

Abstract
We report an unusual case history of Leigh syndrome due to the m.10191T>C mutation in the complex I gene MT-ND3. This mutation has been associated with a spectrum of clinical phenotypes ranging from infant lethality to adult onset. Despite infantile onset and severe symptoms, our patient has survived to early adulthood due to a strict dietary regimen and parental care. This patient is an extreme example of the frequently prolonged course of Leigh syndrome due to this particular mutation.
Keywords: ND3, Leigh syndrome, m.10191T>C, mitochondria, complex I deficiency
Introduction
The term Leigh syndrome (OMIM 256000) was first coined to describe an infant with subacute necrotizing encephalomyopathy affecting symmetrically the basal ganglia, thalamus, and brainstem.1 Since Denis Leigh’s initial case, the syndrome has been expanded to include all progressive encephalomyopathies with radiographic evidence of bilateral focal lesions. Leigh syndrome can result from defects of pyruvate metabolism (pyruvate dehydrogenase complex deficiency) or from a multitude of mitochondrial respiratory chain defects,2 although complex I deficiency underlies approximately one third of these cases.3, 4 Within mitochondria, complex I or nicotinamide adenine dinucleotide:ubiquinone oxidoreductase (EC 1.6.5.3) contributes to the proton gradient via electron transfer, powering 40% of ATP synthesis.5 Complex I encompasses 45 subunits, seven of which are encoded by mitochondrial DNA (mtDNA; genes MT-ND1-6) and the remainder from nuclear DNA. In two studies, nearly 30% of patients with complex I deficiency were found to carry pathogenic mtDNA mutation, and Leigh syndrome was the most common clinical phenotype.6, 7 While there are many genetic causes of both Leigh syndrome and complex I deficiency, mutations in ND genes are likely culprits.
Mutations in MT-ND3 are increasingly being recognized as common causes of complex I deficiency and Leigh syndrome. A recent review summarized 16 of the published patients harboring the m.10191T>C mutation in this gene.8 Surprisingly, the clinical spectrum was quite broad, although most patients had childhood-onset Leigh syndrome. There was poor correlation between degree of heteroplasmy and symptom severity, indicating that nuclear or other mitochondrial risk alleles may contribute to the clinical phenotype.
Here we describe in detail an unusual case of Leigh syndrome due to maternal inheritance of m.10191T>C MT-ND3 mutation in a woman who has reached adulthood despite infantile onset and severe symptoms.
Patient Report
This 25-year-old woman was the first child of non-consanguineous Italian parents with no family history of motor or developmental delay. Following an uncomplicated pregnancy, delivery, and postnatal period, she had motor regression starting at 4–6 months. She stopped gaining weight because of poor feeding and had limited motility including eye movements, which were restricted to upward gaze. By 12 months, she had developed a dystonic posture on the right more than the left side and motor regression had worsened to the point that she could not control her head. At 17 months, she suffered her first focal clonic seizure and was started on phenobarbital. There was transient improvement of motor control followed by further loss of motor skills. Despite medication she continues to suffer from frequent generalized tonic-clonic seizures. Currently, at age 25, she has the physical development of a prepubescent child. Although she showed no improvement on riboflavin, thiamine, biotin, or other vitamins and cofactors, through the years she continued to take phenobarbital, coenzyme Q10, and L-carnitine. The patient’s mother and 23-year-old sister are developmentally normal with normal lactate levels.
Multiple blood lactate values were elevated (5–9 mM; normal <2.2 mM). Computed tomography of the brain in childhood demonstrated bilateral hypodensities in the lateral putamen and caudate nuclei, while magnetic resonance imaging demonstrated involvement of the basal ganglia, thalamus, midbrain, and periaqueductal grey matter. Brain magnetic resonance imaging performed recently showed severe, diffuse cortical and subcortical atrophy with ventricular dilation.
Methods
Tissues
From the proband, her sister, and her mother, we obtained venous blood and urinary sediment from the first morning void. We also obtained muscle tissue and skin fibroblasts from the proband and skin fibroblasts from her sister.
Histochemical and Biochemical Analysis
A right quadriceps muscle biopsy was analyzed with standard histological and histochemical stainings.9 Respiratory chain enzyme activities were measured spectrophotometrically as previously described in a 10% muscle extract.10
Molecular Studies
Total DNA was extracted from blood, urinary sediment, and fibroblasts using Puregene DNA Isolation Kit reagents (Qiagen Sciences, Valencia, California) according to the manufacturer’s recommended protocol. Whole genome amplification was accomplished by REPLI-g mtDNA kit (Qiagen). Whole mitochondrial sequencing was performed by long range polymerase chain reaction using three primer sets that can amplify the entire mtDNA, using 100 ng of input DNA for each reaction. The cycling conditions for all reactions were: 1) 95°C for 2 min; 2) 95°C for 15 s; 3) 68°C for 7 min; 4) repeat step 2 29 times; 5) final extension for 12 min. Direct sequencing of all mitochondrial DNA was performed in an ABI Prism 310 Genetic Analyzer using Big Dye Terminator Cycle Sequencing Reaction Kits (Perkin–Elmer Applied Biosystems, Foster City, California) using appropriate primers. The primer which was used to sequence MT-ND3 initiated at m.10141. For restriction fragment length polymorphism analysis, mtDNA was amplified by polymerase chain reaction using forward and reverse primers initiating at nucleotide positions m.10161 and m.10308. In the presence of the mutation, the mismatched forward primer creates a restriction site for the RsaI endonuclease. Digestion of the 147-bp fragment harboring the mutation yields 2 fragments of 118 bp and 29 bp, whereas the wild type fragment remains uncut. The polymerase chain reaction products were run through a 15% nondenaturing polyacrylamide gel and subjected to SYBR Gold Nucleic Acid Stain (Invitrogen, Carlsbad, California) to quantify the proportion of mutant mitochondrial DNA.
Results
Histochemical analysis of muscle at age 10 months showed fiber size variability and slight increase of perimysial connective tissue. With the modified Gomori trichrome and the nicotinamide adenine dinucleotide-tetrazolium diaphorase stains there was increased intermyofibrillar granularity indicative of mitochondrial proliferation, but no typical ragged-red fibers. Ultrastructural analysis confirmed increased number of mitochondria, some of which were larger than normal and contained irregular cristae.
Biochemical studies of muscle extracts showed a moderate isolated defect of nicotinamide adenine dinucleotide-cytochrome c reductase (complex I) (Table 1).
Table 1.
Respiratory chain biochemistry in the proband’s muscle.
| Enzyme | Proband (nmol/min/mg) | Control range |
|---|---|---|
| Cytochrome c oxidase | 52.4 | 25–75 |
| Succinate cytochrome c reductase | 10.5 | 6–17 |
| Succinate dehydrogenase | 15.1 | 8–20 |
| NADH cytochrome c reductase rotenone sensitive | 2 | 3–9 |
| Cytochrome c oxidase/succinate cytochrome c reductase | 4.99 | 2.5–5.8 |
| Citrate synthase | 146.0 | 80–180 |
Abbreviations: NADH nicotinamide adenine dinucleotide
Since no large-scale mitochondrial DNA deletions were detected (data not shown), we sequenced the entire mitochondrial genome. Of the three mutations identified, only m.10191T>C was predicted to be deleterious by the PolyPhen program (http://genetics.bwh.harvard.edu//pph2/index.shtml) and had been reported in the literature (Figure 1). This change in the coding sequence of MT-ND3 results in the substitution at position 45 of a highly conserved serine residue by proline (p.Ser45Pro). To confirm the mutation, the region was amplified by polymerase chain reaction for both direct Sanger sequencing and for restriction fragment length polymorphism analysis (Figure 2). The proband’s asymptomatic sister and mother were also analyzed, demonstrating maternal inheritance with reduced heteroplasmy. The mutation load was 70% in the proband’s fibroblasts, but only 23% in the sister’s fibroblasts and it was even lower in the sister’s blood and urine (16% and 19%, respectively (Table 2)). The mother had no detectable mutant mtDNA in blood, but 40% mutation load in the urine sediment.
Figure 1.
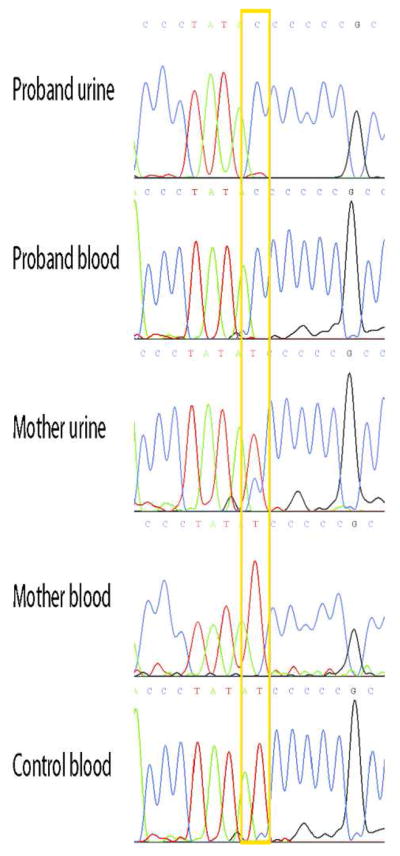
Direct sequence electropherogram of the m.10191T>C mutation. Note the high mutation load as seen by the blue C signal instead of red T signal in the urine and blood of the proband and lesser degree of heteroplasmy in her unaffected mother, compared to control blood.
Figure 2.

Polymerase chain reaction–restriction fragment length polymorphism analysis of the m.10191T>C mutation using a mismatch primer. The mutant sequence introduces an RsaI site yielding additional 118 bp fragment, whereas the wild-type sequence lacks the RsaI site and yields only a 147 bp fragment. Heteroplasmy levels are listed in Table 2.
Table 2.
Heteroplasmy of the m.10191T>C mutation in the proband and family across tissue types, shown as percent of mitochondrial DNA calculated by restriction fragment length polymorphism analysis
| Tissue | Proband | Sister | Mother |
|---|---|---|---|
| Fibroblasts | 70% | 23% | |
| Blood | 16% | 0% | |
| Urine | 19% | 40% |
Discussion
Including our proband, the m.10191T>C mutation has been reported in 22 patients to date (Table 3).6, 8, 11–26 There is a vast spectrum of clinical presentations and ages at onset. Most individuals had abnormal brain imaging consistent with Leigh syndrome, but diagnoses of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like and nonspecific encephalopathy were also given. The most common symptoms were seizures, developmental delay or regression, dystonia or spasticity, and elevated blood and cerebrospinal fluid lactate, indicating that both muscle and brain are typically affected by mitochondrial dysfunction.
Table 3.
Clinical and laboratory features of patients with the m.10191T>C mutation (modified from Nesbitt et al.8).
| Sex | M | F | M | M | U | M | F | M | M | U | F | F | U | F | M | M | M | M | F | M | M | F |
| Age at presentation | 24y | Birth | 20m | Birth | U | 5w | Birth | 21y | 4m | 13m | 5y | 16y | U | 3m | 6m | Birth | 6y | 4–15y | 11 | <1y | 1–16y | 4m |
| Age at death | Alive 42y? | ? | 25y | 4w | ? | 8m | 22m | 27y | 9y | Alive 12y? | Alive 12y? | Alive 24y | 17y | 2y | Alive 9y? | 6w | Alive 16y | ? | Alive 19? | ? | ? | Alive 25y |
| Transmission | Maternal | De novo | De novo | De novo | U | Maternal | Maternal | U | De novo | U | U | De novo | De novo | U | De novo | De novo | De novo | U | U | U | Maternal | |
| Maternal family history | N | N | N | N | U | Miscarriages | U | U | Seizures | U | N | N | U | U | Late onset diabetes | N | N | U | N | U | U | N |
| Developmental delay or regression | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| Cognitive impairment | + | + | + | + | + | + | + | + | ||||||||||||||
| Ataxia | + | + | + | + | + | |||||||||||||||||
| Myoclonus | + | + | + | + | + | + | + | + | + | + | ||||||||||||
| Seizures | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| Hypotonia | + | + | + | + | + | + | + | + | + | |||||||||||||
| Dystonia/spasticity | + | + | + | + | + | + | + | + | + | + | + | |||||||||||
| Abnormal reflexes | + | + | + | + | + | + | + | + | ||||||||||||||
| Weakness | + | + | + | + | + | + | ||||||||||||||||
| Lethargy | + | + | + | + | + | + | ||||||||||||||||
| Respiratory depression | + | + | + | + | + | + | + | + | ||||||||||||||
| Optic atrophy | + | + | + | |||||||||||||||||||
| Nystagmus | + | + | ||||||||||||||||||||
| Gastrointestinal | + | + | + | + | + | + | + | |||||||||||||||
| Failure to thrive | + | + | ||||||||||||||||||||
| Short stature/SGA | + | + | + | + | + | |||||||||||||||||
| MRI/CT finding | + | + | + | U | + | + | + | + | + | + | + | + | U | + | + | + | + | + | + | + | + | |
| CSF/plasma lactate | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| Complex I activity | 40% | U | U | 4% f 8% liver | 25% f 14% m | 38% m | U | 23% m | U | U | 26% m | U | U | 11% f | 20% m | 5% m | 75% bl | Reduced bl | U | Reduced m | Reduced m, f | 0% m |
| Heteroplasmy | 77% m 14% bl | 90% m | 80% m | >98% all tissues | 50% f 90% m | 100% m, bl 50% hair | 97% m, f | 95% m | 69% m 68% bl | U | U | 13% f 73% m 19% bl | 75% m <15% bl | U | 74% m 65% bl | 64% f 87% m, liver | U | U | U | U | U | 70% f |
| Maternal heteroplasmy | 3% bl | ND | ND | ND | U | 5% bl | 36% bl | U | ND | U | U | U | ND | U | ND | ND | ND | U | U | U | U | 0% bl 40% urine |
| Reference | 22 | 23 | 23 | 25 | 26 | 24 | 11 | 6 | 21 | 12 | 13 | 14 | 15 | 16 | 8 | 8 | 19 | 17 | 18 | 20 | 20 | Current |
Abbreviations: M male, F female, U unknown, N none, ND not done, SGA small for gestational age, bl blood, m muscle, f fibroblasts, ? no clinical updated information available
Of the 22 cases with this mutation, updated clinical information was obtained for 12 patients (personal communications from authors) and additional information was available about survival for a total of 17 patients. Nine of those 17 patients were deceased; of these, six died prior to and three died after 10 years of age. Eight patients are either currently alive or were at the time of publication, and 7 out of 8 survived beyond 10 years. One patient had adult onset of symptoms, including epilepsy, strokes, optic atrophy, and cerebellar ataxia.22 Thus only 6/17 or 35% of patients harboring the m.10191T>C mutation died in childhood while two thirds survive into adolescence and occasionally beyond. This survival rate is higher than that of other complex I deficiencies, where the average age of survival is 10 months and 75–85% of patients die prior to 10 years.27, 28
Our patient also has a long survival although she harbors a mutation that causes Leigh syndrome and is often lethal in infancy. Her parents sought multiple medical opinions for diagnosis and symptomatic help. After receiving a diagnosis of celiac disease, they maintained her on a gluten-free, minimal protein diet. As a result of their attentive care, she is still alive at 25 years of age.
The substitution of serine for proline is believed to interrupt a hydrophilic transmembrane alpha helix domain that is crucial on proteomic analysis for the interaction of the major subunits of complex I and therefore for the activity of the complex.29 Analysis of a cybrid line demonstrated that this mutation leads to a 60% reduction in complex I assembled protein, yet the functional capacity is even further reduced, suggesting that ND3 is important both for complex assembly and catalysis.25 This particular mutation may have a better survival curve than other complex I deficiency mutations because it permits residual enzyme activity.25
There are several conditions that may influence the phenotype diversity resulting from this mutation. While mutation loads as measured in muscle and blood do not correlate with symptoms, it is possible that as-yet unmeasured brain heteroplasmy might contribute to phenotype severity.8 Additionally, the m.10191T>C mutation could be modified by other mitochondrial or nuclear gene variants, as is the case for mitochondrial deafness and Leber’s hereditary optic neuritis.30, 31 Whole exome sequencing combined with population-level genetic analysis may soon lead to the discovery of such gene-gene interactions. Finally, infection, toxins, and other metabolic stressors may alter the presentation of the m.10191T>C associated disease symptoms. More detailed patient assessments would be necessary to determine such an effect. To date, none of these factors have been described for the m.10191T>C mutation.
Could the prolonged survival in our patient be due to the life-long administration of coenzyme Q10 and L-carnitine? Although this appears unlikely based on the mild and anecdotal effects of these supplements in other mitochondrial diseases, this question will be answerable only after rigorous double-blind, placebo controlled therapeutic trials of coenzyme Q10 and L-carnitine in mitochondrial diseases will be conducted.
Since some patients appeared to have either a de novo mutation or maternally inherited but vastly increased degree of heteroplasmy, screening for m.10191T>C as a prenatal genetic tool has limited value.21, 23–25 When considering diagnostic screening, however, Leigh syndrome patients with complex I deficiency are more likely to harbor deleterious mutations in mitochondrial than in nuclear genes.6, 26
As our understanding of the genetics and pathophysiology of Leigh syndrome progresses, we will hopefully be able to offer increasing symptomatic treatment and even preventive intervention such as pronuclear or spindle transfer into donor oocytes without mitochondrial mutations.32–34 This latter option, if approved for human application, will be especially useful to our proband’s asymptomatic carrier sister and other female relatives of patients who wish to avoid maternal transmission of Leigh syndrome or other mtDNA-related diseases.
Acknowledgments
Clinical and scientific work was conducted at Columbia University Medical Center in New York, City. The financial support of the Associazione Amici del Centro Dino Ferrari, University of Milan, is gratefully acknowledged.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the National Institutes of Health (HD032062) and from the Marriott Mitochondrial Disorder Clinical Research Fund (MMDCRF).
Footnotes
Author contributions
PG and HOA performed the molecular work documenting the mutation; MS contacted the family and MS and DCD provided clinical care; RJL and SD wrote the paper; SD led the research.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
The research described here was approved by the IRB Office of Columbia University Medical Center.
Contributor Information
Rebecca J. Levy, Email: rjl2127@columbia.edu.
Purificación Gutierrez Ríos, Email: purigu@hotmail.com.
Hasan O. Akman, Email: hoa2101@columbia.edu.
Monica Sciacco, Email: sciaccom@gmail.com.
Darryl C. De Vivo, Email: dcd1@columbia.edu.
Salvatore DiMauro, Email: sd12@columbia.edu.
References
- 1.Leigh D. Subacute necrotizing encephalomyelopathy in an infant. Journal of neurology, neurosurgery, and psychiatry. 1951;14:216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. The New England journal of medicine. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 3.Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Annals of neurology. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 4.Morris AA, Leonard JV, Brown GK, et al. Deficiency of respiratory chain complex I is a common cause of Leigh disease. Annals of neurology. 1996;40:25–30. doi: 10.1002/ana.410400107. [DOI] [PubMed] [Google Scholar]
- 5.Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159. [DOI] [PubMed] [Google Scholar]
- 6.Malfatti E, Bugiani M, Invernizzi F, et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain : a journal of neurology. 2007;130:1894–1904. doi: 10.1093/brain/awm114. [DOI] [PubMed] [Google Scholar]
- 7.Swalwell H, Kirby DM, Blakely EL, et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. European journal of human genetics : EJHG. 2011;19:769–775. doi: 10.1038/ejhg.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nesbitt V, Morrison PJ, Crushell E, et al. The clinical spectrum of the m.10191T>C mutation in complex I-deficient Leigh syndrome. Developmental medicine and child neurology. 2012;54:500–506. doi: 10.1111/j.1469-8749.2012.04224.x. [DOI] [PubMed] [Google Scholar]
- 9.Tanji K, Bonilla E. Optical imaging techniques (histochemical, immunohistochemical, and in situ hybridization staining methods) to visualize mitochondria. Methods in cell biology. 2001;65:311–332. doi: 10.1016/s0091-679x(01)65019-2. [DOI] [PubMed] [Google Scholar]
- 10.DiMauro S, Servidei S, Zeviani M, et al. Cytochrome c oxidase deficiency in Leigh syndrome. Annals of neurology. 1987;22:498–506. doi: 10.1002/ana.410220409. [DOI] [PubMed] [Google Scholar]
- 11.Esteitie N, Hinttala R, Wibom R, et al. Secondary metabolic effects in complex I deficiency. Annals of neurology. 2005;58:544–552. doi: 10.1002/ana.20570. [DOI] [PubMed] [Google Scholar]
- 12.Lee HF, Tsai CR, Chi CS, et al. Leigh syndrome: clinical and neuroimaging follow-up. Pediatric neurology. 2009;40:88–93. doi: 10.1016/j.pediatrneurol.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 13.Lim BC, Park JD, Hwang H, et al. Mutations in ND subunits of complex I are an important genetic cause of childhood mitochondrial encephalopathies. Journal of child neurology. 2009;24:828–832. doi: 10.1177/0883073808331085. [DOI] [PubMed] [Google Scholar]
- 14.Werner KG, Morel CF, Kirton A, et al. Rolandic mitochondrial encephalomyelopathy and MT-ND3 mutations. Pediatric neurology. 2009;41:27–33. doi: 10.1016/j.pediatrneurol.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Chi CS, Lee HF, Tsai CR, et al. Clinical manifestations in children with mitochondrial diseases. Pediatric neurology. 2010;43:183–189. doi: 10.1016/j.pediatrneurol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Goto T, Mori Masato, Yamagata Takanori, Murayama Kei, Ohtake Akira, Momoi MY. A case of Leigh syndrome with Complex I deficiency presenting diffuse necrotizing change in the white matter. XI International Child Neurology Congress; 2010; Cairo, Egypt. 2010. [Google Scholar]
- 17.Ma YY, Wu TF, Liu YP, et al. Mitochondrial respiratory chain enzyme assay and DNA analysis in peripheral blood leukocytes for the etiological study of Chinese children with Leigh syndrome due to complex I deficiency. Mitochondrial DNA. 2013;24:67–73. doi: 10.3109/19401736.2012.717932. [DOI] [PubMed] [Google Scholar]
- 18.Zhao D, Hong D, Zhang W, et al. Mutations in mitochondrially encoded complex I enzyme as the second common cause in a cohort of Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. Journal of human genetics. 2011;56:759–764. doi: 10.1038/jhg.2011.96. [DOI] [PubMed] [Google Scholar]
- 19.Liu YP, Ma YY, Wu TF, et al. Mitochondrial respiratory chain complex I deficiency due to 10191T>C mutation in ND3 gene. Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics. 2012;14:561–566. [PubMed] [Google Scholar]
- 20.Bannwarth S, Procaccio V, Lebre AS, et al. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet. 2013 doi: 10.1136/jmedgenet-2013-101604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bannwarth S, Procaccio V, Rouzier C, et al. Rapid identification of mitochondrial DNA (mtDNA) mutations in neuromuscular disorders by using surveyor strategy. Mitochondrion. 2008;8:136–145. doi: 10.1016/j.mito.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 22.Taylor RW, Singh-Kler R, Hayes CM, et al. Progressive mitochondrial disease resulting from a novel missense mutation in the mitochondrial DNA ND3 gene. Annals of neurology. 2001;50:104–107. doi: 10.1002/ana.1084. [DOI] [PubMed] [Google Scholar]
- 23.Lebon S, Chol M, Benit P, et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J Med Genet. 2003;40:896–899. doi: 10.1136/jmg.40.12.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leshinsky-Silver E, Lev D, Tzofi-Berman Z, et al. Fulminant neurological deterioration in a neonate with Leigh syndrome due to a maternally transmitted missense mutation in the mitochondrial ND3 gene. Biochemical and biophysical research communications. 2005;334:582–587. doi: 10.1016/j.bbrc.2005.06.134. [DOI] [PubMed] [Google Scholar]
- 25.McFarland R, Kirby DM, Fowler KJ, et al. De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency. Annals of neurology. 2004;55:58–64. doi: 10.1002/ana.10787. [DOI] [PubMed] [Google Scholar]
- 26.Bugiani M, Invernizzi F, Alberio S, et al. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Rahman S, Thorburn DR. 189th ENMC international workshop complex I deficiency: diagnosis and treatment 20–22 April 2012, Naarden, The Netherlands. Neuromuscular disorders : NMD. 2013;23:506–515. doi: 10.1016/j.nmd.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Koene S, Rodenburg RJ, van der Knaap MS, et al. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. Journal of inherited metabolic disease. 2012;35:737–747. doi: 10.1007/s10545-012-9492-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galkin A, Meyer B, Wittig I, et al. Identification of the mitochondrial ND3 subunit as a structural component involved in the active/deactive enzyme transition of respiratory complex I. J Biol Chem. 2008;283:20907–20913. doi: 10.1074/jbc.M803190200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo LF, Hou CC, Yang WX. Nuclear factors: roles related to mitochondrial deafness. Gene. 2013;520:79–89. doi: 10.1016/j.gene.2013.03.041. [DOI] [PubMed] [Google Scholar]
- 31.Carelli V, Giordano C, d’Amati G. Pathogenic expression of homoplasmic mtDNA mutations needs a complex nuclear-mitochondrial interaction. Trends in genetics : TIG. 2003;19:257–262. doi: 10.1016/S0168-9525(03)00072-6. [DOI] [PubMed] [Google Scholar]
- 32.Tachibana M, Amato P, Sparman M, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:632–637. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Craven L, Elson JL, Irving L, et al. Mitochondrial DNA diseas: new options for prevention. Hum Mol Genet. 2011;20:R168–R174. doi: 10.1093/hmg/ddr373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paull D, Emmanuele V, Weiss KA, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–637. doi: 10.1038/nature11800. [DOI] [PMC free article] [PubMed] [Google Scholar]