

Abstract
The genetic complexity and heterogeneity of cancer has posed a problem in designing rationally targeted therapies effective in a large proportion of human cancer. Genomic characterization of many cancer types has provided a staggering amount of data that needs to be interpreted to further our understanding of this disease. Forward genetic screening in mice using Sleeping Beauty (SB) based insertional mutagenesis is an effective method for candidate cancer gene discovery that can aid in distinguishing driver from passenger mutations in human cancer. This system has been adapted for unbiased screens to identify drivers of multiple cancer types. These screens have already identified hundreds of candidate cancer-promoting mutations. These can be used to develop new mouse models for further study, which may prove useful for therapeutic testing. SB technology may also hold the key for rapid generation of reverse genetic mouse models of cancer, and has already been used to model glioblastoma and liver cancer.
Introduction
It is becoming increasingly clear that cancer is a very complex and heterogenous disease where each individual cancer type is actually composed of multiple molecular subclasses. Cancer arises from a series of genetic events that result in the corruption of normal cellular development, growth, and proliferation. A variety of sets of genetic events can corrupt these processes, which underlie the variety of molecular subclasses of cancer. In order to develop focused and effective means of treating the disease, greater research is required to further elucidate the cancer-promoting genes that contribute to these subclasses and determine how they function and cooperate in promoting tumorigenesis. Large-scale genomic characterization efforts by The Cancer Genome Atlas (TCGA) network and other groups are revealing staggering numbers of genes mutated, lost, amplified, or dysregulated in human cancer. Some of these alterations have already been identified as recurrent and shown to promote cancer phenotypes, providing insight into the mechanisms of cancer pathogenesis and potential therapeutic strategies, such as EGFR fusions and mutations promoting glioblastoma growth, which may inform the design of clinical trials for EGFR inhibitors for glioblastoma [1]. Unfortunately for many cancers, the confusing heterogeneity of underlying mutations leading to similar cancer phenotypes has precluded the ability to design targeted therapies that are effective in a large percentage of cancer patients. The amount of mutation information becoming available highlights the need for effective methods to distinguish between passenger alterations, which result from genomic instability and have no role in tumorigenesis, and driver alterations, which promote tumor progression and maintenance and, importantly, may serve as effective therapeutic targets or prognostic markers. Models that accurately reflect this genetic heterogeneity and allow it to be understood are desperately needed to identify driver mutations and design rational targeted therapies.
Unbiased screens for cancer promoting mutations provide a means of distinguishing driver from passenger mutations. In transposon-based mutagenesis screens, the random insertion of mutagenic transposons alters normal endogenous genes in the mouse and induces cancer. The genetic changes that drive disease progression can then be identified by the locations of transposon insertions [2–9]. These transposon-based systems, therefore, represent powerful genetic tools for identifying cancer-promoting mutations. This unbiased method of elucidating cancer genes has proven effective. Information derived from these screens and the resulting new cancer models based on this information will contribute greatly towards developing and testing effective therapeutic regimes. Importantly, transposons can be used as both forward and reverse genetic tools to elucidate cancer genes in vivo.
The Sleeping Beauty (SB) transposable element is a synthetic DNA-type transposon belonging to the Tc1/Mariner transposon family that mobilizes in a “cut-and-paste” fashion. It was awakened from millions of years of evolutionary sleep by correcting the mutations responsible for its transposase inactivity [10]. The current SB transposon system consists of two parts: firstly, a transposon vector containing any DNA sequence that is flanked by inverted repeat/direct terminal repeat (IR/DR) sequences and secondly, the SB transposase enzyme that is responsible for excision and reintegration of the transposon placed under the control of a promoter. When both these components are present in a cell, a “cut-and-paste” transposition reaction occurs in which the transposon is excised from its original location and re-integrated at a new location within the genome. The mobilization process is relatively random, although it has the propensity for “local hopping” and the only prerequisite that the transposon reintegrates at a “TA” dinucleotide [11]. SB transposition is active in both transgenic mouse germline and somatic cells [2, 3, 11, 12]. The mutagenic transposon called T2/Onc (Fig. 1A) was designed to cause both gene loss- and gain-of-function insertional mutations, which would be marked by the unique transposon sequences and could be used later to identify cancer genes in solid tumors (Fig. 1B and 1C). T2/Onc combined with transgenes ubiquitously expressing SB transposase in wild-type or cancer predisposed mice induced or accelerated sarcoma and T-cell leukemia [2, 3]. In both cases, the SB-initiated or accelerated tumors were characterized by somatic, tumor-specific transposon insertions that were shown to occur at dozens of recurrently mutated known and novel cancer genes [2, 3]. SB insertion sites are readily cloned and can be characterized rapidly to implicate new genes in solid tumor development using a forward genetic approach. Next-generation sequencing platforms allow for rapid and adequate coverage to identify transposon insertion sites. Sites mutated by insertions significantly more frequently in tumors than predicted by random chance are called common insertion sites (CISs) and are hypothesized to reveal potential driver mutations. This data can be applied to human cancer mutation data to elucidate which alterations found in human tumors may be important cancer drivers (Fig. 2). Comparison between CISs from SB screens and human tumor mutations has already implicated many genes in human disease including colorectal cancer, pancreatic cancer, medulloblastoma, and malignant peripheral nerve sheath tumors [8, 13–16].
Figure 1. Mutagenic transposon vector for gain- and loss- of-function screen.

(A) The mutagenic T2/Onc transposon contains splice acceptors (SAs) followed by polyadenylation (pA) signals in both orientations, which are designed to elicit premature transcript truncation. The 5′-long terminal repeat (LTR) of the murine stem cell virus (MSCV) fused with a splice donor (SD) was placed in between the two SAs to drive misexpression of genes. This MSCV contains enhancer and strong cis-regulatory elements that have been shown to be active and methylation-resistant in pluripotent cells. IR/DR, inverted repeat/direct terminal repeat sequences. (B) Gain-of-function screen for proto-oncogenes. The mutagenic transposon vector can integrate upstream of a proto-oncogene and cause misexpression or create truncated versions by integrating within intronic sequences of the endogenous gene. (C) Loss-of-function screen for tumor suppressor genes. The mutagenic transposon vector can integrate within intronic sequences and disrupt expression of tumor suppressor genes.
Figure 2. Pipeline for selecting candidate cancer genes from SB screens for further study.
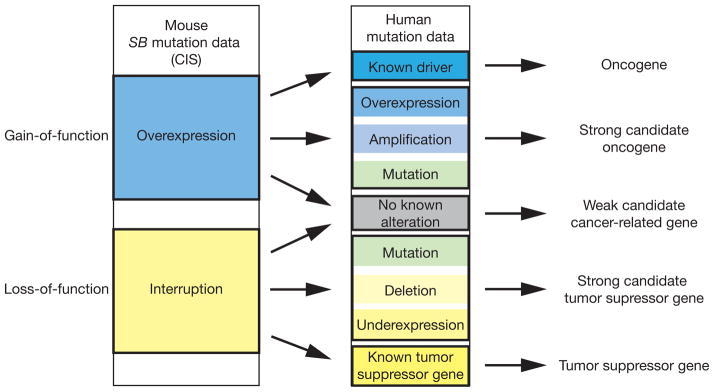
SB insertion mutations can result in gene overexpression or interruption. Human molecular cancer data can likewise reveal potential gain-of-function or loss-of-function alterations. To determine which common insertion sites (CISs) are most likely relevant oncogenes or tumor suppressors in human cancer, human cancer data is analyzed for gain- or loss- of-function alterations in CIS genes. Genes overexpressed by transposon insertions that have gain-of-function alterations in human cancer are considered to be strong candidate oncogenes, and genes interrupted by transposon insertions that have loss-of-function alterations in human cancer are considered strong candidate tumor suppressor genes. These strong candidate cancer genes should be prioritized for further study.
Tissue-specific mutagenesis with SB has developed informative models of various types of human solid tumors. In these studies, mice express the catalytically improved SB transposase, SB11 [17], from the endogenous Rosa26 locus, but only after Cre recombinase has excised a loxP-flanked stop cassette (lsl) separating the SB11 cDNA from the Rosa26 promoter. Thus, using these Rosa26-lsl-SB11 mice, SB mutagenesis can be restricted to tissues expressing the Cre recombinase from a tissue-specific promoter. Conditional SB transposition systems have been successfully used to generate various solid tumors and screen for genes associated with these cancer types [4, 5, 9]. In addition to tissue-specific promoters driving Cre recombinase, predisposed genetic backgrounds can also be incorporated into these screens to elucidate mutations cooperating with common cancer initiating mutations [5, 6]. Variations of this mutagenesis system have been used to reveal both known and novel oncogenes as well as tumor suppressors in solid tumors [2, 4–9]. Conditional mutagenesis systems with hematopoietic cell-specific promoters driving Cre recombinase and predisposed genetic backgrounds have proven useful for modeling liquid tumors such as B-cell precursor acute lymphoblastic leukemia, T-cell acute lymphoblastic leukemia, chronic lymphocytic leukemia, and erythroleukemia [18–21]. This review, however, will focus on solid tumor models.
Cancer-promoting mutations discovered from body wide and tissue specific mutagenesis screens can be compared to human mutation, gene copy number, and gene expression data to identify key cancer-promoting alterations. These alterations may provide novel, effective therapeutic targets and prognostic markers that may inform treatments. In addition, they can be used to create new mouse models of cancer that accurately reflect the many molecular subclasses of the human disease, which could be used for testing novel therapies. Recently, much progress has been made in identifying candidate cancer drivers for many types of cancer using transposable elements [22]. To continue this progress, new mouse models driven by these genes must be developed. Engineered mouse models of several types of cancer including liver cancer [23–25], and neurofibroma/malignant peripheral nerve sheath tumor [26, 27], and glioma [28] that may prove useful for therapeutic testing have been developed. The methods used to generate these models may inform the development of other useful cancer mouse models driven by the genes identified in forward genetic screens.
Mouse models of cancer
Forward genetic screens for gastrointestinal tract cancer genes
Colorectal carcinoma (CRC) is the third most commonly diagnosed cancer for both men and women in the United States, and, with approximately one third of patients dying from this disease, is a leading cause of cancer-related death [29, 30]. It develops through the accumulation of mutations in genes belonging to multiple pathways controlling cell growth, differentiation, and survival [31, 32]. Therefore, it is a highly heterogeneous disease, in that tumors are driven by diverse sets of mutations [31, 32]. Because of this, patients with histologically similar tumors can differ drastically in their response to therapy and survival outcomes [32]. An improved understanding of the molecular basis of CRC development and progression is needed to predict response to current therapies and develop new molecularly targeted therapies in order to improve survival outcomes. Large-scale genomic characterization studies such as those conducted by TCGA network and others have identified many alterations present in human CRC [33–35]. Since CRC is characterized by the accumulation of many mutations and widespread genomic instability [31], however, these studies highlight the challenge of distinguishing passenger alterations from alterations that drive cancer progression and may serve as strong clinical predictive markers or therapeutic targets.
Transposon-based screens are useful for identifying candidate CRC drivers because the cancer-promoting mutations are caused by transposon insertion events rather than genome-wide instability. CRC was modeled using mice carrying the mutagenic T2/Onc2 SB transposons, conditional Rosa26-lsl-SB11 transposase, and villin-Cre to activate transposition specifically in gastrointestinal (GI) tract epithelial cells. This generated intraepithelial neoplasias, adenomas, and adenocarcinomas in the small and large intestines. Transposon insertion site analyses from these tumors identified 77 candidate CRC genes, 60 genes were known to be altered or dysregulated in human CRC. These included well-characterized CRC drivers: adenomatosis polyposis coli (APC), phosphatase and tensin homolog (PTEN) and SMAD family member 4 (SMAD4), as well as novel candidate CRC drivers: R-spondin 2 homolog (Xenopus laevis) (Rspo2), WW domain containing adaptor with coiled-coil (Wac), and protein tyrosine phosphatase, receptor type, K (Ptprk) [9].
Although APC is lost or mutated in about 80% of all CRC, most other mutations in CRC occur at low frequency [36]. Another conditional SB transposon insertional mutagenesis screen was conducted to find low frequency mutations that cooperate with APC mutations to drive CRC. For this screen, mice were generated carrying a mutant allele of Apc (ApcMin) along with the three transgenes used to drive transposon mutagenesis in GI tract epithelial cells described above. ApcMin mice undergoing GI tract transposition developed a severe phenotype with an average lifespan of 85 days and an average of 360 polyps per mouse [37]. The gene most commonly mutated by transposon insertions was Apc, highlighting the importance of loss of the wild-type allele in tumor initiation. Mutations in 32 other loci were also identified as candidate cancer drivers cooperating with loss of Apc, 24 of which were in regions commonly lost or gained in human CRC [37].
Since both sporadic and germline Apc mutations initiate CRC [38–40], another SB-based forward genetic screen was performed in mice with mutant Apc. In this study, SB-driven transposition was used to drive tumorigenesis in the background of either a floxed allele of Apc to model sporadic mutation, or the ApcMin transgene to model germline mutation. For the sporadic model, mice carried transgenes for the mutagenic T2/Onc SB transposon, a conditionally expressed Rosa26-lsl-SB transposase, floxed Apc, and villin-Cre to activate transposition specifically in GI tract epithelial cells [14, 41]. For the germline model, mice carried the mutagenic T2/Onc SB transposon, the Rosa26-SB transposase, and ApcMin [14, 42]. They obtained approximately 65 tumors per mouse in the sporadic and 10 tumors per mouse in the germline Apc-mutant model. These models revealed 867 CIS genes, which were significantly enriched for genes involved in known CRC-associated pathways such as the wingless (Wnt), phosphatidylinositol 3-kinase (Pi3k), rat sarcoma (Ras), transformation related protein 53 (Trp53), and transforming growth factor beta (Tgfb) pathways. Cross-species comparison with human CRC oncogenomic data implicated 234 CIS in human CRC [14]. Together, these screens revealed nearly 1,000 candidate CRC-driving mutations. Although several of these have been tested in human CRC cell lines, new mouse models of CRC driven by these mutations have yet to be developed.
Forward genetic screens for cancer genes involved in malignant peripheral nerve sheath tumorigenesis
Malignant peripheral nerve sheath tumors (MPNSTs) are a rare but an aggressive form of sarcoma associated with extremely poor prognosis. MPNSTs can occur sporadically or in the context of the neurofibromatosis type 1 (NF1) tumor syndrome, an autosomal dominant inherited disease caused by mutations in one copy of the neurofibromin 1 (NF1) gene [43–45]. An understanding of the genetic evolution from a benign neurofibroma to its malignant form in patients with NF1-associated or sporadic MPNSTs currently remains elusive but is expected to be highly complex [43–45]. In NF1 patients, genomic abnormalities other than those at the neurofibromin 1 (NF1) locus have rarely been detected in benign neurofibromas but are numerous in MPNSTs [46]. This suggests that many secondary genetic changes are required for the transformation from a benign neurofibroma to MPNST. Currently, many of the genetic alterations associated with MPNST initiation and progression are unknown, and identification of these genetic changes may have profound clinical benefits.
Using the SB system in a forward genetic screen for genes responsible for sporadic MPNST formation on a predisposed genetic background, loss of Trp53 function and/or overexpression of human epidermal growth factor receptor (EGFR), many candidate mutational drivers of peripheral nerve sheath tumor (PNST) were identified [8]. In this study, the role of forkhead box R2 (FOXR2) was demonstrated as an important oncogene for MPNST development and turning off this gene drastically decreases the growth ability of these tumors [8]. Importantly, phosphatase and tensin homolog (Pten) and neurofibromatosis 1 (Nf1) were amongst the many candidate genes identified in this screen that tended to be co-mutated in the same PNSTs. Pten-regulated pathways have been shown to be major tumor suppressive barriers to PNST progression in Schwann cells in the context of Nf1 loss [26]. Recently, it has also been shown that PTEN and EGFR both play important roles in the initiation of PNSTs [27]. In human MPNSTs, PTEN expression is often reduced, while EGFR expression is often induced [27]. Validation of these candidate drivers of MPNSTs can be done using cell culturing approaches and have been used effectively to date [47]. However, genetically modified mouse models represent a more accurate approach for in vivo validation. Importantly, these mouse models can also be used for testing novel therapeutic regimes.
Reverse genetic mouse models for NF1-associated and sporadic MPNSTs
Based on results from this screen and human mutation data, mouse models that rapidly recapitulate PNST onset and progression to high-grade PNSTs, as seen in both NF1-associated and sporadic MPNST patients have been recently generated. For NF1-associated MPNST, deserthedgehog (Dhh) regulatory element driving Cre recombinase transgenic mice was used to elicit recombination of both conditional floxed alleles of Nf1 [48] and Pten [49], allowing for inactivation of both Nf1 and Pten genes in Schwann cells and/or their precursors. This modeled the co-occurrence of Pten and Nf1 found in PNSTs derived from SB mutagenesis. The loss of both Nf1 and Pten in Schwann cells lead to the rapid onset of high grade PNSTs [26].
For the sporadic MPNST model, transgenic mice carried conditional floxed alleles of Pten under the control of the Dhh promoter driving Cre recombinase, while EGFR was overexpressed under the control of the 2′,3′-cyclic nucleotide 3′ phosphodiesterase (Cnp) regulatory element. Complete loss of Pten and overexpression of EGFR in Schwann cells led to the development of high-grade PNSTs. In vitro experiments using immortalized human Schwann cells also confirmed that loss of PTEN and overexpression of EGFR cooperated to increase cellular proliferation and anchorage-independent colony formation [27]. Importantly, both mouse models can rapidly recapitulate the onset of human neurofibroma tumorigenesis and its progression to PNSTs. These models can be used to accurately recapitulate the human disease and to potentially rapidly test a variety of pharmaceutical compounds in vivo.
Forward genetic screens for liver cancer associated genes
Liver cancer, or hepatocellular carcinoma (HCC), is the sixth most common cancer and the third leading cause of cancer-related mortality worldwide [50]. HCC affects over 700,000 patients per year worldwide and is the most rapidly increasing cause of cancer-related death in developed nations. It is highly aggressive with a dismal prognosis since less than 30% of patients will be eligible for potential curative treatment at the time of diagnosis [51]. The pathogenetic changes underlying the development and progression of HCC are complex and incompletely understood. There are currently many molecular subclasses of human HCCs, further complicating the undertaking of understanding the genetic mechanisms associated with this deadly disease [52, 53].
To specifically model HCC in mice and identify driver mutations, we used the conditional SB transposon system for insertional mutagenesis. Mice were generated carrying the conditional Rosa26-lsl-SB11 transposase transgene, the T2/Onc2 transposon, and the hepatocyte-specific albumin-Cre to limit transposase expression and subsequent transposon mobilization to hepatocytes. In addition, because tumor protein p53 (TP53) is frequently mutated in human HCC, a conditional dominant negative Trp53 transgene was added along with the transposition system. Both the three-transgene mutagenesis system and the mutagenesis system on the Trp53 mutated background produced tumors in mice that modeled all the stages of liver cancer progression including neoplastic nodules, adenomas, HCC, and lung metastasis [5]. These models recapitulated the sex bias seen in humans, with tumors occurring at higher penetrance and lower latency in males than females [5, 23]. This screen uncovered 19 candidate HCC-associated loci including known oncogenes such as EGFR and met proto-oncogene (MET) and novel candidate genes associated with HCC including ubiquitin-conjugating enzyme E2H (Ube2H) and zinc finger and BTB domain containing 20 (Zbtb20) [5]. By examining transposon insertion sites from male and female mouse tumors separately and comparing them to human mutation data, we also identified EGFR as a gene associated with the sex bias of the disease [23]. Mutations in the wingless/catenin (cadherin associated protein), beta 1 (Wnt/Ctnnb1) pathway were found in mice of both sexes, tankyrase, TRF1-interacting ankyrin-related ADP-ribose polymerase 2 (Tnks2) in males and glycogen synthase kinase 3 beta (Gsk3b) in females [23]. The WNT/CTNNB1 pathway has been causatively linked to various cancers, with up to 60% of liver cancers showing active WNT/CTNNB1 signalling. This activation of WNT/CTNNB1 signaling is generally due to mutations in CTNNB1, resulting in its constitutive activation [54–57]. These discoveries have facilitated the development of novel mouse models of HCC [23].
HCC has also been generated in mice using a modified mutagenic transposon, T2/Onc3, in which oncogene over-expression is driven by the cytomegalovirus enhancer/chicken β-actin (CAGGS) promoter, which is more active in epithelial cells than the murine stem cell virus (MSCV) 5′ long-terminal repeat (LTR) promoter used in T2/Onc2 [4]. As expected, body wide mobilization of T2/Onc3 produced more carcinomas [4] than T2/Onc2, which induced mainly hematopoietic malignancies [2]. Of the tumors generated by body wide mobilization of T2/Onc3, liver tumors occurred most frequently [4].
The v-myc avian myelocytomatosis viral oncogene homolog (MYC) oncogene is amplified in 30% and overexpressed in a majority of HCC [58]. To identify genetic alterations cooperating with MYC overexpression, a transposon-based mutagenesis screen was performed on a MYC-induced mouse model of liver cancer. Transgenic mice were generated carrying the mutagenic SB transposon T2/Onc, the ubiquitously expressed Rosa26-SB11 transposase, the tetracycline-repressible MYC, and the liver-specific tet-transactivator transgenes [6]. MYC-overexpressing mice undergoing transposition had an accelerated rate of tumorigenesis, with 65% of mice developing tumors before 16 weeks of age compared to 29% of MYC-overexpressing controls without transposition. Fourteen CISs were identified from tumors in the mice undergoing transposition. Of these, three were validated as tumor suppressor genes by shRNA knockdown in Trp53-null hepatoblasts immortalized by MYC overexpression. Hepatoblasts with nuclear receptor coactivator 2 (Ncoa2), zinc finger protein X-linked (Zfx), or dystrobrevin beta (Dtnb) shRNA knockdown gave rise to tumors when injected into nude mice. Therefore, this study identified 14 candidate MYC-cooperating liver cancer driver mutations, three of which were shown to act as tumor suppressors in context of MYC overexpression [6].
Selecting genes from forward genetic screens for study
These and other transposon-based forward genetic screens have generated a wealth of candidate cancer-promoting mutations with possible relevance for human disease. This can make prioritizing these genes for further study challenging. To select candidate genes for follow-up testing, we propose that SB CIS genes found in mouse models should be compared to human cancer genetic data, when available, to identify the strongest candidate genes. Transposon insertions in the 5′ region of a gene with the MSCV or CAGGS promoter in the forward orientation relative to the gene likely drive overexpression, while transposon insertions scattered throughout a gene likely disrupt expression (Fig. 1). This allows CIS genes to be identified as candidate oncogenes or tumor suppressors based on insertion site profiles [59]. CIS genes without a clear trend of alteration in human cancer should be considered weaker candidate cancer genes, and should be given lower priority for follow-up. CIS genes with known roles as human oncogenes or tumor suppressors are not novel, but can be useful as controls or for proof-of-principle experiments. CIS genes found as candidate oncogenes in SB screens that are amplified, overexpressed, or have activating mutations in human cancer can be considered strong candidate oncogenes that should be prioritized for study. Likewise, candidate tumor suppressor CIS genes from SB screens that are deleted, underexpressed, or have inactivating mutations in human cancer can be considered strong candidate tumor suppressor genes that should be prioritized for further study (Fig. 2). The strong candidate genes can be further tested in vitro for potential roles in tumorigenesis. Based on the results of the cross species oncogenomic comparison and in vitro transformation assays, the strongest candidate cancer genes should be selected to generate novel reverse-genetic mouse models of cancer.
SB-mediated gene delivery for reverse genetic mouse models of cancer
Forward genetic screens and human mutational data are providing a vast number of candidate cancer-associated genes. Reverse genetic models can test the roles of these genes in tumorigenesis. Understanding the mechanism by which these genes drive tumorigenesis may inform the development of novel therapies. Reverse genetic models allow the roles of these genes in tumorigenesis to be tested. Reverse genetic models driven by known agents may also be more useful for testing therapeutic agents targeted at a specific mutation than insertional mutagenesis models, which may not contain the mutation targeted by the therapeutic agent. In addition, reverse genetic models may progress more rapidly than insertional mutagenesis models, as is the case for our models of MPNST [8, 26, 27], making them more amenable to therapeutic testing.
Transgenic mice provide the advantage of allowing development of orthotopic cancer models in immunocompetent mice. Generating transgenic mice, however, is costly and slow, making it an impractical method for addressing the large number of candidate cancer genes becoming available. SB-mediated gene delivery in vivo may provide a more efficient method to generate reverse genetic cancer models. In addition to its use as a mutagen, SB can be used to deliver genes-of-interest or knockdown vectors for stable expression within cells in vivo [25, 28, 60–62]. SB can excise a cDNA sequence for a gene-of-interest or shRNA sequence that is flanked by inverted repeat/direct terminal repeat (IR/DR) sequences from a plasmid and reintegrate it at a random “TA” dinucleotide in the genome [61]. In this way, SB can be used to generate reverse genetic models of cancer, in which specific genes are tested for their role in tumorigenesis.
Reverse Genetic mouse models for liver cancer
A unique mouse model for validating candidate liver cancer genes that uses fumaryl acetoacetate hydrolase (Fah) mutant mice and a SB-based gene delivery system (Fah/SB11) has been established [5, 23–25]. Fah-mutant mice have a defect in the tyrosine metabolic pathway similar to the human hereditary tyrosinemia type I disease and have to be maintained on 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) water. While on NTBC water, these animals are physiologically normal. Candidate liver cancer cDNAs can be introduced into the livers of Fah/SB11 mice for validation along with the Fah cDNA on SB transposon vectors flanked by SB IRDRs (Fig. 3A). The vectors are introduced into the livers of Fah/SB11 mice by hydrodynamic tail vein injection, and then NTBC is removed. As hepatocytes lacking Fah die, the livers of the Fah/SB11 mice are repopulated by hepatocytes in which transposon vectors are integrated by SB and stably express the Fah cDNA and gene-of-interest. This results in generating mice with almost entirely transgenic livers in 6–8 weeks. Candidate genes identified either in forward genetic screens as described above or from human liver cancer data can be validated in vivo using novel gene delivery vectors that carry the Fah cDNA and designed to be compatible with the GateWay system (Life Technologies) (Fig. 3B). Putative oncogene homologues can be purchased commercially and cloned into GateWay pENTR vectors (Life Technologies), followed by clonase reactions into the gene delivery vectors. Short-hairpin RNAs against putative tumor suppressor gene homologues can also be purchased commercially and cloned into SB delivery vectors. This system makes the Fah model even more rapid by allowing efficient generation of gene delivery vectors for candidate oncogenes.
Figure 3. Validation of candidate liver cancer genes in vivo by reverse genetics.

(A) Validation of candidate liver cancer gene using the Fah/SB11 model. Fah/SB11 mice will die of liver failure due to a defect in tyrosine metabolic pathway unless treated with 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC). Gene(s)-of-interest is co-delivered with Fah cDNA by hydrodynamic (high volume/rapid) injection into the tail vein of Fah/SB11 mice and NTBC is removed. Mice are then aged and observed for a liver phenotype. Hepatocytes that do not have the Fah cDNA and/or gene-of-interest will die while hepatocytes that uptake the transgenes will repopulate the liver, mimicking liver disease proliferation. (B) Bi-directional gene delivery plasmid with GateWay cloning system (Life Technologies). CAGGS, cytomegalovirus early enhancer element and chicken beta-actin promoter; cargo, any DNA sequence-of-interest; pA, polyadenylation sequence; attB1 and attB2, GateWay flanking sequences after LR clonase reaction; PGK, phosphoglycerate kinase 1 promoter; IRES, internal ribosome entry sequence; Fah, fumaryl acetoacetate hydrolase cDNA; Luc, firefly luciferase reporter gene; Red arrowheads, SB inverted repeat/direct terminal repeat sequences.
To date, several mouse models for liver cancer have been generated using the Fah/SB11 mouse model for future therapeutic drug testing: neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS) [25], hepatitis B viral X gene (HBx) [24], retrotransposon-like 1 (Rtl1) [63], CTNNB1 [23], and EGFR [23]. Rtl1 was identified as a novel HCC driver in a SB forward genetic screen and found to be overexpressed in human liver cancer samples. It was shown to induce liver tumors in Fah/SB11 mice [63]. EGFR was identified in the conditional SB screen as a potential sex bias associated HCC driver and indeed, induced tumor development with a sex bias in the Fah/SB11 mouse model [23]. CTNNB1 was tested because mutations in the Wnt/Ctnnb1 pathway were found in both male and female mice of both sexes, and it induced tumors with no apparent sex bias in our mouse model [23]. Both CTNNB1 and EGFR liver cancer mouse models represent two human HCC molecular subclasses previously described, namely the CTNNB1 and Poly7, which show the same sex biases in humans observed in our mouse models [52]. This system can be used to efficiently test the role of novel HCC-associated genes uncovered in insertional mutagenesis screens that are altered in human disease to determine their importance as driver mutations.
An advantage of using this animal model is that the cost and effort associated with the generation of germline transgenic animals harboring the gene-of-interest can be avoided. Genes-of-interest can be cloned into SB transposon vectors and introduced into the livers of Fah/SB11 animals by a relatively simple technique involving hydrodynamic tail-vein injection. Live imaging can monitor the pathogenicity of the genes-of-interest by including a luciferase reporter on the gene delivery vector and mice are sacrificed when a phenotype is observed. Most importantly, this approach allows the rapid generation of liver cancer mouse models driven by various genetic alterations.
SB-mediated transgene integration for modeling glioblastoma multiforme
Glioblastoma multiforme (GBM), the most common type of brain tumor in adults, is highly aggressive, invasive, and deadly [64], with an average survival of 12–15 months [65] and 3 year survival rate of only 2.2% [66]. One major factor limiting therapeutic success is the genetic complexity and heterogeneity of GBM, making it difficult to target with single-agent drugs [28]. Various studies have identified 3 to 4 distinct molecular subclasses of GBM based on human tumor DNA, RNA, and protein analyses, which also have distinct histological properties [67–71]. TCGA has revealed hundreds of genes mutated or over-expressed in over 85% of human GBM [67, 72], but their roles in driving GBM have yet to be defined. Many of these common alterations, if shown to drive GBM, could become powerful prognostic markers or therapeutic targets because they are altered in such a large percentage of GBMs. Mouse models would be extremely valuable for evaluating their roles in driving GBM and testing new therapies.
Spontaneous mouse models of GBM in which the tumor develops in context of the host immune system more faithfully recapitulate the human disease than models driven by transplanted tumor cells [28]. Transgenic mouse models can achieve this [73], but are time-consuming and costly to generate making them impractical for testing the many candidate GBM drivers indentified by the large scale molecular analysis of human tumors.
A rapid, relatively inexpensive method to induce spontaneous brain tumors with a variety of molecular drivers has been developed using an SB transposon-based gene delivery strategy [28]. Tumor-promoting cDNA expression constructs are flanked by SB IRDRs and delivered on plasmids as SB transposon vectors along with an SB expression plasmid. Plasmids are delivered to the brain by intracranial injection and transfected into brain cells in vivo with polyethylenimine/plasmid DNA complexes. SB is expressed transiently and stably integrates the transposon vectors, which can then induce tumors. These tumors can be monitored in vivo using fluorescence imaging by including a luciferase transposon vector (Fig. 4).
Figure 4. SB transposon-based gene delivery method for spontaneous brain tumor induction.

Transposon vectors expressing any gene-of-interest can be rapidly generated by the GateWay cloning system (Life Technologies). Plasmid vectors transiently expressing SB and transposon vectors expressing luciferase and any oncogene are complexed with polyethylenimine (PEI/DNA) delivered by intracranial injection into 1-day old perinatal mice. Stable oncogene integration by SB and expression can result in tumor growth, which can be monitored by luciferase imaging. CAGGS, cytomegalovirus early enhancer element and chicken beta-actin promoter; cargo, any DNA sequence-of-interest; pA, polyadenylation sequence; attB1 and attB2, GateWay flanking sequences after LR clonase reaction; PGK, phosphoglycerate kinase 1 promoter; SB, SleepingBeauty transposase enzyme; Luc, firefly luciferase reporter gene; Red arrowheads, SB inverted repeat/direct terminal repeat sequences.
This method has been used to model GBM driven by various combinations of known oncogenes NRAS-G12V, v-akt murine thymoma viral oncogene homolog 1 (AKT1), EGF receptor variant III (EGFRvIII), and RNA short hairpin directed against Trp53. The resulting tumors had similar histological properties to human GBM, being infiltrative with high cell density and areas of necrosis and expressing protein markers seen in human GBM [28]. This model could be easily adapted to test many candidate cancer drivers identified to be altered in human GBM. It can also be used to model GBM driven by a variety of genes and mutations that would prove useful for studying their mechanism of oncogenesis and testing new therapies.
Considerations for reverse genetic model design
When SB transposition is used as a mutagen in forward genetic screens, multiple copies of a transposon designed to maximally disrupt endogenous gene expression like T2/Onc are mobilized within a cell [59]. In contrast to this, when SB transposition is used for gene delivery, the transposons are designed to express cDNAs or short hairpin RNAs, not to maximally disrupt nearby gene expression [25, 28, 60, 62]. Insertional mutagenesis, however, is still a factor that needs to be considered when using SB-mediated gene delivery for reverse genetic studies. SB can be expressed transiently from a plasmid to integrate the transposon vectors with minimal chance of re-mobilization, decreasing the number of insertion events occurring in a cell. This has been done in both the liver and brain [25, 28]. This decreases, but does not completely eliminate, the risk of insertion mutations promoting tumor formation. Experimental design with the appropriate negative controls is important in accounting for this risk. Rates of tumor formation in mice with integrated transposon vectors for cancer gene overexpression or knockdown should be compared to tumor formation rates in mice with similar transposon vectors lacking the oncogenic cargo. This will reveal whether the oncogenic effects observed are due to the transposon cargo or insertional mutagenesis. In the liver and brain cancer models described above, rates of tumor formation in mice with integrated control plasmids are low [24, 28], suggesting that to date, insertional mutations play a minimal role in this type of cancer model.
Summary and future directions
Extensive molecular characterization of many cancer types has revealed copious numbers of alterations and remarkable heterogeneity of the mutations present in cancers, even with similar phenotypes. In order to understand this complex disease, driver mutations need to be identified and characterized. Transposon based insertional mutagenesis is an effective tool for cancer gene discovery and the forward genetic screens in mice using the SB transposon system have generated a vast amount of genetic data that can be used to complement the understanding of the human oncogenome. Due to the complexity and heterogeneity of cancer, candidate genes that are misexpressed in both the mouse models and human disease should be further validated for their roles as driver mutations by either in vitro or in vivo means. Cancer promoting mutations identified in this way has served as the basis for new mouse models to further characterize this disease. The use of the SB transposon system for liver cancer research, for example, has been well developed due to the ability to both perform both forward genetic screens and generate reverse genetic animal models for gene testing.
An important aspect of tumorigenesis is the role of tumor-promoting microenvironments. Microenvironment components including inflammatory cells, extracellular matrix (ECM) component content and organization, and cytokines all influence tumor development [74]. Conditions like obesity, viral infection, and hepatic fibrosis that alter the microenvironment by inducing inflammation, altering the ECM, or altering cytokine signaling can increase the risk of cancer formation [48, 74–78]. Researchers could use the SB transposon system to model cancer in relevant microenvironments by performing SB mutagenesis screens on mice with tumor-predisposing conditions that alter the microenvironment such as diet-induced obesity, chemically-induced fibrosis, or viral infection. This could reveal differences in the mutational profiles driving cancer under these conditions that would be relevant for understanding disease progression or inform therapeutic strategy design.
There is still great need to develop efficient methods to test candidate genes for other cancer types in vivo and to discover and test oncogenes in disease-relevant environments. This will require efficient use of mutation data by analyzing pathways altered rather than only individual mutations and testing sets of potentially cooperating mutations together. While transgenic models will work well for some rapidly progressing tumors, they are often slow and costly to develop. Efficient methods of somatic transgene introduction need to be developed for many tissue types in order to rapidly test candidate oncogenes in multiple cancer types. SB-mediated gene transfer technology may be useful for rapidly generating new mouse models of various types of cancer. Its use for in vivo gene delivery has already been demonstrated in the liver, lung, and brain [25, 28, 60, 62]. In cases where the oncogenic cargo is too large for efficient mobilization by SB, piggyBac transposition may provide an alternative method, since it can mobilize larger transposons efficiently than SB [79]. To model co-alteration of multiple genes, multi-gene vectors can be generated rapidly using the RecWay system [80].
SB transposon-based insertional mutagenesis in mice has proven useful for cancer gene identification. It has facilitated the identification of hundreds of candidate cancer genes and several pathways promoting cancer [4–6, 8, 9, 14, 37]. SB transposon-based gene delivery has also provided an effective method for testing candidate oncogenes in vivo in some mouse models [5, 23, 63]. More mouse models must be developed to elucidate the mechanism of oncogenesis and test new targeted therapies. We believe information obtained from SB transposon insertional mutagenesis screens and reverse genetic studies can serve as the basis to develop the models we need to understand and treat cancer.
Highlights.
Sleeping Beauty (SB) transposon system is a useful genetic tool for cancer gene discovery
SB transposon system used to model many human cancers
Both forward and reverse genetic screens are possible using the SB transposon system
These mouse models of cancers are useful for testing novel therapeutic regimes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–9. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–6. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 3.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–6. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 4.Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, Liu P, et al. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009;69:8150–6. doi: 10.1158/0008-5472.CAN-09-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009;27:264–74. doi: 10.1038/nbt.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Donnell KA, Keng VW, York B, Reineke EL, Seo D, Fan D, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A. 2012;109:E1377–86. doi: 10.1073/pnas.1115433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahrmann EP, Collier LS, Knutson TP, Doyal ME, Kuslak SL, Green LE, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69:4388–97. doi: 10.1158/0008-5472.CAN-08-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahrmann EP, Watson AL, Keng VW, Choi K, Moriarity BS, Beckmann DA, et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat Genet. 2013;45:756–66. doi: 10.1038/ng.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–50. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–10. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 11.Keng VW, Yae K, Hayakawa T, Mizuno S, Uno Y, Yusa K, et al. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods. 2005;2:763–9. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 12.Dupuy AJ, Fritz S, Largaespada DA. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–8. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 13.Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109:5934–41. doi: 10.1073/pnas.1202490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011;43:1202–9. doi: 10.1038/ng.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266–70. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–33. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Dupuy AJ, et al. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol Ther. 2003;8:108–17. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 18.Berquam-Vrieze KE, Nannapaneni K, Brett BT, Holmfeldt L, Ma J, Zagorodna O, et al. Cell of origin strongly influences genetic selection in a mouse model of T-ALL. Blood. 2011;118:4646–56. doi: 10.1182/blood-2011-03-343947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang JZ, Carmichael CL, Shi W, Metcalf D, Ng AP, Hyland CD, et al. Transposon mutagenesis reveals cooperation of ETS family transcription factors with signaling pathways in erythro-megakaryocytic leukemia. Proc Natl Acad Sci U S A. 2013;110:6091–6. doi: 10.1073/pnas.1304234110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, et al. Modeling the evolution of ETV6-RUNX1-induced B-cell precursor acute lymphoblastic leukemia in mice. Blood. 2011;118:1041–51. doi: 10.1182/blood-2011-02-338848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zanesi N, Balatti V, Riordan J, Burch A, Rizzotto L, Palamarchuk A, et al. A Sleeping Beauty screen reveals NF-kB activation in CLL mouse model. Blood. 2013;121:4355–8. doi: 10.1182/blood-2013-02-486035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howell VM. Sleeping beauty--a mouse model for all cancers? Cancer Lett. 2012;317:1–8. doi: 10.1016/j.canlet.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Keng VW, Sia D, Sarver AL, Tschida BR, Fan D, Alsinet C, et al. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology. 2013;57:120–30. doi: 10.1002/hep.26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keng VW, Tschida BR, Bell JB, Largaespada DA. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology. 2011;53:781–90. doi: 10.1002/hep.24091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wangensteen KJ, Wilber A, Keng VW, He Z, Matise I, Wangensteen L, et al. A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology. 2008;47:1714–24. doi: 10.1002/hep.22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keng VW, Rahrmann EP, Watson AL, Tschida BR, Moertel CL, Jessen WJ, et al. PTEN and NF1 Inactivation in Schwann Cells Produces a Severe Phenotype in the Peripheral Nervous System That Promotes the Development and Malignant Progression of Peripheral Nerve Sheath Tumors. Cancer Res. 2012;72:3405–13. doi: 10.1158/0008-5472.CAN-11-4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keng VW, Watson AL, Rahrmann EP, Li H, Tschida BR, Moriarity BS, et al. Conditional Inactivation of Pten with EGFR Overexpression in Schwann Cells Models Sporadic MPNST. Sarcoma. 2012;2012:620834. doi: 10.1155/2012/620834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69:431–9. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 30.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 31.Deschoolmeester V, Baay M, Specenier P, Lardon F, Vermorken JB. A review of the most promising biomarkers in colorectal cancer: one step closer to targeted therapy. Oncologist. 2010;15:699–731. doi: 10.1634/theoncologist.2010-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanthan R, Senger JL, Kanthan SC. Molecular events in primary and metastatic colorectal carcinoma: a review. Patholog Res Int. 2012;2012:597497. doi: 10.1155/2012/597497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nehrt NL, Peterson TA, Park D, Kann MG. Domain landscapes of somatic mutations in cancer. BMC Genomics. 2012;13 (Suppl 4):S9. doi: 10.1186/1471-2164-13-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 36.Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: genetics of development and metastasis. J Gastroenterol. 2006;41:185–92. doi: 10.1007/s00535-006-1801-6. [DOI] [PubMed] [Google Scholar]
- 37.Starr TK, Scott PM, Marsh BM, Zhao L, Than BL, O’Sullivan MG, et al. A Sleeping Beauty transposon-mediated screen identifies murine susceptibility genes for adenomatous polyposis coli (Apc)-dependent intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:5765–70. doi: 10.1073/pnas.1018012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 39.Luchtenborg M, Weijenberg MP, Roemen GM, de Bruine AP, van den Brandt PA, Lentjes MH, et al. APC mutations in sporadic colorectal carcinomas from The Netherlands Cohort Study. Carcinogenesis. 2004;25:1219–26. doi: 10.1093/carcin/bgh117. [DOI] [PubMed] [Google Scholar]
- 40.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–38. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 41.Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, et al. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–46. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 43.Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1–14. doi: 10.1016/j.jaad.2008.12.051. quiz 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1–6. [PubMed] [Google Scholar]
- 45.Rosenfeld A, Listernick R, Charrow J, Goldman S. Neurofibromatosis type 1 and high-grade tumors of the central nervous system. Childs Nerv Syst. 2010;26:663–7. doi: 10.1007/s00381-009-1024-2. [DOI] [PubMed] [Google Scholar]
- 46.Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590–605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson AL, Rahrmann EP, Moriarity BS, Choi K, Conboy CB, Greeley AD, et al. Canonical Wnt/beta-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013;3:674–89. doi: 10.1158/2159-8290.CD-13-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–2. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao A, Yin C, Yang C, Di Cristofano A, Pandolfi PP, Van Dyke T. Somatic induction of Pten loss in a preclinical astrocytoma model reveals major roles in disease progression and avenues for target discovery and validation. Cancer Res. 2005;65:5172–80. doi: 10.1158/0008-5472.CAN-04-3902. [DOI] [PubMed] [Google Scholar]
- 50.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 51.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–17. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 52.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–88. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385–92. doi: 10.1158/0008-5472.CAN-09-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bengochea A, de Souza MM, Lefrancois L, Le Roux E, Galy O, Chemin I, et al. Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. British journal of cancer. 2008;99:143–50. doi: 10.1038/sj.bjc.6604422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, et al. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–21. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–51. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 58.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 59.Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat Rev Cancer. 2010;10:696–706. doi: 10.1038/nrc2916. [DOI] [PubMed] [Google Scholar]
- 60.Belur LR, Frandsen JL, Dupuy AJ, Ingbar DH, Largaespada DA, Hackett PB, et al. Gene insertion and long-term expression in lung mediated by the Sleeping Beauty transposon system. Mol Ther. 2003;8:501–7. doi: 10.1016/s1525-0016(03)00211-9. [DOI] [PubMed] [Google Scholar]
- 61.Ivics Z, Kaufman CD, Zayed H, Miskey C, Walisko O, Izsvak Z. The Sleeping Beauty transposable element: evolution, regulation and genetic applications. Curr Issues Mol Biol. 2004;6:43–55. [PubMed] [Google Scholar]
- 62.Ohlfest JR, Demorest ZL, Motooka Y, Vengco I, Oh S, Chen E, et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the sleeping beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Mol Ther. 2005;12:778–88. doi: 10.1016/j.ymthe.2005.07.689. [DOI] [PubMed] [Google Scholar]
- 63.Riordan JD, Keng VW, Tschida BR, Scheetz TE, Bell JB, Podetz-Pedersen KM, et al. Identification of rtl1, a retrotransposon-derived imprinted gene, as a novel driver of hepatocarcinogenesis. PLoS Genet. 2013;9:e1003441. doi: 10.1371/journal.pgen.1003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85:133–48. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmid RS, Vitucci M, Miller CR. Genetically engineered mouse models of diffuse gliomas. Brain Res Bull. 2012;88:72–9. doi: 10.1016/j.brainresbull.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 66.Smoll NR, Schaller K, Gautschi OP. Long-term survival of patients with glioblastoma multiforme (GBM) J Clin Neurosci. 2013;20:670–5. doi: 10.1016/j.jocn.2012.05.040. [DOI] [PubMed] [Google Scholar]
- 67.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4:e7752. doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hambardzumyan D, Parada LF, Holland EC, Charest A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia. 2011;59:1155–68. doi: 10.1002/glia.21142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kloosterhof NK, de Rooi JJ, Kros M, Eilers PH, Smitt PA, van den Bent MJ, et al. Molecular subtypes of glioma identified by genome-wide methylation profiling. Genes Chromosomes Cancer. 2013 doi: 10.1002/gcc.22062. [DOI] [PubMed] [Google Scholar]
- 71.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weiss WA, Israel M, Cobbs C, Holland E, James CD, Louis DN, et al. Neuropathology of genetically engineered mice: consensus report and recommendations from an international forum. Oncogene. 2002;21:7453–63. doi: 10.1038/sj.onc.1205936. [DOI] [PubMed] [Google Scholar]
- 74.Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, et al. Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res. 2012;72:2473–80. doi: 10.1158/0008-5472.CAN-12-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alzahrani B, Iseli TJ, Hebbard LW. Non-viral causes of liver cancer: Does obesity led inflammation play a role? Cancer Lett. 2013 doi: 10.1016/j.canlet.2013.08.036. [DOI] [PubMed] [Google Scholar]
- 76.Belardi V, Gallagher EJ, Novosyadlyy R, Leroith D. Insulin and IGFs in Obesity-Related Breast Cancer. J Mammary Gland Biol Neoplasia. 2013 doi: 10.1007/s10911-013-9303-7. [DOI] [PubMed] [Google Scholar]
- 77.Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 (Suppl):S2–6. doi: 10.1097/MCG.0b013e3182872f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin Cancer Biol. 2013 doi: 10.1016/j.semcancer.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 79.Kim A, Pyykko I. Size matters: versatile use of PiggyBac transposons as a genetic manipulation tool. Mol Cell Biochem. 2011;354:301–9. doi: 10.1007/s11010-011-0832-3. [DOI] [PubMed] [Google Scholar]
- 80.Moriarity BS, Rahrmann EP, Keng VW, Manlove LS, Beckmann DA, Wolf NK, et al. Modular assembly of transposon integratable multigene vectors using RecWay assembly. Nucleic Acids Res. 2013;41:e92. doi: 10.1093/nar/gkt115. [DOI] [PMC free article] [PubMed] [Google Scholar]