

Abstract
Multiblock, backbone degradable HPMA copolymer-drug conjugates containing gemcitabine and DACH platinum (mP-GEM and mP-DACH Pt), respectively were synthesized by reversible addition fragmentation (RAFT) polymerization and subsequent chain extension by click chemistry. Using combination index analysis, the cytotoxicities of the two multiblock conjugates, as single agents and in combination, were evaluated in vitro in A2780 human ovarian cancer cells, with free drugs as controls. The greatest synergistic cytotoxic effect was observed when A2780 cells were sequentially exposed to mP-GEM for 24 h and mP-DACH Pt for 48 h. In addition, mechanistic studies support the rationale of the synergy between mP-GEM and mP-DACH Pt: mP-GEM pretreatment was able to enhance the platinum-DNA adduct accumulation and inhibit cell proliferation to a higher extent than single mPDACH Pt treatment. These observations are useful for the development of combination macromolecular therapeutics for ovarian cancer based on the second-generation backbone degradable HPMA copolymers.
Keywords: HPMA copolymers, Biodegradable polymers, Combination therapy, Ovarian cancer, gemcitabine, DACH-Pt
1. Introduction
Ovarian cancer is the fifth most common cause of death in women and most lethal cancer of the female reproductive system [1,2]. It caused approximately 16,000 deaths and about 22,000 new cases were diagnosed in the United States in 2012 [1]. The traditional anti-ovarian cancer treatment includes surgical debulking followed by chemotherapy [2]. However, the prognosis of ovarian cancer following treatment is very poor in the majority of ovarian cancer patients, especially the patients at stage III or IV when diagnosed.
To improve the efficacy of chemotherapy, combination therapies of platinum and nonplatinum agents were commonly employed, including the combination of gemcitabine and platinum agent such as oxaliplatin, cisplatin, or carboplatin [2,3]. Gemcitabine, a synthetic nucleoside analog of cytidine, is activated inside the cells into its triphosphate analog (dFdCTP). dFdCTP is an inhibitor of DNA polymerase and can inhibit DNA synthesis by incorporation into DNA [4,5]. Platinum agents, including cisplatin, carboplatin, or oxaliplatin, have been used clinically for the treatment of many types of cancers [6]. Platinum-DNA adducts can cause cell cycle arrest, cell replication arrest and apoptosis [7]. In particular, platinum agents containing DACH ligand (such as carboplatin or oxaliplatin) possess low toxicity and induce DNA lesions which are more resistant to DNA repair pathways than that caused by cisplatin [8]. Because of the synergistic anti-cancer effects as well as the non-overlapping side effects of the two drugs, the combination of gemcitabine and platinum agent such as carboplatin has been approved to treat patients with advanced ovarian cancer [9–11]. Nonetheless, this combination often shows suboptimal anti-cancer response or unfavorable toxicity profile clinically [3,12]. Therefore, strategies to further improve the anti-cancer efficacy and reduce the toxicity of the combination chemotherapies are still of great need.
N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers have been widely used as drug carriers to improve the efficacy and reduce the toxicity of chemotherapeutic agents [13–16]. Nanosized HPMA copolymer-drug conjugates improve the water solubility of chemotherapeutics, increase the circulation time of the drug and lead to enhanced drug accumulation in tumor tissue via the enhanced permeability and retention (EPR) effect [17–19]. Previous studies have shown that HPMA copolymer-DACH platinum (dichloro(1,2-diaminocyclohexane)platinum(II)) conjugate with molecular weight lower than 45 kDa was equally or likely more effective than free Oxaliplatin in treating ovarian cancer patients with excellent tolerability in clinical trials [20]. However, the low molecular weight of the first-generation HPMA copolymer-drug conjugates limits their clinical translation.
Molecular weight and molecular weight distribution are important factors in the design of effective macromolecular therapeutics. The higher the molecular weight of the polymer carrier, the higher the accumulation of polymer-drug conjugates in the tumor tissue with concomitant increase in therapeutic efficacy [17,18,21–23]. However, the renal threshold limits the molecular weight of the first generation (non-degradable) polymeric carriers to below 50 kDa [24]; this lowers the retention time of the conjugate in the circulation with simultaneous decrease in pharmaceutical efficiency. Higher molecular weight drug carriers with a nondegradable backbone deposit and accumulate in various organs, impairing biocompatibility. Clearly, the design of long-circulating backbone degradable HPMA copolymer carriers offers a solution to this problem [21–23,25–27]; it provides a long-circulating polymeric carrier without impairing biocompatibility – the degradation fragments are below the renal threshold.
Recently, backbone degradable multiblock HPMA copolymer drug carriers have been successfully developed through reversible addition-fragmentation chain transfer (RAFT) polymerization and subsequent chain extension by click chemistry [25–27]. The resulting conjugates are composed of alternating low molecular weight HPMA copolymer-drug segments and enzymatically degradable GFLG (glycylphenylalanylleucylglycyl) tetrapeptide segments. The enhanced efficacy of the new, multiblock backbone degradable HPMA copolymer conjugates (when compared to low molecular weight conjugates) has been demonstrated on ovarian cancer xenografts in mice [21,22] and in a rat osteoporosis model [23]. In addition, incorporating free gemcitabine into the multiblock HPMA copolymer carrier helps to stabilize the free drug in circulation. Free gemcitabine can be deactivated in circulation after converting to a uracil metabolite (2′-deoxy-2′-difluorouridine (dFdU)), which limits its efficacy [28].
Previously, we have developed a novel concept of using combination therapy with water-soluble polymer-bound drugs. In vivo combination chemotherapy and photodynamic therapy (PDT) studies on two cancer models, Neuro 2A neuroblastoma induced in A/J mice [29] and human ovarian carcinoma heterotransplanted in nude mice [30–32], demonstrated that macromolecular combination therapy produced tumor cures which could not be obtained with either chemotherapy or PDT alone. Other combination systems were quantitatively evaluated by combination index (CI) analysis in A498 renal carcinoma cells [33] and in OVCAR-3 ovarian carcinoma cells [34]. The results demonstrated synergistic effects of HPMA copolymer-drug (SOS thiophene, doxorubicin, and chlorin e6) conjugate combinations in a wide range of concentrations. Consequently, this manuscript aims to demonstrate that second generation, backbone degradable conjugates have a potential in combination therapy.
To this end, we synthesized high molecular weight HPMA copolymer conjugates with gemcitabine (mP-GEM) and DACH Pt (mP-DACH Pt), respectively using RAFT copolymerization followed by alkyne-azide click chain extension. The design [25–26,35] provides an innovative therapeutic paradigm; moreover, this design has a competitive advantage with simplicity of structure, proven safety of the polymer carrier, and utilization of current effective drugs. Last but not least, the synthesis procedures proposed are versatile; they provide a platform for the preparation of a large variation of polymer-drug conjugates with tailor-made properties, such as predetermined circulation time and composition. The cytotoxicities of the two multiblock conjugates, as single agents and in combination, were evaluated in vitro in A2780 human ovarian cancer cells, with free drugs as controls. The combination effects and possible mechanism of synergy of mP-GEM and mP-DACH Pt were investigated.
2. Materials and Methods
2.1 Materials
Gemcitabine hydrochloride (GEM, ≥99.0%) was purchased from NetQem LLC (Research Triangle Park, NC). DACHPtCl2 and common reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used as received unless otherwise specified. Materials for peptide synthesis (including N-α-Fmoc protected amino acids, resin and coupling reagents) were purchased from AAPPTec Biosciences (Louisville, KY). HPMA [36], N-methacryloylglycylglycine p-nitrophenylester (MA-GG-ONp) [37], N-methacryloylglycylphenylalanylleucylglycyl gemcitabine (MA-GFLG-GEM) [26], 4-cyanopentanoic acid dithiobenzoate [38], and peptide2CTA (Nα,Nε-bis(4-cyano-4-(phenylcarbonothioylthio)pentanoylglycylphenylalanyl-leucylglycyl)lysine) [27] were synthesized as previously described. Initiators, V-65 (2,2′-azobis-(2,4-dimethylvaleronitrile)) was from Wako and V-501 (4,4′-azobis-(4-cyanovaleric acid)) were from Fluka-Sigma-Aldrich. 4,4′-Azobis(N,N′-propargyl-4-cyanopentanamide) (dialkyne-V-501) [25] and Nα,Nδ-(bis(azidobenzoylglycylphenylalanylleucylglycylalanyl)lysine (diazide-GFLGK) were prepared according to described procedures [25].
2.2 Synthesis and characterization of multiblock HPMA copolymer-drug conjugates
2.2.1 Synthesis and characterization of HPMA copolymer-gemcitabine conjugate (mP-GEM)
Polymerizable gemcitabine derivative, MA-GFLG-GEM, was copolymerized with HPMA via RAFT polymerization as previously reported [26] with modifications. Briefly, MA-GFLG-GEM (63.5 mg, 0.09 mmol) and HPMA (130 mg, 0.91 mmol) were dissolved in degassed DMSO and DI H2O under nitrogen atmosphere, respectively. The solutions were transferred into an ampoule via syringe. After addition of peptide2CTA RAFT chain transfer agent, and initiator V-501, the ampoule was sealed, and then kept stirring at 70 °C for 16 h. The polymer was isolated by precipitation into acetone and purified by re-dissolving in methanol and precipitating into acetone two more times. The diblock HPMA copolymer-gemcitabine conjugate (2P-GEM) was obtained as a light pink powder. Mw 62 kDa; Mw/Mn 1.17.
Multiblock backbone degradable HPMA copolymer-gemcitabine conjugate with higher Mw (mP-GEM) were synthesized in two steps: first, 2P-GEM was post-polymerization end-modified with dialkyne-V-501 to produce a telechelic dialkyne conjugate; in the second step the conjugate was chain extended by click reaction with diazide-GFLGK in dimethylformamide (DMF) in the presence of CuSO4 and sodium ascorbate (Figure 1) [21]. The chain extended conjugate was fractionated on a preparative Superose 6 HR 16/60 column using acetate buffer pH 6.5/30% acetonitrile as the mobile phase. The fraction G2 of Mw 139 kDa (Mw/Mn 1.03) was used for further evaluation.
Figure 1.
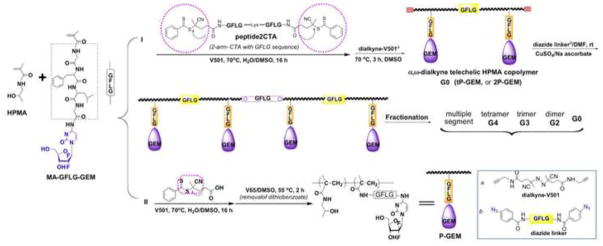
Synthesis and structure of backbone degradable HPMA copolymer-gemcitabine conjugates (mP-GEM).
The molecular weight (weight average, Mw and number average, Mn) and molecular weight distribution of the conjugates were determined by size exclusion chromatography using a Superose 6 HR/16/30 column on an ÄKTA FPLC system (GE Healthcare) equipped with miniDAWN TREOS and OptilabEX detectors (Wyatt Technology, Santa Barbara, CA) with sodium acetate buffer containing 30% acetonitrile (pH 6.5) as mobile phase. HPMA homopolymer fractions were used as molecular weight standards.
Gemcitabine content in the conjugate was estimated by UV spectrophotometry in methanol (ε300 = 5710 L mol−1 cm−1) [26]. Characterization of conjugates is shown in Table 1.
Table 1.
Characterization of the conjugates studied.
| Conjugate | Mw kDa | Mw/Mn | Content of drug wt,-% |
|---|---|---|---|
| mP-GEM | 139 | 1.03 | 5.8 |
| P-GEM | 32 | 1.07 | 7.7 |
| mP-DACH Pt | 365 | 1.06 | 10.0 |
| P-DACH Pt | 38 | 1.08 | 5.1 |
2.2.2 Synthesis and characterization of multiblock HPMA copolymer platinum conjugate (mP-DACH Pt)
Synthesis of MA-GG-diCOOH
Under nitrogen atmosphere, diethyl aminomalonate hydrochloride (800 mg, 3.8 mmol, 1.0 equiv) was dissolved in anhydrous DMF (20 mL). The solution was stirred at 0 °C for 5 min and N,N-diisopropylethylamine (DIPEA; 1.2 g, 9.5 mmol, 2.5 equiv) was added. The solution was stirred in ice bath for 5 min, MA-GG-ONp (1.12 g, 3.6 mmol, 0.95 equiv) was added and the solution was stirred at 25 °C for 24 h. The solvent was removed by rotoevaporation. After addition of EtOAc (150 mL), white solid appeared, and the solution was yellow. The mixture was stirred overnight. The solid was filtered off, washed by EtOAc until color changed to white completely. The solid was washed by ether (2 × 50 mL) and dried to give MA-GG-diCOOEt as white solid in 84% yield (1.07 g, 3.0 mmol).
In the next step, MA-GG-diCOOEt (1.0 g, 2.8 mmol) was dissolved in CH3OH (20 mL), the solution stirred for 5 min in ice bath, then NaOH (1 M, 10 mmol) in CH3OH (25 mL) was added dropwise over 30 min. The solution was stirred in ice bath for 2 h and stirred overnight at ~ 4 °C. After work-up, the solution was stirred in ice bath and acidified to pH = 5.8 with HCl (1 M). The CH3OH was rotoevaporated and the aqueous solution was desalted through a column prepared by placing about 2.5 g C18 sorbent (Sigma, 40–63 μm particle diameter, 60 Å pore size) into a 20-mL polypropylene syringe equipped with a porous disk at the bottom. The collected fraction was lyophilized and yielded the product as a white solid (750 mg, 2.1 mmol, 75%).
Preparation of multiblock, degradable HPMA copolymer – DACH Pt conjugate by RAFT polymerization followed by click reaction (mP-DACH Pt)
Multiblock HPMA copolymer-Pt conjugate (mP-DACH Pt) was synthesized by RAFT polymerization (to produce poly(HPMA-co-MA-GG-diCOOH)), followed by click reaction and Pt complexation (Figure 2).
Figure 2.

Synthesis and structure of backbone degradable HPMA copolymer-DACH platinum conjugates (mP-DACH Pt). (A) Synthesis of N-methacryloylglycylglycine-aminomalonic acid (MA-GG-diCOOH); (B) Synthesis of traditional (low molecular weight) HPMA copolymer – DACH Pt conjugate by RAFT copolymerization; (C) Synthesis of backbone degradable multiblock HPMA copolymer – DACH Pt conjugate by RAFT copolymerization followed by chain extension with alkyne-azide click reaction.
First, HPMA (1.53 g, 10.7 mmol), MA-GG-diCOOH (1.4 g, 4.65 mmol), and N-methacryloyltyrosinamide (38.5 mg, 0.155 mmol; to introduce a site for radioiodination, if needed) in molar ratio 10.7: 4.7: 0.16 were dissolved in 8 mL methanol/water (1:1) and polymerized in an inert atmosphere at 40 °C for 12 h using peptide2CTA (29 mg, 20.5 μmol) as RAFT CTA (chain transfer agent) and VA-044 (4.4 mg, 14 μmol) as the initiator (CTA: VA-044 = 1.5: 1). The copolymer was precipitated into acetone, filtered off and dried. Its solution in DI H2O was dialyzed against water and freeze dried. Yield 1.44 g (48%).
Molecular weight and molecular weight distribution of HPMA copolymers was determined by size-exclusion chromatography (SEC) on an AKTA FPLC system (Pharmacia) equipped with UV, RI detectors as well as miniDAWN TREOS and OptilabEX detectors (Wyatt Technology, Santa Barbara, CA) using a Superose 6 HR/10/30 column with acetate buffer pH 6.5/30% acetonitrile as mobile phase Mw = 113 kDa; Mw/Mn = 1.095.
Chain end modification
The poly(HPMA-co-MA-GG-diCOOH) (above, 400 mg) was reacted with dialkyne-V-501 (71 mg, 0.2 mmol, over 20 times excess) in 3.0 mL deoxygenated CH3OH at 70 °C for 3 h. The polymer was purified by precipitation into acetone twice, resulting in the dialkyne telechelic poly(HPMA-co-MA-GG-diCOOH) as white solid: yield = 93.8%, 375 mg.
Chain extension by click reaction
Dialkyne telechelic poly(HPMA-co-MA-GG-diCOOH) (Mw 113 kDa, 350 mg, about 3.4 μmol), diazide-GFLGK (4.0 mg, 3.4 μmol) and sodium ascorbate (8 mg, 40 μmol) were placed in a vial. The vial was evacuated and refilled with nitrogen three times before adding 1.1 mL of deoxygenated DMF. Then 200 μL deoxygenated solution of CuSO4 (3.2 mg, 20 μmol) was added. The vial was closed and the cloudy mixture was stirred at room temperature for 15 h. Then 1 mL DI H2O was added and the copolymer was precipitated into acetone and dried under vacuum; it was further purified on a Sephadex 25 PD-10 column.
Fractionation
A preparative Superose 6 HR 16/60 column was used for the fractionation of the click product. The flow rate was 1 mL/min acetate buffer pH 6.5/30% acetonitrile and the fractions were collected at 5 min intervals. The different fractions were collected, dialyzed and freeze dried. A fraction of Mw 365 kDa (Mw/Mn = 1.06; 70 mg, yield: 20%) was selected for further evaluation.
Conjugation of platinum to copolymer
A modified procedure from literature was used [39]. The fraction of the multiblock poly(HPMA-co-MA-GG-diCOOH) (64 mg) was dissolved in DI water (1.0 mL). The solution was adjusted to pH 7.4 using 1 M NaOH, and DACHPt(OH2)2 (at least 3 times, excess in 2.0 mL of water) was added. The reaction mixture was readjusted to pH 5.4 and stirred at 5.0–5.4 for 8 h; then Chelex resin (1.5 g) was added and the mixture was stirred for additional 2 h. The resin was filtered off and the resulting solution was diluted to 8 mL, 3 mL buffer solution (containing 0.855 g NaCl, 0.255 g NaH2PO4 and 2.282 g Na2HPO4.7H2O, 27 mL) was added. The pH value was 7.35. The solution was adjusted to pH = 7.42 using 0.1 M NaOH and stirred at 38 °C for 18 h. The mixture was filtered to remove a small amount of precipitate, and the filtrate was purified by dialysis against water (8000 mL× 4) at 4 °C for 18 h and freeze dried, giving colorless powder: yield 88 mg (N,O-chelate). The molecular weight was confirmed by FPLC; Mw 365 kDa.
Pt analysis
3.5 mg polymer was dissolved in 2.5% HNO3 and the volume of solution adjusted to 10 mL; 0.5 mL of this solution was diluted to 10 mL and was used to test the content of Pt in the polymer by Inductively Coupled Plasma Mass Spectrometry (ICP-MS): Pt = 10% (wt%).
The characteristics of all polymer conjugates are shown in Table 1; the synthetic pathway of Pt conjugate is shown in Figure 2.
2.3 Synthesis of traditional (low molecular weight) conjugates
Traditional HPMA copolymer – gemcitabine and HPMA copolymer - DACH Pt conjugates were synthesized similarly by RAFT copolymerization in one step. Their characteristics are shown in Table 1.
2.4 Cell culture
The human ovarian cancer cell line A2780 was obtained from American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in RPMI-1640 medium (Sigma) containing 10 μg/mL insulin (Sigma) and 10% FBS at 37°C, 5% CO2. Cells were routinely passaged when reaching 90% confluency.
2.5 Cytotoxicity study
The cytotoxicity of drugs/conjugates against A2780 cells was determined using the Cell Counting Kit-8 (CCK-8) assay (CCK-8, Dojindo, Japan). In viable cells, the tetrazolium salt WST-8 is converted to an orange-colored formazan by dehydrogenases; the amount of formazan produced is proportional to the number of viable cells. Drug or conjugate stock solutions were prepared in DI water and the working solutions were freshly prepared in growth medium before use. The cells were plated in 96-well plates (10,000 cells/well) and grown for 24 h, followed by exposure to various concentrations of each free drug (gemcitabine, DACH Pt) or each copolymer conjugate (P-GEM, P-DACH Pt, mP-GEM or mP-DACH Pt). After 72 h treatment, the drug solutions were removed and replaced with fresh medium (100 μl). 50 μL of diluted CCK-8 reagent (5 x) was added to each well and the plate was incubated for 60 minutes at 37°C, 5% CO2. The absorbance was read at 450 nm, with 630 nm as reference. Untreated cell were set as 100% viable. IC50 values were determined by nonlinear regression analysis using GraphPad Prism version 5.0 (GraphPad Software Inc., La Jolla, CA).
2.6 Dose-effect analysis and determination of combination index (CI)
In combination studies, A2780 cells were exposed to combination of free drugs (gemcitabine and DACH Pt) or multiblock conjugates (mP-GEM and mP-DACH Pt) sequentially or simultaneously. The summary of the dosing schedules is shown in Figure 3. The ratio of the two drugs or conjugates in a combination was chosen based on their IC50 values and kept constant for all concentration ranges.
Figure 3.
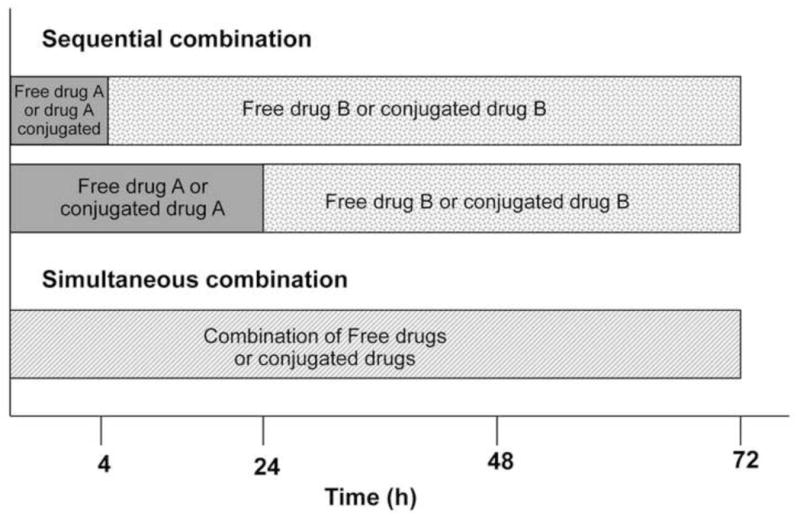
The treatment schedules of the combination study. For sequential combination, cells were exposed to free or multiblock HPMA copolymer conjugated drug A for either 4 h or 24 h, followed by exposure to free or conjugated drug B for 68 h or 48 h. For simultaneous combination, cells were exposed to both drugs (free or multiblock HPMA copolymer conjugated) for a total of 72 h.
Drug interactions and combination index (CI) were determined using the median drug effect analysis according to the method of Chou and Talalay [40,41]. The median-effect equation, , describes dose-effect relationships. fa and fu are the fraction affected [1 − (average absorbance of treatment wells − average absorbance of blanks)/(average absorbance of untreated wells − average absorbance of blanks)] and unaffected (fu = 1 − fa) by the dose or concentration D; Dm is the median-effect dose that inhibits 50% cell growth, and m value is the coefficient signifying the shape [42]. The logarithmic conversion of the above equation yielded the equation, , where m is the slope and Dm is the anti-log of the x-intercept. The plot of x = log(D) versus y = log(fa/fu) is called the median-effect plot.
The Combination Index (CI) describes the interaction between two drugs and quantitates the synergism (CI < 1), antagonism (CI > 1) and additive effects (CI = 1). The CI is determined by the equation where (Dx)1 and (Dx)2 are the doses of drug 1 alone and drug 2 alone that inhibit the cell growth x%, respectively. (D)1 and (D)2 are the doses of each drug in a combination that also inhibit x%. The CI values at different fa’s and the fa-CI plot were constructed using CompuSyn software (ComboSyn Inc., Paramus, NJ).
The dose-reduction index (DRI) is a measure of how many-fold the dose of each drug in a synergistic combination may be reduced at a given effect level compared with the doses of each drug alone [34]. The DRI value was calculated by the equation .
2.7 Dectection of gene-specific platinum intrastrand adducts
Quantitative PCR (QPCR) method was employed to investigate the effect of mP-GEM on the formation of platinum-DNA intrastrand adducts by mP-DACH Pt treatment. This method is based on the ability of drug-DNA adduct to block the progression of DNA polymerase, thus inhibiting the amplification of the damaged DNA template [11,43]. Cells were seeded in 75cm2 cell culture flasks at the density of 2.5 × 106/flask for 16 h before exposure to single or combination of multiblock conjugates. Cells were incubated in the absence or presence of mP-Gem (gemcitabine equivalent 1 μM) for 8 h, followed by incubation with mP-DACH Pt (DACH Pt equivalent 25 μM) for 8 h. After treatment, cells were washed twice and harvested by trypsinization. Total genomic DNA was isolated using PureLink® genomic DNA mini kit (Life technologies, Grand Island, NY) and quantified by NanoDrop spectrophotometer. DNA was then digested with restriction enzyme HindIII at 37°C for 3 h and purified. Quantitative PCR of an 1858-bp fragment in the DHFR gene for different treatment groups was performed using the same amount of purified genomic DNA, primers (sense primer, 5′-AAACGTAGCTCGTCCTCAAA; antisense primer, 5′-TTCCAGTCTACGGGAAGCC) [11] and SYBR® Green PCR master mix (Life technologies, Grand Island, NY). The amount of amplification products was quantified and normalized to the untreated group.
2.8 Cell proliferation assay
Cell proliferation assay was performed using CellTrace™ CFSE (carboxyfluorescein diacetate, succinimidyl ester) Cell Proliferation Kit (Molecular Probes, Eugene, OR) to test the inhibitory effect on A2780 cell proliferation by single and combination of mP-GEM and mP-DACH Pt. Before cell seeding and treatment, A2780 cells were harvested and stained with CFSE reagent. Briefly, cells were counted and resuspended in PBS at the density of 2 × 106 cells/mL. Cells were mixed thoroughly with 1.5 μL DMSO stock solution of CFSE reagent and incubated at 37°C for 10 min. After incubation, cells were washed by adding 5 x volumes of culture medium three times and immediately seeded in 25 cm2 cell culture flask at 4 × 105/flask. Cells immediately after staining were used for flow cytometry parameter settings. After 18 h growth, cells in the combination treatment group were treated with mP-GEM (31.25 nM drug equivalent) for 24 h, followed by mP-DACH Pt treatment (3125 nM drug equivalent) for 48 h. For single conjugate treatment groups, cells were either exposed to mP-GEM (31 nM drug equivalent) for 24 h followed by incubation in fresh growth medium for 48 h, or cultured in fresh medium for 24 h followed by 48 h mP-DACH Pt treatment (3.1 μM drug equivalent). Untreated cells were also included as control. After 72 h treatment, cells were harvested and resuspended in 400 μL PBS for flow cytometry.
2.9 Data analysis
Results are expressed as the mean ± S.D. of n experiments. The data were analyzed using one-way analysis of variance to compare more than two groups, with p-value<0.05 considered to be significant.
3. Results and discussion
3.1 Cytotoxicity of Gemcitabine, DACH platinum and their conjugates against A2780 cells
The cell growth inhibitory effects of gemcitabine, DACH Pt, P-GEM, P-DACH Pt, mP-GEM or mP-DACH Pt were evaluated on A2780 cells after exposure to drugs/conjugates for 72 h by CCK-8 assay. The dose response curves and IC50 values are shown in Figure 4 and Table 2. The IC50 value for gemcitabine was lower than for DACH Pt. This agrees with previously published data; it was reported that A2780 was more sensitive to gemcitabine than to platinum agent [9,10]. When comparing the free drug with their conjugated form, the IC50 values (Table 2) for P-GEM (9.22 nM) and mP-GEM (6.53 nM) were very similar to that of gemcitabine (6.17 nM). This was mostly due to the hydrolytic release of free gemcitabine from both of the conjugates within the 72 h incubation time period [44]. Similar IC50 values of free drug and polymer-drug conjugates when the drug is bound via an ester bond and the cells are incubated with the conjugates for extended time periods is a generally known phenomenon. For example, IC50 doses of free GEM and HPMA copolymer-GEM conjugates were similar in agreement with the kinetics of hydrolytic cleavage of GEM from the polymer carrier [44]. However, P-DACH Pt and mP-DACH Pt showed 8.3 and 9.9 times higher IC50s than free DACH Pt. This phenomenon can be explained by the different mechanisms of cell entry between HPMA copolymer-drug conjugates and free drugs. HPMA copolymers are internalized into cells through pinocytosis or endocytosis [45]; whereas free platinum could be uptaken by passive diffusion and endocytosis-independent membrane translocation [46,47]. Despite the differences in cytotoxicity between the free and conjugated DACH Pt in vitro, P-DACH Pt and especially mP-DACH Pt could offer superior therapeutic benefits over free platinum formulations in vivo. Indeed, the traditional HPMA copolymer-DACH Pt conjugate delivered significantly higher amount of platinum in the active form into tumor cells and to the target DNAs than free oxaliplatin [20]. It is conceivable that the high molecular weight mP-DACH Pt would show better therapeutic effects due to the significantly prolonged circulation time in vivo [18,21].
Figure 4.
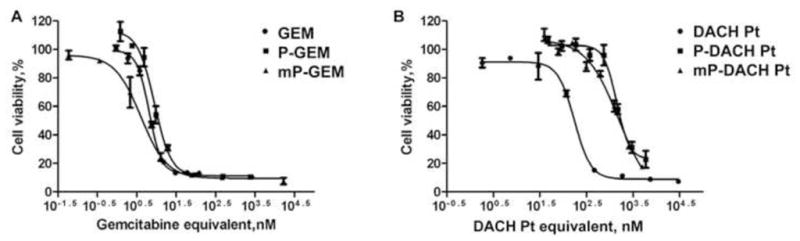
Dose response curve of A2780 cells treated with drug or conjugate for 72 h. (A) Gemcitabine, P-GEM, mP-GEM; (B) platinum, P-DACH Pt, and mP-DACH Pt.
Table 2.
Summary of IC50 Values of Gemcitabine, P-Gem, mP-Gem, DACH Platinum, P-DACH Pt, mP-PGFLG-DACH platinum against A2780 cells for 72 h.
| Drugs | Mw (kDa) | IC50 (nM) | ||
|---|---|---|---|---|
| Gemcitabine.HCl | 0.3 | 6.17 ± 0.45 |
|
|
| P-GEM | 32 | 9.22 ± 1.73 | ||
| mP-GEM | 139 | 6.53 ± 1.56 | ||
| DACH Platinum | 0.195 | 161.3 ± 14.6 |
|
|
| P-DACH Pt | 38 | 1340.7 ± 81.9 | ||
| mP-DACH Pt | 365 | 1592.3 ± 272.4 | ||
IC50 is the concentration that inhibits 50% cells growth in treated cells when compared to control cells. Mw is the weight average molecular weight. Values are mean ± SD of at least three independent experiments.
represents the statistically significant difference between the two groups (one-way ANOVA, p<0.01).
3.2 Combination effects and combination index analysis of mP-GEM and mP-DACH Pt in A2780 cells
To investigate the combination effects of multiblock conjugates by combination index analysis, A2780 cells were exposed to the conjugates sequentially or simultaneously. Combination of free gemcitabine and DACH platinum served as controls. Dose ratios were chosen based on the IC50 values of drugs or conjugates when used as single agent for the same exposure durations as used in the combination. The summary of combination index values at IC50 for different dosing schedules was shown in Figure 5. In general, the combinational effects followed the similar trend between free drug and multiblock drug conjugate combinations across several different dosing schedules, indicating that multiblock HPMA copolymer drug carrier can deliver the free drugs at their site of action. For both free drug and multiblock drug conjugate combination, sequential treatment of (free or conjugated) gemcitabine for 24 h and (free or conjugated) DACH platinum for 48 h showed the most synergistic cytotoxic effect against A2780 cells. Free or conjugated gemcitabine pretreatment for a shorter period (4 h) resulted in additive to mild synergistic cytotoxic effects, possibly due to inadequate exposure of cells to gemcitabine. In addition, treatment of A2780 cells by the reversal sequence (DACH platinum 24 h first, followed by gemcitabine 48 h) resulted in mild synergistic to additive cytotoxic effect. In contrast, the simultaneous combination of free or conjugated gemcitabine and DACH platinum were antagonistic.
Figure 5.
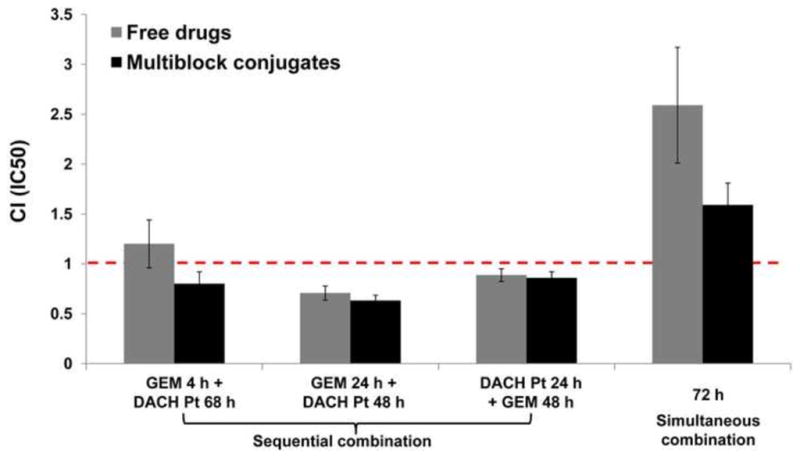
Summary of combination index at IC50 (fa=0.5) for combination treatments of A2780 cells with free gemcitabine + DACH Pt, and mP-GEM + mP-DACH Pt, under different treatment schedules.
The synergism between gemcitabine and platinum agents has been widely observed experimentally and clinically in ovarian cancer [9–11] as well as in other tumors [48,49]. However, the drug interactions largely depend on dosing duration and sequence, as well as the dose ratios of the individual agent [9,50]. Synergism was observed more often in the cases when cells were pretreated with gemcitabine followed by platinum agents, whereas the results for simultaneous combinations and sequential combinations with platinum pretreatments were often suboptimal [47,48]. Our results for both free and conjugated drug combinations agreed with the above observations, which could potentially be explained by the mechanisms of drug interactions. When cells were pretreated with gemcitabine, its incorporation into DNA 1) may cause DNA distortion which facilitates more platinum-DNA adduct formation [10]; 2) inhibit the extent of DNA damage repair caused by platinum-DNA intra-strand and inter-strand adducts [10,11]. In addition, gemcitabine diphosphate inhibits ribonucleotide reductase [51], thereby contributing to the inhibition of DNA damage repair.
The combinational effects of the sequential treatment (24 h free or conjugated gemcitabine pretreatment followed by 48 h free or conjugated DACH Pt treatment) were analyzed in more detail. Here the combination of gemcitabine and cisplatin was included as an additional control, since this combination has been proven to be effective against ovarian cancer cells [9–11]. The Dm values of free drugs and multiblock conjugates both in combination and in single treatments are shown in Table 3. These data suggest that the doses of each drug or multiblock conjugate that inhibit 50% of cell growth were significantly lower than the doses of the same drug or conjugate when used as a single agent to inhibit 50% cell growth (comparing columns six and eight, seven and nine). These results were therapeutically meaningful, since it indicated that the doses of each drug or conjugate could be reduced if used in combination compared to the doses used as a single agent, to achieve the same level of therapeutic effect. The dose reduction index (DRI) values at fa=50% were shown in Table 4. DRIs for gemcitabine and DACH platinum in both free and multiblock conjugate combinations were similar, which were higher than the DRIs for gemcitabine and cisplatin combination.
Table 3.
IC50 doses for drugs and conjugates in sequential combination or single treatments in A2780 cells.
| Drug combination | Dose ratio | Dm (nM)a | D (nM)b in combination | Dm (nM) as single treatment | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Drug A | Drug B | Drug A | Drug B | Drug A | Drug B | |||
| Drug A 24 h→Drug B 48 h | Gemcitabine | Cisplatin | 1:50 | 634.9 ± 44.4 | 12.5 | 622.5 | 38.2 ± 0.6 | 1187.4 ± 98.4 |
| Gemcitabine | DACH platinum | 1:50 | 571.2 ± 56.9 | 11.2 | 560.0 | 45.4 ± 4.0 | 1311.7 ± 47.7 | |
| mP-GEM | mP-DACH Pt | 1:50 | 6905.2 ± 982.1 | 135.4 | 6769.8 | 607.2 ± 259.1 | 17195.6 ± 2281.6 | |
Dm represents the combined dose of two single agents in the combination that inhibits the cell growth by 50%.
D represents the dose of each drug or conjugate in a combination that inhibits the cell growth by 50%.
Table 4.
Dose-reduction index values at 50% growth inhibition level of sequential combination treatments in A2780 cells
| Drug combination | Dose-reduction index (DRI) | |||
|---|---|---|---|---|
|
| ||||
| Drug A | Drug B | Drug A | Drug B | |
| Drug A 24h → drug B 48 h | Gemcitabine | Cisplatin | 3.1 ± 0.3 | 1.9 ± 0.3 |
| Gemcitabine | DACH platinum | 4.1 ± 0.5 | 2.4 ± 0.3 | |
| mP-GEM | mP-DACH Pt | 4.4 ± 1.2 | 2.6 ± 0.1 | |
The combination index (CI) versus fraction affected (fa) plot for sequential combinations was shown in Figure 6. When cells were pretreated with mP-GEM followed by mP-DACH Pt, moderate to strong synergistic effect was observed at fa higher than 0.2. Combination of free gemcitabine and DACH platinum showed moderate to strong synergistic effect at fa higher than 0.4, but antagonistic to additive effect at fa lower than 0.4. In addition, combination of gemcitabine and cisplatin exhibited additive to synergistic cytotoxic effect. The combination index at fa between 0.5 and 0.9 for both the combinations of free and conjugated gemcitabine and DACH platinum was significantly lower than that of gemcitabine and cisplatin combination (p<0.05). To conclude, the synergistic cytotoxic interaction between gemcitabine and DACH platinum was retained after incorporating free drugs into the multiblock HPMA copolymer drug carriers. The multiblock HPMA copolymers were able to deliver gemcitabine and DACH-platinum to their site of action at the subcellular level with favorable kinetics. Based on the above in vitro evaluations, sequential administration of mP-GEM followed by mP-DACH Pt could possibly be the best dosing regimen for future in vivo evaluations.
Figure 6.
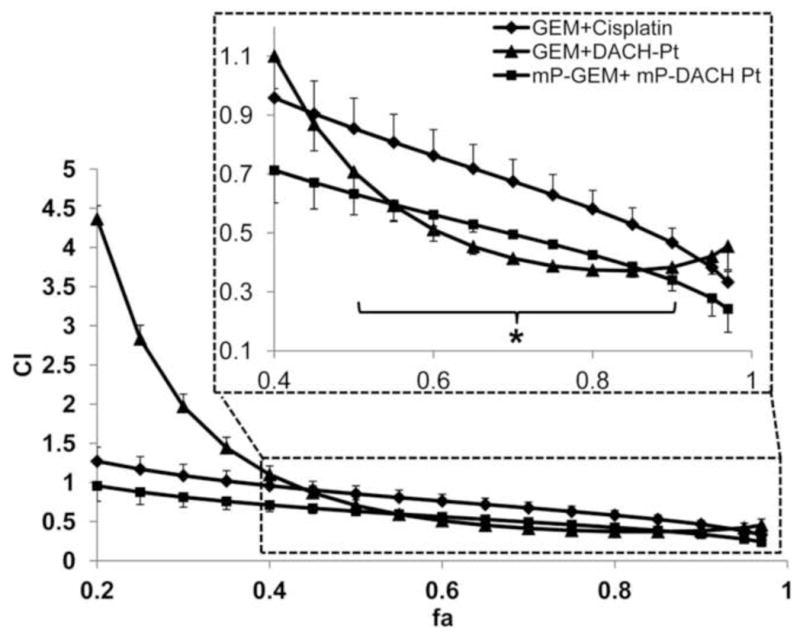
Combination Index (CI) - Fraction affected (fa) plot of sequential combination treatments of free drugs or multiblock conjugates. “*” represents statistically significant difference between (mP-GEM + mP-DACH Pt) and (GEM + DACH Pt) groups, and between (GEM + DACH Pt) and (GEM + cisplatin) groups, at all the data points between fa of 0.5 and 0.9 (p<0.05).
3.3 Accumulation of DNA intrastrand adducts following sequential treatment of mP-GEM and mP-DACH Pt
To further investigate the possible mechanism of the observed synergy of sequential combination of mP-GEM and mP-DACH Pt, we evaluated the effect of mP-GEM on the accumulation of platinum-DNA intrastrand adducts in a constitutively expressed gene DHFR induced by mP-DACH Pt treatment. The relative amount of DHFR gene-specific platinum-DNA intrastrand adducts was estimated based on the quantity of PCR amplification product. The more adducts formed, the more inhibition on the replication of the affected gene fragment, thus leading to fewer gene amplification products. This method was used previously to measure the DNA intrastrand adducts induced by free platinum agent [11,43]. In this experiment, the cells were treated with single or combination of the multiblock conjugates for a shorter period of time, in order to minimize the hydrolytic release of the free drug from the conjugate into the culture medium. As shown in Figure 7, A2780 cells treated with only mP-DACH Pt (DACH platinum 25 μM) for 8 h showed slightly decreased PCR amplification compared to untreated control cells. Treatment of cells with mP-GEM followed by mP-DACH Pt significantly decreased the amount of PCR amplified product when compared to the product in both untreated cells and mP-DACH Pt treated cells. This observation demonstrated that mP-GEM pretreatment enhanced platinum-DNA intrastrand adduct accumulation in A2780s cells. The same mechanism has been observed in the studies of free drug combination (gemcitabine and platinum agent) on ovarian cancer cells [11].
Figure 7.
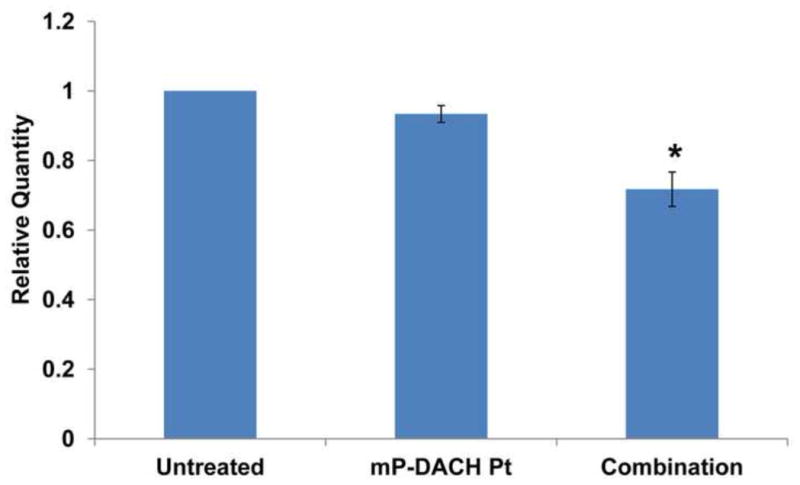
Relative quantity of DHFR gene-specific PCR amplification product in A2780 cells following single and combination treatment of multiblock conjugates. *, p<0.05.
3.4 Inhibition of cell proliferation following sequential treatment of mP-GEM and mP-DACH Pt
We have observed synergistic effect of sequential treatment of mP-GEM followed by mP-DACH Pt, especially when the cell growth inhibition was greater than 20% on A2780 cells. Both gemcitabine and DACH platinum exhibit anti-cancer effects by incorporating into cancer cell DNA, potentially arresting cell cycle progression and cell division. Here we investigated the effect of multiblock conjugates, single or in combination, on cell proliferation at the concentration causing less than 20% of cell death. As seen in Figure 8, the A2780s cells without treatment proliferated rapidly, leading to the dramatic decrease of mean CFSE signal intensity from above 105 on the day of seeding to around 102 on day 4. The CFSE signal in cells treated with single mP-GEM or single mP-DACH Pt on day 4 was slightly higher than untreated cells on day 4, demonstrating certain degree of inhibition on cell proliferation (Figure 8); while the most significant inhibition on cell proliferation was observed following the sequential combination treatment of mP-GEM and mP-DACH Pt. Thus, mP-GEM and mP-DACH Pt can act collaboratively to halt the ovarian cancer cell division.
Figure 8.
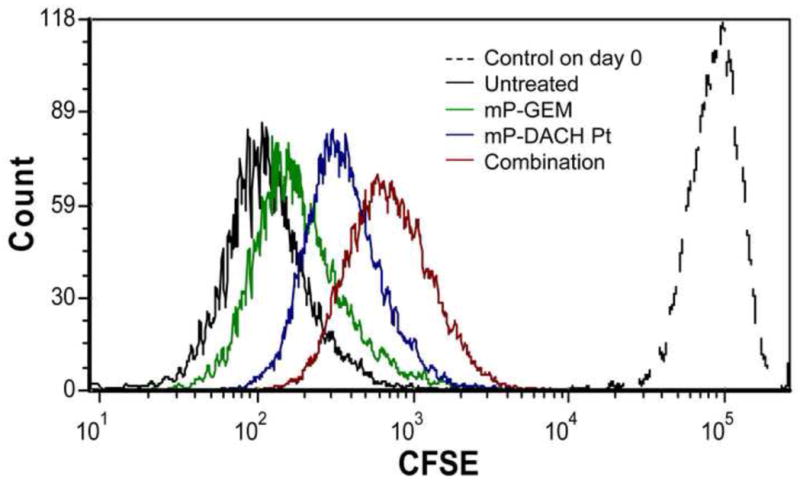
Inhibition on A2780 cell proliferation following single and combination treatment of multiblock conjugates. Dashed line: Untreated control on day 0; black solid line: untreated control on day 4; green solid line: mP-GEM treatment group on day 4; blue solid line: mP-DACH Pt treatment group on day 4; red solid line: sequential combination of mP-GEM (24 h) and mP-DACH Pt (48 h) group on day 4.
4. Conclusions
Backbone degradable multiblock HPMA copolymer-gemcitabine and HPMA copolymer-DACH platinum conjugates were synthesized and characterized. The greatest synergistic cytotoxic effect was observed when A2780 cells were sequentially exposed to mP-GEM for 24 h and mP-DACH Pt for 48 h. In addition, mechanistic studies support the rationale of the synergy between mP-GEM and mP-DACH Pt: mP-GEM pretreatment was able to enhance the platinum-DNA adduct accumulation and inhibit cell proliferation to a higher extent than single mP-DACH Pt treatment. These observations are useful for the development of combination macromolecular therapeutics for ovarian cancer based on the second-generation backbone degradable HPMA copolymers. This study also provides useful information for the design of the in vivo combination dosing regimen for ovarian cancer treatment. Sequential administration of mP-GEM followed by mP-DACH Pt may be the most beneficial strategy to inhibit ovarian cancer growth in vivo. Moreover, our recent study investigating another combination therapy of backbone degradable HPMA copolymer-drug conjugates showed that it exhibited superior anti-ovarian cancer effect than the combination therapy using traditional low molecular weight drug conjugates. Due to the prolonged circulating time of the backbone degradable conjugates and more sustained exposure of the cancer cells to both therapeutic agents, it may be expected that the combination of mP-GEM and mP-DACH Pt would be more effective than the combination of the free drugs or the first-generation drug conjugates.
Acknowledgments
The research was supported in part by NIH grants R01 CA51578 and R41 CA156933 from the National Cancer Institute and by the University of Utah Research Foundation. We also acknowledge support in conjunction with grant P30 CA042014 awarded to the Huntsman Cancer Institute, University of Utah.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mould T. An overview of current diagnosis and treatment in ovarian cancer. Int J Gynecol Cancer. 2012;22:S2–S4. doi: 10.1097/IGC.0b013e318251c8e3. [DOI] [PubMed] [Google Scholar]
- 2.Roett MA, Evans P. Ovarian cancer: an overview. Am Fam Physician. 2009;80:609–616. [PubMed] [Google Scholar]
- 3.Meriggi F, Zaniboni A. Gemox: a widely useful therapy against solid tumors-review and personal experience. J Chemother. 2010;22:298–303. doi: 10.1179/joc.2010.22.5.298. [DOI] [PubMed] [Google Scholar]
- 4.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17:v7–v12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 5.Ueno H, Kiyosawa K, Kaniwa N. Pharmacogenomics of gemcitabine: can genetic studies lead to tailor-made therapy? Br J Cancer. 2007;97:145–151. doi: 10.1038/sj.bjc.6603860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 8.Cabral H, Nishiyama N, Okazaki S, Koyama H, Kataoka K. Preparation and biological properties of dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt)-loaded polymeric micelles. J Control Release. 2005;101:223–232. doi: 10.1016/j.jconrel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 9.Bergman AM, Ruiz van Haperen VW, Veerman G, Kuiper CM, Peters GJ. Synergistic interaction between cisplatin and gemcitabine in vitro. Clin Cancer Res. 1996;2:521–530. [PubMed] [Google Scholar]
- 10.van Moorsel CJ, Pinedo HM, Veerman G, Bergman AM, Kuiper CM, Vermorken JB, van der Vijgh WJ, Peters GJ. Mechanisms of synergism between cisplatin and gemcitabine in ovarian and non-small-cell lung cancer cell lines. Br J Cancer. 1999;80:981–990. doi: 10.1038/sj.bjc.6690452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moufarij MA, Phillips DR, Cullinane C. Gemcitabine potentiates cisplatin cytotoxicity and inhibits repair of cisplatin-DNA damage in ovarian cancer cell lines. Mol Pharmacol. 2003;63:862–869. doi: 10.1124/mol.63.4.862. [DOI] [PubMed] [Google Scholar]
- 12.Kalykaki A, Papakotoulas P, Tsousis S, Boukovinas I, Kalbakis K, Vamvakas L, Kotsakis A, Vardakis N, Papadopoulou P, Georgoulias V, Mavroudis D. Hellenic Oncology Research Group, Gemcitabine plus oxaliplatin (GEMOX) in pretreated patients with advanced ovarian cancer: a multicenter phase II study of the hellenic oncology research group (HORG) Anticancer Res. 2008;28:495–500. [PubMed] [Google Scholar]
- 13.Lu ZR, Shiah JG, Sakuma S, Kopečková P, Kopeček J. Design of novel bioconjugates for targeted drug delivery. J Control Release. 2002;78:165–173. doi: 10.1016/s0168-3659(01)00495-3. [DOI] [PubMed] [Google Scholar]
- 14.Kopeček J, Kopečková P. HPMA copolymers: origins, early developments, present, and future. Adv Drug Deliv Rev. 2009;62:122–149. doi: 10.1016/j.addr.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Ponyer R, Boivin C, Hesslewood S, Twelves C, Blackie R, Schätzlein A, Jodrell D, Bissett D, Calvert H, Lind M, Robbins A, Burtless S, Duncan R, Cassidy J. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal carcinoma. Int J Oncol. 2009;34:1629–1636. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- 16.Kopeček J, Kopečková P. Design of polymer-drug conjugates. In: Kratz F, Senter P, Steinhagen H, editors. Drug Delivery in Oncology. 2. Wiley-VCH; Weinheim: 2012. pp. 485–512. [Google Scholar]
- 17.Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164:138–144. doi: 10.1016/j.jconrel.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 18.Shiah JG, Dvořák M, Kopečková P, Sun Y, Peterson CM, Kopeček J. Biodistribution and antitumour efficacy of long-circulating N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in nude mice. Eur J Cancer. 2001;37:131–139. doi: 10.1016/s0959-8049(00)00374-9. [DOI] [PubMed] [Google Scholar]
- 19.Kopeček J, Kopečková P, Minko T, Lu ZR. HPMA copolymer-anticancer drug conjugates: design, activity, and mechanism of action. Eur J Pharmaceutics Biopharm. 2000;50:61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 20.Nowotnik DP, Cvitkovic E. ProLindacTM (AP5346): a review of the development of an HPMA DACH platinum polymer therapeutic. Adv Drug Deliv Rev. 2009;61:1214–1219. doi: 10.1016/j.addr.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Zhang R, Luo K, Yang J, Sima M, Sun Y, Janát-Amsbury MM, Kopeček J. Synthesis and evaluation of a backbone degradable multiblock HPMA copolymer nanocarriers for delivery of paclitaxel. J Control Release. 2013;166:66–74. doi: 10.1016/j.jconrel.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan H, Sima M, Yang J, Kopeček J. Synthesis of long-circulating backbone degradable HPMA copolymer-doxorubicin conjugates and evaluation of molecular weight dependent antitumor efficacy. Macromol Biosci. 2013;13:155–160. doi: 10.1002/mabi.201200353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan H, Sima M, Miller SC, Kopečková P, Yang J, Kopeček J. Promotion of bone formation in ovariectomized rats by high molecular weight backbone degradable HPMA copolymer – prostaglandin E1 conjugate. Biomaterials. 2013;34:6528–6538. doi: 10.1016/j.biomaterials.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seymour LW, Duncan R, Strohalm J, Kopeček J. Effect of molecular weight (Mw) of N-(2-hydroxypropyl)methacrylamide copolymers on body distribution and rate of excretion after subcutaneous, intraperitoneal, and intravenous administration to rats. J Biomed Mater Res. 1987;21:1341–1358. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]
- 25.Luo K, Yang J, Kopečková P, Kopeček J. Biodegradable multiblock N-(2-hydroxypropyl)methacrylamide copolymers via reversible addition-fragmentation chain transfer polymerization and click chemistry. Macromolecules. 2011;44:2481–2488. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of biodegradable multiblock copolymers by click coupling of RAFT-generated heterotelechelic polyHPMA conjugates. Reactive Functional Polym. 2011;71:294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan H, Yang J, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition-fragmentation chain transfer polymerization and thiol-ene coupling reaction. Biomacromolecules. 2011;12:247–252. doi: 10.1021/bm101254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: a mechanism of self-potentiation. Cancer Res. 1992;52:533–539. [PubMed] [Google Scholar]
- 29.Krinick NL, Sun Y, Joyner D, Spikes JD, Straight RC, Kopeček J. A polymeric drug delivery system for the simultaneous delivery of drugs activatable by enzymes and/or light. J Biomater Sci Polym Ed. 1994;5:303–324. doi: 10.1163/156856294x00040. [DOI] [PubMed] [Google Scholar]
- 30.Shiah JG, Sun Y, Kopečková P, Peterson CM, Straight RC, Kopeček J. Combination chemotherapy and photodynamic therapy of targetable N-(2-hydroxypropyl)methacrylamide copolymer – doxorubicin/mesochlorin e6 – OV-TL16 antibody immunoconjugates. J Control Release. 2001;74:249–253. doi: 10.1016/s0168-3659(01)00325-x. [DOI] [PubMed] [Google Scholar]
- 31.Peterson CM, Lu JM, Sun Y, Peterson CA, Shiah JG, Straight RC, Kopeček J. Combination chemotherapy and photodynamic therapy with N-(2-hydroxypropyl)methacrylamide copolymer-bound anticancer drugs inhibit human ovarian carcinoma heterotransplanted in nude mice. Cancer Res. 1996;56:3980–3985. [PubMed] [Google Scholar]
- 32.Shiah JG, Sun Y, Peterson CM, Straight RC, Kopeček J. Antitumor activity of HPMA copolymer-meso chlorin e6 and adriamycin conjugates in combination treatments. Clin Cancer Res. 2000;6:1008–1015. [PubMed] [Google Scholar]
- 33.Hongrapipat J, Kopečková P, Prakongpan S, Kopeček J. Enhanced antitumor activity of combinations of free and HPMA copolymer-bound drugs. Int J Pharmaceutics. 2008;351:259–270. doi: 10.1016/j.ijpharm.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hongrapipat J, Kopečková P, Liu J, Prakongpan S, Kopeček J. Combination chemotherapy and photodynamic therapy with Fab’ fragment targeted HPMA copolymer conjugates in human ovarian carcinoma cells. Mol Pharmaceutics. 2008;5:696–709. doi: 10.1021/mp800006e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan H, Yang J, Kopečková P, Luo K, Kopeček J. Polymeric drug delivery conjugates and methods of making and using thereof. Appl 24U03.1-500. US Patent. Filed 03/08/2010.
- 36.Kopeček J, Bažilová H. Poly[N-(2-hydroxypropyl)methacrylamide]. 1. Radical polymerization and copolymerization. Europ Polym J. 1973;9:7–14. [Google Scholar]
- 37.Rejmanová P, Labský J, Kopeček J. Aminolyses of monomeric and polymeric p-nitrophenyl esters of methacryloylated amino acids. Makromol Chem. 1977;178:2159–2168. [Google Scholar]
- 38.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules. 2001;34:2248–2256. [Google Scholar]
- 39.Sood P, Thurmond KB, II, Jacob JE, Waller LK, Silva GO, Stewart DR, Nowotnik DP. Synthesis and characterization of AP5346, a novel polymer-linked diaminocyclohexyl platinum chemotheraputic agent. Bioconj Chem. 2006;17:1270–1279. doi: 10.1021/bc0600517. [DOI] [PubMed] [Google Scholar]
- 40.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 41.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 42.Chou TC. Derivation and properties of Michaelis-Menten type and Hill type equations for reference ligands. J Theor Biol. 1976;59:253–276. doi: 10.1016/0022-5193(76)90169-7. [DOI] [PubMed] [Google Scholar]
- 43.Ponti M, Forrow SM, Souhami RL, D’Incalci M, Hartley JA. Measurement of the sequence specificity of covalent DNA modification by antineoplastic agents using Taq DNA polymerase. Nucleic Acids Res. 1991;19:2929–2933. doi: 10.1093/nar/19.11.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larson N, Yang J, Ray A, Cheney DL, Ghandehari H, Kopeček J. Biodegradable multiblock poly(N-2-hydroxypropyl)methacrylamide gemcitabine and paclitaxel conjugates for ovarian cancer cell combination treatment. Int J Pharmaceutics. 2013;454:435–443. doi: 10.1016/j.ijpharm.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Bauer H, Callahan J, Kopečková P, Pan H, Kopeček J. Endocytic uptake of a large array of HPMA copolymers: elucidation into the dependence on the physicochemical characteristics. J Control Release. 2010;143:71–79. doi: 10.1016/j.jconrel.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Binks SP, Dobrota M. Kinetics and mechanism of uptake of platinum-based pharmaceuticals by the rat small intestine. Biochem Pharmacol. 1990;40:1329–1336. doi: 10.1016/0006-2952(90)90400-f. [DOI] [PubMed] [Google Scholar]
- 47.Beretta GL, Righetti SC, Lombardi L, Zunino F, Perego P. Electron microscopy analysis of early localization of cisplatin in ovarian carcinoma cells. Ultrastruct Pathol. 2002;26:331–334. doi: 10.1080/01913120290104610. [DOI] [PubMed] [Google Scholar]
- 48.Wang S, Zhang H, Cheng L, Evans C, Pan CX. Analysis of the cytotoxic activity of carboplatin and gemcitabine combination. Anticancer Res. 2010;30:4573–4578. [PMC free article] [PubMed] [Google Scholar]
- 49.Faivre S, Raymond E, Woynarowski JM, Cvitkovic E. Supraadditive effect of 2′,2′-difluorodeoxycytidine (gemcitabine) in combination with oxaliplatin in human cancer cell lines. Cancer Chemother Pharmacol. 1999;44:117–123. doi: 10.1007/s002800050955. [DOI] [PubMed] [Google Scholar]
- 50.Mayer LD, Harasym TO, Tardi PG, Harasym NL, Shew CR, Johnstone SA, Ramsay EC, Bally MB, Janoff AS. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5:1854–1863. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]
- 51.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol. 1990;38:567–572. [PubMed] [Google Scholar]