

Abstract
Myeloid-derived suppressor cells (MDSCs) accumulate in the glioma microenvironment during tumor progression and promote immunosuppression. Interleukin-12 (IL-12) immunogene therapy can alter MDSCs toward an antigen-presenting cell phenotype and these mature cells can have a central role in antigen presentation. It remains unclear, however, how MDSC depletion can affect glioma immunotherapy. In this study, we generated a replication-deficient adenoviral vector, Ad.5/3.cRGD-mIL12p70, that transduces the GL261-based murine glioma cell line, resulting in the induction of biologically active, murine IL12p70 expression. Ex vivo, IL-12 expressed by GL261 cells induced interferon-γ synthesis in CD8 +T cells (P<0.001), CD4 +T cells (P =0.009) and natural killer cells (P =0.036). When injected 1 week after tumor implantation, Ad.5/3.cRGD-mIL12p70 successfully prolonged the survival of glioma-bearing mice. Sixty percent of animals treated with IL-12 immunotherapy were long-term survivors over 175 days, whereas all the control group animals expired by 40 days after tumor implantation (P =0.026). Mice receiving Ad.5/3.cRGD-mIL12p70 also accumulated 50% less MDSCs in the brain than the control group (P =0.007). Moreover, in the IL-12 group, MDSCs significantly overexpressed CD80 and major histocompatibility complex class II molecules (P =0.041). Depletion of MDSCs with Gr1 +antibody had no survival benefit induced by IL-12-mediated immunotherapy. Of note, IL-12 therapy increased the presence of myeloid dendritic cells (mDCs) in the glioma microenvironment (P =0.0069). Ultimately, the data show that in the context of IL-12 immunogene therapy, MDSCs are dispensable and mDCs may provide the majority of antigen presentation in the brain.
Keywords: interleukin-12, myeloid-derived suppressor cells, myeloid dendritic cells, plasmacytoid dendritic cells, immunotherapy, glioblastoma
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common brain tumor and is associated with the worst prognosis.1 Even under the most aggressive treatment regimens, GBM recurs with a median patient survival of 14.6 months.2,3 A major impediment to successful targeting of GBM is the highly immunosuppressive microenvironment. Therefore, overcoming the immune suppression and generating an effective immune response against GBM has been a long-standing goal in the field of immunotherapy. This approach relies on re-engineering the properties of the immune system from that which is tolerant to—or promoting of—GBM progression to one that induces cytotoxic T-cell-mediated GBM cell clearance.4–6 Most of the data we have in glioma immunotherapy has been generated in rodent models. Mouse GL261 orthotopic glioma is one of the best-studied models in immunotherapy because it is a reliable, reproducible model in immunocompetent C57/Bl6 mice.7 The GL261 microenvironment, similar to human GBM, contains cells with immunosuppressive potential, such as regulatory T cells and myeloid-derived suppressor cells (MDSCs).8,9 The role of regulatory T cells (CD4 +CD25 +FoxP3 +) in GBM has been studied extensively in glioma and their depletion is well known to improve survival across a variety of murine glioma models.10 On the other hand, the role of MDSCs in glioma, especially during immunotherapy, is an area of active research. Recently, it has been shown that MDSC depletion prolongs mouse survival,11 while blocking chemotactic CCL2 signaling in GL261 reduces recruitment of MDSCs and tissue-associated macrophages.12 Nevertheless, we know little about the interplay between MDSCs and other immune cells or immunotherapies in the context of the glioma environment. Therefore, a better characterization of the role MDSCs have in well-defined immunotherapeutic approaches, such as interleukin-12 (IL-12) gene therapy, is required before targeting these cells. Successful depletion of MDSCs in cancer models by using a monoclonal antibody to Gr1 +has increased survival of animals but not always.11,12 In fact, depleting these subsets in certain contexts has the opposite desired effect. Previous work has demonstrated that the depletion of MDSCs during IL-12-mediated immunotherapy in models of cancer outside of the central nervous system results in the dissipation of its benefits.13,14 To date, it remains to be determined how IL-12-mediated immunotherapy can be combined with strategies that target MDSCs in glioma.
IL-12 is a well-defined cytokine associated with cancer immunotherapy, including GBM. The efficacy of IL-12 therapy has been attributed to its propensity to induce lymphocyte proliferation, a Th1 cytotoxic phenotype, and to enhance interferon-γ (IFN-γ) secretion.15,16 However, recent work has shown that, in non-central nervous system cancers, the ability of IL-12 to induce an effective antitumor immune response requires the alteration of MDSC programming toward a phenotype associated with antigen presentation.13,14 In a mouse model of gastrointestinal cancer, Medina-Echeverez et al.13 showed that the depletion of intratumoral MDSCs inhibited the survival benefit associated with IL-12-mediated gene therapy. However, whether this unexpected effect is generalizable to other tumors, such as GBM, remains to be tested.
Here, we studied the effects of IL-12 gene therapy in the antitumor immune response in the GL261 orthotopic glioma model. We focused our investigation on the immunosuppressive MDSCs and the antigen-presenting cells: myeloid (mDCs) and plasmacytoid (pDCs) dendritic cells. In addition, we tested the hypothesis that MDSCs are required for the survival benefit during IL-12-mediated immunotherapy in an orthotopic glioma model. We show that our IL-12-expressing replication-incompetent adenoviral vector simultaneously induces a cytotoxic immune response while decreasing the number of MDSCs in the brain and altering their phenotype. When IL-12 is delivered in the GL261 orthotopic glioma, it prolongs survival irrespective of whether MDSCs are depleted or not. Finally, we show that IL-12 therapy increases the presence of mDCs in the tumor and these cells can provide the antigen presentation required for effective antiglioma immune responses.
MATERIALS AND METHODS
Cell lines and adenoviral vectors
The mouse glioma cell line GL261 was received from the NCI (National Cancer Institute, Frederick, MD, USA) and grown in Dulbecco’s modified Eagle’s medium (from Cellgro, Mediatech, Manassas, VA, USA) with 10% fetal bovine serum (Atlanta Biologicals, Lawerenceville, GA, USA), 100 μg ml−1 penicillin and 100 μg ml −1 streptomycin (Cellgro). The OVA surface antigen cDNA was obtained from Origene (Rockville, MD, USA). The GL261-OVA cell line, generated based on the protocol provided by Origene, was kept in puromycin-containing media. Mouse neural stem cells (SCR029) were purchased from Millipore (Billerica, MA, USA) and grown in their recommended media (SCM003). Mouse mesenchymal stem cells were isolated from the bone marrow of 5–7 weeks old mice, as described elsewhere.17 All cells were grown in a humidified atmosphere, with 5% CO2 and 37 °C conditions. Cells were subcultured using 1 ml per 106 cells of a 0.25% trypsin/2.21 mmol l −1 ethylenediaminetetraacetic acid solution (Cellgro). Trypsin activity was quenched using the appropriate media for each cell type, then washed at 300 relative centrifugal forces and finally plated at the indicated densities.
The replication-deficient adenoviral vector Ad.5/3.cRGD-mIL12p70.GFP construct is shown in Supplementary Figure S1A. First, the polymerase chain reaction (PCR) fragment flanked with BglII–NheI sites and coding the full open reading frame of mouse IL gene, porf-mIL12 from Invivogen (San Diego, CA, USA), was cloned into adenoviral pShuttle-IRES-hrGFP-2 vector (Stratagene, Santa Clara, CA, USA). After sequence validation, the selected clone was recombined with pAdEasy-based backbone containing the 5/3-cRGD modification in the fiber region.18 The resulting vector, mIL12GFP-5/ 3cRGD or GFP-5/3cRGD, plasmids were then used to rescue replication-deficient adenoviruses, Ad.5/3.cRGD-mIL12p70.GFP (Ad.mIL12) and Ad.5/ 3.cRGD-GFP (Ad.GFP), respectively, using a standard protocol (Stratagene).
ELISA for IL-12
The supernatant of previously in vitro Ad.mIL12-infected cells was analyzed for the presence of IL12p70 content. We relied on the Ebioscience enzyme-linked immunosorbent assay (ELISA) kit for IL12p70 quantification. Samples were read in a Microplate Reader (ELx800, BioTek Instruments, Winooski, VT, USA) as described in the instruction sheet. As a control, we used non-infected and Ad.GFP-infected cells.
Antibodies and reagents
The antibodies anti-mouse CD3-APC (clone 17A2), CD4-PE (clone GK1.5), CD8-Pacific Blue (clone 53-6.7), NK1.1 (clone PK136), CD11b-PE (clone M1/70), Gr1-Pacific Blue and Biotin (clone RB6-8C5), PDCA1-PE Cy7 (clone eBio927), major histocompatibility complex class II (MHCII)-PE Cy7 (clone M5/114.15.2), CD45-Pacific Blue (clone 30-F11), anti-IFNγ-PerCP Cy7 and immunoglobulin controls were purchased from Ebioscience (San Diego, CA, USA). CD11c was purchased from BioLegend (San Diego, CA, USA), CD28 and CD49d from BD Biosciences (San Jose, CA, USA) and CD80-PerCP Cy 5.5 was obtained from Invitrogen (Grand Island, NY, USA).
Leukocyte activation cocktail, containing phorbol myristate acetate, ionomycin and Golgi-Plug, was purchased from BD Biosciences. OVA peptide (323–339; RP10610) was purchased from GenScript (Piscataway, NJ, USA).
Depletion of circulating MDSCs
The depletion of circulating Gr1+was based on previously published protocols.19 The depleting antibody to Gr1+, clone RB6-8C5, was purchased from Ebioscience. The antibody (0.25 mg dose) was delivered systemically by intraperitoneal injection, two times weekly for a total of four injections.11 The control group received intraperitoneal injection of purified rat immunoglobulins (Jackson Immunoresearch, West Grove, PA, USA).
Flow cytometry
Brain, cervical (draining) lymph node and spleen cell suspensions were prepared by passing the tissue through 70 nm cell strainers. Red blood cells were removed by treatment with ACK (Ammonium-Chloride-Potassium) Lysis Buffer (Lonza, Walkersville, MD, USA) for 4 min at 4 °C. Inflammatory cells were isolated via a Percoll gradient, as described below. The mononuclear cell layer and cells from lymph nodes and spleen were suspended in 1% fetal bovine serum in phosphate-buffered saline (PBS) and then counted using a tabletop cell counter TC20 (Bio-Rad, Hercules, CA, USA). When indicated, cells were incubated with phorbol myristate acetate (50 ng ml −1) and ionomycin (500 ng ml −1) for 4 h in the presence of Golgi-Plug (1 μl ml−1). Surface staining was performed while keeping cells on ice for 30 min. For intracellular detection of IFNγ, the cells were permeabilized, fixed and stained on ice using the Cytofix/Cytoperm buffer (BD Biosciences) according to the manufacturer’s instructions. Data were acquired and analyzed in BD FACSCanto with CellQuest (Becton Dickinson, San Jose, CA, USA) and FlowJo (TreeStar, Ashland, OR, USA) software. Experiments were performed two times independently, in triplicate.
Isolation of mononuclear cells from the animals’ brain
Animals were sacrificed according to the recommendations of The University of Chicago Institutional Animal Care and Use Committee. Mouse brain tissue was passed through a cell strainer with 70 μm pores. After treatment with ACK buffer for 5 min, peripheral blood mononuclear cells underwent a standard Percoll gradient isolation protocol.
Quantitative RT-PCR for transcription levels in MDSCs
Cell suspensions of mouse brains containing GL261 gliomas injected with Ad.mIL12 or Ad.GFP were prepared as described above. MDSCs (CD11b +Gr1+CD45+) were isolated by cell sorting using BD FACSAria. As MDSCs comprise only 0.5–1% of the peripheral blood mononuclear cell layer collected from the Percoll gradient of brain tissues, sorting has a poor yield. Therefore, four animals were pooled so that one sample could get at least 1 × 104 cells needed for RNA analysis. Total cellular RNA was isolated using an RNeasy Micro kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol and purified mRNA was reverse transcribed to complementary DNA using the iScript cDNA conversion kit (Bio-Rad). Primer sequence for arginase-1 and inducible nitric oxide synthase was coded from the available literature.20 Quantitative PCR was conducted using the SYBR Green quantitative PCR kit (Invitrogen) for all experiments. Optimization of annealing temperatures for each transcript was first conducted. Each transcript of interest was amplified in triplicate at its proper annealing temperature and products were analyzed using the Opticon 2 software (Bio-Rad). Relative expression was evaluated using the ΔCT method (ΔCT = CT gene of interest − CT GAPDH).
Animal experiments
Animals were cared for according to a study-specific animal protocol approved by The University of Chicago Institutional Animal Care and Use Committee. Seven- to eight-week-old C57/Bl6 male mice (Jackson Laboratories, Bar Harbor, ME, USA) were injected intracranially with GL261 mouse glioma cells to establish orthotopic tumors. In brief, mice were anesthetized with an intraperitoneal injection of a cocktail containing ketamine hydrochloride (25 mg ml−1)/xylazine (2.5 mg ml−1). For intracranial injection, a midline incision was made, and a 1-mm burr hole centered 2 mm posterior to the coronal suture and 2 mm lateral to the sagittal suture was made. Animals were placed in a stereotactic frame and 2 × 105 GL261 cells, diluted in 2.5 μl of PBS, were injected via a 26-G Hamilton needle, 3 mm deep into the brain. After 7 days, animals were injected intracranially, in the same location, with PBS, Ad.GFP or Ad.mIL12. Depletion of MDSCs was achieved as described above. Animals were sacrificed at the indicated time points. The long-term survivors were rechallenged with intracranial implantation of glioma cells. All mice brains that did not develop signs of disease were examined for the presence of tumors.
Statistical analysis
Unpaired Student’s t-test was used to determine the statistical significance of the difference between means of two groups. One-way analysis of variance was used to compare means among three or more independent groups. A P-value of <0.05 was considered statistically significant. Statistical analysis was performed using SigmaPlot version 12.2 from Systat Software (San Jose, CA, USA).
RESULTS
Adenoviral vector Ad.5/3.cRGD-mIL12p70 induces secretion of active IL-12 from glioma cells
The first step in adenoviral transduction is attachment of the viral fiber to the coxsackie-adenovirus receptor on the surface of target cells. Mouse glioma cells lack coxsackie-adenovirus receptor but express high levels of integrin αVβ3.21 Thus, we used an adenoviral construct with 5/3.cRGD fiber modification (Supplementary Figure S1A) that would provide enhanced transduction efficiency in murine cells.
We first assessed the ex vivo efficacy of the adenoviral construct to induce secretion of mIL-12p70 in mouse GL261 glioma cells, mesenchymal stem cells and neural stem cells. All cells were infected with increasing concentrations of adenoviral infectious units (IU) and the supernatant was analyzed for IL-12 secretion at 72 h. Glioma cells secreted IL-12 after infection with as low as 10 IU per cell (Figure 1a). In contrast, both mesenchymal and neural stem cells required a minimum of 100 IU for producing an equivalent amount of IL-12 (Supplementary Figures S1B and C). Moreover, infection of glioma cells with the adenovirus induced secretion of mIL-12p70 for at least 1 week (Figure 1b).
Figure 1.

In vitro characterization of the gene therapy vector Ad5/3.cRGD.mIL-12 to secrete functional interleukin-12 (IL-12). (a) Enzyme-linked immunosorbent assay (ELISA) was performed to detect secreted interleukin levels from mouse glioma cells. mIL12p70 levels were dependent on the initial quantity of the viral infectious units present in the environment/media. (b) Assessment of the time interval that murine glioma cells are capable of secreting mIL12p70 was performed via an ELISA. (c–e) Functionality assessment of the secreted IL-12 from glioma cells. Supernatant from adenovirus-infected glioma cells was used to incubate splenocytes for 72 h. Induction of interferon-γ (IFN-γ) in CD8 (c), CD4 (d) and natural killer (NK) (e) cells was assessed by flow cytometry after blocking cytokine secretion with Golgi-Plug for 4 h. As a positive control, we used recombinant mouse IL12p70 (rmIL12p70). ND, none detected; *P<0.05.
Next, we evaluated the functionality of the mIL-12p70 secreted from glioma cells by quantifying the IFN-γ secretion induced by splenocytes, ex vivo. Splenocytes were incubated with the supernatant from Ad.mIL12- or control Ad.GFP-transduced GL261 cells. We found that the supernatant of Ad.mIL12-infected cells induced IFN-γ in all effector cells analyzed, including CD8 +T cells (P<0.001), CD4 +T cells (P =0.009) and natural killer (NK) cells (P =0.036) (Figures 1c–e, respectively). The amount of effector immune cells producing IFN-γ more than doubled after incubation with media from the mIL-12p70-infected glioma cells. Collectively, these data support the hypothesis that our GL261 cells transduced with the Ad.mIL12 adenoviral construct were able to produce biologically active IL-12.
Intracranial injection of Ad.5/3.cRGD-mIL12p70 prolongs mouse survival and reduces the infiltration of MDSCs in a mouse model of glioma
To test the in vivo efficacy of IL-12-mediated immunotherapy, we established intracranial GL261 gliomas, and a week later, injected intratumorally PBS, Ad.GFP vector control or Ad.mIL12. Mice that received intracranial PBS or Ad.GFP had a median survival of 31 and 33 days, respectively; with all animals expiring within 40 days of glioma establishment. In contrast, 60% of the mice that received Ad.mIL12 were long-term survivors of >175 days (Figure 2a; *P =0.026). After demonstrating the efficacy of the IL-12 immunogene therapy in prolonging survival of mice bearing orthotopic gliomas, we focused on the effects of how this expression would affect MDSCs. In the control-treated group, greater number of MDSCs were recruited to the tumor site by day 10 compared with day 3 (Figure 2b; *P =0.0004). However, by day 10 after immunotherapy, Ad.mIL12 treatment significantly decreased the recruitment of MDSCs compared with the control group. The percent of MDSCs in the IL-12-treated group was <50% of the vector control group (Figure 2b; *P =0.007).
Figure 2.

The survival benefit of Ad.mIL12 gene therapy is associated with alterations in the myeloid-derived suppressor cell (MDSC) population. (a) Intratumoral delivery of Ad.mIL12 1 week after GL261 mouse glioma establishment results in tumor clearance and long-term survivors. (b) Flow cytometry shows that interleukin-12 (IL-12) immunotherapy reduces MDSCs’ presence in the brains of glioma-bearing mice (representative flow plot depicted below the graph). (c–e) Phenotypic changes of MDSCs after IL-12 immunotherapy. (c) Flow cytometry detects increased expression of antigen-presenting and costimulatory molecules such as major histocompatibiltiy complex (MHC) class II and CD80 (representative flow plot depicted below the graph). Quantitative reverse transcription-polymerase chain reaction (RT-PCR) shows reduction of mRNA transcripts for immunosuppressive factors, such as arginase-1 (d) and inducible nitric oxide synthase (iNOS) (e). mRNA transcription was expressed as the percent of control-treated group. *P<0.05.
Induced IL-12 expression in murine brain tumors had multiple effects on MDSCs, including the alteration of their phenotype. Both MHCII and CD80 expression was increased in MDSCs after Ad.mIL12 injection (Figure 2c; P =0.0414). Interestingly, mRNA levels for arginase-1 (Figure 2d; *P =0.0315) and inducible nitric oxide synthase (Figure 2e; *P =0.006), enzymes associated with promoting immunosuppressive function, were decreased by 50% after administration of IL-12 gene therapy. Collectively, these data suggest that IL-12 expression by glioma cells simultaneously decreases the recruitment of MDSCs and transcriptional programming that governs immunosuppressive functions.
The role of MDSCs as antigen-presenting cells is dispensable in a mouse model of glioma
Given the previous observations demonstrating a difference in MDSC phenotype after IL-12 gene therapy, we sought to determine whether these alterations were significant enough to affect the in vivo induced immune response. For this, we depleted MDSCs by intraperitoneal injection of Gr1 monoclonal antibody based on previously established protocols and tested whether depletion of MDSC affects the survival benefit provided by IL-12-mediated immunotherapy. Intratumoral injection of Ad.mIL12 resulted in more than 60% of long-term survivors, irrespective of systemic MDSC depletion (Figure 3a; P>0.05). To assess whether this survival effect was associated with immunological memory, the long-term survivors were rechallenged with GL261-OVA at 80 days after initial tumor establishment. All animals that were rechallenged survived without showing signs of disease for another 80 days (Table 1). Immunological response among animals that received Ad.mIL12 in the presence or absence of MDSC depletion was also assessed by flow cytometry. The recruitment of CD8 T cells (Figure 3bi; 9.17±1.14 vs 9.32±2.49) and induction of IFN-γ in these cells (Figure 3bii; 0.16±0.16 vs 0.19±0.09) was similar in both groups (P>0.05). Collectively, the data suggest that depletion of circulating MDSCs has no effect on inducing or maintaining immunological memory through IL-12-mediated immunotherapy.
Figure 3.
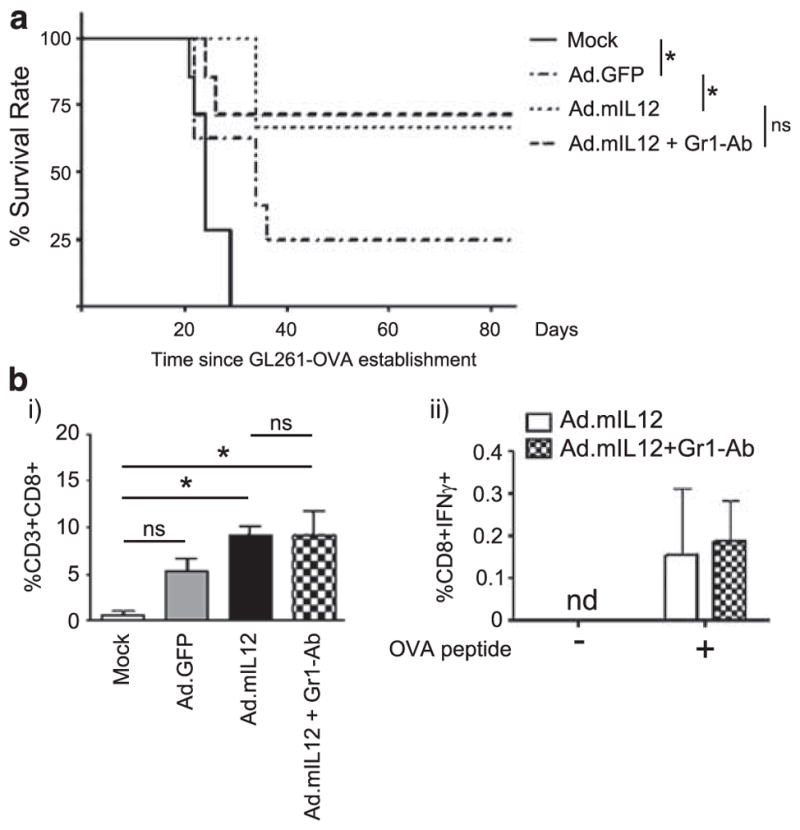
Depletion of myeloid-derived suppressor cells (MDSCs) by systemic delivery of Gr1 antibody does not alter the effects of intracranial interleukin-12 (IL-12) immunotherapy. (a) MDSC depletion does not affect the survival of animals treated with IL-12 gene therapy. One week after establishing GL261-OVA tumors, mice were injected intracranially with phosphate-buffered saline (PBS), Ad.GFP or Ad.mIL12 with or without MDSC depletion. (b) The effect of Gr1-Ab depletion on induction of CD8 T-cell recruitment in the brain (i) and quantification of immune memory response to OVA in intracranial CD8 T cells (ii). Immune cells from animals bearing orthotopic GL261-OVA glioma and treated with IL-12 in the presence or absence of MDSC depletion were segregated from whole brain homogenate via a Percoll gradient and stimulated as shown in the figure with either control (Sc peptide) or OVA peptide in the presence of costimulatory signals from CD28/CD49b antibodies. Interferon-γ (IFN-γ) induction was quantified by flow cytometry. ND, none detected; NS, nonsignificant difference; *P<0.05.
Table 1.
Mice survival after rechallenge with GL261-OVA
| Groups | Animals rechallenged with GL261-OVA | Animals surviving at 80 days after rechallenge |
|---|---|---|
| Ad.GFP | 2 | 2/2 |
| Ad.mIL12 | 4 | 4/4 |
| Ad.mIL12+GR1 | 5 | 5/5 |
Myeloid DCs provide the majority of antigen presentation during IL-12 immunotherapy
Depletion of MDSCs did not inhibit the benefits of Ad.mIL12-mediated immunotherapy in the GL261-based murine glioma model. To better understand the role of MDSCs in acting as antigen-presenting-like cells, we quantified the presence of DCs in the glioma microenvironment. We found that in control-treated animals, MDSCs and mDCs were present in similar numbers (4292±788 and 4483±440, respectively), whereas pDCs were four times less frequent (1003±550 per animal) (Figure 4a). Following Ad.mIL12 immunotherapy, the level of MDSCs was decreased by 50% (*P =0.007), whereas the number of mDCs doubled (Figures 4a and bi; *P =0.0069), resulting in fourfold more mDCs than MDSCs in the glioma microenvironment. In contrast, pDC frequency decreased after IL-12 gene therapy (Figure 4ci; *P =0.045). Furthermore, IL-12-mediated immunotherapy induced the upregulation of MHCII expression by DCs. However, this was only statistically significant in pDCs (Figure 4cii; *P =0.0068). Notably, the expression of MHCII in mDCs was high even in control animals that were injected Ad.GFP and did not change during IL-12 immunotherapy (72.8±0.9% vs 77.1±8.4%, respectively; P>0.05). Ultimately, the data suggest that mDCs are present in higher numbers in IL-12-treated tumors, constitutively express high levels of MHCII expression and have an important role in antigen presentation in IL-12 immunotherapy.
Figure 4.

Myeloid dendritic cells (mDCs) infiltrate glioma at greater numbers after interleukin-12 (IL-12) immunotherapy. (a) Quantification of DC numbers in comparison with myeloid-derived suppressor cells (MDSCs) was carried out by flow cytometry and cell counting. (b) Ad.mIL12 increases the intracranial presence of mDCs (i) without altering their major histocompatibility complex (MHC) class II expression (ii); representative flow plot depicted below the graph). (c) Ad.mIL12 decreases the presence of pDCs (i) while increasing their MHC class II expression (ii; representative flow plot depicted below the graph). NS, nonsignificant difference; *P<0.05.
DISCUSSION
Gene delivery-based immunotherapy for the treatment of solid tumors, including glioma, remains one of the most extensively studied preclinical approaches.4 The immune environment and its responses rely on a continuous equilibrium between the immunostimulatory and immunosuppressive arms.
The role of MDSCs in glioma is an area of active research. Preclinical studies have shown that MDSCs contribute to glioma progression and rely on chemotactic stimuli for recruitment into the GL261 glioma microenvironment.11,12 Considering the diversity among tumor models and methods used in immunotherapy, we chose GL261 model as it offers several advantages: (1) cells can be passaged in vitro while retaining their tumor establishing potential; (2) they do not induce an immune response when injected in the murine brain; (3) they generate tumors and therefore die within a shorter time as compared with genetic models. These characteristics have made GL261 widely used for immunotherapy including IL-12-based gene therapy.22 It is well known that IL-12 immunotherapy prolongs survival in this model by inducing cytotoxic immune responses. However, there are no studies evaluating the effects of such a therapy in specific groups of cells such as MDSCs. It has been proposed in other types of cancer that IL-12 converts MDSCs into antigen presenting -like cells and requires this new subset to prolong survival.13,14 To explore whether the same happens in glioma, we generated a replication-deficient adenoviral vector encoding mIL12p70 with tropism for mouse glioma cells (Supplementary Figure S1A). After determining that this vector prolongs survival of mice with glioma, we focused on whether MDSC depletion inhibits this effect. Overall, we found that IL-12 immunotherapy does not rely on MDSCs to achieve a survival benefit in the GL261 orthotopic glioma model.
First, we characterized the gene therapy vector engineered to induce in situ production of IL-12. The replication-deficient adenoviral vector (Supplementary Figure S1A) was engineered based on a modified 5/3.cRGD adenoviral backbone for better transduction of mouse glioma cells. The secreted mIL12p70 was quantified via an ELISA in the supernatant of infected GL261 glioma cells. We found that as low as 10 IU of Ad.mIL12 were enough to produce detectable levels of mIL12p70 in GL261 supernatant (Figure 1a). Greater virus concentration during infection produced even higher levels of cytokine production. The level of cytokine production peaked on day 3 after infection and remained at a maximum for a week, until plated cells overgrew and detached. On the other hand, when the same loading protocol was used for stem cells, we were able to detect mIL12p70 only at infections with 1000 IU per cell, which is 100 times higher than for GL261 (Figure 1a and Supplementary Figures S1B and C). Also, the levels produced by stem cells did not reach the quantity generated by glioma cells. These comparative findings are important because intratumoral production of IL-12 can be achieved by different approaches. We could either inject the virus directly in the brain tumor or infect stem cells with known tumor tropic potential ex vivo and then deliver them intracranially.22–24 Both approaches have their advantages and pitfalls. Direct virus delivery into the tumor with needle injection is limited by the distribution of the virus within tumor tissue, which is mostly localized along the needle tract.25 Instead, delivery via neural or mesenchymal stem cells suffers from poor engraftment rates following systemic delivery. In our study, considering the very low efficacy of our construct to produce mIL12p70 in stem cells, we decided to infect glioma cells directly through intratumoral injection for in vivo studies.
Before advancing to in vivo studies, we evaluated the functionality of our IL-12 construct in vitro (Figures 1c–e). Supernatants from GL261 glioma cells infected with Ad.mIL12 or Ad.GFP vector control were used as conditioned media to incubate mouse splenocytes. We found that the supernatant of GL261 infected with Ad.mIL12 induced higher levels of IFNγ expression in cytotoxic cells as compared with the control Ad.GFP.
Intracranial delivery of our IL-12-expressing vector, Ad.mIL12, prolonged the survival of animals bearing orthotopic glioma. Injection at 1 week after GL261 implantation resulted in three out of five animals surviving after immunotherapy (Figure 2a). The benefits of IL-12 immunotherapy have been shown to depend on the time of injection after the tumor establishment. Early expression of IL-12 may preclude tumor establishment completely; instead, IL-12 immunotherapy benefits are limited or even dissipated in big size tumors.26 Therefore, intracranial injection at 1 week after tumor establishment provided the window to test for increase or dissipation of benefits in combined therapy with depletion of MDSCs.
The lineage-negative Lin −CD11b +Gr1 +MDSCs act mainly through induction of immunosuppressive pathways via generation of reactive oxygen species, or expression of arginase-1. Intracranial injection of Ad.mIL12 reduced MDSCs’ presence in glioma by half and altered their phenotype: increased MHCII/CD80 expression and reduced arginase-1 and inducible nitric oxide synthase transcription. This is a novel finding in glioma that recapitulates similar observations in tumor models outside the central nervous system.27 In solid tumors IL-12 not only reduces the recruitment of MDSCs but also alters their phenotype. MDSCs treated with IL-12 immunotherapy lose their immunosuppressive potential and show signs of maturation, with increased expression of MHCII molecules, suggesting that these cells may have a role in antigen presentation.13,27 Medina-Echeverz et al.13 proposed that these altered MDSCs have a very important role in the IL-12-generated immune response. Most importantly, they found that intratumoral MDSC depletion precluded the benefits of immunotherapy. These findings are a new trend in the approach toward MDSCs, which until recently has focused on a search for therapies to eradicate such cells.28 If immunotherapies were to rely on MDSCs, it would be detrimental to deplete them. Henceforth, we evaluated how depletion of MDSCs via Gr1 +antibody would affect IL-12 therapy in the GL261 orthotopic intracranial glioma model.
MDSCs were depleted through intraperitoneal injection of anti-Gr1+antibody based on a previously published protocol.19 We found that depletion of MDSCs did not alter the survival benefit of IL-12 immunotherapy. Both groups receiving intracranial Ad.mIL12, with or without MDSC depletion, had more than 60% long-term survivors (Figure 3a) and showed similar recruitment of cytotoxic CD8 T cells (Figure 3bi).
In addition, when the long-term survivors were rechallenged with GL261-OVA, all animals survived (Table 1). Therefore, our study, in contrast with findings in the gastric cancer model, shows that MDSCs are dispensable for the full benefits of IL-12 immunotherapy. However, there are some differences that need to be discussed. First, is the methodological difference in depleting MDSCs While we depleted MDSCs by intraperitoneal injection of Gr1 monoclonal antibody, Medina-Echeverz et al.13 injected the same antibody directly into the tumor.13 Second, we relied on an orthotopic glioma model, and it is well known that the brain remains an immune privileged location due to the blood–brain barrier. Instead, the dorsal chamber location of the gastric cancer model offers no barriers to circulating cells and mature MDSCs may have a more important role as antigen-presenting cells in such models.
The major antigen-presenting cells in glioma are tumor-associated macrophages, mDCs and pDCs.29 Their antigen presentation properties have been explored as potential immuno-therapies in various tumors, including glioma. The IL-12 immunotherapy has already been shown to increase tumor-associated macrophages and their activation.30 To understand the potential contribution of mDCs, pDCs and MDSCs in glioma antigen presentation, we evaluated their presence before and after IL-12 immunotherapy. We found that after IL-12 immunotherapy MDSCs are reduced in half (Figures 2b and 4a), pDCs reduced fourfold (Figure 4ci), while mDCs more than doubled (Figure 4a and bi). At the same time, the effects of Ad.mIL12 on MHCII upregulation were noticed across all the three groups analyzed. Whether mDCs have the major antigen presentation role in glioma, it remains to be studied in the future. It is well established that mDCs increase 100-fold during gliomagenesis, but how immunotherapies affect antigen-presenting cells remains understudied.29 However, improving antigen presentation in glioma immunotherapy would provide at least an additive benefit to current approaches.31
In conclusion, our study shows that IL-12 gene-based therapy remains a good model to study the immune response during glioma eradication. We show that Ad.mIL12 immunotherapy reduces the presence of MDSCs and alters their phenotype in an orthotopic mouse glioma model. Furthermore, we demonstrate that depletion of MDSCs does not have any detrimental effect on IL-12 immunotherapy. This allows for combination of IL-12 immunotherapy with MDSC-depleting drugs. At the same time, we reveal that IL-12 immunotherapy increases recruitment of mDCs that may have a major role in glioma antigen presentation. Finally, this study emphasizes one more time the immunological uniqueness of the central nervous system and the potential pitfalls we face when generalizing systemic findings to brain tumors.
Supplementary Material
Acknowledgments
This work was supported by the NCI (R01CA122930, R01CA138587) and the National Institute of Neurological Disorders and Stroke (U01NS069997, F32NS073366, K99NS082381).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on Cancer Gene Therapy website (http://www.nature.com/cgt)
References
- 1.Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13:916–926. doi: 10.1016/S1470-2045(12)70265-6. [DOI] [PubMed] [Google Scholar]
- 2.Becker KP, Yu J. Status quo—standard-of-care medical and radiation therapy for glioblastoma. Cancer J. 2012;18:12–19. doi: 10.1097/PPO.0b013e318244d7eb. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Wainwright DA, Nigam P, Thaci B, Dey M, Lesniak MS. Recent developments on immunotherapy for brain cancer. Expert Opin Emerg Drugs. 2012;17:181–202. doi: 10.1517/14728214.2012.679929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8 +T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic–polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szatmari T, Lumniczky K, Desaknai S, Trajcevski S, Hidvegi EJ, Hamada H, et al. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006;97:546–553. doi: 10.1111/j.1349-7006.2006.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev. 2007;7:880–887. doi: 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Andaloussi A, Han Y, Lesniak MS. Prolongation of survival following depletion of CD4 +CD25 +regulatory T cells in mice with experimental brain tumors. J Neurosurg. 2006;105:430–437. doi: 10.3171/jns.2006.105.3.430. [DOI] [PubMed] [Google Scholar]
- 11.Fujita M, Kohanbash G, Fellows-Mayle W, Hamilton RL, Komohara Y, Decker SA, et al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71:2664–2674. doi: 10.1158/0008-5472.CAN-10-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neuro-Oncol. 2011;104:83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medina-Echeverz J, Fioravanti J, Zabala M, Ardaiz N, Prieto J, Berraondo P. Successful colon cancer eradication after chemoimmunotherapy is associated with profound phenotypic change of intratumoral myeloid cells. J Immunol. 2011;186:807–815. doi: 10.4049/jimmunol.1001483. [DOI] [PubMed] [Google Scholar]
- 14.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–4757. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson MJ, Ritz J. Interleukin 12: basic biology and potential applications in cancer treatment. Oncologist. 1996;1:88–97. [PubMed] [Google Scholar]
- 16.Xu M, Mizoguchi I, Morishima N, Chiba Y, Mizuguchi J, Yoshimoto T. Regulation of antitumor immune responses by the IL-12 family cytokines, IL-12, IL-23, and IL-27. Clin Dev Immunol. 2010;2010 doi: 10.1155/2010/832454. pii832454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soleimani M, Nadri S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat Protocols. 2009;4:102–106. doi: 10.1038/nprot.2008.221. [DOI] [PubMed] [Google Scholar]
- 18.Tyler MA, Ulasov IV, Borovjagin A, Sonabend AM, Khramtsov A, Han Y, et al. Enhanced transduction of malignant glioma with a double targeted Ad5/3-RGD fiber-modified adenovirus. Mol Cancer Therap. 2006;5:2408–2416. doi: 10.1158/1535-7163.MCT-06-0187. [DOI] [PubMed] [Google Scholar]
- 19.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukocyte Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 20.Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol. 2007;7:140–151. doi: 10.1016/j.intimp.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 21.Davis FB, Tang HY, Shih A, Keating T, Lansing L, Hercbergs A, et al. Acting via a cell surface receptor, thyroid hormone is a growth factor for glioma cells. Cancer Res. 2006;66:7270–7275. doi: 10.1158/0008-5472.CAN-05-4365. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Ehtesham M, Samoto K, Wheeler CJ, Thompson RC, Villarreal LP, et al. In situ adenoviral interleukin 12 gene transfer confers potent and long-lasting cytotoxic immunity in glioma. Cancer Gene Ther. 2002;9:9–15. doi: 10.1038/sj.cgt.7700399. [DOI] [PubMed] [Google Scholar]
- 23.Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, Yu JS. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002;62:5657–5663. [PubMed] [Google Scholar]
- 24.Ryu CH, Park SH, Park SA, Kim SM, Lim JY, Jeong CH, et al. Gene therapy of intracranial glioma using interleukin 12-secreting human umbilical cord blood-derived mesenchymal stem cells. Hum Gene Ther. 2011;22:733–743. doi: 10.1089/hum.2010.187. [DOI] [PubMed] [Google Scholar]
- 25.Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508–2518. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- 26.Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65:7194–7204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steding CE, Wu ST, Zhang Y, Jeng MH, Elzey BD, Kao C. The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology. 2011;133:221–238. doi: 10.1111/j.1365-2567.2011.03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerkar SP, Restifo NP. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012;72:3125–3130. doi: 10.1158/0008-5472.CAN-11-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiu TL, Wang MJ, Su CC. The treatment of glioblastoma multiforme through activation of microglia and TRAIL induced by rAAV2-mediated IL-12 in a syngeneic rat model. J Biomed Sci. 2012;19:45. doi: 10.1186/1423-0127-19-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava MK, Zhu L, Harris-White M, Kar UK, Huang M, Johnson MF, et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLoS One. 7:e40677. doi: 10.1371/journal.pone.0040677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.