

SUMMARY
Protein interactions between acyl carrier proteins (ACP’s) and trans-acting acyltransferase domains (trans-AT’s) are critical for regioselective extender unit installation by many polyketide synthases. Yet, little is known regarding the specificity of these interactions, particularly for trans-AT’s with unusual extender unit specificities. Currently, the best-studied trans-AT with non-malonyl specificity is KirCII from kirromycin biosynthesis. Here, we developed a new assay to probe ACP interactions based on leveraging the extender unit promiscuity of KirCII. The assay allows us to identify residues on the ACP surface that contribute to specific recognition by KirCII. This information proved sufficient to modify a non-cognate ACP from a different biosynthetic system to be a substrate for KirCII. The findings form a foundation for further understanding the specificity of trans-AT:ACP protein interactions, and for engineering modular polyketide synthases to produce analogues.
INTRODUCTION
The scaffolds of a variety of clinically relevant natural products are biosynthesized by polyketide synthases (PKS’s). Type I PKS’s are organized in a modular fashion, whereby each module is responsible for the installation of an extender unit acyl-coenzyme A (CoA) into the growing polyketide (Staunton and Weissman, 2001). The growing chain and all other substrates are covalently tethered to the phosphopantetheinyl prosthetic arm of acyl carrier proteins (ACP’s), located within each module. Acyltransferase (AT) domains are required to select extender units and attach them to an ACP, while ketosynthase (KS) domains catalyze C-C bond formation to extend the growing polyketide. Modifications to this basic paradigm can be found in natural biosynthetic systems. For example, fungal PKS’s are type I PKS’s that act iteratively, whereby a given module catalyzes several extender unit installations (Cox and Simpson, 2009). In most type I PKS’s, the AT domains are embedded within each module and are cis-acting. In contrast, a growing number of PKS’s are being discovered that house AT’s external to the modules (Cheng, et al., 2003; Piel, 2010). Such transacting AT’s (trans-AT’s) at the genetic level can be easily transferred between biosynthetic systems. Subsequently, given their discrete nature, trans-AT’s might constitute a valuable alternative to strategies which involve cis-AT domain swapping, and there is much interest in developing trans-AT’s for use as tools in synthetic biology platforms aimed at the regioselective modification of polyketides (Kumar, et al., 2003; Walker, et al., 2013). For example, promiscuity of KS’s from the 6-deoxyerythronolide synthase (DEBS) towards diverse extender units (Koryakina, et al., 2013; Sundermann, et al., 2013) could be harnessed by trans-AT’s that display acyl-CoA specificity orthogonal to that of the DEBS cis-AT domains. Yet, one critical barrier to the use of trans-AT’s as tools for polyketide diversification is our poor understanding of extender unit and ACP specificity. For example, the native substrate for many trans-AT’s is malonyl-CoA (Figure 1), and these enzymes are unlikely to display activity towards other acyl-CoA’s (Koryakina, et al., 2013). Furthermore, while some trans-AT’s that use malonyl-CoA install the extender unit at multiple positions within the resulting polyketide and are ACP promiscuous (Wong, et al., 2010), others install unusual extenders at limited positions in the polyketide and are likely ACP specific (Figure 1) (Liu, et al., 2009; Musiol, et al., 2011; Zhao, et al., 2010). Thus, the ACP specificity of trans-AT’s needs to be better understood so that specificity can be manipulated. Although evidence of the ability to manipulate ACP interactions with other domains is emerging (Haines, et al., 2013; Kapur, et al., 2011; Kapur, et al., 2012), little is known regarding trans-AT:ACP interactions. Indeed, including the malonyl-CoA-ACP transacylase (MCAT) that primarily serves ACP’s from type II fatty acid synthases, only two trans-AT structures are available (Keatinge-Clay, et al., 2003; Wong, et al., 2011), in addition to that of a bi-functional trans-AT/decarboxylase (Lohman, et al., 2013). Docking models have helped propose the ACP interaction epitopes of MCAT and at least one genuine malonyl-CoA specific trans-AT (Arthur, et al., 2009; Keatinge-Clay, et al., 2003; Wong, et al., 2011), and NMR studies have also contributed to this picture (Arthur, et al., 2009).
Figure 1.

Substrate specificity of trans-AT’s. (A) Examples of polyketides biosynthesized via trans-AT’s. The contribution of each trans-AT, and the corresponding substrate are shown color-coded. (B) Examples of acyl-thioester substrates for trans-AT’s.
Recently, we probed the promiscuity of several trans-AT’s towards a panel of acyl-CoA’s (Koryakina, et al., 2013). One target, KirCII from kirromycin biosynthesis (Figure 2), is responsible for installation of the C28 ethyl moiety of kirromycin, via ethylmalonyl-CoA (Musiol, et al., 2011). KirCII has the distinction of being the only characterized trans-AT to naturally utilize a non-malonyl acyl-CoA substrate (Musiol and Weber, 2012). All other unusual trans-AT extender units are introduced into polyketides linked to ACP’s (Figure 1). Our study led to the discovery that in addition to ethylmalonyl-CoA, KirCII can utilize other acyl-CoA’s, making KirCII the most promiscuous trans-AT known. Kirromycin biosynthesis involves the action of another trans-AT (KirCI) responsible for malonyl installation onto ACP’s within 11 unique modules of the kirromycin biosynthetic assembly line (Musiol, et al., 2013). According to this biosynthetic logic, it is likely that the acyl-CoA and ACP specificity of KirCII is orthogonal to that of KirCI. This feature would present a valuable opportunity to elucidate the molecular basis for ACP specificity of a uniquely acyl-CoA promiscuous trans-AT. Here, we describe a cycloaddition assay to rapidly characterize and probe KirCII:ACP interactions, which led to characterization of the ACP specificity of KirCII, and promoted identification of the KirCII:ACP interaction epitope. This information allowed us to successfully convert a non-cognate ACP into a substrate for KirCII via a single amino acid mutation. The insight into trans-AT:ACP interactions obtained here will contribute to improving trans-AT based strategies for polyketide diversification.
Figure 2.

Biosynthesis of kirromycin. The putative biosynthetic contribution of KirCI (malonyl-CoA, red) and KirCII (ethylmalonyl-CoA, purple) to the final structure of kirromycin is highlighted.
RESULTS
Cycloaddition Assay for Probing KirCII:ACP Interactions
The ability of KirCII to utilize extender unit acyl-CoA’s modified with azide/alkynyl-functionality could enable the quantification of KirCII activity via bioorthogonal ligation chemistry and in-gel fluorescence detection (Figure 3A). In-gel fluorescence detection of KirCII-catalyzed trans-acylation of holo-ACP5Kir was evaluated using azidoethylmalonyl-CoA (AzEM-CoA) as substrate and strain-promoted azide-alkyne cycloaddition using a commercially available dibenzocyclooctyne (DIBO)-Alexa Fluor 647 conjugate. Upon fluorescence visualization of a SDS-PAGE gel, a distinct band was observed at a position corresponding to the loaded ACP5Kir (Figure 3B). In the absence of KirCII, acyl-CoA, or cyclooctyne-fluorophore, only background signal was detected. As expected, activity was not detected when apo-ACP5Kir was used in place of holo-ACP5Kir. Furthermore, Sfp was used to transfer the azido-functionalized phosphopantetheine moiety from AzEM-CoA to apo-ACP5Kir, resulting in strong labeling with the cyclooctyne-fluorophore (Figure 3B). Next, the fluorescence intensity of the labeled ACP bands was quantified by in-gel fluorescence analysis. Inclusion of the MCAT from Streptomyces coelicolor or the malonyl-CoA trans-AT from the disorazole synthase (DSZS), which cannot utilize AzEM-CoA, in place of KirCII failed to produce labeled ACP (Figure 3C). In addition, the fluorescence intensity was shown to be dependent on the KirCII reaction incubation time (Figure 3D) and the concentration of KirCII (Figure 3E). Cumulatively, this data shows that AzEM-CoA and cycloaddition enables the rapid and efficient quantification of KirCII activity.
Figure 3.

Cycloaddition assay for KirCII:ACP interactions. (A) KirCII-catalyzed trans-acylation and subsequent cycloaddition. (B) In-gel fluorescence detection of a series of KirCII control assays. Coomassie Blue image shows ACP loading in each reaction. (C) Fluorescence intensity of labeled ACP5Kir in a series of control reactions using various trans-ATs or no enzyme. (D) Fluorescence intensity of KirCII-labeled ACP5Kir as a function of time. (E) Fluorescence intensity of labeled ACP5Kir as a function of KirCII concentration. Error bars represent the standard deviation of the mean (n=3)
ACP Specificity of KirCII
Previous studies led to the conclusion that KirCII is responsible for installation of the C29-ethyl moiety in kirromycin (Musiol, et al., 2011). Given the presence of another trans-AT in the kirromycin biosynthetic gene cluster, we set out to investigate whether KirCII displays specificity for ACP5Kir, or whether KirCII could potentially interact with other ACP’s from kirromycin biosynthesis. Each kirromycin ACP was sub-cloned into an expression plasmid as an N-terminal His6-tagged fusion protein, fully converted to the holo-ACP, and subsequently purified to homogeneity (see SI Methods). The conversion of each apo-ACP to the holo-form was monitored by LC-MS analysis of the reaction mixtures, which indicated that in every case, conversion was 100% (Figure S1). Each holo-ACP was then tested as a substrate for KirCII-catalyzed trans-acylation using AzEM-CoA. In this assay, ACP5Kir supported the highest activity with KirCII, although ACP0Kir, ACP3Kir, and ACP7Kir supported basal activity at 15–25% of ACP5Kir. The remainder of the ACP’s tested displayed activities that were <10% of ACP5Kir. This data suggests that ACP5Kir is the preferred interaction partner for KirCII, and other kirromycin ACP’s are poor substrates for KirCII. The ACP specificity of KirCII supports a role for programmed protein interactions in maintenance of kirromycin biosynthetic fidelity.
Mapping the KirCII:ACP5Kir Interaction Epitope
The ACP specificity of KirCII led us to speculate that ACP5Kir interacts with KirCII through a set of unique residues at the ACP surface. Accordingly, we set out to map this epitope via alanine scanning mutagenesis. In order to identify residues predicted to be surface exposed, a homology model for ACP5Kir was constructed using the Swiss-Model workspace and the apo-D-alanyl carrier protein as template (pdb: 1DV5) (see Experimental Procedures for details). A total of 61 (non-Ala/Gly) surface residues were then individually mutated to Ala, expressed, subjected to Sfp-mediated phosphopantetheinylation, and purified. Notably, three of these mutants, D64A, L66A and I83A, could not be completely converted to the holo-form, while L52A, L58A, Y79A, F88A and T93A could not be expressed in soluble form. D64 and L66 are immediately adjacent to the presumed phosphopantetheinylation site (S65), and mutations at this position in other ACP’s are known to hinder modification by Sfp (Weissman, et al., 2006). The I83A mutant is largely unfolded, as judged by analysis of the corresponding CD spectra. Thus, these eight mutants were omitted from further analysis. The CD spectra of a representative panel of the ACP5Kir mutants matched very closely to that of wild-type (WT) ACP5Kir, indicating similar secondary structures (Figure S2). Next, each ACP5Kir mutant was tested as a substrate for KirCII using AzEM-CoA and the cycloaddition assay. Many of the mutants displayed activity similar to WT ACP5Kir, indicating that mutation to Ala at these positions does not significantly impact interaction with KirCII (Figure 5A). However, substitution at five positions (H44, L45, R51, D60, and R70) resulted in activities <20% than that of the WT ACP5Kir with KirCII (Figure 5A). The locations of these five positions were mapped onto the ACP5Kir model (Figure 5B), which for the first time begins to define at least part of the interaction epitope. In addition to those residues identified from alanine scanning mutagenesis, the well-known and highly conserved ‘DSL’ motif comprising ACP5Kir residues D64, S65, and L66 (see Table S1 for a list of carrier protein residues involved in various protein interactions) can likely be included in this epitope, given the phosphopantetheinylation site is embedded within this motif, and previous experimental evidence (Weissman, et al., 2006).
Figure 5.

Mapping the KirCII:ACP interaction epitope by alanine scanning mutagenesis. (A) Trans-acylation rates of holo-ACP5Kir alanine mutants with KirCII. Rates are expressed as a percentage of the activity with wild-type ACP5Kir. Wild-type positions that were Ala/Gly were not mutated. Mutants that displayed <20% the activity of the wild-type holo-ACP5Kir are highlighted red. (B) Trans-acylation activities of ACP5Kir alanine mutants mapped onto an ACP5Kir homology model ribbon diagram (left) and its computed surface (right). Red, <20% activity; green, 20–80% activity; yellow, >80% activity; grey, not determined. Error bars represent the standard deviation of the mean (n=3).
Computational Docking
To provide further insight into the molecular basis of the KirCII ACP specificity, a homology model for KirCII was obtained using the Swiss-Model workspace (Arnold, et al., 2006). Interestingly, the natively cis-acting [KS3][AT3] di-domain from DEBS (34% identity to KirCII) was selected by Swiss-Model as the template. Although KirCII functionally resembles the trans-ATs DSZS and MCAT, the crystal structures of which are known (Keatinge-Clay, et al., 2003; Wong, et al., 2011), these two structures were not selected by Swiss-Model, likely due to poor sequence identity with KirCII (27% and 22% sequence identity to KirCII, respectively). This is in agreement with phylogenetic analyses, which showed that KirCII is more closely related to cis-AT’s than to malonate-specific trans-ATs (Musiol and Weber, 2012). In addition to the large and small AT subdomains typical of trans-AT’s, the modeled KirCII structure also includes a KS-AT linker domain that is usually associated with cis-acting AT domains from type I PKSs (Liew, et al., 2012; Tang, et al., 2006). This prediction is in agreement with the sequence length of KirCII (which is longer than that of DSZS and MCAT), homology between the N-terminal portion of KirCII and various cis-AT’s (Figure S3), in addition to secondary structure similarity between the predicted KirCII KS-AT linker and structure of a known linker domain (Figure S4). The KS-AT linker domain was included in our subsequent protein interaction studies, given the possibility that the linker region participates in ACP recognition by cis-acting AT domains. Furthermore, the KS-AT linker domain appears important for solubility of KirCII, given a truncated version of KirCII that lacks the linker could not be expressed in soluble form. The PatchDock (Schneidman-Duhovny, et al., 2005) and the ClusPro (Kozakov, et al., 2013) servers were then used for molecular docking simulations using the ACP5Kir and KirCII models. Gratifyingly, both servers converged on almost identical docking results, without any physical constraints. One of the top models was chosen on the basis of the alanine scanning mutagenesis data described above (Figure 6A). Overall, the docking model resembles that of the iterative type I PKS DynE8 AT and ACP (Liew, et al., 2012), and also that of the AT3-ACP3 model from DEBS (Wong, et al., 2010). ACP5Kir is predicted to sit in a large cleft formed by parts of the KS-AT linker domain, as well as the small and large subdomains. Gratifyingly, this model places the ACP phosphopantetheine attachment site (S65) ~20 Å away from the presumed active site catalytic Ser (S203) of KirCII, and an active site tunnel can be identified that could accommodate the requisite ACP phosphopantetheine. This model positions each of the five residues identified from alanine scanning mutagenesis (H44, L45, R51, D60, and R70) intimately at the interface between the ACP and KirCII. For example, the carboxylate side chain of D60 of ACP5Kir is predicted to make a salt bridge with the terminus of the R410 side chain of KirCII (Figure 6A). Interestingly, docking models of the DynE8 AT:ACP (Liew, et al., 2012), DEBS AT4:ACP4 (Tang, et al., 2006), and fungal non-reducing PksA:ACP complexes (Bruegger, et al., 2013), also implicate an Asp equivalent to D60 of ACP5Kir at the interface (Table S1). Notably, D60, along with the nearby D64, appear to form a negatively charged group of residues in loop-I (L1) of ACP5Kir that interacts with three consecutive Arg residues from KirCII (R408–R410). Positively charged residues of other ATs are often involved in ACP interactions with an Asp residue analogous to ACP5Kir D64 (Keatinge-Clay, et al., 2003; Liew, et al., 2012; Tang, et al., 2007; Tang, et al., 2006). In addition, ACP5Kir R51 and R70, both judged as important for interaction with KirCII by alanine scanning mutagenesis, are positioned to interact with D98 and E395 of KirCII, respectively. Further, the main chain nitrogen of ACP5Kir A48 is positioned for hydrogen bonding with the hydroxyl of KirCII Y426. Another key predicted interaction involves E245 and R69 from KirCII and ACP5Kir, respectively (Figure 6A). To test the accuracy of this model, a series of KirCII mutants was constructed and assayed by using the cycloaddition assay and ACP5Kir as substrate. The secondary structure of each mutant matched closely to that of WT KirCII, as judged by CD spectroscopy (Figure S5). The KirCII mutants R408A, R409A, and R410A supported trans-acylation at slightly lower rates compared to WT KirCII with ACP5Kir as substrate (Figure 6B). Moreover, substitution of each Arg with Glu further reduced activity of each mutant. In addition, a triple mutant where all three Arg residues (R408–R410) were mutated to Ala displayed less than 10% trans-acylation activity, compared to wild-type KirCII, while no activity was detected for when all three arginines were mutated to Glu (Figure 6B). These results suggest that KirCII R408–R410 plays a significant role in the interaction with ACP5Kir. Similarly, mutation of D98 of KirCII to Ala reduced activity to just 30% of the WT KirCII activity (Figure 6B). Mutation of E245 to Ala reduced activity in comparison to WT KirCII, while activity could not be detected when the same residue was substituted with Arg (Figure 6B). Substitution of KirCII E395 with Ala resulted in loss of activity, while substitution with Arg completely abolished activity. Similarly, the predicted importance of Y426 of KirCII was confirmed by mutagenesis (Figure 6B). This data is consistent with the electrostatic contributions of these residues predicted on the basis of the docking model, and suggests that this set of residues are important for controlling interactions between KirCII and ACP5Kir. Finally, two surface residues more distant from the predicted interface were also targeted. Substitution of D123 with Ala resulted in a slight increase in activity with ACP5Kir as substrate, while substitution of the same residue with Arg resulted in ~50% trans-acylation activity, compared to WT KirCII (Figure 6B). Meanwhile, substitution of E170 with Ala resulted in small decrease in activity with ACP5Kir as substrate, and substitution of E170 with Arg resulted in ~50% trans-acylation activity, compared to WT KirCII, (Figure 6B). Thus, substitution of negatively charged residues distal from the predicted interface with a neutral amino acid has a small effect compared to equivalent substitutions at predicted interface residues. Similarly, substitution at these distal sites with oppositely charged residues results in only ~2-fold decreased activity, compared to wild-type KirCII, whereas such substitutions at residues predicted to be involved in the KirCII epitope (e.g. R408E, R409E, R410E and E245R) lead to dramatic decreases in activity, compared to wild-type KirCII. Cumulatively, this data suggests that several electrostatic interactions contribute to molecular recognition between KirCII and its cognate ACP. Electrostatic surface maps of KirCII and ACP5Kir highlight the charge complementarity between several regions of the predicted interface (Figure 6C).
Figure 6.

Docking model of the KirCII:ACP5Kir interaction. (A) A homology model for ACP5Kir was used as a ligand to dock with a homology model for KirCII. See Experimental Procedures for details regarding generation of the homology models. KirCII large subdomain is shown in light orange, the small subdomain of KirCII is shown in light pink, KirCII linker domain is green, and ACP5Kir is shown in teal. The HII’ portion of LI of ACP5Kir is highlighted red. Phosphopantetheinylation site of ACP5Kir and the KirCII catalytic Ser are shown as spheres. For clarity, not every highlighted residue is labeled. (B) Mutational analysis of KirCII putative interface residues. Error bars represent the standard deviation of the mean (n=3). (C) Electrostatic surface potential maps of KirCII and ACP5Kir are calculated using the PDB2PQR server. Colors range from blue (positive) to white (neutral) to red (negative). Four key electrostatic contacts are shown boxed, the KirCII contacts are A (D98), B (R410) C (R409), and D (E245), while those of ACP5Kir are A′ (R51), B′ (D60), C′ (D64), and D′ (R69). The asterisk indicates the position of the KirCII active site Ser and the ACP5Kir phosphopantetheinylation site. The surfaces are represented so that if ACP5Kir is rotated 180°, the letters from each surface would match.
Mutagenesis of a Non-Cognate ACP
The docking model and mutagenesis data described above suggests that a portion of L1, several C-terminal residues of helix-I (HI), and N-terminal residues of helix-II (HII) are each involved in the KirCII interaction epitope, while loop-II (LII) and helix-III (HIII) of ACP5Kir do not make significant contributions to the interaction with KirCII. To test this hypothesis, and to further define the ACP sequence elements required for ACP specificity of KirCII, chimeras between ACP5Kir and a non-cognate ACP from kirromycin biosynthesis were constructed (Figures 7A/B). ACP10Kir was selected to contribute to these chimeras, since this ACP shares the highest amino acid sequence identity (35%) to ACP5Kir among the kirromycin ACP’s. Notably, the entire N-terminal half (L0-HI-LI) of ACP5Kir is sufficient to provide robust recognition by KirCII when fused with the C-terminal portion (HII-LII-HIII-LIII) of ACP10Kir. The corresponding chimera, ACP10Kir-NA5, supported 50% activity compared to the WT ACP5Kir, a 4-fold improvement in efficiency compared to the non-cognate ACP (Figure 7B). In contrast, the reverse chimera that includes the N-terminal portion of ACP10Kir and the C-terminal portion of ACP5Kir displays no detectable activity with KirCII (compare ACP10Kir NA5 and ACP10Kir CA5, Figure 7B). This data suggests that the majority of the ACP5Kir:KirCII recognition features are located in the N-terminal L0-HI-LI portion of ACP5Kir, in agreement with the docking model and alanine scanning mutagenesis results. In an attempt to further refine the putative interaction epitope, a series of chimeras was designed that exchanged individual secondary structure elements. Exchange of ACP10Kir HI or LI with the corresponding structural element from ACP5Kir each led to a 2-fold improvement in activity, compared to the WT ACP10Kir (Figure 7B), and could not therefore recapitulate the full activity of the ACP10Kir NA5 chimera that contained the entire N-terminal portion of ACP5Kir. Interestingly, preceding HI of each kirromycin ACP is a short sequence labeled L0 that is predicted to form a random coil (Figure S6). As judged by sequence homology, this sequence element is not equivalent to the docking domains postulated to play some role in protein interactions between other trans-AT’s and ACP’s (Tang, et al., 2004) (data not shown). Substitution of the ACP10Kir L0 region with that from ACP5Kir failed to significantly alter ACP specificity (see ACP10Kir L0A5, Figure 7B), while deletion of L0 from ACP5Kir resulted in an insoluble protein.
Figure 7.

Probing the ACP:KirCII interaction epitope by mutagenesis of a non-cognate ACP. (A) Sequence logo for the kirromycin ACP’s. Shown in grey and orange, respectively, are the amino acids at selected positions of ACP5Kir and ACP10Kir. Asterisk indicates the phosphopantetheinylation site. Positions discussed in the text are highlighted. Boundaries between each secondary structure element are also shown. For brevity, N- and C-terminal loop regions (L0 and LIII) are not shown. L0 is the region preceding HI and LIII is the region following HIII. (B) Scheme showing contribution of ACP5Kir and ACP10Kir to each chimera or mutant (left). Activities of WT, chimeric, and mutant kirromycin ACP’s with KirCII (right). Rates are expressed as a percentage of the activity with WT holo-ACP5Kir. (C) Activities of WT and mutant ACP6DEBS with KirCII, expressed as a percentage of the activity with WT holo-ACP5Kir. See Experimental Procedures for reaction and assay conditions. Error bars represent the standard deviation of the mean (n=3).
Interestingly, many residues within HI, LI, and HII of ACP5Kir/ACP10Kir, are poorly conserved among kirromycin ACP’s, as judged by alignment of their amino acid sequences (Figure 7A and Figure S7). Moreover, five of these poorly conserved positions correspond to residues that were previously highlighted by alanine scanning mutagenesis of ACP5Kir (H44, L45, R51, D60, and R70). Accordingly, we hypothesized that some of these poorly conserved residues, perhaps including some from the alanine scanning mutagenesis set, might dictate the ACP specificity of KirCII. To test the contribution of these poorly conserved positions in controlling specificity, a panel of 13 mutants was constructed that substituted each selected residue within HI, LI, and HII of ACP10Kir with the corresponding residue from ACP5Kir (Figure 7A). With respect to positions highlighted by alanine scanning mutagenesis, substitution at I37 and E53 in ACP10Kir (equivalent to H44 and D60 in ACP5Kir, Figure 7A) provided only small changes to the trans-acylation activity, compared to wild-type ACP10Kir (see I37H and E53D, Figure 7B). In contrast, the ACP10Kir mutants A44R and N63R (corresponding to positions R51 and R70 in ACP5Kir, Figure 7A) each display approximately 2-fold higher activity with KirCII, compared to wild-type ACP10Kir, underscoring the importance of these two previously highlighted positions for recognition by KirCII. Interestingly, substitution of ACP10Kir Q62 with Arg (equivalent to R69 in ACP5Kir) failed to improve activity with KirCII, compared to WT ACP10Kir, even though this residue is adjacent to the equivalent of R70 in ACP5Kir. This result is entirely consistent with the alanine scanning mutagenesis data, which shows that of the R69/R70 pair in ACP5Kir, only substitution at R70 is detrimental to activity with KirCII. Presumably, R70 in ACP5Kir contributes more to the interaction with KirCII than the neighboring R69, even though R69 is predicted to interact with KirCII via E245 (Figures 6A–C). Perhaps mutation to Ala at R69 can be better compensated by other side chain rearrangements than Ala substitution at R70. Notably, aside from ACP5Kir, Arg is never found at residue 69 among the kirromycin ACP’s, and this residue is most frequently Leu or Val (Figure 7A and Figure S7). Similarly, at residue 70, Arg is found only three times among all 15 kirromycin ACP sequences, and is most often the oppositely charged Glu (Figure 6A and Figure S7). The most active ACP10Kir single amino acid exchanges were those at positions F51 and Y54 (equivalent to L58 and V61 in ACP5Kir), and do not correspond to positions highlighted by our alanine scanning mutagenesis. Substitution at these two positions with the equivalent residue from ACP5Kir, Leu and Val, respectively, supported activity 5-fold higher than WT ACP10Kir (Figure 7B). Consistent with this data, mutagenesis of the ACP5Kir residue corresponding to ACP10Kir Y54 resulted in a mutant that showed only 21% the activity of WT ACP5Kir (ACP5Kir V61A, Figure 5A). Intriguingly, the ACP5Kir residue equivalent to ACP10Kir F51 (L58) was not expressed in soluble form when mutated to Ala as part of the previous surface mutagenesis, and was therefore not highlighted by the surface mutagenesis data. Interestingly, ACP5Kir L58 and V61 are not well conserved among the kirromycin ACP’s (Figure 7A and Figure S7). For example, residue 58 is most often Phe (as in ACP10Kir F51), and is Leu in only four of the kirromycin ACP sequences. Residue 61 is usually Leu or Tyr (as in ACP10Kir Y54) among the kirromycin ACP’s and is Val only in ACP5Kir. Combination of both F51L and Y54V in ACP10Kir actually resulted in a slight decrease in trans-acylation activity compared to either the F51L or Y54V mutant (Figure 7B). However, exchange of a portion of a LI sequence in ACP10Kir that included F51 and Y54 with that from ACP5Kir (HII’, corresponding to V57-D60 in ACP5Kir), resulted in a chimera with 5-fold the activity of wild-type ACP10Kir (ACP10Kir-H*, Figure 7B). Thus, the precise identities of residues within this short helix are critical for maintaining maximal activity with KirCII. Together, this data suggests that in ACP5Kir, Arg residues at positions 51/70, and Leu/Val at residues 58/61, respectively, are required but not totally sufficient for recognition by KirCII.
Reprogramming KirCII Interactions Toward a Non-Cognate ACP From The Erythromycin PKS
Some trans-ATs have been shown to be promiscuous towards the ACP substrate, and can utilize ACP’s from different biosynthetic pathways (Walker, et al., 2013; Wong, et al., 2010). The conclusion that Arg is required at positions 51/70 of ACP5Kir, and that a Leu and Val are required at positions 58 and 61, led us to predict whether ACP’s from other biosynthetic systems would serve as substrates for KirCII. For example, ACP6 from DEBS includes Asp and Gly at positions equivalent to 51/70 of ACP5Kir, while at residues equivalent to ACP5Kir 58 and 61, Phe and Leu are present in the ACP6DEBS, respectively. Accordingly, we predicted that ACP6DEBS would not serve as an efficient substrate for KirCII. This hypothesis was tested by carrying out the cycloaddition assay using AzEM-CoA, KirCII, and holo-ACP6DEBS. As predicted, WT holo-ACP6DEBS was not a detectable substrate for KirCII (Figure 7C). Given that the ACP10Kir mutation F51L led to improved trans-acylation activity with KirCII (Figure 7B), compared to the wild-type ACP10Kir, and that the equivalent residue in ACP6DEBS is also Phe (F38), we proposed that introduction of leucine at position 38 of ACP6DEBS would improve the trans-acylation efficiency of this non-cognate ACP with KirCII. To test this notion, the mutant ACP6DEBS F38L was constructed, converted to the holo-form and assayed with KirCII. Remarkably, and in complete contrast to wild-type ACP6DEBS, the F38L mutant was a detectable substrate for KirCII, and this substitution improved trans-acylation activity at least 14-fold compared to WT ACP6DEBS (Figure 7C). To the best of our knowledge, this is the first time that the activity of a trans-AT has been rationally engineered towards a non-cognate carrier protein.
DISCUSSION
An improved understanding of protein interactions is required to overcome limitations related to the ACP specificity of trans-AT’s. Although structural studies of trans-AT’s that utilize malonyl-CoA have slowly emerged (Keatinge-Clay, et al., 2003; Wong, et al., 2011), nothing is known regarding the structure and ACP interactions of trans-AT’s with unusual acyl-CoA specificities. In this study, we harnessed the acyl-CoA promiscuity of KirCII (Koryakina, et al., 2013) to rapidly probe its interaction with a series of ACP’s. The expected cognate protein, ACP5Kir, was found to be the preferred substrate for KirCII, while the enzyme was capable of utilizing other kirromycin ACP’s to a lesser extent. This data suggests that programmed protein interactions could play a role in controlling extender unit incorporation into kirromycin, but do not rule out the role of downstream substrate specificity or editing functions.
The specificity of KirCII for ACP5Kir, coupled with the relatively high sequence homology between the other kirromycin ACP’s, provided an opportunity to identify ACP elements that contribute to trans-AT specificity. Alanine scanning mutagenesis identified several ACP residues that when mutated to Ala, almost completely abolished activity with KirCII. Interestingly, this result contrasts with alanine scanning mutagenesis of the malonyl-CoA specific, yet ACP promiscuous trans-AT, DSZS (Wong, et al., 2011), which revealed only two positions that were sensitive to substitution. Computational prediction of the interaction between ACP5Kir and KirCII provided a model that was consistent with the alanine scanning mutagenesis data. The KS-AT linker domain, the large AT subdomain, and small AT subdomain of KirCII are all predicted to be involved in ACP recognition. The KS-AT linker domain of KirCII may contribute to the strict ACP specificity of KirCII, given that structures of the ACP promiscuous trans-AT’s MCAT and DSZS do not include this domain. In fact, R51 of ACP5Kir, which interacts with the linker domain in the model (Figure 6A), is most often Glu or Ala in other ACP’s. Furthermore, the position equivalent to ACP5Kir R51 in other ACP’s is not predicted to be involved in interactions with trans-AT’s or other interacting domains (Table S1). Additional ACP5Kir residues that appear to contribute to recognition by KirCII that are also not identified by other protein interaction studies include R56 and R70. Notably though, some predicted KirCII:ACP5Kir interactions involve ACP residues that have been implicated in other protein interaction studies. For example, D64 is part of the highly conserved DSL motif, and is predicted to be involved in diverse interactions between various PKS domains. Another key residue, D60 is often Asp or Glu in other ACP’s, and has been implicated in protein interactions between other AT’s. Curiously, D60 locates to a short helical turn (HII’ in Figures 5B and 6A) in the middle of LI of ACP5Kir. Intriguingly, short helical turns similar to HII’ have been proposed to contribute to other ACP interactions. For example, a short helix located within LII has been proposed to flag ACP’s for β-branching in polyketide biosynthesis (Haines, et al., 2013). In another example, a similarly short helical turn was implicated in the specific interaction between the curacin A ACPI and halogenase embedded within the CurA module (Busche, et al., 2012). Further, in the non-reducing PKS PksA ACP, an aspartate equivalent to ACP5Kir D60, located in a small helical turn, is predicted to make a key electrostatic interaction with the KS domain of PksA (Bruegger, et al., 2013). Notably, we found that recognition by KirCII was very sensitive to amino acid substitution in the HII’ region of ACP5Kir. Individual substitutions at two key hydrophobic sites in this region were sufficient to improve the activity of ACP10Kir 5-fold with KirCII. Moreover, we established that amino acid substitution at one site within this region of an ACP from an entirely different biosynthetic system was sufficient to convert a completely inactive ACP into a robust substrate. Thus, the overall shape, position of D60, or dynamics of the HII’ region may be important for specifying the activity of a given ACP with KirCII. In fact, hydrophobic packing within a similar short helical turn of ACP-mupA3a from mupirocin biosynthesis was recently found to contribute significantly to recognition by the cognate trans-acting β-branching domain, MupH (Haines, et al., 2013). In addition to our extensive mutagenesis of the kirromycin ACP’s, the role of several KirCII residues was probed by substituting key charged residues with neutral or oppositely charged amino acids. The results were consistent with the docking model and suggested that charged residues at the KirCII:ACP5Kir interface contribute to the strict ACP specificity of KirCII by forming key electrostatically matched interactions. Furthermore, this data suggests that KirCII harnesses a larger number of electrostatic interactions with its cognate ACP than that of ACP promiscuous trans-AT’s such as MCAT and DSZS. Thus, a picture is emerging whereby the ACP specificity of KirCII is a consequence of a large and unique set of charged and hydrophobic residues at the ACP5Kir surface. Together, this data for the first time describes the interaction epitope between a non-malonyl utilizing trans-AT and cognate ACP.
Critically, those ACP5Kir residues involved in recognition by KirCII would have been difficult to identify from sequence analysis alone. For example, several charged ACP5Kir residues that are poorly conserved among the kirromycin ACP’s are not important for KirCII specificity (e.g. R34, D35, R38). Similarly, a pair of Arg residues in HII of ACP5Kir (R73/R74) are quite conspicuous and might have been assumed to be important for KirCII recognition by comparison to other kirromycin ACP’s. Yet neither residue was found to be important herein.
Cumulatively, our data provides a platform for harnessing the acyl-CoA promiscuity of KirCII for diversification of polyketides. For example, with knowledge of the ACP5Kir:KirCII epitope in hand, ACP’s from other biosynthetic systems might now be identified by sequence comparison that could be recognized by KirCII. In addition, the insight obtained here will enable strategies for further engineering trans-AT:ACP interactions. It is envisioned that residues in non-cognate ACP’s that correspond to those recognized by KirCII (or other trans-ATs) could be targeted for mutagenesis and screening or selection, by utilizing the cycloaddition assay in combination with cell surface display.
SIGNIFICANCE
Trans-AT’s offer exciting possibilities for polyketide diversification, especially given the potential extender unit promiscuity of KS domains (Koryakina, et al., 2013). However, little is known regarding the ACP or extender unit specificity of many trans-AT’s. Putative ACP recognition features have emerged only with those trans-AT’s that naturally utilize malonyl-CoA, with limited potential to introduce other extender units into polyketides. Understanding the features of ACP’s that allow recognition by a cognate trans-AT is a crucial first step that may enable manipulation of trans-AT ACP specificity. We proposed that the trans-AT KirCII only interacts with a single ACP within the parent kirromycin biosynthetic assembly line, and this would provide an opportunity to probe the ACP features required for recognition by a uniquely acyl-CoA promiscuous trans-AT. We recognized that the ability of KirCII to utilize substrates modified with chemical handles for click chemistry could be leveraged to provide a rapid and convenient assay for measuring the activity of KirCII. Accordingly, the activity of KirCII towards each kirromycin ACP was measured, providing evidence that KirCII interacts specifically with the expected cognate ACP. A combination of ACP surface mutagenesis, docking simulations, KirCII mutagenesis, and substitutions of amino acids and structural elements, led to the identification of several key electrostatic interactions that define the ACP5Kir:KirCII interaction epitope. Remarkably, this insight enabled engineering a completely non-cognate ACP into a detectable substrate for KirCII via the introduction of a single amino acid substitution. Our data combined with previous studies highlight the importance of small helical turns located within loop regions that connect the larger helices that form the basis of all ACP structures. In the long term, an improved understanding of the features that allow recognition of an ACP by trans-AT’s can be used as a foundation for designing strategies that allow the diversification of polyketides.
Experimental Procedures
General
Unless otherwise stated, all materials and reagents were of the highest grade possible and purchased from Sigma (St. Louis, MO). Isopropyl β-D-thiogalactoside (IPTG) was from Calbiochem (Gibbstown, NJ). Bacterial strain Escherichia coli (E. coli) BL21(DE3) competent cells was from Promega (Madison, WI). Primers were ordered from Integrated DNA Technologies (Coralville, IA). Analytical HPLC was performed on a Varian ProStar system.
Cloning and Expression of Kirromycin ACP’s
The genes encoding kirromycin ACP’s were amplified from cosmids 1C24 and 2C23 (Weber, et al., 2008) using HotStar HighFidelity polymerase (Qiagen) and the primers listed in Table S2. Fragments were cloned in pET30Ek/LIC (Novagen/Merck Millipore) using the manufacturer’s protocol. Plasmids harboring each kirromycin ACP were co-transformed with plasmid pSU20-Sfp into E. coli BL21(DE3). A single colony was then used to inoculate 3 mL of LB (Luria Burtani) medium containing 34 μg/mL chloramphenicol and 50 μg/mL kanamycin which was cultured overnight at 37 °C with shaking at 250 rpm. The overnight culture was used to inoculate 300 mL of LB media containing the same antibiotics and was then incubated at 37 °C at 250 rpm until the OD600 had reached ~0.6. Protein expression was induced by the addition of 0.5 mM IPTG and was then incubated at 18 °C at 250 rpm for 18 h. MS analysis of initial trials indicated that in vivo phosphopantetheinylation yielded significant but incomplete conversion to the holo-form. To drive phosphopantetheinylation to completion, an in vitro conversion to the holo-form was also performed using cleared cell extract from each ACP over-expression. Briefly, cleared cell extract prepared from 300 mL cell culture was mixed with 40 μL of 50 mM CoA and was incubated at room temperature for 4 h then kept at 4 °C overnight. The proteins were then purified by Ni-NTA chromatography largely as previously described (Musiol, et al., 2013), except the batch purification method was used. MS analysis showed full conversion to the holo ACP for every ACP (Figure S1).
Cloning and Expression of Sfp, KirCII, DSZS and MCAT
Cloning and expression of Sfp, KirCII, DSZS and MCAT were performed as previously described (Koryakina, et al., 2013).
Chemo-Enzymatic Synthesis of Azidoethylmalonyl-CoA
Chemo-enzymatic synthesis of azidoethylmalonyl-CoA was performed in 400 μL of reaction mixture containing 100 mM sodium phosphate (pH 7), MgCl2 (2 mM), ATP (6 mM), coenzyme A (3 mM), azidoethyl malonate (15 mM) (Koryakina, et al., 2013), and mutant MatB (20 μg) at 25 °C. Production of azidoethylmalonyl-CoA was confirmed by HPLC. Aliquots were removed after overnight incubation, quenched with an equal volume of ice-cold methanol, and centrifuged at 10,000g for 10 min and cleared supernatants were used for HPLC analysis on a Varian ProStar HPLC system. A series of linear gradients was developed from 0.1% TFA (A) in water to methanol (HPLC grade, B) using the following protocol: 0–32 min, 80% B; 32–35 min, 100% A. The flow rate was1 mL/min, and the absorbance was monitored at 254 nm using Pursuit XRs C18 column (250 mm × 4.6 mm, Varian Inc.).
General Procedure for Strain-Promoted Azide-Alkyne Cycloaddition Assay
The strain-promoted azide-alkyne cycloaddition assay was performed in a total volume of 6 μL and contained 4 μL of KirCII assay mixture containing 20 μM ACP5Kir and 2 μL of600 μM of DIBO-Alexa Fluor 647 (Life Technologies). Reaction was incubated in dark at room temperature with gentle agitation for 1 h. Reactions were boiled after addition of protein loading dye for 5 min before analysis by SDS-PAGE. The gels were scanned using a Typhoon 7000 phosphoimager to determine the intensity of DIBO labeled proteins bands. The bands were quantified by ImageQuant TL software (GE Life Sciences) and rubber-band background subtraction method was applied.
KirCII Titration Assay
KirCII titration assay was performed in 10 μL of reaction mixture containing 50 mM Tris-HCI (pH 7.5), 50 mM MgCI2, 300 μM of azidoethylmalonyl CoA, 50 μM holo ACP5Kir, and KirCII at room temperature for 20 min. 4 μL of KirCII reaction mixture was then removed for strain-promoted azide-alkyne cycloaddition assay.
KirCII Time Course Assay
KirCII time course assay was performed in 70 μL of reaction mixture containing 50 mM Tris-HCI (pH 7.5), 50 mM MgCI2, 300 μM of azidoethylmalonyl CoA, 30 μM holo ACP5Kir, and 1 μM KirCII at room temperature. Aliquots were removed at different time points and stored and −80 °C before used for strain-promoted azide-alkyne cycloaddition assay.
Alanine Scanning Mutagenesis of ACP5Kir
Primers for alanine scanning mutagenesis of ACP5Kir were designed with lengths between 25 and 30 bp, while melting temperatures were between 70 and 75 °C. Site directed mutagenesis was performed following the QuikChange protocol (Agilent Technologies, Inc.).
KirCII assay using each ACP5Kir alanine scanning mutant was performed in 10 μL of reaction mixture containing 50 mM Tris-HCI (pH 7.5), 50 mM MgCI2, 300 μM of azidoethylmalonyl CoA, 30 μM holo ACP5Kir, and 3 μM KirCII at room temperature for 1 h. An aliquot (4 μL) of the reaction mixture was then removed and assayed using the strain-promoted azide-alkyne cycloaddition procedure.
Circular Dichroism
Circular dichroism (CD) measurements were performed with a JASCO 810 CD Spectropolarimeter. Samples for CD were buffer exchanged into CD buffer (10 mM potassium phosphate, 50 mM sodium sulfate, pH 7.4) and the concentration of samples was 0.2 mg/ml. Spectra from 190 nm to 260 nm were scanned at a step of 0.5 nm at 20 °C in a 0.1 cm cuvette, with ten repeats. The scan speed was 100 nm/min.
Homology Model of ACP5Kir and KirCII
Homology models for ACP5Kir and KirCII were generated using the automated mode of SWISS-MODEL Workspace (http://swissmodel.expasy.org/). The template used for ACP5Kir was 1DV5, sequence identity between the template and ACP5Kir was 26%. The template for KirCII was 2QO3, sequence identity between template and KirCII was 34%. Based on QMEAN z-scores (Benkert, et al., 2011), the quality for the homology models is good.
Site-Directed Mutagenesis and Chimeragenesis of ACP’s and KirCII
Plasmid pET30-KirACP10 was used as a template for mutagenesis of ACP10Kir. For mutagenesis of KirCII, plasmid pET52-KirCII was used (Koryakina, et al., 2013). ACP and KirCII amino acid substitutions were performed using the QuikChange protocol (Agilent Technologies, Inc.). Multiple-template-based sequential PCR (Shan and Lynch, 2010) was used for construction of ACP chimeras. E. coli DH5α was used for plasmid amplification. All mutations were confirmed by DNA sequencing. Specific primer sequences used for construction of ACP10Kir/ACP5Kir chimeras can be found in the supplemental information (Table S3).
Supplementary Material
Figure 4.
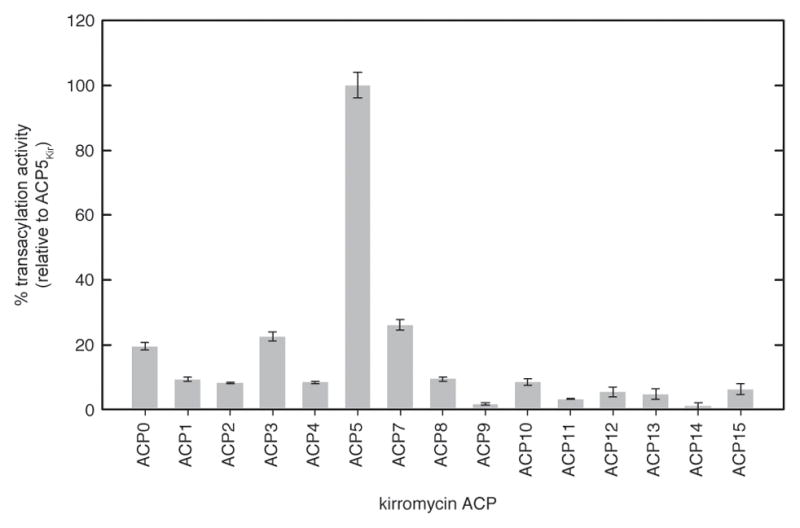
ACP specificity of KirCII. Activity of 3 μM KirCII with 30 μM each kirromycin ACP and 300 μM AzEM-CoA was determined by the cycloaddition assay. Error bars represent the standard deviation of the mean (n=3).
Highlights.
Little is known regarding how trans-acting acyltransferases interact with PKS’s
A detailed picture of how KirCII interacts with its cognate ACP is presented
A non-cognate ACP was converted into a substrate for KirCII
This work will enable engineering the biosynthesis of polyketide analogues
Acknowledgments
This study was supported by an NSF CAREER Award (CHE-1151299, G.J.W), NIH Grant GM104258-01 (G.J.W), and a Faculty Research and Professional Development Award from NC State University (G.J.W). T.W. was supported by the German Center for Infection Research. The authors would like to thank Christopher Ladner for helpful discussions, and the Biological Core Instrumentation Facility in the Department of Chemistry at NC State.
Footnotes
Supplemental Information includes seven figures and three tables, and can be found online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Arthur CJ, Williams C, Pottage K, Ploskon E, Findlow SC, Burston SG, Simpson TJ, Crump MP, Crosby J. Structure and malonyl CoA-ACP transacylase binding of streptomyces coelicolor fatty acid synthase acyl carrier protein. ACS Chem Biol. 2009;4:625–636. doi: 10.1021/cb900099e. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruegger J, Haushalter B, Vagstad A, Shakya G, Mih N, Townsend CA, Burkart MD, Tsai SC. Probing the selectivity and proteinprotein interactions of a nonreducing fungal polyketide synthase using mechanism-based crosslinkers. Chem Biol. 2013;20:1135–1146. doi: 10.1016/j.chembiol.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche A, Gottstein D, Hein C, Ripin N, Pader I, Tufar P, Eisman EB, Gu L, Walsh CT, Sherman DH, et al. Characterization of molecular interactions between ACP and halogenase domains in the Curacin A polyketide synthase. ACS Chem Biol. 2012;7:378–386. doi: 10.1021/cb200352q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YQ, Tang GL, Shen B. Type I polyketide synthase requiring a discrete acyltransferase for polyketide biosynthesis. Proc Natl Acad Sci USA. 2003;100:3149–3154. doi: 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RJ, Simpson TJ. Fungal type I polyketide synthases. Methods Enzymol. 2009;459:49–78. doi: 10.1016/S0076-6879(09)04603-5. [DOI] [PubMed] [Google Scholar]
- Haines AS, Dong X, Song Z, Farmer R, Williams C, Hothersall J, Ploskon E, Wattana-Amorn P, Stephens ER, Yamada E, et al. A conserved motif flags acyl carrier proteins for beta-branching in polyketide synthesis. Nat Chem Biol. 2013;9:685–692. doi: 10.1038/nchembio.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Chen AY, Cane DE, Khosla C. Molecular recognition between ketosynthase and acyl carrier protein domains of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci USA. 2011;107:22066–22071. doi: 10.1073/pnas.1014081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Lowry B, Yuzawa S, Kenthirapalan S, Chen AY, Cane DE, Khosla C. Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation. Proc Natl Acad Sci USA. 2012;109:4110–4115. doi: 10.1073/pnas.1118734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatinge-Clay AT, Shelat AA, Savage DF, Tsai SC, Miercke LJ, O’Connell JD, 3rd, Khosla C, Stroud RM. Catalysis, specificity, and ACP docking site of Streptomyces coelicolor malonyl-CoA:ACP transacylase. Structure. 2003;11:147–154. doi: 10.1016/s0969-2126(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Koryakina I, McArthur J, Randall S, Draelos MM, Musiol EM, Muddiman DC, Weber T, Williams GJ. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem Biol. 2013;8:200–208. doi: 10.1021/cb3003489. [DOI] [PubMed] [Google Scholar]
- Koryakina I, McArthur JB, Draelos MM, Williams GJ. Promiscuity of a modular polyketide synthase towards natural and non-natural extender units. Org Biomol Chem. 2013;11:4449–4458. doi: 10.1039/c3ob40633d. [DOI] [PubMed] [Google Scholar]
- Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, Vajda S. How good is automated protein docking? Proteins. 2013;82:2159–2166. doi: 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Koppisch AT, Cane DE, Khosla C. Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J Am Chem Soc. 2003;125:14307–14312. doi: 10.1021/ja037429l. [DOI] [PubMed] [Google Scholar]
- Liew CW, Nilsson M, Chen MW, Sun H, Cornvik T, Liang ZX, Lescar J. Crystal structure of the acyltransferase domain of the iterative polyketide synthase in enediyne biosynthesis. J Biol Chem. 2012;287:23203–23215. doi: 10.1074/jbc.M112.362210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Huang Y, Shen B. Bifunctional acyltransferase/decarboxylase LnmK as the missing link for beta-alkylation in polyketide biosynthesis. J Am Chem Soc. 2009;131:6900–6901. doi: 10.1021/ja9012134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman JR, Bingman CA, Phillips GN, Jr, Shen B. Structure of the bifunctional acyltransferase/decarboxylase LnmK from the leinamycin biosynthetic pathway revealing novel activity for a double-hot-dog fold. Biochemistry. 2013;52:902–911. doi: 10.1021/bi301652y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiol EM, Greule A, Hartner T, Kulik A, Wohlleben W, Weber T. The AT(2) domain of KirCI loads malonyl extender units to the ACPs of the kirromycin PKS. ChemBioChem. 2013;14:1343–1352. doi: 10.1002/cbic.201300211. [DOI] [PubMed] [Google Scholar]
- Musiol EM, Hartner T, Kulik A, Moldenhauer J, Piel J, Wohlleben W, Weber T. Supramolecular templating in kirromycin biosynthesis: the acyltransferase KirCII loads ethylmalonyl-CoA extender onto a specific ACP of the trans-AT PKS. Chem Biol. 2011;18:438–444. doi: 10.1016/j.chembiol.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Musiol EM, Weber T. Discrete acyltransferases involved in polyketide biosynthesis. Medchemcomm. 2012;3:871–886. [Google Scholar]
- Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2010;27:996–1047. doi: 10.1039/b816430b. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Lynch JW. Chimera construction using multiple-template-based sequential PCRs. J Neurosci Meth. 2010;193:86–89. doi: 10.1016/j.jneumeth.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. Enzyme-Directed Mutasynthesis: A Combined Experimental and Theoretical Approach to Substrate Recognition of a Polyketide Synthase. ACS Chem Biol. 2013;8:443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- Tang GL, Cheng YQ, Shen B. Leinamycin biosynthesis revealing unprecedented architectural complexity for a hybrid polyketide synthase and nonribosomal peptide synthetase. Chem Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen AY, Kim CY, Cane DE, Khosla C. Structural and mechanistic analysis of protein interactions in module 3 of the 6-deoxyerythronolide B synthase. Chem Biol. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YY, Kim CY, Mathews II, Cane DE, Khosla C. The 2.7-angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci U S A. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MC. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341:1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Laiple KJ, Pross EK, Textor A, Grond S, Welzel K, Pelzer S, Vente A, Wohlleben W. Molecular analysis of the kirromycin biosynthetic gene cluster revealed beta-alanine as precursor of the pyridone moiety. Chem Biol. 2008;15:175–188. doi: 10.1016/j.chembiol.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Weissman KJ, Hong H, Popovic B, Meersman F. Evidence for a protein-protein interaction motif on an acyl carrier protein domain from a modular polyketide synthase. Chem Biol. 2006;13:625–636. doi: 10.1016/j.chembiol.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wong FT, Chen AY, Cane DE, Khosla C. Protein-protein recognition between acyltransferases and acyl carrier proteins in multimodular polyketide synthases. Biochemistry. 2010;49:95–102. doi: 10.1021/bi901826g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FT, Jin X, Mathews II, Cane DE, Khosla C. Structure and Mechanism of the trans-Acting Acyltransferase from the Disorazole Synthase. Biochemistry. 2011;50:6539–6548. doi: 10.1021/bi200632j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Coughlin JM, Ju J, Zhu D, Wendt-Pienkowski E, Zhou X, Wang Z, Shen B, Deng Z. Oxazolomycin biosynthesis in Streptomyces albus JA3453 featuring an “acyltransferase-less” type I polyketide synthase that incorporates two distinct extender units. J Biol Chem. 2010;285:20097–20108. doi: 10.1074/jbc.M109.090092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.