl diphosphate, electrophoretic mobility shift, membrane association using subcellular fractionation or immunofluorescence of intact cells, [3H]mevalonic acid labeling, followed by immunoprecipitation and SDS-PAGE, and in vitro transcription, translation and prenylation in reticulocyte lysates. These protocols require from 1 day (enzyme assays) to up to 3 months (autoradiography of [3H]-labeled proteins).
INTRODUCTION
Techniques to investigate the effects of FTIs and GGTIs on protein prenylation
Protein prenylation, a universal post-translational modification (PTM) is required for the cancer-causing ability of some proteins, among which the Ras superfamily of small GTPases is the most prominent. This has prompted the development of FTIs and GGTIs as potential anticancer drugs and chemical probes. Despite the conceptual advances made over the last decade, the major challenge in the prenylation field still is to identify the prenylated proteins, the inhibition of which is responsible for mediating the antitumor effects of prenyl transferase (PT) inhibitors (PTIs). Given the heterogeneity of cancer, these proteins most likely will vary from one cancer cell type to another, and will largely depend on the tumor-or cell type–specific mutations and the vulnerabilities arising from these mutations. We predict that the identification of critical FTI or GGTI targets will ultimately lead to the design of inhibitors of specific farnesyl transferase (FT)/geranylgeranyl transferase (GGT)-1 substrates rather than FT or GGT-1 enzymes themselves. Pursuing such a strategy would also avoid inhibiting those prenylated proteins that have tumor suppressor function, and might pave way for designing more effective/less toxic cancer therapies.
In this article, we describe techniques used to characterize protein prenylation and the effects of FTIs and GGTIs on prenylation. We provide detailed procedures for in vitro methods, intact cells and biopsies from animal or patient tumor tissue or peripheral blood mononuclear cells (PBMCs). In brief, we describe quantitative methods to determine FT or GGT enzymatic activities and the degree of prenylation of individual proteins. The enzymatic activity assays can serve two purposes: the first, which is done with pure or partially purified enzymes, is to assess the potency and selectivity of a novel inhibitor, or the quality of a known inhibitor that has been stored for an extended time. The second purpose is to determine the levels of FT or GGT-1 activity in cells or tissue extracts before and after PTI treatment. Both of these approaches use [3H]farnesyl diphosphate (FPP) or [3H]geranylgeranyl diphosphate (GGPP) as prenyl donor. The other two methods involving radioactivity (in vitro transcription/translation and labeling of cells with [3H]MVA) are designed to establish whether or not a candidate PT substrate undergoes prenylation in vitro and in intact cells, respectively, and to determine the effects of PTIs on this prenylation. We also describe two other assays that take advantage of the fact that, for most proteins, inhibition of prenylation results in slower electrophoretic mobility or, alternatively, relocation to cellular membranes. These techniques can be used to answer, either directly or indirectly, the following important biological questions facing the area of prenylation research. Is a (novel) protein of interest a substrate for FT and/or GGT-1? Can this protein be modified by prenylation in vitro1–3 and in whole cells4? This is important, because for reasons discussed above, it is very likely that not all proteins regulated by prenylation have been identified yet.
Can PTIs be developed either as chemical probes or potential anticancer agents? To function as chemical probes, PTIs have to meet the criterion that they inhibit selectively their intended target. This is initially established using in vitro PT activity assays with purified or partially purified enzymes5–8. To function as anticancer agents, they must successfully pass a number of tests, which include inhibiting their targets in whole cells7–10 or tissue11–13, inhibiting anchorage-dependent or anchorage-independent cell proliferation7,9,14 or angiogenesis15,16, and/or inducing10,17,18 or sensitizing to cell death or apoptosis19–21. In this context, it is important to correlate the changes in these cancer hallmarks in response to PTIs with their effects on the target proteins.
Which proteins mediate the anticancer effects of FTIs or GGTIs? Is the antitumor efficacy of FTIs or GGTIs correlated with inhibition of certain prenylated proteins10,14,22–24?
Finally, are FT and/or GGT-1 activities elevated in cancer cells versus ‘normal’ cells25? Do elevated FT or GGT-1 activities in cancer correlate with cancer progression and metastasis? Are FT or GGT-1 activities decreased in response to therapy with FTIs or GGTIs? This issue has been investigated in several FTI clinical trials, either directly by measuring FT26,27 enzyme activities or indirectly by determining the prenylation of HDJ-2 and/or other proteins28,29. The assays described here could easily be adapted to answer these questions by using cell lysates from animal or human tumors as the source of enzyme or prenylated protein. An example is our recent phase 2 clinical trial, in which we compared FT activities before and after treatment with the FTI tipifarnib in fresh human breast tumor biopsies30.
General aspects of protein prenylation
Protein prenylation, a universal and irreversible PTM, affects many proteins involved in a variety of cellular processes including survival, proliferation and migration. It consists of the enzymatically catalyzed covalent attachment of an isoprenyl lipid, namely a C15 farnesyl or a C20 geranylgeranyl moiety, to the sulphydryl group of a cysteine residue located within four residues from the carboxy terminus in the target protein via thioether linkage. In eukaryotes, the three enzymes catalyzing these lipid modifications are FT, GGT-1 and GGT-2. Proteins undergoing prenylation appear to share conserved consensus recognition motifs, which are C-terminal and share the pattern CaaX, CXC or CC, where C is cysteine, ‘a’ often but not always is an aliphatic amino acid and X is any amino acid. Both FT and GGT-1 are active on CaaX motifs, whereas GGT-2 appears to modify exclusively Rab GTPases and is active on CXC or CC motifs. The nature of the X in the C-terminal CaaX motif specifies whether a protein is a substrate for FT or GGT-1. Whereas FT prefers X to be methionine, serine, glutamine or cysteine, GGT-1 prefers X to be leucine or isoleucine (for reviews see refs. 31–33). This can be exploited to generate mutant proteins that either switch from being an exclusive FT substrate to an exclusive GGT-1 substrate, or vice versa, or that are prenylation resistant (see EXPERIMENTAL DESIGN and Table 1). However, as several lines of evidence suggest, these rules are not absolute; for instance, a CaaX-protein with a C-terminal phenylalanine can be farnesylated or geranylgeranylated34. In addition, although GGT-1 clearly prefers X to be leucine35,36, some CaaL motifs can also be prenylated by FT37. Furthermore, at least one protein, RhoB, is naturally both farnesylated and geranylgeranylated38. In addition, some proteins such as K-Ras are naturally only farnesylated (CaaX box: CVIM), but can become geranylgeranylated once FT is inhibited in cells39. Prenylated proteins can undergo up to three more PTMs, all of which serve to enhance the hydrophobicity and membrane anchoring of the target protein (reviewed in refs. 40–42).
TABLE 1.
Useful mutants in prenylation research.
| Isoform/mutant | C-terminal sequence | Substrate for |
|---|---|---|
| Wild type | Caa(MSGC) | FT |
| Switched substrate specificity | CaaL | GGT-1 |
| Prenylation resistant | Saa(MSGC) | None |
| Wild type | Caa(LI) | GGT-1 |
| Switched substrate specificity | CaaM | FT |
| Prenylation resistant | Saa(LI) | None |
As described in the text and summarized here, proteins undergoing prenylation typically bear a C-terminal CaaX consensus sequence. Mutants that either convert the protein of interest from an FT into a GGT-1 substrate (or vice versa) or render them resistant to prenylation are of great value for prenylation research. To name just one example, prenylation-resistant mutants can be used to determine whether prenylation is required for a phenotype associated with the protein in question. See Figure 5 for an example.
Physiological role(s) of protein prenylation
Protein prenylation is necessary for membrane localization and proper function of otherwise cytosolic proteins. The exact number of proteins being prenylated in vivo (i.e., the prenylome) is unknown43,44; however, although more than 100 proteins have experimentally been shown to undergo prenylation32, contemporary reviews estimated several hundred proteins to be subject to prenylation33,45. A search of the most recent UniProtKB/SwissProt database (released 20 April 2010) has returned 587 human genes that potentially encode proteins bearing a C-terminal CXXX motif (N.B. and S.M.S., unpublished results), suggesting that many prenylated proteins have not been identified yet. Not surprisingly then, genetic disruption of FT has further suggested a critical role for farnesylation: FT knockout mice die early in embryonic development46. However, in conditional knockout mice, FT is dispensable for postnatal development and adult tissue homeostasis. Furthermore, FT is not required for tumor initiation, but for tumor progression and maintenance. Perhaps most importantly, in the conditional absence of FT, the FT substrate protein H-Ras is still associated with cellular membranes46, contradicting earlier studies in whole cells demonstrating that farnesylation is required for H-Ras membrane association47, and a non-prenylatable mutant of H-Ras exclusively resides in the cytosolic fraction48. However, these results have been challenged by two more recent studies. First, the FT null allele generated by Mijimolle et al.46 may have been ‘leaky’, allowing for expression of partially active FT49. Second, in mice with a conditional FT deletion, H-Ras fails to associate with membranes50, thus confirming a variety of earlier studies in whole cells48,51,52. Although conditional GGT-1 knockout studies were reported (see below), whether constitutive ablation of GGT-1 in mammals is also embryonically lethal has not been determined yet. However, studies conducted in both Saccharomyces cerevisiae53 and Drosophila54 suggest that the GGT-1 β-subunit is an essential protein.
After the discovery that Ras is farnesylated2 and that farnesylation is required for its transforming activity55, FT and GGT became major targets for developing anticancer drugs. Besides Ras, many PT substrates are involved in regulating cell growth, proliferation and survival, as well as migration, invasion and metastasis56. These include small GTPases of the Rac, Rho, Rheb and Ral sub-families, as well as transducin, the γ subunits of trimeric G protein and lamins. Ras can activate at least 11 signal transduction pathways, some of which are well documented to contribute to malignant transformation, including the Raf-MEK-ERK57, phosphoinositide 3-kinase/Akt58, Ral GDS-Ral, TIAM1-Rac and Rho-ROCK pathways (reviewed in refs. 59, 60). Thus, it is not surprising that many of these prenylated proteins promote and/or maintain malignant transformation.
Preclinical and clinical activity of farnesyltransferase inhibitors
In well-defined systems in which mutant H-Ras drives the transformation of NIH 3T3 cells, FTIs inhibit the farnesylation of H-Ras, prevent its plasma membrane association and inhibit downstream signal transduction pathways, such as Raf-MEK-ERK, leading to inhibition of tumor growth47,61. Similarly, FTIs induce breast tumor regression in mutant H-Ras transgenic mice, but only tumor-growth inhibition in mutant K-Ras transgenic mice62,63. In human tumor cell lines with multiple genetic alterations, FTIs inhibit proliferation and/or induce apoptosis (reviewed in refs. 45, 64, 65), and in most cell lines this is associated with mitotic arrest66–68, but occasionally with G1 phase accumulation69. Consistent with the fact that K-Ras is geranylgeranylated in FTI-treated cells and is fully functional39, the sensitivity of human cancer cells to FTIs does not depend on K-Ras mutation status9, suggesting that FTIs have at least one other critical farnesylated target that is responsible for limiting tumor cell proliferation and/or survival. Inhibition of RhoB farnesylation, which results in accumulation of geranylgeranylated RhoB has been suggested as a potential contributor14,70, but the fact that both farnesylated and geranylgeranylated RhoB have tumor-suppressive activity argues against this71. Inhibition of Rheb, a GTPase that stimulates mammalian target of rapamycin (mTOR), has also been suggested as a critical FTI target24, but more direct evidence for this is required. In mice, the antitumor activity of FTIs is evident when used as single agents, but enhanced in combination with other antisignaling agents such as inhibitors of MEK, CDKs or Bcr-Abl kinase, as well as cytotoxic agents such as paclitaxel, cisplatin and 5-fluorouracil (reviewed in ref. 65). However, clinically, FTIs have little activity except in a few cases (reviewed in ref. 65). For example, in phase 3 clinical trials, the FTI tipifarnib is not active in pancreatic cancer72, colorectal cancer73 or AML74. However, in more recent phase 2 clinical studies with locally advanced breast cancer patients, the addition of tipifarnib to standard chemotherapy (adriamycin and cyclophosphamide) improved the pathological complete response rate of these patients75.
Geranylgeranyltransferase 1 inhibitors
The development of GGTIs as potential anticancer agents was prompted by the fact that K-Ras becomes geranylgeranylated under FTI pressure and that in some human malignancies such as pancreatic cancer, pathways mediated by geranylgeranylated proteins such as RalA and RalB may be more relevant to onco-genesis than those mediated by MEK or Akt76,77. However, unlike FTIs, human cancer cells treated with GGTIs undergo G1 phase arrest78–80. Consistent with this, GGTIs induce p21 expression, inhibition of CDKs and hypophosphorylation of pRb78. Furthermore, GGTIs induce the accumulation of p27 in the nucleus by a mechanism that involves inhibition of CDK2 phosphorylation of Thr187 in p27 (ref. 18). GGTI-mediated induction of apoptosis may be associated with their ability to reduce phospho-Akt and survivin levels81. Furthermore, the ability of GGTIs to induce apoptosis and inhibit anchorage-independent growth has been suggested to depend on their ability to inhibit the geranylgeranylation of RalB and RalA, respectively3.
In mice, conditional GGT-1 deficiency82 or conditional simultaneous knockout of both FT and GGT-1 (ref. 50) markedly reduces the formation of K-Ras–induced lung cancer and also extends the life span of these mice. Using pharmacological inhibition of GGT-1, our laboratory has recently demonstrated that GGTI-2418 effectively prevents tumor growth in nude mice and results in breast tumor regression in transgenic mice expressing ErbB2 (ref. 18). GGTI-2418 has recently entered phase 1 clinical trials83.
Experimental design
In Figure 1, we present a flow diagram that presents the key steps of the methods described below. As we describe several alternative methods to characterize protein prenylation, our protocol basically consists of only two major steps. The first is the preparation of the source material ((A) treated or untreated cultured cells; (B) pure enzyme or postmicrosomal supernatants as a source of partially purified FT or GGT1; (C) normal and tumor tissue biopsies; (D) cultured cells labeled with [3H] mevalonic acid (MVA); or (E) cDNA encoding a candidate PT substrate). The second is the selection and performance of subsequent analytical procedures ((A) enzyme assay; (B) electrophoretic mobility shift assay (EMSA); (C) subcellular fractionation; (D) immunoprecipitation of [3H]-labeled protein of interest; or (E) in vitro transcription- translation-prenylation). The procedures of Step 2 fall into two broad categories: either determination of FT and GGT-1 enzymatic activities (option A) or the prenylation state of individual proteins (options B–E). Three of the analytical methods in Step 2A–C can easily be performed with cultured cells, PBMCs or tumor tissue from mice or humans.
Figure 1.

Linear flow diagram summarizing the alternative protocols designed to assess protein prenylation and the effects of PTIs on prenylation. The first step of the procedure consists of five different options to prepare biological samples for analysis of protein prenylation. In the second part, we describe five different methods to either quantify enzymatic activity of FT or GGT-1 (option A) or to assess the prenylation state of individual proteins (options B–E). Dual-colored steps (options 2A–C) indicate that these methods are suitable for cultured cells, PBMCs, normal tissue or tumor biopsies. Red arrows show methods that require handling of radiochemicals. Steps 2A–C are designed to determine either PT activity or the prenylation state of select proteins in response to prior exposure to a PTI. Steps 2D–E are primarily designed to determine whether or not a candidate protein undergoes prenylation in intact cells (2D) or in vitro (2E), but they can also be done in the presence or absence of previous drug treatment.
In the first part of Step 2 (Step 2A in Fig. 1), we describe two conceptually distinct methods for determining PT enzymatic activity. The first method is designed to measure the inhibitory activity of PTIs, including their IC50 values, using purified FT or GGT-1 (both of which are commercially available). The second method is a variation of the first that is designed to quantify PT enzymatic activity in cultured cells, organs, tumors or blood cells from animals or humans that have been treated with a PTI. We have used this technique with cultured cells84, PBMCs28 and tumor biopsies from mice 80 or humans30. Furthermore, this assay can be adapted to measure the activity of PTIs using FT or GGT-1 isolated from untreated normal cells, cancer cells or tumors. The concentration range of the PTI investigated should initially be broad and/or based on results obtained from prior high-throughput screens. We typically cover the low-nanomolar to mid-micromolar range, using concentrations from 1 nM up to 300 μM, in Steps 1–3 and 10–30 (and so on). This parameter may have to be optimized further. We have used this protocol to evaluate CaaX peptidomimetics85,86, non-peptide Ras-CaaX mimetics87, FTI-244 (ref. 88), FTI 276/277 and GGTI-286/287 (refs. 47, 89), whereas others have used the same or similar assays to characterize the potency and specificity of the FTIs Lonafarnib66 and BMS-214662 (ref. 90). In both cases, [3H]-labeled FPP and GGPP are used as the farnesyl or geranylgeranyl donors. To measure FT activity, we routinely use purified recombinant wild-type H-Ras, which has the C-terminal tetrapeptide sequence CVLS. To measure GGT-1 activity, we use a mutant H-Ras in which the C-terminal serine is replaced by leucine; this is followed by expression in Escherichia coli91 and purification of the recombinant mutant H-Ras protein. These recombinant protein substrates can be made in-house, but they are also commercially available.
The second part of Step 2 of this protocol describes assays for the protein prenylation state of individual proteins (Fig. 1) from cultured cells (options B–D) and from PBMCs, normal tissue and tumor biopsies (options B–C). In contrast to protein phosphor-ylation, antibodies recognizing unique prenylation motifs are not available. Therefore, many of the current techniques rely on more indirect methods or the use of radiochemicals to investigate protein prenylation. Three approaches are possible:
Western blotting to demonstrate change in apparent Mr upon inhibition of prenylation (EMSA, Step 2B in Figure 1).
Subcellular fractionation, followed by western blot to demonstrate association with cell membranes upon prenylation, or association with the cytosolic fraction upon inhibition of prenylation (Step 2C in Fig. 1).
Immunoprecipitation, SDS-PAGE and autoradiography to demonstrate 3H incorporation upon labeling of cells with [3H]MVA (Step 2D in Fig. 1).
The first approach takes advantage of the fact that most proteins migrate faster and decrease their apparent Mr upon prenylation. This would be the method of choice, because it is straightforward and takes the least amount of time. The even easier use of prenylation state-specific antibodies in western blots would also fall into this category. Antibodies to non-farnesylated prelamin A have been described92, but to our knowledge the only antibody that is specific for an unprocessed PT substrate and commercially available is sc-1482 from Santa Cruz, which recognizes unprocessed Rap1A. As other PTMs such as phosphorylation are associated with changes in apparent Mr, it is important not to rely solely on this approach to determine whether a protein is prenylated. This is especially true when studying novel candidates for prenylation. We have used this technique, also termed EMSA, with all three isoforms of Ras47,61,89,93, Rap1A11,21,78,93,94, RhoA79, RalA and RalB3, as well as HDJ-2 (refs. 18, 95), whereas others have also demonstrated a mobility shift of K-Ras in response to L-778,123 (ref. 96).
The second strategy is based on the fact that prenylation often leads to association of the protein being prenylated with cellular membranes. This strategy is recommended in cases in which a shift in apparent Mr upon prenylation is either lacking or too small to resolve by standard electrophoretic procedures, e.g., for RhoA18,79. This approach consists of preparing membrane and cytosolic fractions by subcellular fractionation techniques, followed by western blotting for the protein being studied. A previous exposure of the cells to a PTI should thus result in a cytosolic accumulation of the protein of interest. Membrane association or the lack thereof can alternatively be demonstrated by immunofluorescence3,57,97 or fluorescence resonance energy transfer98,99. As these procedures are well established and not unique to prenylated proteins, we have not described them in detail here.
As controls for the two strategies above, we recommend the following. For a novel PTI, one should ideally use a closely related molecule that has been demonstrated to be inactive in vitro as a negative, and a ‘tried and tested’ PTI as a positive control. Regardless of the identity of the protein being studied, one should probably test the effects of FTIs or GGTIs on the behavior of HDJ-2 (exclusively farnesylated) and Rap1 (exclusively geranylgeranylated) proteins in the final read-out.
The third approach relies on labeling cells with [3H]MVA, which is a C6 precursor of C5 isopentenyl diphosphate. Isopentenyl diphosphate is then sequentially polymerized to yield C10 geranyl, C15 farnesyl and C20 geranylgeranyl diphosphates, the last two of which are the precursors of protein-bound isoprenyl groups. To ensure maximum labeling with [3H]MVA, it is advisable to deplete unlabeled endogenous MVA with an inhibitor of hydroxymethyl glutaryl–coenzyme A reductase, the enzyme generating MVA from hydroxymethyl glutaryl. In addition, using medium containing dialyzed serum will aid in further reducing the endogenous MVA pool. Provided an antibody capable of immunoprecipitating the native protein of interest is available, this would permit the investigation of endogenously expressed proteins. In case such antibodies are unavailable, an alternative approach is to ectopically express the protein carrying an N-terminal epitope tag. The immuno-precipitated protein is then separated by SDS-PAGE, followed by electrotransfer to PVDF membranes and autoradiography. This approach, although it is the most elaborate and possibly most time consuming because of potentially lengthy exposure times of membranes to X-ray film, is the most direct evidence for prenylation in intact cells and is most suitable to establish that a protein previously unknown to undergo prenylation is subject to this PTM. In 1990, this approach has been successfully used to show that K-Ras is isoprenylated55. More recently, variations of this technique have been used to determine that in intact cells, lamin B is farnesylated9 and the F-box protein FBXL2 is geranylgeranylated4.
In all of the methods of the second part, the following experimental parameters may need to be optimized, because they are dependent on the cell line, the protein or the effects of the PTI being studied: size of cell culture plate, initial cell density, incubation time and PTI concentrations. The determining factor here is to obtain enough samples to detect the protein of interest. Many laboratories, including our own typically, conduct other assays designed to measure cellular phenotypes in response to PTIs, e.g., cell viability, cell death and cell proliferation. These assays have to be set up in parallel sister cultures and will not be covered here.
A fourth and last approach is a method to assess the degree of prenylation of a protein of interest using in vitro transcription-translation systems (Step 2E, Fig. 1). This method is suitable for investigating the prenylation of candidate FT or GGT-1 substrates in situations in which the cDNAs encoding such substrates, but not the purified proteins themselves or cognizant antibodies, are available. This approach is of particular interest if investigators want to determine whether a protein is a substrate for FT or GGT-1 or both. We have used this technique with RhoB97, as well as RalA and RalB3. To determine whether a protein is farnesylated or geranylgeranylated or both, the cDNA encoding the protein under investigation is being incubated in separate tubes with commercially available transcription-translation systems and [3H]FPP or [3H]GGPP. A third reaction is carried out in the presence of [35S]methionine in order to assess the protein levels generated during the translation reaction.
Finally, we emphasize the significance of farnesylation-, geranylgeranylation- and prenylation-resistant mutants as valuable research tools in this field (Table 1). Although they are not covered in detail in the protocols described below, these mutants enable us to ask important questions. For example, if one mutates a GGT-1 substrate into an FT substrate, prenylation of such a mutant should be resistant to GGTIs and ectopic expression of such mutant should rescue cells from GGTI effects. Similarly, an FT substrate protein mutated into a GGT-1 substrate should be resistant to FTIs and should rescue cells from FTI-induced effects. These exclusively farnesylated or exclusively geranylgeranylated proteins are generated by converting the C-terminal residue into methionine (for FT) or leucine (for GGT-1). For an example of how we used these mutants in defining the roles of RalA and RalB in transformation, see reference 3. Finally, rendering a protein prenylation resistant is crucial to link prenylation to a cellular phenotype. These mutants are generated by replacing the prenylatable cysteine with serine. This approach was used to demonstrate that farnesylation is required for the transforming activity of K-Ras55.
Limitations of the protocols
In summary, although the methods described here were developed more than 15 years ago, they are still appropriate for quantifying PT enzyme activities and for studying prenylation events of single proteins. To our knowledge, there are no alternative methods available to the PT activity assays we describe, except mass spectrometry (see below). The strategies described here are invaluable to establish that a given PTI has inhibited its primary target, i.e., either FT or GGT-1, or prevented the prenylation of select FT or GGT-1 substrates. Because PTIs affect possibly hundreds of PT substrates, a general disadvantage of these methods is that they cannot unequivocally establish a link between the biochemical effect on a certain protein and the effect on a cellular phenotype of a given PTI. One possible approach to overcome this conundrum is to use small interfering RNAs silencing prenylated proteins that are suspected to be responsible for mediating the PTI effects.
A specific disadvantage of the EMSA and the subcellular fractionation assay is that the former only works for proteins in which the prenylation status changes their apparent Mr, and the latter only works for proteins that accumulate in the cytosol in response to a PTI. For newly identified PT substrates, this needs to be established first in order to take advantage of these methods. A key drawback of the method designed to investigate candidate proteins for prenylation (Steps 1D and 2D) is the length of time required for autoradiography. Provided the sequence of the candidate protein is known, a nonradioactive alternative would be to subject the immunoprecipitated candidate protein to proteolytic digestion and to analyze the resulting C-terminal peptides by mass spectrophotometry.
The most important limitation of these methods is that they appear unsuitable to investigate prenylated proteins on a larger, maybe even proteome-wide scale, in a timely manner. Such approaches are urgently needed in order to identify the subsets of prenylated proteins that are affected by FTIs and/or GGTIs, which in turn should help to link the physiological effects of various PTIs to their molecular targets, and thus design improved clinical trials. Maurer-Stroh et al. recently made an important step toward this goal when they developed a software that predicts whether a protein can potentially be prenylated. This takes into account the requirement for specific residues within the CaaX box, evolutionary conservation of the prenylation motif across phyla and physicochemical constraints extending up to 11 residues upstream of the prenylatable Cys43,44. The same laboratory also described in vitro prenylation assays based on incubation of target proteins expressed in rabbit reticulocyte lysates with 3H-labeled prenyl precursors and subsequent analysis by thin-layer chromatography, which bypasses the long exposure times required for 3H-autoradiography100. Another encouraging approach to identify prenylated proteins is to use modified prenyl donors containing antigenic or chemoselective groups that can be exploited for subsequent detection. Such a technique, incorporating the use of a synthetic azido-farnesyl analog and affinity purification by a biotinylated phosphine capture reagent, led to the proteomic identification of 18 farnesylated proteins101. This so-called ‘tagging-via-substrate’ approach has recently been developed further by Onono et al.102, who labeled cells with anilinogeraniol, an alcohol precursor of an unnatural prenyl donor, i.e., 8-anilinogeranyl diphosphate. Cell lysates were then analyzed by 2D-PAGE and western blotting with antibodies to anilinogeranyl, thus identifying the farnesylated prenylome102. Another significant advance was presented by Nguyen et al.103, who used biotinylated geranyl diphosphate (BGPP) as a lipid donor for GGT-2. As neither FT nor GGT-1 accepts BGPP as a substrate, the authors modified the prenyl donor site of both enzymes, enabling them to recognize BGPP. Using either wild-type GGT-2 or modified FT and GGT-1 together with BGPP, protein substrates can be biotinylated, isolated from cell lysates by streptavidin beads and identified by proteolytic digestion and analysis by tandem mass spectrometry103. These novel techniques and others104–106 have recently been reviewed in ref. 107.
MATERIALS
REAGENTS
Step 1A and 1D (mammalian cell culture)
• Complete tissue culture medium appropriate for the cells being studied (various suppliers)
• FBS (heat-inactivated at 56 °C, triple 0.1 μm–filtered; Atlanta Biologicals, cat. no. S11150H)
• Antibiotics ‘pen-strep’ (10,000 μ ml −1 penicillin, 10 mg ml −1 streptomycin; Invitrogen, cat. no. 15140)
• FTIs, GGTIs (we use inhibitors mostly synthesized in-house, but some can also be purchased from commercial sources)
• DMSO (sterile-filtered, hybridoma-tested; Sigma, cat. no. D2650): most common solvent used for preparing stock solutions of PTIs
Step 1B (Preparation of cell extracts for PT activity assays)
• Trizma hydrochloride (Tris/HCl, Sigma, cat. no. T5941)
• EDTA (Sigma, cat. no. E5134)
• EGTA (Sigma, cat. no. E4378)
• DTT (Sigma, cat. no. D5545)
• Complete protease inhibitor cocktail tablets (Roche, cat. no. 04 693 132 001)
• PMSF (Sigma, cat. no. P7626; prepare a 100 mM stock solution in isopropanol) ! CAUTION PMSF powder is hazardous. Skin/eye contact or inhalation can cause tissue damage and/or skin burns. Prepare stock solutions using personal protective equipment in a fume hood.
Step 1C (preparation of tissue or tumor biopsy extracts)
• Tissue Protein Extraction Reagent (T-PER, Thermo Scientific, Pierce, cat. no. 78510)
• Ethanol (Pharmco, cat. no. 111ACS200)
Step 1D ([3H]MVA labeling of cells)
• Lovastatin (mevinolin; Sigma, cat. no. M2147) ! CAUTION Lovastatin may cause irritation to eyes, skin or mucous membranes and upper respiratory tract. It may be harmful if ingested. Wear protective gloves, eye and face protection and a lab coat.
• Mevalonic acid (Sigma, cat. no. 41288) or, alternatively, Mevalonolactone (Sigma, cat. no. M4677) ! CAUTION Mevalonic acid causes serious eye damage. It may be harmful if inhaled, absorbed through skin or ingested. Wear protective gloves, eye and face protection and a lab coat.
• RS-[5-3H]mevalonic acid (MVA), triethylammonium salt (1 mCi ml −1, American Radiolabeled Chemicals, cat. no. 0334), supplied in ethanol ! CAUTION [5-3H]MVA is radioactive. As 3H is a weak β-emitter, no special protective shielding is necessary, but it is a radiation hazard when inhaled, ingested via food, water or absorbed through the skin. A lab coat, gloves and protective eyewear should be worn. Wipe tests of the working area should be conducted after the completion of each experiment.
Step 1D (preparation of cDNA-encoding candidate PT substrates)
• Optional: AccuPrime Taq DNA polymerase system (Invitrogen, cat. no. 12339-016)
• Optional: QIAQuick PCR purification kit (Qiagen, cat. no. 28106). These two reagents are only necessary if the cDNA of interest is only available in linear form in tiny quantities.
Step 2 General reagents
• Trizma hydrochloride (Sigma, cat. no. T5941)
• EDTA (Sigma, cat. no. E5134), Steps 2A and 2D only
• EGTA (Sigma, cat. no. E4378), Steps 2A, 2B and 2D only
• DTT (Sigma, cat. no. D5545), Step 2A only
• Complete protease inhibitor cocktail tablets (Roche, cat. no. 04 693 132 001), Steps 2A, 2B and 2D only
• Phenylmethylsulphonylfluoride (PMSF; Sigma, cat. no. P7626; prepare a 100 mM stock solution in isopropanol), Steps 2A, 2B and 2D only ! CAUTION PMSF powder is hazardous. Skin/eye contact or inhalation can cause tissue damage and/or skin burns. Prepare stock solutions using personal protective equipment in a fume hood.
• Bio-Rad protein assay reagent (Bio-Rad, cat. no. 500-0006), Steps 2A, 2C and 2D only
• Potassium chloride (KCl; Sigma, cat. no. P9333), Steps 2A, 2B, 2C and 2D only
• Magnesium chloride (MgCl2; Sigma, cat. no. M8266), Steps 2A and 2B only
Step 2A (PT activity assay)
• Sodium orthovanadate (Na3VO4, ≥99.98; Sigma-Aldrich, cat. no. 450243)
• 4-Nitrophenylphosphate (Sigma, cat. no. P4744)
• Zinc chloride (ZnCl2; Sigma, cat. no. 39059). Prepare a 0.1 M solution in H2O.
• Purified recombinant human H-Ras wild type (CVLS; prepared in-house85). This protein serves as a substrate for FT activity measurements and is commercially available from CalBiochem (cat. no. 553325). Store at −80 °C.
• Purified recombinant human H-Ras-CVLL (prepared in-house91), this protein serves as a substrate for GGT-1 activity measurements and is also commercially available from CalBiochem (cat. no. 553322). Store at −80°C
• [3H]FPP (0.5mCi ml −1, 15–30 Ci mmol −1, 17–33 μM; Perkin-Elmer, cat. no. NET1042001MC) ! CAUTION [3H]FPP is radioactive. As 3H is a weak β-emitter, no special protective shielding is necessary, but it is a radiation hazard when inhaled, ingested via food, water or when absorbed through the skin. A lab coat, gloves and protective eyewear should be worn. Wipe tests of the working area should be conducted after the completion of each experiment.
• [3H]GGPP (0.5mCi ml −1, 10–30 Ci mmol −1, 17–50 μM; Perkin-Elmer, cat. no. NET1052250UC) ! CAUTION [3H]GGPP is radioactive. As 3H is a weak β-emitter, no special protective shielding is necessary, but it is a radiation hazard when inhaled, ingested via food, water or absorbed through the skin. A lab coat, gloves and protective eyewear should be worn. Wipe tests of the working area should be conducted after the completion of each experiment.
• SDS (Bio-Rad, cat. no. 161-0302). Prepare as a 10% (wt/vol) stock solution ! CAUTION SDS powder is hazardous. Prepare stock solution using personal protective equipment in a fume hood. Required throughout Step 2.
• Trichloroacetic acid (TCA; Sigma, cat. no. T9159). To prepare a 100% (wt/vol) TCA stock solution, add 227 ml of H2O to a bottle containing 500 g of TCA. Store at 4 °C ! CAUTION TCA is very hazardous in case of skin or eye contact, ingestion or inhalation.
• Scintillation fluid Ecolite (MP Biomedicals, cat. no. 882475)
Step 2B (cell lysis)
• CelLytic MT Cell Lysis Reagent (Sigma, cat. no. C3228)
• HEPES (Fisher Scientific, cat. no. L-12213)
• Sodium fluoride (NaF; Fisher Scientific, cat. no. S299)
• Triton X-100 (Fisher Scientific, cat. no. BP151)
Steps 2B–D (cell lysis)
• Trypsin-EDTA (0.05% (wt/vol); Invitrogen, cat. no. 25300)
• Sodium chloride (NaCl; Fisher Scientific, cat. no. L-11621)
• Disodium hydrogen phosphate, anhydrous (Na2HPO4; Fisher Scientific, cat. no. S374-1)
• Potassium dihydrogen phosphate, anhydrous (KH2PO4; Fisher Scientific, cat. no. P286-1)
• Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Scientific, Pierce, cat. no. 89826), only required for option C
• Glycerol (Sigma, cat. no. G5516)
Steps 2B–E (electrophoresis, electrotransfer and western blotting)
• 2-Mercaptoethanol (Fisher Scientific, cat. no. O3446I)
• Bromophenol blue (Sigma, cat. no. B6131)
Step 2D (immunoprecipitation of [3H]-labeled proteins)
• Igepal CA-360 (NP-40 substitute; Sigma, cat. no. I3021)
• Sodium deoxycholate (Pierce, cat. no. 89904)
Step 2E (in vitro transcription-translation-prenylation)
• TnT reticulocyte lysate system (Promega, cat. no. L4600, L4610, L4950, L5010, L5020) M CRITICAL Which system to purchase depends on the nature of the RNA polymerase promoter used in the plasmid of interest.
• Rnasin ribonuclease inhibitor (Promega, cat. no. N2111)
• Nuclease-free water (Promega, cat. no. P1193)
• l-[35S]Met (1 mCi; Perkin-Elmer, cat. no. NEG709A001MC) ! CAUTION l-[35S]Met is radioactive. 35S is a relatively weak β-emitter, and thus no special protective shielding is necessary, but it is a radiation hazard when inhaled, ingested via food, water or absorbed through the skin. A lab coat, gloves and protective eyewear should be worn. A particular danger associated with 35S is that this isotope is extremely volatile. Therefore, predetermined amounts of 35S should be removed from the radioactive vial in a chemical hood dedicated to radioactive work. In addition, sterile aerosol tips should be used to avoid contamination of pipettes. Wipe tests of the working area including any incubators should be conducted after the completion of each experiment.
EQUIPMENT
• Precision balance (many suppliers)
• Parafilm or equivalent laboratory film
• Vortexer (many suppliers)
• Nitrogen gas source
• Vacuum concentrator (e.g., SpeedVac)
• Water baths (several suppliers) Steps 1A, 1D, 2A and 2E
• Biological safety cabinet (Thermo Electron, Forma Class II A2) Steps 1A, 1D
• Incubator (Thermo Forma Series II water jacketed CO2 incubator, model 3310) Steps 1A, 1D
• Cell culture plates or dishes (many suppliers) Steps 1A, 1D
• Refrigerated tabletop centrifuge for ≤50-ml tubes (Eppendorf, model 5810R) Steps 1A, 1C, 2B and C
• Refrigerated tabletop centrifuge for ≤2.0-ml tubes (Eppendorf, model 5417R) Steps 1B and C, 2B and C
• Plastic cuvettes (10 mm × 4 mm × 45 mm; Sarstedt, cat. no. 67.742) Steps 2A–C
• PowerGen* Model 125 homogenizer (Fisher Scientific, cat. no. 14-359-251) Step1C only
• PCR machine (MJ Research, model PTC-200 Peltier Thermo Cycler) Step 1E only
• Heatblock Digtl 2-BLK (VWR, cat. no. 12259-052)
• Autoradiography film (MidSci, Classic film BX, 5 × 7 or 8 × 10 in, depending on size of gels, cat. no. B×57 or B×810) Steps 2B–D only
• X-ray film processer Mini-Medical (AFP ImageWorks, cat. no. 9992305300) Steps 2B–D only
• Kodak BioMax MS film (Carestream Health, 8 × 10 in, cat. no. 829-4985) Steps 2D and E only
• Kodak TranScreen LE intensifying screen (Carestream Health, 8 × 10 in, cat. no. 162-2034) Steps 2D and E only
• Rotator for incubating immunoprecipitation reactions (Boekel Scientific Instruments, Orbitron Rotator II) Step 2D only
Step 1B only
• Branson sonifier 450 (VWR, cat. no. 33995-590)
• Ultracentrifuge Optima L-90K (Beckman Coulter, cat. no. 365670)
• SW 55-Ti rotor (Beckman Coulter, cat. no. 342194)
• Centrifuge tubes, Ultra-Clear 13 × 51 mm (Beckman Coulter, cat. no. 344057)
Step 2A only
• GF/B glass fiber filters (25 mm diameter; Whatman, cat. no. 1821-025)
• Sampling manifold 12-hole (Millipore, cat. no. XX2702550)
• Liquid scintillation counting system (Beckman, model LS 6500)
Steps 2B–E only
• PVDF membrane Immobilon-P (Millipore, cat. no. IPVH00010) Steps 2B–E
• Rocking platform (many suppliers)
REAGENT SETUP
FTI and GGTI Most PTIs we have investigated are soluble in DMSO up to concentrations of 100 mM. Ideally, weigh as little drug powder as possible ( < 10 mg) in a 1.7-ml tube, and add DMSO to arrive at a final concentration of 100 mM. Store in small aliquots at − 20 °C . The stability of each compound has to be determined empirically.
? TROUBLESHOOTING
Cell sonication buffer Cell sonication buffer is prepared using 50 mM Tris/HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA and 1 mM DTT. Store at 4 °C indefinitely. Just before use, add protease inhibitor cocktail tablets and 2 mM PMSF.
Tissue lysis buffer (T-PER reagent) Store T-PER reagent at room temperature (RT; 21 °C) indefinitely. Just before use, add 2 mM Na3VO4, 2 mM PMSF and 6.4 mg ml −1 4-nitrophenylphosphate to T-PER reagent protease inhibitor cocktail tablets.
Tris-buffered saline Tris-buffered saline (TBS) is prepared using 20 mM Tris/HCl (pH 7.4) and 0.9% (wt/vol) NaCl. Store at RT for up to 1 month.
PT reaction buffer (3×) Mix the following components to the indicated final concentrations: 100 mM Tris-Cl (pH 7.5), 150 μM ZnCl2, 60 mM KCl, 9 mM
MgCl2 and 3 mM DTT. Store at 4 °C.
6% (wt/vol) TCA/2% (wt/vol) SDS Mix 0.6 ml of 100% (wt/vol) TCA, 2 ml of 10% (wt/vol) SDS and 7.4 ml of ddH O to a final volume of 10 ml.
Store at RT idefinitely.
PBS (pH 7.4) PBS is prepared using 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4. For 1 liter of PBS, dissolve 8.00 g of NaCl, 0.20 g of KCl, 1.42 g of Na2HPO4 and 0.27 g of KH2PO4 in 800 ml of ddH2O. Adjust the pH to 7.4 with HCl. Add ddH2O to 1 liter. Store at RT for up to 3 months.
CelLytic MT cell lysis reagent Store at RT indefinitely. Just before use, add protease inhibitor cocktail tablets, 2 mM Na3VO4, 2 mM PMSF and 6.4 mg ml −1 4-nitrophenylphosphate.
Lysis buffer (2×) Mix 60 mM HEPES (pH 7.5), 20 mM NaCl, 10 mM MgCl2, 50 mM NaF, 2 mM EGTA, 2% (vol/vol) Triton X-100 and 20% glycerol. Store at 4 °C for up to 3 months.
SDS sample buffer (4×) Combine 40% (vol/vol) glycerol, 240 mM Tris/HCl (pH 6.8), 8% (wt/vol) SDS, 0.04% (wt/vol) bromophenol blue and 5% (vol/vol) 2-mercaptoethanol . For 10 ml, mix 4 ml 100% (vol/vol) glycerol, 2.4 ml 1 M Tris/HCl (pH 6.8), 0.8 g SDS, 4 mg of bromophenol blue, 0.5 ml 2-mercaptoethanol and 3.1 ml H2O. Store in small aliquots at − 20 °C indefinitely.
Tris-buffered saline For convenience, prepare a 10× stock solution consisting of 200 mM Tris/HCl (pH 7.4) and 9% (wt/vol) NaCl. Store at RT for up to 6 months. Before use, dilute 1:10 with ddH2O. For probing immunoblots, prepare just before use TBS-T, i.e., TBS containing 0.2% (vol/vol) Tween-20.
Lovastatin (10 mM) Dissolve 101 mg of lovastatin in 1.8 ml of ethanol at 55 °C. Add 0.9 ml of 0.6 M NaOH and 18 ml of ddH2O. Incubate for 30 min at RT. Adjust to pH 8 with HCl and add ddH2O to a final volume of 25 ml. Store indefinitely at − 20 °C in 500-μl aliquots108.
RIPA buffer Mix the following components to the indicated final concentrations: 50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1% (vol/vol) NP-40, 0.1% (wt/vol) SDS, 0.1 mM EDTA and 0.1 mM EGTA. Store at 4 °C for up to 6 months. Just before use, add protease inhibitor cocktail tablets, 2 mM Na3VO4 2 mM PMSF and 6.4 mg ml−1 4-nitrophenylphosphate.
Immunoprecipitation wash buffer Mix the following components to the indicated final concentrations: 50 mM Tris/HCl (pH 7.5), 100 mM NaCl, 0.2% (vol/vol) NP-40 and 0.5% (vol/vol) sodium deoxycholate. Store at 4 °C for up to 6 months.
PROCEDURE
1 Select and prepare a source material suitable for the question addressed (see EXPERIMENTAL DESIGN). Either grow mammalian cells of interest (option A), prepare cell extracts containing partially purified FT and GGT1 (option B), prepare extracts from a biopsy of interest (option C), label cells with [3H]MVA (option D) or generate cDNA encoding a candidate PT substrate protein (option E).
(A) Growth of cells in culture ● TIMING 3–4 d including the experimental drug treatment
(i) On day 1, plate the cells of interest in an appropriate medium containing 10% (vol/vol) FBS and antibiotics at 37 °C and 5% (vol/vol) CO2 atmosphere, typically using 60-mm plates if you plan to prepare cell extracts for PT assays (Step 1B), and 100-mm dishes if you plan to prepare cytosolic and membrane fractions (Step 2C) or label cells with [3H]MVA (Step 1D).
▲ CRITICAL STEP Perform Steps 1A(i) and 1A(ii) using sterile techniques.
▲ CRITICAL STEP If you plan to perform enzyme activity assays (Step 1B), or prepare cell lysates for the EMSA (Step 2B) or subcellular fractionation (Step 2C), consider that the initial plating density should be optimized for each cell line in such a way that by the end of the incubation period (≥72 h) the cells will not have reached confluency. In most cases, 60-mm plates are big enough to yield sufficient protein for western blot analysis, but occasionally, 100-mm dishes may be required.
▲ CRITICAL STEP If you plan to label cells with [3H]-MVA (Step 1D), it is advisable to set up cells at two split ratios and choose the culture closest to 70% to 80% confluency for labeling. The cultures should contain between 0.5 × 106 and 0.5 × 107 cells. Note that the required number of cells to be plated and therefore the type of plate to be used depend on the abundance of the protein of interest and the efficiency of the subsequent immunoprecipitation step.
(ii) (optional) On day 2, remove medium and in fresh medium add the PTI under investigation and incubate the cells for 24 to 72 h under the same conditions as in Step 1A(i). As negative controls, include untreated cells and cells treated with the vehicle to dissolve the drug. As positive control, add a known PTI at a concentration that inhibits prenylation by 50% to 100%.
Note that PTI concentrations necessary to inhibit PT in cells may be an order of magnitude higher than those inhibiting PT in vitro.
Note that this step can be omitted if proceeding to Step 1D below.
? TROUBLESHOOTING
(iii) To prepare extracts for PT activity assays, proceed with Step 1B. To label cells with [3H]MVA, proceed with Step 1D. To prepare extracts for mobility shift assay or perform subcellular fractionations, proceed with Step 2B or 2C, respectively.
(B) Preparation of cell extracts for PT activity assay ● TIMING ~4 h for 10–20 samples
(i) Harvest cultured cells as follows: aspirate the cell culture medium, add 1 ml of cell sonication buffer and transfer to 1.7-ml microcentrifuge tubes.
Note that if preparing PT from PBMCs the method is identical, except use only ≤500 μl sonication buffer28.
▲ CRITICAL STEP Perform Steps 1B(i–v) on ice or at 4 °C.
(ii) Sonicate the cells using a sonifier such as the Branson sonifier 450. If using PBMCs and performing western blots, following sonication proceed directly to Step 2B(v) below.
(iii) Centrifuge the lysate at 12,000g for 30 min.
(iv) Transfer the clear supernatants to new prelabeled 3.5-ml ultracentrifuge tubes.
(v) Centrifuge the supernatant at 60,000g for 1 h. The resulting postmicrosomal supernatant is the source of FT or GGT-1.
(vi) Proceed with Step 2A.
(C) Preparation of tissue or tumor biopsy extracts ● TIMING ~3 h
(i) By using sterile technique, measure the weight of the tumor biopsies (ranging from 50 mg to 1 g); quickly return the tumor to ice by placing it into a prelabeled, precooled 50-ml sterile plastic tube. If you wish to perform PT activity assays (Step 2A) or mobility shift assays (Step 2B), add tissue lysis buffer. If you wish to perform subcellular fractionations (Step 2C), add TBS to normal or tumor tissue biopsies in a ratio of 20 ml per gram.
! CAUTION For experiments involving human patients, informed consent must be obtained from all subjects.
! CAUTION For experiments involving animals, national and institutional guidelines on animal welfare must be followed throughout.
(ii) Cut the tissue into small pieces with a clean razor blade.
(iii) By using the PowerGen* Model 125 homogenizer, homogenize the tumor biopsies, keeping tubes containing biopsies on ice during the homogenization. Wash blades of the homogenizer with 70% (vol/vol) ethanol 2–3 times, followed by one wash with ddH2O before and between samples.
(iv) After homogenization, to perform enzyme activity assays (Step 2A) or mobility shift assays (Step 2B), centrifuge the sample tubes at 200g for 1 min at 4 °C to collect samples at the bottom of the tube. To pursue subcellular fractionation, centrifuge the homogenate at 1,000g for 5 min at 4 °C, discard the supernatant and proceed with Step 2C(iii).
(v) Transfer the homogenates from 50-ml tubes to 1.7-ml sterile prelabeled microcentrifuge tubes.
(vi) Centrifuge samples at 12,000g for 30 min at 4 °C.
(vii) To perform PT activity assays, proceed with Step 1B(iv)80 or to perform EMSAs proceed with Step 2B(vii)18.
(D) Metabolic labeling of cells with [3H]MVA L TIMING ~28 h
(i) On the day of the experiment, replace the medium with 5 ml of medium containing 5% (vol/vol) dialyzed serum and 15 ìM lovastatin. Preincubate the cells in this medium for ≤2 h at 37 °C. When using this approach to study novel candidate PT substrates, be sure to include parallel cultures to examine a positive control, such as H-Ras (exclusively farnesylated) and/or Rap1A (exclusively geranylgeranylated).
M CRITICAL STEP Perform Steps 1D(i–iv) using sterile techniques.
(ii) Transfer enough 1 mCi ml −1 R-[5-3H]MVA to label all samples at a concentration of 50 to 200 μCi ml −1 into a micro-centrifuge tube. Concentrate the radioactive reagent by removing organic solvent with a very gentle stream of nitrogen. Alternatively, the organic solvent may be removed in a SpeedVac evaporator. ! CAUTION MVA must not be heated above RT because it is unstable.
(iii) Add the concentrated [3H]MVA at the desired concentration to 2 ml of culture medium containing 5% (vol/vol) dialyzed serum and 15 μM lovastatin. Incubate for up to 24 h at 37°C.
CRITICAL STEP The most appropriate concentration of the radiolabeled compound and duration of incubation should be experimentally determined for each cell type.
? TROUBLESHOOTING
(iv) Proceed with Step 2D.
(E) Generation of cDNAs encoding candidate PT substrates ● TIMING ~1 d
(i) Use purified circular plasmid DNA if possible, as best results are obtained with this form. If this is not available, amplify the linear cDNA encoding the candidate PT substrate protein via PCR using AccuPrime Taq polymerase and 1 μg plasmid DNA.
(ii) Subsequently, isolate cDNA using a QIAquick PCR purification kit.
▲ PAUSE POINT The purified cDNA can be stored at − 20 °C until being further processed.
(iii) Proceed with Step 2E.
2 Select an analytical procedure suitable for the question addressed (see EXPERIMENTAL DESIGN). If you wish to perform PT activity assay using cell or tissue extracts, perform option A. If you wish to perform an electrophoretic mobility shift assay, follow option B. If you wish to compare membrane and cytosolic fractions, follow option C. If you wish to label cells with [3H]MVA, follow option D. If you wish to analyze in vitro prenylation, follow option E.
(A) PT activity assay in lysates from cultured cells, PBMCs, tissue or tumor biopsies ● TIMING ~5 h
(i) After the last centrifugation step, transfer the clear supernatant to a new prelabeled 1.7-ml tube.
(ii) Measure the protein concentration in each sample using the Bradford assay (Bio-Rad kit) 109. Adjust the protein concentration to approximately 1–10 μg μl −1 by adding sonication buffer. The concentration needed for Step 2A(iv) may need to be optimized for the different sources of the FT and GGT-1 (cultured cells, PBMCs, organs or tumor tissue).
(iii) Prepare a mixture of two parts 3× PT reaction buffer and one part 1:10 diluted [3H]FPP or [3H]GGPP (‘hot mix’), using volumes sufficient to accommodate the number of samples to be processed.
(iv) Transfer 20 μl of enzyme from Step 2A(ii) to 12 × 75 mm glass tubes. If you wish to characterize novel PTIs in vitro with purified enzymes or to determine the inhibitory activity of PTIs in samples from untreated cells or tumors, mix the PTI of interest with purified FT or GGT-1 (or alternatively the postmicrosomal supernatant prepared in Step 1B) in a final volume of 20 μl for each sample. Preincubate the two components for 5 min on ice. Prepare a set of blanks without the inhibitor.
▲ CRITICAL STEP The PTI concentrations used should cover several orders of magnitude. This may have to be optimized depending on the properties of the inhibitor tested.
(v) Add 30 μl of ‘hot mix’ to each enzyme sample.
(vi) Start the reaction by adding 10 μl of FT substrate Ras-CVLS (60 μM) or GGT-1 substrate Ras-CVLL (60 μM) to each sample, resulting in a final volume of 60 μl. To the blanks, add 10 μl ddH2O instead.
(vii) Vortex samples briefly to mix the contents, and then incubate in a water bath equilibrated at 37 °C for 30 min. ! CAUTION Seal the lids of tubes with Parafilm to prevent radioactive spills.
(viii) Remove samples from incubator and add 0.5 ml 4% (wt/vol) SDS, and then add 0.5 ml 30% (wt/vol) TCA to each sample.
(ix) Vortex and incubate on ice for 45 min (to maximize TCA precipitation of proteins).
(x) During Step 2A(ix), presoak glass fiber filters in ddH2O.
(xi) After completion of incubation, add 2 ml per sample of 6% (wt/vol) TCA/2% (wt/vol) SDS solution.
(xii) Filter samples onto presoaked Whatman GF/B glass fiber filters using a Millipore filter manifold. Wash each sample tube twice with 2 ml per wash with 6% (wt/vol) TCA/2% (wt/vol) SDS solution.
(xiii) Wash each filter five times with 2 ml 6% (wt/vol) TCA; dry filters.
(xiv) Transfer filters to scintillation vials; add 5 ml of scintillation fluid and count in a liquid scintillation counter (e.g., Beckman model LS6500).
(xv) The counts per minute (c.p.m.) obtained from samples without Ras (blanks) should be subtracted from those containing Ras (experimental samples). Express data as percentage of control.
(B) EMSA ● TIMING ~28 h for minigels, ~2 d for standard-sized gels
(i) Harvest the cells by trypsinization: add ~0.04 ml of 0.05% (wt/vol) trypsin-EDTA per cm2 growth surface area and incubate at 37 °C until the cells detach.
▲ CRITICAL STEP Perform Steps 2B(i–vi) on ice or at 4 °C.
▲ CRITICAL STEP Note that the protocol to prepare lysates suitable for SDS-PAGE varies slightly for different source materials.
(ii) Add three volumes of complete medium + FBS to neutralize trypsin and transfer suspended cells to a fresh sterile 15-ml plastic tube.
(iii) Centrifuge the cells for 5 min at 300g. Carefully aspirate the medium.
(iv) Wash the cells once in PBS.
■ PAUSE POINT Pelleted cells can be stored for short periods of time at − 20 °C until they are further processed by SDS-PAGE.
(v) Carefully aspirate the PBS and add ~0.1 ml CelLytic lysis buffer per 10 cm2 growth surface area. Transfer the cell lysate to a fresh 1.7-ml tube. If you are working with PBMCs, mix the sonicated cells from Step 1B above with 1 volume of 2× lysis buffer28.
▲ CRITICAL STEP This rule applies to plates with nearly confluent cells. If any treatment has resulted in markedly lower cell density, reduce the volume of lysis buffer accordingly, or use larger cell culture plates.
(vi) Incubate on ice for 30 min while vortexing every 10 min. Centrifuge lysate at 12,000g for 5 min to remove insoluble material.
(vii) Determine the protein concentration using the Bradford assay109.
(viii) Add one-third of the sample volume of 4× SDS sample buffer to each sample; e.g., if the sample volume is 90 μl, add 30 μl of 4× SDS sample buffer and heat the samples at 100 °C for 5–10 min. Centrifuge the samples briefly to collect the contents at the bottom of the tubes.
■ PAUSE POINT Boiled samples can be stored at − 20 °C until being further processed by SDS-PAGE.
(ix) Determine the apparent Mr of the protein(s) in question by SDS-PAGE, followed by western blotting with the appropriate antibodies.
▲ CRITICAL STEP The mobility shift assay can serve two purposes: it can be used to determine the degree of PTI-mediated PT inhibition, or the degree of PTI-mediated inhibition of specific proteins.
In the first case, we recommend that the apparent Mr of well-known substrates be verified for farnesylation or geranylgeranylation, namely HDJ-2 and Rap1A, respectively. The mobility shift of HDJ-2 (Mr = 45 kDa) can conveniently be detected by 7.5% (wt/vol) SDS-PAGE minigels by allowing the 37 kDa Mr marker protein to migrate into the lower third of the gel, whereas the lack of prenylation of Rap1A can be detected by an antibody specific for the unprocessed Rap1A (see EXPERIMENTAL DESIGN), using a 15% (wt/vol) gel.
To determine whether a PTI has prevented the prenylation of a specific protein other than HDJ-2 or Rap1A, it is very important to select an acrylamide concentration that is optimal for the molecular mass of the protein being studied. Although minigels are much more convenient to handle and are far less time consuming, the resolution afforded by them may not be sufficient to unequivocally demonstrate a shift in electrophoretic mobility and apparent Mr in response to a drug. When in doubt, we recommend that a second experiment be carried out using standard-sized or oversized gels (16 or 20 cm length).
? TROUBLESHOOTING
(C) Preparation and analysis of membrane and cytosolic fractions ● TIMING ~34 h if using minigels for SDS-PAGE and western blotting
(i) Isolate up to 5 × 106 cells per sample by centrifuging harvested cell suspensions at 850g for 2 min in 1.7-ml microcentrifuge tubes. Wash pelleted cells once in PBS.
▲ CRITICAL STEP Perform Steps 2C(i–vi) on ice or at 4 °C.
(ii) Carefully remove and discard the supernatant.
(iii) For Steps 2C(iii–vi), use the Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (see REAGENTS), which consists of Reagents A, B and C (because these reagents are proprietary, their composition is unknown). Add 150 μl of Reagent A to the cell pellet. Pipette up and down to obtain a homogeneous cell suspension. Incubate for 10 min at RT with occasional vortexing. Note that white, flocculent debris should appear upon addition of Reagent A.
▲ CRITICAL STEP To check the cell lysis efficiency, spot 5 μl of cell lysate onto a glass slide, place cover slip and view under a light microscope. Compare with 5 μl of the same number of intact cells in 150 μl of PBS.
? TROUBLESHOOTING
(iv) Place lysed cells on ice.
(v) Dilute two parts of Reagent C with one part of Reagent B, making sufficient mixture for each sample to receive 450 μl (e.g., for ten extractions, combine 3.33 ml of Reagent C with 1.67 ml of Reagent B). Keep Reagents B and C at 4 °C or on ice at all times.
(vi) Add 450 μl of diluted Reagent C to each tube of lysed cells and vortex. Incubate tubes on ice for 30 min, vortexing every 5 min.
(vii) Centrifuge tubes at 10,000 g for 3 min at RT. Transfer the supernatant to new tubes and incubate for 10 min in a 37 °C water bath to separate the membrane protein fraction. To enhance phase separation, increase incubation time to 20 min.
(viii) Centrifuge tubes for 2 min at 10,000g at RT to isolate the hydrophobic fraction (i.e., the fraction containing the membrane protein of interest) from the hydrophilic fraction.
(ix) Carefully remove the hydrophilic phase (top layer) from the hydrophobic protein phase (bottom layer) and save in a new tube. Perform phase separations as quickly as possible, because the interface between the layers slowly disappears at RT.
(x) Place the separated fractions on ice. The majority of membrane protein will be in the lower hydrophobic fraction, which can be used for membrane protein analysis.
(xi) Determine protein concentration of each sample using the Bradford assay (Bio-Rad kit) 109.
(xii) Add one-third volume of 4× SDS sample buffer to each sample. Heat the samples at 100 °C for 5–10 min. Centrifuge the samples briefly to collect the contents at the bottom of the tubes.
■ PAUSE POINT Boiled samples can be stored at − 20 °C until being further processed by SDS-PAGE.
(xiii) Determine the subcellular distribution of the protein in question by running them on a 12.5% (wt/vol) SDS-PAGE gel, followed by western blotting with the appropriate antibody.
▲ CRITICAL STEP To assess the purity of the membrane and cytosolic fractions, it is crucial to probe for proteins that are known to be exclusively cytosolic, such as GAPDH or membrane bound, such as E-cadherin110.
? TROUBLESHOOTING
(D) Immunoprecipitation of [3H]labeled proteins ● TIMING ≤ 1.5 d until autoradiography, up to 3 months for autoradiography
(i) Place the cells on ice and aspirate the medium. Wash the cells twice with ice-cold PBS. ! CAUTION Radioactive medium and washes must be disposed of appropriately.
(ii) Lyse cells by adding 1 ml of RIPA buffer for immunoprecipitation.
(iii) Immunoprecipitate protein of interest by standard procedures. Note that optimum conditions must be established prior to these radiolabeling experiments.
(iv) Solubilize the immunoprecipitate in 25–50 μl of 1× SDS-PAGE sample buffer and heat the samples at 100 °C for 5–10 min. Centrifuge the samples briefly to collect the contents at the bottom of the tubes.
! CAUTION Take care to prevent popping of lids as these might lead to spills of radioactive material.
■ PAUSE POINT Boiled samples can be stored at − 20 °C until being further processed by SDS-PAGE.
(v) Separate the immunoprecipitate by SDS-PAGE. The appropriate percentage of acrylamide/bisacrylamide depends on the molecular mass of the protein being studied.
(vi) To allow detection by direct autoradiography, electrotransfer the proteins to Immobilon-P membranes using standard procedures.
We prefer this approach because it has the added advantage that, unlike dried gels that are to be examined by fluorography, membranes do not crack and can be used to perform other assays, such as western blotting4.
(vii) Mount the completely dried membrane in an X-ray film cassette, place an intensifying screen between the membrane and a sheet of BioMax MS film.
(viii) Expose membranes to film at RT for up to 1 month.
! CAUTION Exposure times of up to 3 months may sometimes be required.
(E) In vitro prenylation using in vitro transcription-translation systems ● TIMING 6 h for Steps 2E(i–iv), up to 3 months for Step 2E(v)
(i) Accorcding to Promega Technical Bulletin 126 assemble the components of the transcription-translation reaction mix on ice. These include the reticulocyte lysate, reaction buffer, the appropriate RNA polymerase, amino acid mix minus leucine or methionine, as well as a RNase inhibitor. Use 12.5 μl rabbit reticulocyte lysate along with either 5 μCi [3H]FPP, 5 μCi [3H]GGPP or 10 μCi [35S]methionine in the presence or absence of appropriate concentrations of the PTI to be tested. Add nuclease-free water to a final reaction volume of 25 μl. Start the reaction by adding between 0.5 and 1.0 μg of plasmid DNA.
(ii) Incubate the reaction mixture in a water bath equilibrated at 30 °C for 120 min.
(iii) Stop the reaction by adding one-third volume of 4× SDS sample buffer and heat at 80 °C for 4–5 min; briefly centrifuge the samples to collect the contents at the bottom of the tube.
! CAUTION Because of the high protein concentration, heating at 100 °C may cause precipitation; therefore, lower the denaturing temperature to 80 °C. Take care to prevent the popping of lids as these might lead to spills of radioactive material.
J PAUSE POINT Denatured samples can be stored at − 20 °C until being further processed by SDS-PAGE.
(iv) Use between 5 and 20% of the 35S-labeled samples and 100% of the 3H-labeled samples to separate the proteins by SDS-PAGE gels. The appropriate percentage of acrylamide/bisacrylamide depends on the molecular mass of the protein being studied.
(v) Follow Steps 2D(vi–viii) as described above.
? TROUBLESHOOTING
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 4.
TABLE 4.
Troubleshooting table.
| Step | Problem | Possible Reason | Possible solution |
|---|---|---|---|
| Reagent setup | Solubility of PTIs in aqueous buffers | Hydrophobicity | Try vortexing, incubation at 37 °C, and/or sonication |
| If this fails, try different vehicles or formulation | |||
| If this fails, lower the concentration of the compound | |||
| 1A(ii) | No detectable change in prenylation | Half-life of protein is too long | Incubate cells for up to 120 h in the presence of PTI |
| 1D(iii) | Cells are dying or do not grow | Lovastatin is toxic to the cells being studied | Decrease the concentration of or the time of exposure to lovastatin |
| 2B(ix) | No shift in apparent Mr in response to PTI detectable | Protein becomes cross-prenylated by the PT not affected | Treat cells with a combination of FTI and GGTI |
| Insufficient resolution of the chosen acrylamide concentration | Perform subcellular fractionation instead (Step 2C) | ||
| 2C(iii) | Insufficient cell lysis | Too many cells | Increase volume of or incubation time with Reagent A |
| 2C(xiii) | No relocation to cytosol in response to PTI detectable | Protein becomes cross-prenylated by the PT not affected | Treat cells with a combination of FTI and GGTI |
| Protein is not prenylated but associates with other organelles or matrices | Perform EMSA instead (Step 2B) |
● TIMING
Step 1A, Growth of mammalian cells including drug treatment: 3–4 d
Step 1B, Preparation of cell extracts for PT activity assays: ~4 h
Step 1C, Preparation of tumor biopsy extracts containing PT: ~3 h
Step 1D, Metabolic labeling of cells with [3H]MVA: ~28 h
Step 1E, Generation of cDNA-encoding candidate PT substrate (optional): ~1 d
Step 2A, PT activity assay in lysates from cultured cells, PBMCs, tissue or tumor biopsies: ~5 h
Step 2B, EMSA: ~28 h for mini gels, ~2 d for standard-sized gels
Step 2C, Preparation and analysis of membrane and cytosolic fractions: ~34 h
Step 2D, Metabolic labeling of cells with [3H]MVA and analysis: Steps 2D(i–vi), immunoprecipitation and SDS-PAGE/electrotransfer: ≤ 1.5 d, Steps 2D(vii–viii), autoradiography: up to 3 months
Step 2E, Prenylation assay using in vitro transcription-translation systems: transcription-translation assay, 4 h; SDS-PAGE and electrotransfer, 4 h; autoradiography, up to 3 months
ANTICIPATED RESULTS
The experiments described in the above protocol are designed to yield results that are easily interpretable. Figure 2 shows two variations of PT activity assays that we routinely use. One of the first steps of a typical drug discovery project is to evaluate the potency and selectivity of novel lead compounds. As already discussed in EXPERIMENTAL DESIGN, it is important to determine the IC50 value and the selectivity of the compound for the targeted PT18 (Fig. 2a). When treating cells, animals or human patients with an FTI or GGTI, it is critical to determine whether the drug has affected its intended target. Figure 2b shows the results from a recent clinical trial suggesting that therapy with tipifarnib significantly inhibited FT in PBMCs from patients with myelodys-plastic syndrome28. The data also show that this in itself is no guarantee for a tumor response.
Figure 2.

Typical results from enzyme activity assays described in Step 2A. (a) An important step in the evaluation of novel PTIs is to estimate IC50 values for FT and GGT-1. In this example, the novel GGTI-2418 was shown to be ~5,600-fold more selective for GGT-1 over FT (reproduced from ref. 18). (b) When treating cells, animals or, as in this example, humans, it is crucial to determine the enzyme activity targeted by the given drug. This figure shows that FT activity in PBMCs from patients with myelodysplastic syndrome is inhibited after treatment with the FTI tipifarnib. PBMCs from patients before (0) and during treatment (on days 2 and 7, for example) were prepared as described in Step 1B, and FT activity was measured as described in Step 2A. CR, complete remission; HI, hematological improvement; NR, no response; PD, progressive disease; PR, partial remission. There appears to be no obvious correlation between the response and the degree of FT inhibition. GGT-1 activity, which was measured concurrently, was not inhibited by tipifarnib (reproduced from ref. 28, this study was conducted in accordance with institutional internal review board policy after obtaining written informed consent from participating patients).
Both the EMSA and the subcellular fractionation described in Steps 2B and 2C are the most frequently conducted experiments in a laboratory interested in prenylation. To determine whether a PTI has inhibited the prenylation of its intended target, we routinely determine the prenylation status of FT and GGT-1 substrates, namely, the farnesylated HDJ-2 and geranylgeranylated Rap1A. The demonstration of a slower migrating band and a higher apparent Mr in response to an FTI (GGTI) would thus suggest that (i) the protein in question is a substrate for FT (GGT-1) and (ii) the prenyltransferase inhibitor has affected its intended target (Fig. 3). Note that it cannot be taken for granted that all PT substrates increase their electrophoretic mobility upon prenylation. When studying novel candidate PT substrates, this has to be established by [3H]MVA labeling of intact cells in the absence or presence of a PTI, followed by immunoprecipitation of the protein of interest. For proteins that do not change their mobility, e.g., RhoA, their prenylation can be assessed by studying their distribution between membrane and cytosolic fractions79 (Fig. 4). Finally, in Figure 5, we present the results of an in vitro transcription-translation- prenylation experiment3.
Figure 3.

Typical results from EMSAs described in Step 2B. After treatment of cells with PTIs, the protein under investigation will accumulate in the unprocessed (unprenylated) form (U), at the expense of the prenylated species (P). To verify whether a PTI has functioned properly in a given experiment, we recommend checking the prenylation status of the FT substrate HDJ-2 and the GGT-1 substrate Rap1A. This can be detected by a change in electrophoretic mobility (HDJ-2) or with antibodies specific for the unprenylated form (Rap1A), using 7.5 or 15.0% (wt/vol) acrylamide gels. Note that substrates for FT, GGT-1 or both PTs react differently to experimental drug treatment. After treatment of cells with an FTI, a GGTI or a combination of the two, one can envision three different outcomes: exclusively farnesylated proteins (HDJ-2) respond to an FTI, exclusively geranylgeranylated proteins (Rap1A) respond to a GGTI, whereas alternatively prenylated proteins (K-Ras, reproduced from ref. 61) only respond to inhibition of both enzymes.
Figure 4.
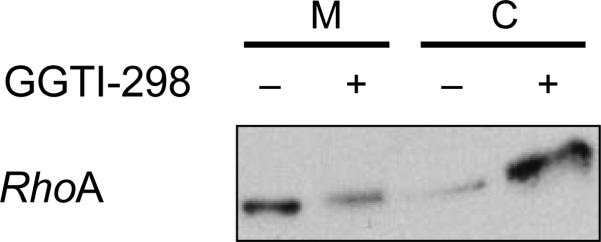
Typical results from subcellular fractionation experiments described in Step 2C. With some proteins, e.g., RhoA, it is notoriously difficult to demonstrate a mobility shift upon PTI treatment. In this case, we recommend preparing cytosolic and membrane fraction, followed by western blotting. The figure (from ref. 79) shows accumulation of RhoA in the cytosol following treatment of CaLu-1 cells with GGTI-298.
Figure 5.

Typical results from in vitro transcription-translation-prenylation assays described in Step 2E. In the example shown, wild-type RalA (CaaX box: CCIL) and GTP-locked mutant RalA-Q72L (CaaX box: CCIL) were geranylgeranylated, but not farnesylated in vitro. However, mutants with a C-terminal CCIS motif were farnesylated instead, whereas mutants with a C-terminal SCIL motif were resistant to prenylation (reproduced from ref. 3).
TABLE 2.
Tried and tested antibodies in prenylation research.
| Protein | PT | Antibody | Reactivity | Suitable for2 | Company | Cat. no. |
|---|---|---|---|---|---|---|
| Cdc42 | GGT-1 | Mouse mAb | M, R, H | WB, IP, IF, IHC | Santa Cruz | sc-8401 |
| HDJ-2 | FT | Mouse mAb | H, M, R | WB, IP, IF, IHC | Thermo Scientific | MS-225 |
| MYPT3 | FT | Rabbit pAb | H, M, Rb | WB | Millipore | 07-470 |
| Rac1 | GGT-1 | Rabbit pAb | M, R, H | WB, IP, IF | Santa Cruz | sc-217 |
| RalA | GGT-1 | Mouse mAb | H, M, R | WB, IF | BD | 610222 |
| RalB | GGT-1 | Mouse mAb | H, M, R | WB | Millipore | 04-037 |
| Rap1A | GGT-1 | Goat pAb | M, R, H | WB, IP, IF | Santa Cruz | sc-1482a |
| H-Ras | FT | Rabbit pAb | M, R, H | WB, IP | Santa Cruz | sc-520 |
| K-Ras | FT, GGT-1 | Mouse mAb | H, M, R | WB | Calbiochem | OP24 |
| N-Ras | FT, GGT-1 | Rabbit pAb | H, M, R | WB, IP, IF | Santa Cruz | sc-519 |
| Rheb | FT | Rabbit pAb | H, M, R | WB, IHC | Cell Signaling | 4935 |
| RhoA | GGT-1 | Mouse mAb | M, H, R | WB, IP | Santa Cruz | sc-418 |
| RhoB | GGT-1 | Rabbit pAb | M, R, H | WB, IP | Santa Cruz | sc-180 |
| RhoC | GGT-1 | Goat pAb | M, R, H | WB, IF | Santa Cruz | sc-12116 |
H, human; IF, immunofluorescence; IHC, immunohistochemistry; IP, immunoprecipitation; M, mouse; R, rat; Rb, rabbit; WB, western blotting; mAb, monoclonal antibody; pAB, polyclonal antibody.
Antibodies to the most frequently studied prenylated proteins. We use these antibodies routinely in the laboratory and find them of outstanding or sufficient quality. Some antibodies react with antigens from additional species.
This antibody is specific to the unprenylated form of Rap1A.
TABLE 3.
Secondary antibodies.
| Antibody | Company | Cat. no. |
|---|---|---|
| Goat anti-mouse IgG | Jackson Immuno Research | 115-035-003 |
| Goat anti-rabbit IgG | Jackson Immuno Research | 111-035-003 |
| Rabbit anti-goat IgD | Jackson Immuno Research | 305-035-003 |
We use these secondary antibodies in combination with those Listed in Table 2.
ACKNOWLEDGMENTS
This work was supported by grants from the NIH CA067771 (S.M.S) and CA098473 (S.M.S.). We thank A.D. Hamilton and his group for a wonderful and highly productive collaboration and M.A. Blaskovich and A. Kazi for their excellent suggestions. We also thank the past and present members of the Sebti lab for their contributions to this work, particularly J. Adnane, M.E. Balasis, M.A. Blaskovich, C. Bucher, P.M. Campbell, A.E. Carie, Z. Chen, N.C.Crespo, F. Delarue, S.C. Falsetti, K. Forinash, K. Jiang, A. Kazi, E.C. Lerner, T.F. McGuire, M. Nigam, S. Paquette, R.Patel, J. Sun, Y. Sun, V. Thai, A. Vogt, D. Wang, A. Tecleab, H. Yang and K. Zhu.
Footnotes
AUTHOR CONTRIBUTIONS Both N.B. and S.M.S. jointly wrote the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 2.Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA. 1989;86:8323–8327. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falsetti SC, et al. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis, and RalA to inhibit anchorage-independent growth. Mol. Cell. Biol. 2007;27:8003–8014. doi: 10.1128/MCB.00057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell. 2005;18:425–434. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Kohl NE, et al. Protein farnesyltransferase inhibitors block the growth of ras-dependent tumors in nude mice. Proc. Natl. Acad. Sci. USA. 1994;91:9141–9145. doi: 10.1073/pnas.91.19.9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop WR, et al. Novel tricyclic inhibitors of farnesyl protein transferase. Biochemical characterization and inhibition of Ras modification in transfected Cos cells. J. Biol. Chem. 1995;270:30611–30618. doi: 10.1074/jbc.270.51.30611. [DOI] [PubMed] [Google Scholar]
- 7.Sun J, et al. Antitumor efficacy of a novel class of non-thiol-containing peptidomimetic inhibitors of farnesyltransferase and geranylgeranyltransferase I: combination therapy with the cytotoxic agents cisplatin, taxol, and gemcitabine. Cancer Res. 1999;59:4919–4926. [PubMed] [Google Scholar]
- 8.End DW, et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001;61:131–137. [PubMed] [Google Scholar]
- 9.Sepp-Lorenzino L, et al. A peptidomimetic inhibitor of farnesyl:protein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer Res. 1995;55:5302–5309. [PubMed] [Google Scholar]
- 10.Zheng H, et al. Ras homologue enriched in brain is a critical target of farnesyltransferase inhibitors in non-small cell lung cancer cells. Cancer Lett. 2010;297:117–125. doi: 10.1016/j.canlet.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Qian Y, Hamilton AD, Sebti SM. Ras CAAX peptidomimetic FTI 276 selectively blocks tumor growth in nude mice of a human lung carcinoma with K-Ras mutation and p53 deletion. Cancer Res. 1995;55:4243–4247. [PubMed] [Google Scholar]
- 12.Mangues R, et al. Antitumor effect of a farnesyl protein transferase inhibitor in mammary and lymphoid tumors overexpressing N-ras in transgenic mice. Cancer Res. 1998;58:1253–1259. [PubMed] [Google Scholar]
- 13.Cohen-Jonathan E, et al. The farnesyltransferase inhibitor L744,832 reduces hypoxia in tumors expressing activated H-ras. Cancer Res. 2001;61:2289–2293. [PubMed] [Google Scholar]
- 14.Du W, Lebowitz PF, Prendergast GC. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol. Cell. Biol. 1999;19:1831–1840. doi: 10.1128/mcb.19.3.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han JY, et al. Hypoxia-inducible factor 1alpha and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J. Natl. Cancer Inst. 2005;97:1272–1286. doi: 10.1093/jnci/dji251. [DOI] [PubMed] [Google Scholar]
- 16.Kim CK, et al. The farnesyltransferase inhibitor LB42708 suppresses vascular endothelial growth factor-induced angiogenesis by inhibiting ras-dependent mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signal pathways. Mol. Pharmacol. 2010;78:142–150. doi: 10.1124/mol.110.063586. [DOI] [PubMed] [Google Scholar]
- 17.Sun SY, Zhou Z, Wang R, Fu H, Khuri FR. The farnesyltransferase inhibitor Lonafarnib induces growth arrest or apoptosis of human lung cancer cells without downregulation of Akt. Cancer Biol. Ther. 2004;3:1092–1098. doi: 10.4161/cbt.3.11.1176. discussion 1099–1101. [DOI] [PubMed] [Google Scholar]
- 18.Kazi A, et al. Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol. Cell. Biol. 2009;29:2254–2263. doi: 10.1128/MCB.01029-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moasser MM, et al. Farnesyl transferase inhibitors cause enhanced mitotic sensitivity to taxol and epothilones. Proc. Natl. Acad. Sci. USA. 1998;95:1369–1374. doi: 10.1073/pnas.95.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoover RR, Mahon FX, Melo JV, Daley GQ. Overcoming STI571 resistance with the farnesyl transferase inhibitor SCH66336. Blood. 2002;100:1068–1071. doi: 10.1182/blood.v100.3.1068. [DOI] [PubMed] [Google Scholar]
- 21.Zhu K, et al. Farnesyltransferase inhibitor R115777 (Zarnestra, tipifarnib) synergizes with paclitaxel to induce apoptosis and mitotic arrest to inhibit tumor growth of multiple myeloma cells. Blood. 2005;105:4759–4766. doi: 10.1182/blood-2004-11-4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Punt CJ, van Maanen L, Bol CJ, Seifert WF, Wagener DJ. Phase I and pharmacokinetic study of the orally administered farnesyl transferase inhibitor R115777 in patients with advanced solid tumors. Anticancer Drugs. 2001;12:193–197. doi: 10.1097/00001813-200103000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Castro AF, Rebhun JF, Clark GJ, Qilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J. Biol. Chem. 2003;278:32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 24.Mavrakis K, et al. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008;22:2178–2188. doi: 10.1101/gad.1690808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caruso MG, et al. Increased farnesyltransferase activity in human colorectal cancer: relationship with clinicopathological features and K-ras mutation. Scand. J. Gastroenterol. 2003;38:80–85. doi: 10.1080/00365520310000483. [DOI] [PubMed] [Google Scholar]
- 26.Ryan DP, et al. Phase I clinical trial of the farnesyltransferase inhibitor BMS-214662 given as a 1-hour intravenous infusion in patients with advanced solid tumors. Clin. Cancer Res. 2004;10:2222–2230. doi: 10.1158/1078-0432.ccr-0980-3. [DOI] [PubMed] [Google Scholar]
- 27.Kurzrock R, et al. Phase I study of alternate-week administration of tipifarnib in patients with myelodysplastic syndrome. Clin. Cancer Res. 2008;14:509–514. doi: 10.1158/1078-0432.CCR-07-1532. [DOI] [PubMed] [Google Scholar]
- 28.Kurzrock R, et al. Farnesyltransferase inhibitor R115777 in myelodysplastic syndrome: clinical and biologic activities in the phase 1 setting. Blood. 2003;102:4527–4534. doi: 10.1182/blood-2002-11-3359. [DOI] [PubMed] [Google Scholar]
- 29.Adjei AA, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003;21:1760–1766. doi: 10.1200/JCO.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 30.Sparano JA, et al. Phase II trial of tipifarnib plus neoadjuvant doxorubicin-cyclophosphamide in patients with clinical stage IIB-IIIC breast cancer. Clin. Cancer Res. 2009;15:2942–2948. doi: 10.1158/1078-0432.CCR-08-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 32.Lane KT, Beese LS. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J. Lipid Res. 2006;47:681–699. doi: 10.1194/jlr.R600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Sala D. Protein isoprenylation in biology and disease: General overview and perspectives from studies with genetically engineered animals. Front. Biosci. 2007;12:4456–4472. doi: 10.2741/2401. [DOI] [PubMed] [Google Scholar]
- 34.Carboni JM, et al. Farnesyltransferase inhibitors are inhibitors of Ras but not R-Ras2/TC21, transformation. Oncogene. 1995;10:1905–1913. [PubMed] [Google Scholar]
- 35.Moores SL, et al. Sequence dependence of protein isoprenylation. J. Biol. Chem. 1991;266:14603–14610. [PubMed] [Google Scholar]
- 36.Roskoski R, Jr., Ritchie P. Role of the carboxyterminal residue in peptide binding to protein farnesyltransferase and protein geranylgeranyltransferase. Arch. Biochem. Biophys. 1998;356:167–176. doi: 10.1006/abbi.1998.0768. [DOI] [PubMed] [Google Scholar]
- 37.Boutin JA, et al. Chromatographic assay and peptide substrate characterization of partially purified farnesyl- and geranylgeranyltransferases from rat brain cytosol. Arch. Biochem. Biophys. 1998;354:83–94. doi: 10.1006/abbi.1998.0678. [DOI] [PubMed] [Google Scholar]
- 38.Reid TS, Terry KL, Casey PJ, Beese LS. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substate selectivity. J. Mol. Biol. 2004;343:417–433. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 39.Whyte DB, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 40.Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer. 2005;5:405–412. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- 41.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 42.Baekkeskov S, Kanaani J. Palmitoylation cycles and regulation of protein function (Review). Mol. Membr. Biol. 2009;26:42–54. doi: 10.1080/09687680802680108. [DOI] [PubMed] [Google Scholar]
- 43.Maurer-Stroh S, Eisenhaber F. Refinement and prediction of protein prenylation motifs. Genome Biol. 2005;6:R55. doi: 10.1186/gb-2005-6-6-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maurer-Stroh S, et al. Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput. Biol. 2007;3:e66. doi: 10.1371/journal.pcbi.0030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sebti SM. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell. 2005;7:297–300. doi: 10.1016/j.ccr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Mijimolle N, et al. Protein farnesyltransferase in embryogenesis, adult homeostasis, and tumor development. Cancer Cell. 2005;7:313–324. doi: 10.1016/j.ccr.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Lerner EC, et al. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic ras signaling by inducing cytoplasmic accumulation of inactive ras-raf complexes. J. Biol. Chem. 1995;270:26802–26806. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 48.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583–586. doi: 10.1038/310583a0. [DOI] [PubMed] [Google Scholar]
- 49.Yang SH, et al. Caution! Analyze transcripts from conditional knockout alleles. Transgenic Res. 2009;18:483–489. doi: 10.1007/s11248-008-9237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu M, et al. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc. Natl. Acad. Sci. USA. 2010;107:6471–6476. doi: 10.1073/pnas.0908396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Willumsen BM, Norris K, Papageorge AG, Hubbert NL, Lowy DR. Harvey murine sarcoma virus p21 ras protein: biological and biochemical significance of the cysteine nearest the carboxy terminus. EMBO J. 1984;3:2581–2585. doi: 10.1002/j.1460-2075.1984.tb02177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butrynski JE, Jones TL, Backlund PS, Jr., Spiegel AM. Differential isoprenylation of carboxy-terminal mutants of an inhibitory G-protein alpha-subunit: neither farnesylation nor geranylgeranylation is sufficient for membrane attachment. Biochemistry. 1992;31:8030–8035. doi: 10.1021/bi00149a037. [DOI] [PubMed] [Google Scholar]
- 53.Ohya Y, et al. Yeast CAL1 is a structural and functional homologue to the DPR1 (RAM) gene involved in ras processing. J. Biol. Chem. 1991;266:12356–12360. [PubMed] [Google Scholar]
- 54.Therrien M, et al. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 55.Jackson JH, et al. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc. Natl. Acad. Sci. USA. 1990;87:3042–3046. doi: 10.1073/pnas.87.8.3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004;14:105–114. doi: 10.1016/j.semcancer.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 57.Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez-Viciana P, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 59.Appels NMGM, Beijnen JH, Schellens JHM. Development of farnesyltransferase inhibitors: a review. Oncologist. 2005;10:565–578. doi: 10.1634/theoncologist.10-8-565. [DOI] [PubMed] [Google Scholar]
- 60.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun J, Qian Y, Hamilton AD, Sebti SM. Both farnesyltransferase and geranylgeranyltransferase I inhibitors are required for inhibition of oncogenic K-Ras prenylation but each alone is sufficient to suppress human tumor growth in nude mouse xenografts. Oncogene. 1998;16:1467–1473. doi: 10.1038/sj.onc.1201656. [DOI] [PubMed] [Google Scholar]
- 62.Kohl NE, et al. Inhibition of farnesyltransferase induces regression of mammary salivary carcinomas in ras transgenic mice. Nat. Med. 1995;1:792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 63.Omer CA, et al. Mouse mammary tumor virus-Ki-rasB transgenic mice develop mammary carcinomas that can be growth-inhibited by a farnesyl: protein transferase inhibitor. Cancer Res. 2000;60:2680–2688. [PubMed] [Google Scholar]
- 64.Sebti SM, Der CJ. Searching for the elusive targets of farnesyltransferase inhibitors. Nat. Rev. Cancer. 2003;3:945–951. doi: 10.1038/nrc1234. [DOI] [PubMed] [Google Scholar]
- 65.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications: farnesyl transferase inhibitors. J. Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 66.Ashar HR, et al. Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J. Biol. Chem. 2000;275:30451–30457. doi: 10.1074/jbc.M003469200. [DOI] [PubMed] [Google Scholar]
- 67.Crespo NC, Ohkanda J, Yen TJ, Hamilton AD, Sebti SM. The farnesyltransferase inhibitor, FTI-2153, blocks bipolar spindle formation and chromosome alignment and causes prometaphase accumulation during mitosis of human lung cancer cells. J. Biol. Chem. 2001;276:16161–16167. doi: 10.1074/jbc.M006213200. [DOI] [PubMed] [Google Scholar]
- 68.Crespo NC, et al. The farnesyltransferase inhibitor, FTI-2153, inhibits bipolar spindle formation during mitosis independently of transformation and Ras and p53 mutation status. Cell Death Differ. 2002;9:702–709. doi: 10.1038/sj.cdd.4401023. [DOI] [PubMed] [Google Scholar]
- 69.Sepp-Lorenzino L, Rosen N. A farnesyl-protein transferase inhibitor induces p21 expression and G1 block in p53 wild type tumor cells. J. Biol. Chem. 1998;273:20243–20251. doi: 10.1074/jbc.273.32.20243. [DOI] [PubMed] [Google Scholar]
- 70.Prendergast GC. Actin’ up: RhoB in cancer and apoptosis. Nat. Rev. Cancer. 2001;1:162–168. doi: 10.1038/35101096. [DOI] [PubMed] [Google Scholar]
- 71.Chen Z, et al. Both farnesylated and geranylgeranylated RhoB inhibit malignant transformation and suppress human tumor growth in nude mice. J. Biol. Chem. 2000;275:17974–17978. doi: 10.1074/jbc.C000145200. [DOI] [PubMed] [Google Scholar]
- 72.Van Cutsem E, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 73.Rao S, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J. Clin. Oncol. 2004;22:3950–3957. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 74.Harousseau JL, et al. A randomized phase 3 study of tipifarnib compared with best supportive care, including hydroxyurea, in the treatment of newly diagnosed acute myeloid leukemia in patients 70 years or older. Blood. 2009;114:1166–1173. doi: 10.1182/blood-2009-01-198093. [DOI] [PubMed] [Google Scholar]
- 75.Sparano JA, et al. Targeted inhibition of farnesyltransferase in locally advanced breast cancer: A phase I and II trial of Tipifarnib plus dose-dense doxirubicin and cyclophosphamide. J. Clin. Oncol. 2006;24:3013–3018. doi: 10.1200/JCO.2005.04.9114. [DOI] [PubMed] [Google Scholar]
- 76.Hamad NM, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–2057. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim K-H, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–545. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 78.Vogt A, Sun JZ, Qian YM, Hamilton AD, Sebti SM. The geranylgeranyltransferase-I inhibitor GGTI-298 arrests human tumor cells in G0/G1 and induces p21WAF1/CIP1/SDI1 in a p53-independent manner. J. Biol. Chem. 1997;272:27224–27229. doi: 10.1074/jbc.272.43.27224. [DOI] [PubMed] [Google Scholar]
- 79.Sun J, et al. The geranylgeranyltransferase I inhibitor GGTI-298 induces hypophosphorylation of retinoblastoma and partner switching of cyclin-dependent kinase inhibitors. A potential mechanism for GGTI-298 antitumor activity. J. Biol. Chem. 1999;274:6930–6934. doi: 10.1074/jbc.274.11.6930. [DOI] [PubMed] [Google Scholar]
- 80.Sun J, et al. Geranylgeranyltransferase I inhibitor GGTI-2154 induces breast carcinoma apoptosis and tumor regression in H-ras transgenic mice. Cancer Res. 2003;63:8922–8929. [PubMed] [Google Scholar]
- 81.Dan HC, et al. Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene. 2004;22:706–715. doi: 10.1038/sj.onc.1207171. [DOI] [PubMed] [Google Scholar]
- 82.Sjogren A-K, et al. GGTase-1 deficiency reduces tumor formation and improves survival in mice with K-Ras-induced lung cancer. J. Clin. Invest. 2007;117:1294–1304. doi: 10.1172/JCI30868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O'Dwyer PJ, Gallagher M, Nguyen B, Waddell MJ, Chiorean EG. Phase I accelerated dose-escalating safety and pharmacokinetic (PK) study of GGTI-2418, a novel geranylgeranyltransferase I inhibitor in patients with refractory solid tumors. Ann. Oncol. 2010;21(Suppl 2):ii42. [Google Scholar]
- 84.Vogt A, et al. Burkitt lymphoma Daudi cells contain two distinct farnesyltransferases with different divalent cation requirements. Biochemistry. 1995;34:12398–12403. doi: 10.1021/bi00038a037. [DOI] [PubMed] [Google Scholar]
- 85.Nigam M, Seong CM, Qian Y, Hamilton AD, Sebti SM. Potent inhibition of human tumor p21ras farnesyltransferase by A1A2-lacking p21ras CA1A2X peptidomimetics. J. Biol. Chem. 1993;268:20695–20698. [PubMed] [Google Scholar]
- 86.Qian Y, et al. Design and structural requirements of potent peptidomimetic inhibitors of p21ras farnesyltransferase. J. Biol. Chem. 1994;269:12410–12413. [PubMed] [Google Scholar]
- 87.Vogt A, et al. A non-peptide mimetic of Ras-CAAX: selective inhibition of farnesyltransferase and Ras processing. J. Biol. Chem. 1995;270:660–664. doi: 10.1074/jbc.270.2.660. [DOI] [PubMed] [Google Scholar]
- 88.McGuire TF, et al. CAAX peptidomimetic FTI-244 decreases platelet-derived growth factor receptor tyrosine phosphorylation levels and inhibits stimulation of phosphatidylinositol 3-kinase but not mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 1995;214:295–303. doi: 10.1006/bbrc.1995.2287. [DOI] [PubMed] [Google Scholar]
- 89.Lerner EC, Qian Y, Hamilton AD, Sebti SM. Disruption of oncogenic K-Ras4B processing and signaling by a potent geranylgeranyltransferase I inhibitor. J. Biol. Chem. 1995;270:26770–26773. doi: 10.1074/jbc.270.45.26770. [DOI] [PubMed] [Google Scholar]
- 90.Hunt JT, et al. Discovery of (R)-7-cyano-2,3,4, 5-tetrahydro-1- (1H-imidazol-4-ylmethyl)-3- (phenylmethyl)-4-(2-thienylsulfonyl)-1H-1, 4-benzodiazepine (BMS-214662), a farnesyltransferase inhibitor with potent preclinical antitumor activity. J. Med. Chem. 2000;43:3587–3595. doi: 10.1021/jm000248z. [DOI] [PubMed] [Google Scholar]
- 91.Cox AD, Hisaka MM, Buss JE, Der CJ. Specific isoprenoid modification is required for function of normal, but not oncogenic, Ras protein. Mol. Cell. Biol. 1992;12:2606–2615. doi: 10.1128/mcb.12.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sinensky M, Fantle K, Dalton M. An antibody which specifically recognizes prelamin A but not mature lamin A: application to detection of blocks in farnesylation-dependent protein processing. Cancer Res. 1994;54:3229–3232. [PubMed] [Google Scholar]
- 93.Lerner EC, et al. Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene. 1997;15:1283–1288. doi: 10.1038/sj.onc.1201296. [DOI] [PubMed] [Google Scholar]
- 94.Vogt A, Qian Y, McGuire TF, Hamilton AD, Sebti SM. Protein geranylgeranylation, not farnesylation, is required for the G1 to S phase transition in mouse fibroblasts. Oncogene. 1996;13:1991–1999. [PubMed] [Google Scholar]
- 95.Delarue FL, et al. Farnesyltransferase and geranylgeranyltransferase I inhibitors upregulate RhoB expression by HDAC1 dissociation, HAT association and histone acetylation of the RhoB promoter. Oncogene. 2007;26:633–640. doi: 10.1038/sj.onc.1209819. [DOI] [PubMed] [Google Scholar]
- 96.Brunner TB, et al. Pancreatic cancer cell radiation survival and prenyltransferase inhibition: the role of K-Ras. Cancer Res. 2005;65:8433–8441. doi: 10.1158/0008-5472.CAN-05-0158. [DOI] [PubMed] [Google Scholar]
- 97.Wang D-A, Sebti SM. Palmitoylated cysteine 192 is required for RhoB tumor-suppressive and apoptotic activities. J. Biol. Chem. 2005;280:19243–19249. doi: 10.1074/jbc.M411472200. [DOI] [PubMed] [Google Scholar]
- 98.Bivona TG, et al. PKC regulates a farnesyl-electrostatic switch on K-ras that promotes its association with Bcl-Xl on mitochondria and induces apoptosis. Mol. Cell. 2006;21:481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 99.Bivona TG, Quatela S, Philips MR. Analysis of Ras activation in living cells with GFP-RBD. Methods Enzymol. 2006;407:128–143. doi: 10.1016/S0076-6879(05)07012-6. [DOI] [PubMed] [Google Scholar]
- 100.Benetka W, Koranda M, Maurer-Stroh S, Pittner F, Eisenhaber F. Farnesylation or geranylgeranylation? Efficient assays for testing protein prenylation in vitro and in vivo. BMC Biochem. 2006;7:6. doi: 10.1186/1471-2091-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kho Y, et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. USA. 2004;101:12479–12484. doi: 10.1073/pnas.0403413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Onono FO, et al. A tagging-via-substrate approach to detect the farnesylated proteome using two-dimensional electrophoresis coupled with Western blotting. Mol. Cell. Proteomics. 2010;9:742–751. doi: 10.1074/mcp.M900597-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nguyen UT, et al. Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat. Chem. Biol. 2009;5:227–235. doi: 10.1038/nchembio.149. [DOI] [PubMed] [Google Scholar]
- 104.Troutman JM, Roberts MJ, Andres DA, Spielmann HP. Tools to analyze protein farnesylation in cells. Bioconjug. Chem. 2005;16:1209–1217. doi: 10.1021/bc050068+. [DOI] [PubMed] [Google Scholar]
- 105.Chan LN, et al. A novel approach to tag and identify geranylgeranylated proteins. Electrophoresis. 2009;30:3598–3606. doi: 10.1002/elps.200900259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Degraw AJ, et al. Evaluation of alkyne-modified isoprenoids as chemical reporters of protein prenylation. Chem. Biol. Drug Des. 2010;6:460–471. doi: 10.1111/j.1747-0285.2010.01037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hannoush RN, Sun J. The chemical toolbox for monitoring protein fatty acylation and prenylation. Nat. Chem. Biol. 2010;6:498–506. doi: 10.1038/nchembio.388. [DOI] [PubMed] [Google Scholar]
- 108.Kita T, Brown MS, Goldstein JL. Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J. Clin. Invest. 1980;66:1094–1100. doi: 10.1172/JCI109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 110.Berndt N, et al. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 2010;17:1795–1804. doi: 10.1038/cdd.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]