

Abstract
α-Conotoxin MII (α-CTxMII) is a 16 amino acid peptide with the sequence GCCSNPVCHLEHSNLC containing disulfide bonds between Cys2-Cys8 and Cys3-Cys16. This peptide, isolated from the venom of the marine cone snail Conus magus, is a potent and selective antagonist of neuronal nicotinic acetylcholine receptors (nAChRs). To evaluate the impact of channel-ligand interactions on ligand binding affinity, homology models of the heteropentameric α3β2-nAChR were constructed. The models were created in MODELLER using crystal structures of the Torpedo marmorata-nAChR (Tm-nAChR, PDB ID: 2BG9) and the Aplysia californica-acetylcholine binding protein (Ac-AChBP, PDB ID: 2BR8) as templates for the α3 and β2 subunit isoforms derived from rat neuronal nAChR primary amino acid sequences. Molecular docking calculations were performed with AutoDock to evaluate interactions of the heteropentameric nAChR homology models with the ligands acetylcholine (ACh) and α-CTxMII. The nAChR homology models described here bind ACh with commensurate binding energies to previously reported systems, and identify critical interactions that facilitate both ACh and α-CTxMII ligand binding. The docking calculations revealed an increased binding affinity of the α3β2-nAChR for α-CTxMII with ACh bound to the receptor, which was confirmed through two-electrode voltage clamp experiments on oocytes from Xenopus laevis. These findings provide insights into the inhibition and mechanism of electrostatically driven antagonist properties of the α-CTxMIIs on nAChRs.
Keywords: AutoDock, Conotoxin, Homology Modeling, Nicotinic Acetylcholine Receptor, Two-Electrode Voltage Clamp
Introduction
Nicotinic acetylcholine receptors (nAChRs)[1] are heteropentameric members of the ligand-gated ion channel superfamily. To date, ten α(α1-10) and four β (β1-4) nAChR subunits have been identified with a number of functional combinations that play significant regulatory roles throughout the brain and in the autonomic nervous system.[2] Specific nAChR subtypes have been implicated in a number of neurological disorders including Parkinson’s Disease (PD) and Alzheimer’s disease (AD), drug addiction, and pain.[3] Therefore, a more complete understanding of these nAChRs, and the actions of antagonists on these receptors is of considerable interest as they represent a potential target for intervention in these disorders.[4] For example, studies indicate that nAChRs containing α6 subunits, which are found predominantly in the brain, appear to play a major role in striatal dopamine (DA) release, which is deficient in PD.[5] Similarity in sequence and structure between α3 and α6 subunits has prompted the investigation of generic ligand binding paradigms in α3β2-nAChR systems. A reason for the widespread appearance of the α3β2-nAChR studies in literature is due to an established system of study in vitro.[6] Heteropentameric α3β2-nAChR can be expressed in Xenopus oocytes in a 2:3 ratio of α:β subunits,[2a–e, 7] to study the actions of agonists and antagonists.
Homology models offer an in silico method to investigate nAChRs, with the goal of accurately predicting ligand binding determinants. To that end considerable effort has been expended to generate structural models for nAChR isoforms using X-ray data obtained from the acetylcholine binding protein (AChBP) of various invertebrates, including Lymnaea stagnalis (Ls), Bulinus truncates (Bt), and Aplysia californica (Ac). The structure of the full-length ACh-binding protein Ls-AChBP first appeared in the literature in 2001 with a structure resolved to 2.7 Å by X-ray crystallography.[8] The Ls-AChBP structure gained considerable recognition as a model for more complex membrane bound nAChRs for several reasons. First, it has a high degree of similarity in the ligand binding domain with mammalian nAChRs. Second, ligands such as nicotine, epibatine, (+)-tubocurarine, and α-bungarotoxin that act on nAChRs are also potent on Ls-AChBP. Third, each of these ligands target highly conserved amino acid binding sites identified through mutagenesis for nAChRs.[9] More recently, the X-ray determined structures of the Bt-AChBP and Ac-AChBP have become available and together present dynamic templates to investigate the influence of agonist and antagonist docking to the ligand binding domain of the receptor.[10] Importantly, several native α-conotoxins (α-CTxs) and their analogs have been elucidated primarily by NMR techniques including α-CTxImI (PDB ID: 1IMI, 1CNL, 1E74),[11] α-CTxOmIA (PDB ID: 2GCZ),[12] and α-CTxPnIA (PDB ID: 1PEN).[13] Interest in these ligands as selective antagonists of nAChRs has led to X-ray structures for the conotoxins bound to Ac-AChBPs that include α-CTxIMI (PDB ID: 2C9T, 2Byp),[10a, 10c] α-CTxPnIA (PDB ID: 2BR8),[10b] and α-CTxTxIA (PDB ID: 2UZ6).[10d] These protein database files provide a molecular snapshot of potential interactions critical for antagonist binding.[10a, 10c, 10d, 14]
Conotoxins are small peptides, isolated from cone snail venom, that provide a possible approach for the design of molecular probes capable of distinguishing between nAChR isoforms, Figure 1.[15] α-CTxs are disulfide rich polypeptides that typically act as competitive inhibitors of nAChRs[16] with some exceptions such as α-CTxImII[17] and ψ-CTxPIIIE[18]. α-CTxMII, isolated from Conus magus, is comprised of 16 amino acids (GCCSNPVCHLEHSNLC) with disulfide bridges between Cys2-Cys8 and Cys3-Cys16. This peptide inhibits the α3β2-nAChR isoform with an IC50 of 0.5 nM.[6] In 2002, Schapira et al. created a homology model of the human neuronal nAChR α3β2 subunit combination using the Ls-AChBP structure as the template.[9e] The α3β2-nAChR model provided valuable information with regard to the competitive binding nature of α-CTxMII versus the endogenous ligand, acetylcholine (ACh), Figure 1. Homology models of the rat α3β2-nAChR, which allows for a direct comparison to the Xenopus oocyte experimental model, have been created based on the α1-nAChR and AChBP from Lymnaea stagnalis.[7b] This homology model has enabled mechanistic insights into the structure, function, and receptor interactions of the atypical α-CTxLtIA. However, as is the case with the majority of nAChR homology models, the molecular level studies do not include the full pentameric channel, focus on only the α-β gap, and do not take into account the interactions between ACh and the antagonist ligand. Here we present new in silico findings that explore ligand binding paradigms for ACh and α-CTxMII ligands to pentameric α3β2-nAChR homology models constructed from Ac-AChBP and Torpedo Marmorata nicotinic acetylcholine receptor (Tm-nAChR) templates.
Figure 1.
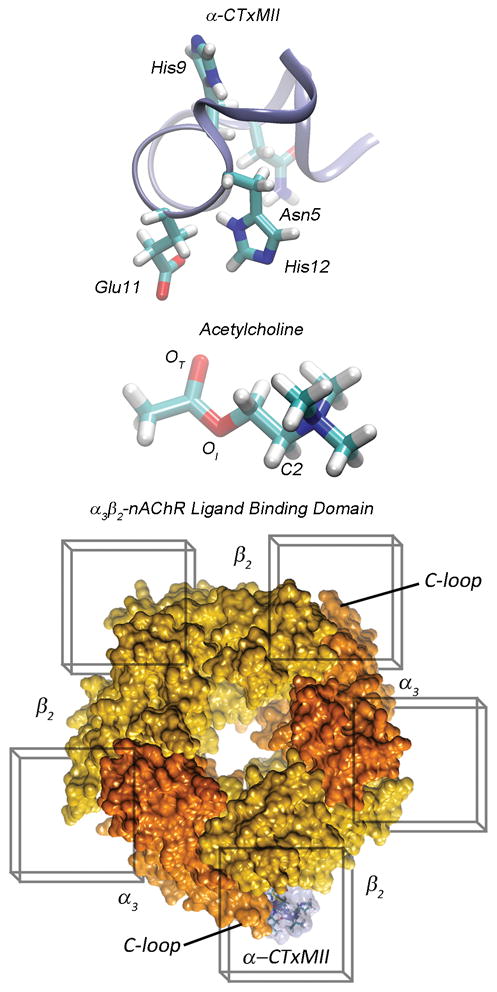
(Top) Ribbon representation of α-CTxMII backbone with licorice representations of critical amino acids at positions 5, 9, 11, and 12. (Middle) Licorice representation of acetylcholine, with the carbonyl oxygen labeled as OT, the ester linkage oxygen as OI and the carbon bound to the trimethyl ammonium group as C2. (Bottom) Top view of the α3β2-nAChR ligand binding domain in surface representation with docking grid boxes around each of the 5 C-loop binding regions, the channel subunits are color coded with orange representing α3 and yellow representing β2 subunits. The α-CTxMII is represented bound to a C-loop region between α3 and β2 subunits (transparent blue surface with amino acids in licorice).
Evaluation of the available experimental structural data and the various created homology models has identified a structurally conserved 10 Å wide hydrophobic binding pocket for ACh at the interface between α3 and β2 subunits, which is composed of several loops important for the binding process (Figure 1). The interface is characterized by loops A–C on the primary subunit (+) and loops D–F on the complementary subunit (−) (Figure 2).[19] The C-loop is of particular importance as it has a large range of motion and the closure of this loop has been associated with successful agonist binding and channel gating.[3b] α-CTxMII is ~15 Å long with peripheral bulky side chains that prevent it from binding in the same pocket and in the same manner as ACh.
Figure 2.

(Left) Side view of the homology model for the α3β2-nAChR (ribbon) depicting the overall structural components of the transmembrane ligand-gated ion channel. (Right) Side view of the subunit dimer complex composed of a primary (+) α and a complementary (−) β subunit designating specific loop regions including the C-loop (i.e., where ACh binds) and the Cys-loop (i.e., characteristic of this family of channels).
The Tm-nAChR structure is the only available experimentally determined structure for heteropentameric nAChRs and has been successfully used as a template for modeling heteropentameric nAChRs.[20] The Ac-AChBP template contains a homopentameric binding protein that has a large toxin (i.e. α-CTxPnIA) bound to the subunits.[10b] The presence of the large toxin presents Ac-AChBP as an ideal template with a more open C-loop in the binding protein. In this study, we have created two homology models using the Ac-AChBP and Tm-nAChR as templates to obtain structures of the rat neuronal α3β2 heteropentamer that offer different states of the C-loop[21] (open vs. closed C-loop, respectively). It is noted that throughout this work we are characterizing the state of the C-loop as open or closed and not the state of the channel pore. The homology models were then utilized in docking calculations individually with ACh or α-CTxMII, and in sequence with ACh binding followed by α-CTxMII exposure to investigate the fundamental interactions driving binding affinity and specificity at all subunit interfaces. Wash-out two-electrode voltage clamp experiments were conducted on oocytes from Xenopus laevis that express α3β2-nAChRs to evaluate binding affinities of ACh, α-CTxMII, and the combination of ACh and α-CTxMII predicted by our computational results. Together, these studies provide important molecular-level details into ligand-receptor binding and the impact of agonist binding on subsequent antagonist binding.
Results and Discussion
Homology Model Results
The creation of homology models for complex systems like the heteropentameric α3β2-nAChR relies on homologous primary sequences that have been structurally characterized. Presented here are the results of two nAChR homology models created from the Tm-nAChR (closed C-loop, PDB ID: 2BG9) and Ac-AChBP (open C-loop, PDB ID: 2BR8) templates and the rat α3β2-nAChR primary amino acid sequences. The amino-acid sequence alignments of the α3 and β2 subunits with their corresponding Tm-nAChR template subunits are shown in Figure 3, in which the conserved, semi-conserved residues and regions with conserved secondary structures are highlighted. The α3 and β2 subunits of the nAChR consist of 2 main structural entities - the trans-membrane region and the ligand binding region. The trans-membrane region is characterized by four α-helices connected together by short loops (TM1 to TM4 in Figure 3). The ligand binding region is characterized by one α-helix (H1 in Figure 3) at the top and a number of β-sheets (β1-β10 in Figure 3) that contribute to the binding domain. It should be noted that, apart from the regions corresponding to the above mentioned structural motifs (TM1-TM4, H1 and β1-β10), even the looped regions have highly conserved sequences, thus indicating significant homology and suggesting possible biological significance.
Figure 3.

Pairwise sequence alignments for the α3 (top) and β2 (bottom) amino acids of the target sequence (rat nAChR) with the template sequence (2BG9 – Tm-nAChR). An asterisk (*) is used to represent conserved residues, colon (:) for similar amino acids, and a period (.) for dissimilar residues. The boxed segments indicate regions of the protein sequences that share the same secondary structure. The alignment was created based on the Needleman-Wunsch alignment algorithm using the Matchmaker module in the UCSF Chimera package.
The nAChR assemblies and the combinations of subunits present in the homology model and the templates have been depicted in Figure 4. Each nAChR subunit is denoted as A, B, C, D or E. For the Tm-nAChR X-ray structure (PDB ID: 2BG9), subunit combinations consist of two α (purple), one β (green), one δ (blue), and one γ (red) polypeptide (Figure. 4; far left) in the following order (A=α) - (B=β) - (C=δ) - (D=α) - (E=γ). The corresponding order of arrangement of subunits in α3β2-nAChR homology model is (A=α3) - (B=β2) - (C=β2) - (D=α3) - (E=β2). The α3 and β2 subunits are colored gray and turquoise respectively (Figure 4; middle three images). The structure on the far right shows the overlap between the homology model and the template structure.[20c] In the case of the Ac-AChBP (2BR8), the template structure is a homopentamer where all subunits have the same monomeric amino acid sequence (magenta) (Figure 4; left). The sequence alignment with the conserved, semi-conserved residues and regions with conserved secondary structures for the Ac-AChBP are shown in Supplemental Figure S1.
Figure 4.

Homology modeling results for the two α3β2-nAChR systems. (Top) Structural and sequence alignment results for template Tm-nAChR. (Bottom, left to right) X-ray structure of Ac-AChBP, model depiction of the subunits, resulting homology model, and the overall alignment of the homology model to the X-ray structure template.
The root mean square deviations (RMSDs) between these two homology models and the templates (2BG9 and 2BR8) after superimposition are also listed in Figure 4. RMSD values of 0.406 Å and 0.612 Å indicate good structural conservation between the templates and the Tm-nAChR and Ac-AChBP homology models, respectively. As an additional validation for our Tm-nAChR based α3β2-nAChR model, we obtained the PDB file for the homology model of the heterodimeric α3β2-nAChR from Dutertre’s lab,[21a] and calculated a RMSD between their structure and ours in Chimera, with an RMSD = 0.577 Å.[22] The low RMSD value (≤ 2.0 Å) indicated that our model is consistent with previously published results.[21a] The MolProbity analysis of the homology model reveals a MolProbity score of around 1.5 indicating a high quality structure.[23]
Open versus Closed C-Loop Regions
An outstanding issue concerning the docking of ligands to the α3β2-nAChR is whether the C-loop regions should be classified as open or closed. To address this issue, two homology models were constructed, one to represent the C-loop closed state (based on the Tm-nAChR) and one to represent the C-loop open state (based on the Ac-AChBP). The open and closed C-loop homology models have distinctly different geometric parameters with respect to residues at the apex of the C-loop (+) and the β-sheet on the adjacent subunit (−), where the (+) indicates the primary chain on which the currently observed C-loop resides and (−) represents the complementary chain overlapped by the currently observed C-loop. For example, for the α3-β2 gap, the α3 will be the (+) and the β2 will be the (−); for the β2-α3 gap the β2 will be the (+) and the α3 will be the (−).[24] To quantify measurements in the open versus closed C-loop state, the distance between the loop region and the (−) β-sheet wall was calculated. The distance calculation consisted of selecting an apex atom (sulfur in the Cys192 side chain on the α3 C-loop or the central carbon in the D402 side chain on the β2 C-loop) and growing its van der Waal (VDW) radius until the outer most surface engulfed residues on the (−) β-sheet wall. The resulting value indicates the approximate accessible distance between the loop region and the (−) β-sheet wall (see Table 1). Upon comparison between the open and closed C-loop homology models, the resulting distances indicate a significant difference in the available space between the C-loop and the (−) β-sheet wall. A span of 10 Å can be measured between the R-groups for the α3-β2 gap in the open C-loop model, which is 2.6 Å larger than that observed in the closed C-loop model. A slightly larger C-loop opening distance of 14 Å was measured for the β2-α3 open C-loop homology model, compared to 10.1 Å in the closed C-loop model. Unlike the other two gap regions, the β2-β2 gaps have the opposite trend; the gap distances are very similar (9.0 Å (open C-loop model) and 9.8 Å (closed C-loop model).
Table 1.
Distance between C-loops (+) and the adjacent β-sheets (−) with the standard deviations in parenthesis.
| Subunit Gap | Model | Distance (Å) |
|---|---|---|
| (+)α3 (−)β2 | closed | 7.4 ± 0.3 |
| open | 10.0 ± 1.0 | |
| (+)β2 (−)α3 | closed | 10.1 ± 0.5 |
| open | 14.0 ± 1.0 | |
| (+)β2 (−)β2 | closed | 9.8 |
| open | 9.0 |
ACh Binding Pocket
Previous studies indicate that the pentameric nAChR ion channel’s binding specificity for ACh is primarily due to a cluster of residues located in the aromatic binding pocket behind the C-loop, which stabilize ACh binding through cation-π, ionic, reinforced ionic (Pharmacophore description of charge-charge interactions that also have hydrogen bonding characteristics) and hydrophobic interactions.[24–25] In 2004, Dutertre et al.[24] identified the conserved aromatic residues of the binding pocket for the α7 homopentameric nAChR as (+) W143, (+) Y89, (+) Y185, (+) Y192, (−) W53, and (−) Y164. For subunit interfaces in heteropentameric nAChRs, the residues critical for ACh binding have been previously identified to be (+) W149, (+) Y93, (+) Y190, (+) Y198, (−) E189, (−) T119, (−) W57[26] and are described to be located on loops A–C on the primary subunit (+) and on loops D–F on the complimentary subunit (−). Table 2 compares these critical residues in the α3 and β2 subunits with the reference subunits of Tm-nAChR. The majority of these critical residues were conserved in our homology model. Tyrosine, tryptophan and glutamic acid residues were conserved in both α3 and β2 subunits of the homology model, indicating a binding pocket topology that is similar in α3-β2, β2-α3, β2-β2 subunit interfaces. As expected, the cysteine residues of the C-loop region were not conserved in the β subunits. Although the threonine on the E-loop was not conserved on the β subunit, this did not impact the docking results since the observed ACh binding poses revealed the closest interactions with residues on loops A, B and F (see Docking Calculations section). While it is widely accepted that ACh binds mainly to α subunit interfaces, it has been suggested that ACh may also bind at non-α subunit interfaces.[27]
Table 2.
Residues critical to ACh binding and their corresponding homologous residues in the α3 and β2 subunits of the heteropentameric nAChR homology model.
| Primary Subunit | Reference Structure α Subunit of Tm-nAChR | α3 Subunit | β2 Subunit |
|---|---|---|---|
| A-loop | Y93 | Y93 | Y95 |
| B-loop | W149 | W149 | W151 |
| C-loop | Y190-C192-C193-Y198 | Y190-C192-C193-Y197 | P191-D192-D193-Y196 |
| Complimentary Subunit | Reference structure δ Subunit of Tm-nAChR | α3 Subunit | β2 Subunit |
|---|---|---|---|
| D-loop | E189 | E175 | E177 |
| E-loop | T119 | T117 | F119 |
| F-loop | W57 | W55 | W57 |
Our models identified important amino acid interactions that stabilize ACh binding in α3-β2 gap, β2-α3 gap and β2-β2 gap. In addition to the hydrophobic interactions, we also found that charged residues appear to have stabilizing electrostatic interactions with ACh and/or α-CTxMII ligands. These findings, described below, are consistent with previously reported ACh binding motifs.[9e, 28] It is noted that although ACh binding at non-α subunit interfaces was observed in this study, this does not necessarily indicate that the binding results in channel gating. The role of ACh in the gating mechanism of nAChRs is beyond the scope of the docking studies presented in this paper and is currently being investigated in a separate study. For a discussion of the “conformational wave change” hypothesis of ligand binding in Cys-loop receptors, the reader is encouraged to read the review by Cederholm et al. (2009).[29]
α3-β2 gap
In our homology models, the α3-β2 gap contains a hydrophobic binding pocket, deep in the C-loop region, which consists of conserved aromatic residues (+) W149, (+) Y93, (+) Y190, (+) Y197, (−) W57, (−) F172 (Figure 5). In the closed C-loop homology model, the aromatic side chains of (−) F119 and (−) Y114 are oriented toward the C-loop binding pocket, and thus may contribute to favorable cation-π interactions when the ACh ligand is bound (Figure 5).
Figure 5.

Acetylcholine binding in the aromatic pocket for the open homology model depicting the α3-β2 (Top), β2-α3 (Middle) and β2-β2 (Bottom) gaps. The figure has been color-coded with the α3 subunit structures and neutral residues (gold), β2 subunit structures and neutral residues (orange), negatively charged residues (red) and positively charged residues (blue). The binding pose for α3-β2 represents the traditional binding pose for ACh, while the β2-α3 and β2-β2 gaps depict alternate ACh binding poses that were within kBT of the more favorable traditional binding pose.
In the open homology model, the amino acid side chains in the binding pockets show less steric hindrance due to the increased distance between the C-loop and the (−) β-sheet wall. This reduction in steric hindrance between side chains results in less conformational deviation from gap to gap (i.e. when compared to β2-α3 and β2-β2 gaps). The observation of conformational flexibility for the hydrophobic residues in the open versus closed C-loop models indicates that dynamic conformational re-orientations of the residues may take place to accommodate a ligand as well as to respond to the movement of the C-loop region.
The above discussion focused exclusively on the hydrophobic region of the ACh binding pocket and the resulting aromatic interactions (e.g. cation-π). However, there also exist a number of charged residues in the C-loop binding region, enabling the possibility for ionic and reinforced ionic interactions. The critical charged residues that may contribute to the overall binding energy and specificity are (+) E195, (+) E194, and (+) K189 on the C-loop, (+) K145, (+) K153, (+) D152, and (+) D199 which reside deep in the aromatic binding pocket, (−) D170 and (−) D171 which are directly below the binding pocket (towards the trans-membrane portion of the channel), and (−) E61, (−) D165, and (−) K163, which are on the outer edge of the ACh binding region. The residues on the outer edge of the binding pocket are believed to be in proximity and contribute to charge-charge interactions with ligand in or near the ACh binding pocket (Figure 5).
β2-α3 gap
In the β2-α3 gaps, the pattern for the conserved ACh binding pocket corresponds to (+) W151, (+) Y95, (+) Y196, (−) W55, and (−) W171 (Figure 5). It is observed that the β2-α3 gaps have three major differences as compared to the α3-β2 gaps. First, there is no aromatic (+) Y190 equivalent in the β2-α3 gap. Second, the aromatic (−) W171 replaces (−) F172. Third, in the β2-α3 gap, the side chains of (+) Y196 and (−) W170 occupy different orientations depending on the state of the loop region (open versus closed C-loop), again highlighting conformational flexibility in the binding pocket. While the hydrophobic pocket in the β2-α3 gap is similar to that found in the α3-β2 gap, the charged residues there were found to be quite different. The β2-α3 gap is characterized by the absence of (+) K189 and the presence of (+) D193 and (+) D192 at the end of the β2 C-loop, instead of the two cysteine residues present on the α3 C-loop in the α3-β2 gap. Similar to the α3-β2 gap, the β2-α3 gap has residues (+) D154, (+) R149, (+) D198 and (+) K147, which are near the ACh aromatic binding pocket. Additionally, the charged residues (−) D169, (−) E172, and (−) K168 are below the ACh binding pocket (see Figure 5).
β2-β2 gap
In the β2-β2 gap, the pattern for the conserved ACh binding pocket residues corresponds to (+) W151, (+) Y95, (+) Y196, (−) W57, (−) F172, and (−) F119 (see Figure 5). As observed for the β2-α3 gap, there is no aromatic (+) Y190 equivalent residue while there is orientation flexibility seen for the side chains (+) W151, (+) Y196, and (−) F172 between the open and closed C-loop conformations.
In the β2-β2 gap the charged interactions are a combination of the two previously described gaps. As was the case with β2-α3 gaps, there is no (+) K189, while (+) D192 and (+) D193 reside at the apex of the C-loop; (+) K147, (+) R149, (+) D154 and (+) D198 are deep in the aromatic binding pocket; (−) D170 and (−) D171 are directly below the binding pocket, while (−) E61, (−) E63, (−) D115, (−) K163 and (−) D165 are on the outer edge of the ACh binding pocket. As was the case for the α3-β2 and β2-α3 gaps, these residues are near enough to the binding pocket that they may interact in a favorable ionic (charge-charge) fashion, with ligands such as ACh and α-CTxMII.
Docking Calculations
ACh binding to nAChR
The docking calculations between ACh and the open and closed C-loop nAChR models did not reveal a statistical preference for ACh binding to either α3-β2 or the β2-α3 subunit pairs of the pentameric nAChR (Table 3). The exception is the β2-β2 subunit pair, which unlike the other gaps has a very similar gap opening distance between the open and closed C-loop homology models, < 1 Å (Table 1); the slightly larger gap corresponded to the closed C-loop homology model. The open model (closed C-loop region) has a reduced binding energy for ACh as compared to the closed model (open C-loop region) due to the reduced gap opening distance for the open structure, and the presence of increased unfavorable steric interactions (see Table 3). It is noted that due to the presence of only one β2-β2 gap there is no measure of statistical significance on the energies associated with this particular gap. Thus, while an energetic difference is observed, in the absence of variance it is not possible to comment on the statistical significance of the difference, and therefore it remains uncertain whether or not a significant preference of ACh for any particular gap exists. It is evident that ACh has favorable interactions with both active subunit pairs (α3-β2), while the state of the C-loop (open versus closed C-loop) has little impact on the overall favorable binding energy. This result is not surprising given the small size of ACh (~6 Å long and ~3 Å wide) relative to the C-loop to (−) β-sheet distance (~10 to 14 Å) (Table 1). The relatively narrow distribution of binding energies between ACh and α3β2-nAChR clearly indicate that there exists no energetically favored predisposition of the ACh for a particular subunit pair, with the possible exception of the β2-β2 gap as previously discussed.
Table 3.
The lowest binding energies for the independent and competitive docking results of ACh and α-CTxMII to open and closed C-loop homology models for α3β2-nAChR.
| nAChR Subunit | Model | ΔEbinding (Kcal/mole)
|
||
|---|---|---|---|---|
| ACh | α-CTxMII | ACh + α-CTxMII | ||
| α3 to β2 | closed | −4.9 ± 0.2 | −2.8 ± 0.1 | −3.0 ± 0.1 |
| open | −5.0 ± 0.1 | −2.9 ± 0.3 | −4.9 ± 0.7 | |
| β2 to α3 | closed | −5.2 ± 0.1 | −2.6 ± 0.2 | −2.6 ± 0.2 |
| open | −5.1 ± 0.2 | −4.5 ± 0.5 | −5.8 ± 0.1 | |
| β2 to β2 | closed | −5.4 | −5.6 | −5.2 |
| open | −6.1 | −3.0 | −6.1 | |
Evaluation of the docking interactions reveals that the most favorable docking pose for ACh to the closed C-loop model corresponds to ACh bound near the aromatic pocket (see Table 3 and Figure 5). However, there are also secondary binding positions that are less favorable, located outside this pocket. The secondary site does possess favorable ionic interactions between ACh’s quaternary ammonium ion and several residues on the (−) β-sheet wall. In contrast to the closed C-loop model, the open model consistently docks ACh in the aromatic pocket deep in the binding loop region, in agreement with the binding poses reported in literature.[9e, 28] This result further supports the conclusion that our open C-loop model has a more accessible binding pocket than the closed homology model. A common binding motif observed in the docking calculations was the cation-π pharmacophore interactions deep within the binding pocket between the quaternary ammonium group of ACh and the aromatic residues.
α-CTxMII binding to nAChR
The docking calculations reveal that α-CTxMII binding with the open and closed C-loop nAChR models have favorable binding energies for all gaps (Table 3), with the open β2-α3 and open β2-β2 having the most favorable interaction energies. The state of the α3 C-loop (i.e. the α3-β2 gap with open versus closed C-loop) has little impact on the binding energy, while the state of the β2 C-loop (β2-α3, β2-β2; open versus closed C-loop) significantly influences the binding energy of α-CTxMII. Specifically, α-CTxMII prefers the open C-loop state of the β2-α3 subunit pair compared to the closed conformation, and much like the ACh binding results, has a strong preference for the β2-β2 subunit pair when the loop is open. A comparison of the docking energies and the distance between the C-loops and the wall of the (−) β-sheet indicates very similar binding energies for α3-β2 and β2-α3 when the opening is <10.1 Å, but an increased favorable binding energy when the opening is larger than 10.1 Å. This difference may indicate a criterion for delineating between an open versus closed C-loop conformation.
Evaluation of the docking interactions reveals a diverse hydrogen bonding network that is not conserved between the open or closed C-loop models or between subunit gaps as shown in Supplemental Table S1. When evaluating all docking clusters (not just the lowest binding energy configuration) several binding poses for α-CTxMII are found with the ligand either above or below the C-loop (data not shown). One consistent observation is that α-CTxMII does not completely bind under the C-loop for the closed C-loop model, most likely due to steric hindrance. For example, the largest loop opening is ~10 Å, whereas the length, width, and height of α-CTxMII are estimated to be 15 × 12 × 11 Å respectively.
However, a consistent pattern found in all α-CTxMII and ACh binding poses was the prevalence of charge-charge interactions. An important electrostatic interaction was observed between the N-terminus of α-CTxMII and several negatively charged amino acids in the nAChR. For example, >60% of docking clusters have the toxin N-terminus interacting with negatively charged residues. This finding is further supported by the observation that when the N-terminus of the toxin binds, it often forms a reinforced ionic bond. For the best binding poses (i.e. lowest binding energy conformation) in the α3-β2 gaps, the N-terminus is found to reside between either (−) D171 and (−) D170 or (+) E194 and (+) E195 in the open C-loop structure and between (−) E61 and (−) E63 in the closed structure. For the β2-α3 gaps, (+) D192 and (+) D193 or (+) D198 are found to be the major contributors for the open C-loop structure while (−) E34 or (+) E189 are found in the closed orientation. In the β2-β2 gaps, the N-terminus of the toxin interacts with (−) D115 in the open C-loop structure or (+) D192 and (+) D193 in the closed structure. In describing any of the α-CTxMII binding poses, charge-charge interactions, both ionic and reinforced ionic, appear to be the predominant factor impacting ligand binding.
In addition to the N-terminal charge-charge interactions there are also charge interactions seen that involve E11 on α-CTxMII. In the α3-β2 gaps, several binding poses reveal interactions between E11 and (−) D115, (+) K153 and (−) K163 while in the β2-α3 and β2-β2 gaps, (−) K163 and (−) K168 are involved in charge-charge interactions with that toxin residue. α-CTxMII’s E11 and complementary receptor atoms are at a greater distance than those seen for the N-terminal cation and therefore are believed to be less of a contributing factor to binding. It is also observed that the overall dipole of α-CTxMII aligns with the electrostatic field lines emanating out of the C-loop binding region (data not shown). This alignment of the dipole to the receptor macromolecule electrostatic field lines coincides with the ability of α-CTxMII’s E11 to promote favorable interactions with positively charged residues such as (+) K153 and also (−) K163 (α3-β2 gap). In addition to the charge interactions, the α-CTxMII side chains of H9 and H12 form favorable π-π interactions with the nAChR, in addition to several non-conserved hydrogen bonds (Supplemental Table S1). In the case of H9, favorable π-π interactions are seen with (−) F119 in the α3-β2 gap and (−) W55 in the β2-α3 gap, whereas H12 displayed no such trends.
α-CTxMII binding to ACh bound nAChR
A very interesting result is observed from the docking calculations between α-CTxMII and the nAChR channel that already has ACh bound. The presence of ACh in the C-loop favorably impacts the binding energy of α-CTxMII and enhances the preference for binding of α-CTxMII to the open C-loop state of the nAChRs. In the presence of bound ACh, the overall dipole of α-CTxMII aligns with the electrostatic field lines emanating from the C-loop binding region. For example, in the case of the α3-β2 gap, alignment of the dipole to the receptor macromolecule electrostatic field lines coincide with a reorientation of α-CTxMII’s E11 from interacting with (+) K153 on the C-loop to a position between two charged residues namely, (+) K153 on the C-loop and (−) K163 on the β-sheet wall. Coalescence of α-CTxMII binding poses post-ACh binding suggests there may be a significant overall electrostatic interaction leading to agonist amplification of antagonist binding, i.e. enhancement of α-CTxMII binding due to the presence of bound ACh. It is clear that in any gap, the binding of ACh is possible and severely changes the local electrostatic environment.
Review of our docking conformations again displays the affinity of the α-CTxMII’s N-terminal cation for strong electronegative patches on the receptor. However, the binding event of ACh appears to modulate the local electrostatic environment, thereby influencing α-CTxMII to bind in a different pose than it would in the absence of ACh. Interestingly, this shift in binding conformation allows α-CTxMII to gain other, more favorable interactions including stronger hydrogen bonds, more intimate π-π and cation-π interactions, and potential reinforcement of the already dominant ionic interactions of its N-terminus. Binding of α-CTxMII after ACh in α3-β2 gaps puts its N-terminus closer to (+) E194 and (+) E195 of the open C-loop structure (Figure 6) and to (−) E61 of the closed structure. For β2-α3 gaps, the N-terminus is near (+) D192 and (+) D193 in the open C-loop structure (Figure 6) and near (−) E172 in the closed structure. In β2-β2 gaps, the N-terminus is found to interact with (+) D154 in the open (Figure 6) and (−) D170 and (−) D171 in the closed C-loop structure. In the above observations the average distance change in the ionic interaction of toxin to receptor due to the presence of ACh is around 1 Å. In addition to the N-terminus charge-charge interactions there are also secondary interactions such as π-π and cation-π that are important for the aromatic binding pocket.[30] It is these interactions, especially those involving H9 and H12 of α-CTxMII, that contribute to toxin binding by forming cation-π or π-π interactions with either positively charged residues or aromatic residues in the binding pocket, C-loop region, and/or (−) β-sheet wall. Also important is the role that α-CTxMII E11 plays in modifying the electrostatic environment and generating a dipole that promotes binding to α3β2-nAChRs. Lastly, intermolecular interactions critical to the efficient binding of α-CTxMII, are also found to involve S4, N5 and N14 in combination with the cysteine sulfur bonds to the C-loop. Examples in literature report that the mutation of α-CTxMII’s H12 results in ~2,700-fold decrease in binding affinity to α3β2-nAChRs, further displaying the importance of the π-π, cation-π and hydrogen bonding interactions in this system.[31] We hypothesize that after ACh binds, the interaction between the receptor cavity and α-CTxMII is enhanced, leading to an increased inhibition of receptor channel.
Figure 6.

Conotoxin binding poses in presence of ACh in the Ac-AChBP (open C-loop) homology model. Panels on the left are front views while the panels on the right are top views. The conotoxin is depicted by the grey translucent surface representation and ACh is represented by a licorice representation and critical amino acids are represented by lines. Alternate energetically accessible ACh binding poses are represented for the β2-α3 and β2-β2 gaps. These are only representations of alternate configurations while the traditional configuration was utilized for all α-CTxMII binding + ACh docking experiments.
When comparing the various docking experiments with ACh, α-CTxMII, and ACh + α-CTxMII, a complete picture of the overall docking process becomes apparent. If ACh is bound first, α-CTxMII forms stronger hydrogen bonds with the nAChR as compared to α-CTxMII binding in the absence of ACh (Table 3). It is also noted that α-CTxMII seems to form a random hydrogen bonding network with the receptor, while some interactions are conserved between ACh bound first and not at all (Supplemental Table S1). These observations, coupled with the absolute binding energies (Table 3), indicate that ACh docking alters the electrostatics of the binding region such that α-CTxMII binds tighter to the C-loop region. The difference in orientation of α-CTxMII, between poses when ACh is not bound and when ACh is bound first, is generally a rotational repositioning. However, this repositioning does not seem to have a significant impact on the overall binding energy.
The binding of ACh is a critical factor in the binding of α-CTxMII, and should be considered a standard when comparing computational data to in vitro or in vivo experiments. It is postulated based on these results that the binding of ACh impacts the binding of α-CTxMII favorably and therefore should be included when comparing computational models to experimental data in an attempt to more directly explain the mechanisms of α-CTx interactions with nAChRs.
Two-electrode voltage clamp
The observation of the various binding poses and the shift in α-CTxMII docking energies in the presence of ACh as compared to α-CTxMII binding without ACh, led us to the hypothesis that ACh favorably impacts the binding affinity of α-CTxMII for the α3β2-nAChR. To test this hypothesis two-electrode voltage clamp experiments were conducted to evaluate the impact of ACh, α-CTxMII, and ACh + α-CTxMII on Xenopus oocytes expressing the α3β2-nAChR. Initially, short pulses of ACh were applied to establish a control response of 0.5 to 4 μA. The oocytes were then treated for 5 min with 300 μM ACh, 100 nM α-CTxMII, or both agonist and antagonist simultaneously. During the perfusion that followed each treatment tests for ACh response were monitored for up to 24 min (Figure 7).
Figure 7.
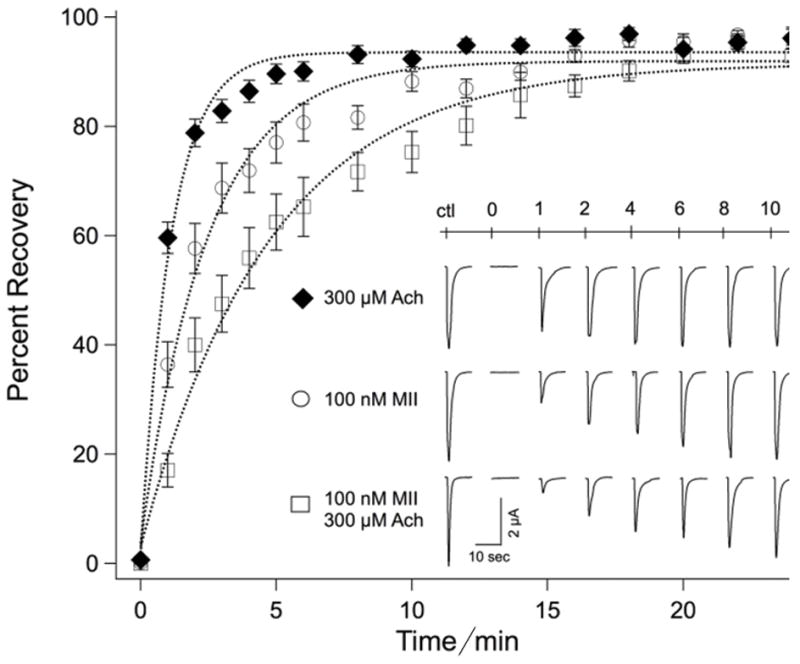
Kinetics of recovery from ACh receptor desensitization (black diamond), α-CTxMII (open circles) or both ACh + α-CTxMII (open squares). Responses to local application of ACh are shown in the inset for each treatment for times indicated, and are plotted as percent recovery. Values represent the mean ± standard error of the mean for 15 to 25 experiments.
The prolonged exposure to ACh results in desensitization of the receptor. Therefore, when comparing the recovery of receptors treated with ACh and α-CTxMII to that observed for receptor treated with α-CTxMII alone, it was important to quantify recovery from desensitization, as shown by the plot for recovery from ACh alone in Figure 7. From this curve it is apparent that during the first few minutes of recovery some of the differences in recovery for receptors treated with agonist and antagonist together, compared to antagonist alone, is a consequence of the desensitization of receptors to ACh. However, since recovery from desensitization was complete by 8 min, the observation that receptors treated with agonist and antagonist exhibited a lesser extent of recovery from 8 min to 18 min compared to those treated with antagonist alone demonstrates that the presence of agonist provides a more effective binding of antagonist. Specifically, receptor block was prolonged subsequent to recovery from desensitization, when α-CTxMII was applied with ACh compared to application of α-CTxMII in the absence of ACh.
Several previous studies have functionally characterized the binding of α-CTxMII to α3β2-nAChRs using Xenopus oocyte expression systems.[4a] Our wash-out experiments with 100 nM α-CTxMII are consistent with those of McIntosh et al.[32] and Luo et al.[7b], in which a slow recovery from toxin block was observed. Interestingly, in the latter study a second toxin LtIA from Conus litteratus was studied; pre-treatment of oocytes with this toxin accelerates the recovery from block by α-CTxMII, suggesting overlapping binding sites in this region. Our studies suggest that the presence of ACh modifies the local conformation and electrostatic environment of the receptor, leading to an increased affinity of conotoxin for its binding site. It is well established that point mutations of the receptor in this region have marked effects on toxin binding, and our results suggest that electrostatic and orientational perturbations are also an important consideration with respect to binding of conotoxins.
Conclusion
Presented in this study is the creation of two heteropentameric α3β2-nAChR models based on Ac-AChBP (open, PDB ID: 2BR8) or Tm-nAChR (closed C-loop, PDB ID: 2BG9) template and the rat α3β2-nAChR primary sequence. These two models differ in the state of the C-loop regions and have been used to investigate, for the first time, all subunit gaps in the binding of ACh and α-CTxMII using a complete heteropentameric mammalian α3β2-nAChR homology model. The various docking calculations presented indicate that ACh binds with relatively the same binding energy to the α3-β2, β2-α3, and β2-β2 gaps, with the state of the C-loop (open versus closed C-loop) having little or no impact on binding energies. These results reveal that ACh non-specifically binds to all of the subunit gaps, although its involvement in the channel gating mechanism remains to be investigated. The binding of α-CTxMII to the α3β2-nAChR homology models also showed non-specificity when the respective loops were in the closed C-loop conformation. However, when the C-loop regions are in the open C-loop conformation it is found that the β2-α3 and β2-β2 gaps bind α-CTxMII more favorably than the α3-β2 gap, which showed no preference for the open or closed C-loop region. The most striking observation was that presence of ACh in the binding pocket had the effect of increasing the favorability of α-CTxMII binding. The predicted enhancement of α-CTxMII binding was confirmed by voltage clamp experiments that revealed a slowed wash-out of toxin when ACh was present. In all binding calculations for both ACh and α-CTxMII it is found that the quaternary ammonium group and the N-terminal cation dominate and guide the binding poses.
Experimental Section
Homology Models
α3β2-nAChR
Homology modeling was used to predict the three dimensional (i.e. tertiary and quaternary) structure of the α3β2-nAChR. The procedure outlined below is based on the homology modeling method that has been successfully used for in silico studies of a nAChR[20b] and a glycyl radical enzyme.[33] The amino acid sequences of α3 and β2 subunits of the rat neuronal nAChR were obtained from the UniProt database (UniProtKB accession numbers P04757.1 (α3) and P12390.2 (β2)).[34] The α3 and β2 subunits of the rat nAChR were chosen because they share more than 94% sequence identity with the corresponding α3 and β2 subunits of the human nAChR, their plasmids are readily available, they express robustly in Xenopus oocytes, and are therefore considered to be good models for human neuronal nAChR. Structure template files with high primary sequence homology to rat α3 and β2 sequences were identified using Basic Local Alignment Search Tool (BLAST).[35] BLAST results indicated that the α3 subunit of rat nAChR shares 56% sequence identity with chains A and D (α subunit) of the Tm-nAChR, while the β2 subunit shares 48% sequence identity with chain B (β subunit) of the same nAChR. The heteropentameric Tm-nAChR, characterized using electron microscopy at a resolution of around 4 Å (PDB ID: 2BG9),[20c] served as the template for the creation of the first homology model. The homology models of the pentameric nAChR were created using DockoMatic[36] and MODELLER v9.9[34] packages. The template search and target-template sequence alignment was performed with DockoMatic, while the sequence alignment was viewed using UCSF Chimera.[22, 36a] To allow proper sequence alignment to the target structure, the cytoplasmic portions of the α3 and β2 rat sequences were removed. MODELLER was then used to generate 100 models, which were ranked based on DOPE scores[34] and GA341 scores[37]. The best ranking model was submitted to PROCHECK and Verify3d for side chain geometry and sequence validation.[38] Side chains were amended using the rotamer library in UCSF Chimera package.[22] Residues with poor stereochemical quality were identified using Ramachandran plot analysis and inaccurate dihedral angles were subsequently corrected. Energy minimization (1000 steps) was then performed on the structure, and the side-chains of residues with clashes were reoriented to refine the structure. The quality of the final structure was analyzed using the MolProbity server.[39]
α3β2-AChBP
In addition to the Tm-nAChR homology model, a homology model based on the Ac-AChBP crystal structure was also created.[10b] This template was selected due to the presence of a relatively large toxin bound in the region enclosed by the loop that connects beta sheets 9 and 10, commonly referred to as the C-loop.[21] The presence of the large toxin results in this C-loop being more distant from the adjacent complementary (−) subunit, hence creating a wider opening for entry into the ACh binding pocket. This ‘open C-loop’ homology model was constructed using the crystal structure of α-CTxPnIA bound Ac-AChBP with 2.4 Å resolution (PDB ID: 2BR8) as the template. The α-CTxPnIA ligand was removed from the receptor template before the creation of the homology model. Since the Ac-AChBP template consists of the N-terminal extracellular portion of the nAChR, the trans-membrane and cytosolic regions of the rat nAChR sequence were not included for the creation of the α3β2-nAChR homology model from this template. Additionally, since the Ac-AChBP is a homomeric pentamer, the monomeric subunit of Ac-AChBP was used as the template for both α3 and β2 subunits. The subunits, in a ratio of 2α3 to 3β2, were oriented to provide correct α3 and β2 overlap for binding. The procedure for obtaining and refining the homology model was the same as that mentioned for Tm-nAChR model.
Based on the geometric orientation of the C-loop, the homology models created from the Ac-AChBP and Tm-nAChR templates are referred to as the open C-loop model and the closed C-loop models respectively. Since there are currently no geometric criteria to definitively describe the state of the C-loop, and because the C-loop region has a wide range of movement,[10a] the terms open and closed have been used in a relative sense to isolate two points on the C-loop continuum specific to this study. Further consideration on the open versus closed nature of the C-loop can be found in the ‘open versus closed C-loop region’ section in Results & Discussion.
Docking Calculations
AutoDock v4.2[40] was used for docking calculations of ACh and/or α-CTxMII ligands with the homology models for the heteropentamer α3β2-nAChR isoform. AutoDockTools[40] (ADT) was used to prepare the input files and analyze the output of docking simulations. In all docking calculations ACh and α-CTxMII ligands were fully flexible consisting of a total of 4 and 28 active torsions with the torsion tree root residing on the C2 carbon (carbon of the CH2 group bound to the trimethylammonium group of ACh) and the α-carbon of Cys2 in α-CTxMII, respectively (Figure 1). The ligands were prepared by adding polar hydrogen atoms, merging all non-polar hydrogen atoms, and adding Gasteiger charges. The coordinates for ACh and α-CTxMII molecules were obtained from the PDB data bank (PDB ID: ACH and 1M2C, respectively).[41] We used our α3β2-nAChR homology models as the receptor structure. The nAChR receptor macromolecule structures were initialized using the standard ADT protocol for generating input macromolecule PDBQT files (e.g. incorporation of Gasteiger charges). In all docking calculations the receptor macromolecule was rigid while the ligand was allowed to sample the specified torsional parameters. The grid box was created in ADT and was sufficiently large to encompass each of the loop regions of the α3β2-nAChR macromolecule resulting in five sets of grid box files, one for each of the five C-loop regions (Figure 1). AutoGrid 4 was used to produce grid maps for the α3β2-nAChR atom types with a default grid spacing of 0.375 Å. The docking calculations for the ligand and the receptor macromolecule used the standard AutoDock force field and the Lamarckian Genetic Algorithm (LGA) to search for the lowest energy docked ligand conformers. Each docking experiment consisted of 100 independent LGA runs with a population size of 150 and a random initial geometry for the ligand. The maximum number of energy evaluation for each LGA run was set at 2,500,000 with a maximum number of 27,000 generations. The maximum number of top individuals that automatically survived was set to 1, the mutation rate was set to 0.02, the crossover rate was set to 0.8, the translational step size was set to 2 Å, and the quaternions and torsion step size was set to 50°. For the analysis of the docking calculations, 100 conformers were considered for each ligand-macromolecule complex, and the resulting docking clusters were calculated with a 2.0 Å root mean squared deviation (RMSD) tolerance on the heavy atoms.
In this study, all the C-loop regions were investigated in an effort to identify the binding affinity and binding poses of ligands for all putative accessible binding regions. Although the current hypothesis for agonist and antagonist binding of nAChRs stresses the importance of the α3β2 interface, our results suggest that three other binding interfaces, two β2α3 and one β2β2, may also make important contributions to ligand binding.
α3β2-nAChR expression in Xenopus laevis oocytes
The rat isoforms of neuronal nAChR subunits α3 and β2 in vectors pSP64 and pSP65 respectively were generously provided by Dr. Steven Heinemann (Salk Institute, San Diego, CA). Plasmids were linearized with EcoRI (α3) or Hind III (β2), and mRNA was generated using SP6 RNA polymerase (mMESSAGE mMACHINE kits, Ambion, Austin TX). Transcripts were co-injected into oocytes taken from adult Xenopus laevis frogs according to IACUC guidelines of the Animal Use and Care Committee at Idaho State University. Oocytes were cultured in medium containing NaCl (96 mM), KCl (2 mM), CaCl2 (1.8 mM), MgCl2 (1 mM), HEPES (5 mM) and sodium pyruvate (2.5 mM), with gentamicin sulfate (100 mg/L) and 4% horse serum (Hyclone Laboratories, Fisher Scientific, Pittsburgh, PA, USA) at pH 7.4, for 3 to 7 days prior to recording. All salts and antibiotics were obtained from Sigma Chemical Co., St. Louis, MO, USA.
Two-electrode voltage clamp
To record the response to ACh, oocytes were impaled with glass electrodes containing KCl (3 M) for two-electrode voltage clamping. Oocytes were placed in a 300 μL recording chamber and perfused at 1.5 mL/min with ND-96 bath solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, and 1 mM MgCl2), containing atropine sulfate (1 μM) and bovine serum albumin (0.1 mg/mL). Voltage clamp was performed with an OC-725C amplifier (Warner Instruments, Hamden, CT) with data Kacquisition using an ITC-18 interface and PatchMaster 2.35 software (HEKA Instruments, Bellmore, NY). Oocytes were held at −70 mV between trials, and at −80 mV during trials of ACh application.
Responses to ACh application were normalized as percent control prior to treatment, and the resulting curves fit with an exponential function for recovery from desensitization, toxin block, or both.
| (1) |
where I is current amplitude at time t, K0 is the asymptote, K1 is the initial amplitude of current, and τH is the time constant of recovery. After 5 min of exposure to ACh (300 μM), the receptors recovered from desensitization with a time constant of 1.12 min, and recovery reached its asymptote at approximately 8 min. With 5 min exposure to α-CTxMII (100 nM), the receptors recovered from block with a time constant of 2.46 min and with an asymptote at approximately 16 min. We tested the prediction from docking studies that ACh promotes greater binding efficiency of the toxin by monitoring the recovery of receptors after 5 min exposure to both ACh (300 μM) and α-CTxMII (100 nM). With that treatment, receptors recovered with a time constant of 4.72 min, and with an asymptote of approximately 20 min. Thus, receptors pre-treated with both agonist and antagonist showed a slowed recovery compared to receptors pre-treated with antagonist alone.
During perfusion, a 35 to 50 ms pulse of ACh chloride (0.01 M, Sigma) in bath solution was locally applied from a glass capillary tube positioned above the oocyte, with pressure ejection performed with a PicoSpritzer II valve controller (General Valve Corporation, Fairfield, NJ). Once a reliable response was obtained, the perfusion was stopped, and the chamber filled with ACh (300 μM), α-CTxMII (100 nM), or both, for 5 min. Synthetic α-CTxMII was purchased from C. S. Bio (Menlo Park, CA) in folded form as a white powder. Peptide purity and quantity were validated by nuclear magnetic resonance spectroscopy. After 5 min the perfusion was resumed and ACh pulses were applied every 1 min for 6 min, and every 2 min thereafter. Current amplitudes at each time point were normalized against the control response to ACh pulses, and plotted against time following application of agonist and/or antagonist treatment.
Supplementary Material
Acknowledgments
Research Corporation Cottrell Scholar program, St. Luke’s Mountain States Tumor Medical Research Institute, NIH Idaho Idea Network of Biomedical Research Excellence (NIH Grant P20 RR016454), ISU Faculty Research Grant AS11-2U. Computational support and resources were provided in part by Colorado Schools of Mines Campus Computing, Communications, and Information Technologies and the Golden Energy Computing Organization at the Colorado School of Mines.
Footnotes
Supporting information for this article is available.
Contributor Information
Prof. Owen M. McDougal, Email: owenmcdougal@boisestate.edu.
Prof. C. Mark Maupin, Email: cmmaupin@mines.edu.
References
- 1.Quik M, Wonnacott S. Pharmacol Rev. 2011;63:938–966. doi: 10.1124/pr.110.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Cooper E, Couturier S, Ballivet M. Nature. 1991;350:235–238. doi: 10.1038/350235a0. [DOI] [PubMed] [Google Scholar]; b) Anand R, Conroy WG, Schoepfer R, Witing P, Lindstorm J. J Biol Chem. 1991;266:11192–11198. [PubMed] [Google Scholar]; c) Role LW, Berg DK. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]; d) Wada E, Wada K, Boulter J, Deneris E, Heinemann S. J Comp Neurol. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]; e) Le Novere N, Grutter T, Changeux JP. Proc Natl Acad Sci. 2002;99:3210–3215. doi: 10.1073/pnas.042699699. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yeh JJ, Yasuda RP, Davila-Garcia MI, Xiao Y, Ebert S, Gupta T, Kellar KJ, Wolfe BB. J Neurochem. 2001;77:336–346. doi: 10.1046/j.1471-4159.2001.t01-1-00259.x. [DOI] [PubMed] [Google Scholar]
- 3.a) Dray A. Br J Anaesth. 2008;101:48–58. doi: 10.1093/bja/aen107. [DOI] [PubMed] [Google Scholar]; b) Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Michel PP, Toulorge D, Guerreiro S, Hirsch EC. FASEB J. 2013;27:3414–3423. doi: 10.1096/fj.12-220418. [DOI] [PubMed] [Google Scholar]
- 4.a) Azam L, McIntosh JM. Acta Pharmacol Sin. 2009;30:771–783. doi: 10.1038/aps.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lloyd GK, Williams M. The Journal of pharmacology and experimental therapeutics. 2000;292:461–467. [PubMed] [Google Scholar]; c) Dwoskin LP, Crooks PA. The Journal of pharmacology and experimental therapeutics. 2001;298:395–402. [PubMed] [Google Scholar]
- 5.a) Bordia T, Grady SR, McIntosh JM. Mol Pharmacol. 2007;72:52–61. doi: 10.1124/mol.107.035998. [DOI] [PubMed] [Google Scholar]; b) Quik M, McIntosh JM. J Pharmacol Exp Ther. 2006;316:481–489. doi: 10.1124/jpet.105.094375. [DOI] [PubMed] [Google Scholar]; c) McCallum SE, Parameswaran N, Bordia T, McIntosh JM, Grady SR. Mol Pharmacol. 2005;68:737–746. doi: 10.1124/mol.105.012773. [DOI] [PubMed] [Google Scholar]
- 6.Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- 7.a) Nicke A, Wonnacott S, Lewis RJ. Eur J Biochem. 2004;271:2305–2319. doi: 10.1111/j.1432-1033.2004.04145.x. [DOI] [PubMed] [Google Scholar]; b) Luo S, Akondi KB, Zhangsun D, Wu Y, Zhu X, Hu Y, Christensen S, Dowell C, Daly NL, Craik DJ, Wang CI, Lewis RJ, Alewood PF, Michael McIntosh J. J Biol Chem. 2010;285:12355–12366. doi: 10.1074/jbc.M109.079012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 9.a) Hansen SB, Talley TT, Radić Z, Taylor P. J Biol Chem. 2004;279:24197–24202. doi: 10.1074/jbc.M402452200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Curtis L, Chiodini F, Spang JE, Bertrand S, Patt JT, Westera G, Bertrand D. Eur J Pharmacol. 2000;393:155–163. doi: 10.1016/s0014-2999(00)00053-4. [DOI] [PubMed] [Google Scholar]; c) Karlin A. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]; d) Sine SM. J Neurobiol. 2002;53:431–446. doi: 10.1002/neu.10139. [DOI] [PubMed] [Google Scholar]; e) Schapira M, Abagyan R, Totrov M. BMC Struct Biol. 2002;2:1. doi: 10.1186/1472-6807-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Celie PHN, Kasheverov IE, Mordvintsev DY, Hogg RC, van Nierop P, van Elk R, van Rossum-Fikkert SE, Zhmak MN, Bertrand D, Tsetlin V, Sixma TK, Smit AB. Nat Struct Mol Biol. 2005;12:582–588. doi: 10.1038/nsmb951. [DOI] [PubMed] [Google Scholar]; c) Ulens C, Hogg RC, Celie PH, Bertrand D, Tsetlin V, Smit AB, Sixma TK. Proc Natl Acad Sci. 2007;103:3615–3620. doi: 10.1073/pnas.0507889103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dutertre S, Ulens C, Büettner R, Fish A, van Elk R, Kendel Y, Hopping G, Alewood PF, Schroeder C, Nicke A, Smit AB, Sixma TK, Lewis RJ. EMBO J. 2007;26:3858–3867. doi: 10.1038/sj.emboj.7601785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Maslennikov IV, Shenkarev ZO, Zhmak MN, Ivanov VT, Methfessel C, Tsetlin VI, Arseniev AS. Febs Lett. 1999;444:275–280. doi: 10.1016/s0014-5793(99)00069-1. [DOI] [PubMed] [Google Scholar]; b) Gehrmann J, Daly NL, Alewood PF, Craik DJ. J Med Chem. 1999;42:2364–2372. doi: 10.1021/jm990114p. [DOI] [PubMed] [Google Scholar]; c) Rogers JP, Luginbuhl P, Pemberton K, Harty P, Wemmer DE, Stevens RC. J Mol Biol. 2000;304:911–926. doi: 10.1006/jmbi.2000.4247. [DOI] [PubMed] [Google Scholar]
- 12.Chi SW, Kim DH, Olivera BM, McIntosh JM, Han KH. Biochem Biophys Res Commun. 2006;345:248–254. doi: 10.1016/j.bbrc.2006.04.099. [DOI] [PubMed] [Google Scholar]
- 13.Hu SH, Gehrmann J, Guddat LW, Alewood PF, Craik DJ, Martin JL. Structure. 1996;4:417–423. doi: 10.1016/s0969-2126(96)00047-0. [DOI] [PubMed] [Google Scholar]
- 14.a) Celie PH, Klaassen RV, van Rossum-Fikkert SE, van Elk R, van Nierop P, Smit AB, Sixma TK. J Biol Chem. 2005;280:26457–26466. doi: 10.1074/jbc.M414476200. [DOI] [PubMed] [Google Scholar]; b) Talley TT, Olivera BM, Han KH, Christensen SB, Dowell C, Tsigelny I, Ho KY, Taylor P, McIntosh JM. J Biol Chem. 2006;281:24678–24686. doi: 10.1074/jbc.M602969200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Livett BG, Sandall DW, Keays D, Down J, Gayler KR, Satkunanathan N, Khalil Z. Toxicon. 2006;48:810–829. doi: 10.1016/j.toxicon.2006.07.023. [DOI] [PubMed] [Google Scholar]; b) McDougal OM, Granum DM, Swartz M, Rohleder C, Maupin CM. J Phys Chem B. 2013;117:2653–2661. doi: 10.1021/jp3117227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Olivera BM, Walker C, Cartier GE, Hooper D, Santos AD, Schoenfeld R, Shetty R, Watkins M, Bandyopadhyay P, Hillyard DR. Ann NY Acad Sci. 1999;870:223–237. doi: 10.1111/j.1749-6632.1999.tb08883.x. [DOI] [PubMed] [Google Scholar]; b) Arias HR. Neurochem Int. 2000;36:595–645. doi: 10.1016/s0197-0186(99)00154-0. [DOI] [PubMed] [Google Scholar]
- 17.Kasheverov IE, Zhmak MN, Fish A, Rucktooa P, Khruschov AY, Osipov AV, Ziganshin RH, D’Hoedt D, Bertrand D, Sixma TK, Smit AB, Tsetlin VI. J Neurochem. 2009;111:934–944. doi: 10.1111/j.1471-4159.2009.06359.x. [DOI] [PubMed] [Google Scholar]
- 18.Shon KJ, Grilley M, Jacobsen R, Cartier GE, Hopkins C, Gray WR, Watkins M, Hillyard DR, Rivier J, Torres J, Yoshikami D, Olivera BM. Biochemistry. 1997;36:9581–9587. doi: 10.1021/bi970235w. [DOI] [PubMed] [Google Scholar]
- 19.Lemoine D, Jiang R, Taly A, Chataigneau T, Specht A, Grutter T. Chem Rev. 2012 doi: 10.1021/cr3000829. [DOI] [PubMed] [Google Scholar]
- 20.a) Haddadian EJ, Cheng MHY, Coalson RD, Xu Y, Tang P. J Phys Chem B. 2008;112:13981–13990. doi: 10.1021/jp804868s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cheng XL, Lu BZ, Grant B, Law RJ, McCammon JA. J Mol Biol. 2006;355:310–324. doi: 10.1016/j.jmb.2005.10.039. [DOI] [PubMed] [Google Scholar]; c) Unwin N. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 21.a) Dutertre S, Lewis RJ. Eur J Biochem. 2004;271:2327–2334. doi: 10.1111/j.1432-1033.2004.04147.x. [DOI] [PubMed] [Google Scholar]; b) Itier V, Bertrand D. Febs Lett. 2001;504:118–125. doi: 10.1016/s0014-5793(01)02702-8. [DOI] [PubMed] [Google Scholar]
- 22.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Davis IW, Murray LW, Richardson JS, Richardson DC. Nucleic Acids Res. 2004;32:W615–619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutertre S, Nicke A, Tyndall JDA, Lewis RJ. J Mol Recog. 2004;17:339–347. doi: 10.1002/jmr.683. [DOI] [PubMed] [Google Scholar]
- 25.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty D. Proc Natl Acad Sci. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arias HR. Brain Res Rev. 1997;25:133. doi: 10.1016/s0165-0173(97)00020-9. [DOI] [PubMed] [Google Scholar]
- 27.Chatrenet B, Kotzba-Hibert F, Mulle C, Changeux JP, Goeldner MP, Hirth C. Mol Pharmacol. 1992;41:1100–1106. [PubMed] [Google Scholar]
- 28.Meltzer RH, Thompson E, Soman KV, Song XZ, Ebalunode JO, Wensel TG, Briggs JM, Pedersen SE. Biophys J. 2006;91:1302–1314. doi: 10.1529/biophysj.106.081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cederholm JM, Schofield PR, Lewis TM. Eur Biophys J. 2009;39:37–49. doi: 10.1007/s00249-009-0452-y. [DOI] [PubMed] [Google Scholar]
- 30.Blum AP, Lester HA, Dougherty DA. Proc Natl Acad Sci. 2010;107:13206–13211. doi: 10.1073/pnas.1007140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everhart D, Cartier GE, Malhotra A, Gomes AV, McIntosh JM, Luetje CW. Biochemistry. 2004;43:2732–2737. doi: 10.1021/bi036180h. [DOI] [PubMed] [Google Scholar]
- 32.McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- 33.Bharadwaj VS, Dean AM, Maupin CM. J Am Chem Soc. 2013;135:12279–12288. doi: 10.1021/ja404842r. [DOI] [PubMed] [Google Scholar]
- 34.Sali A, Blundell TL. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 35.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.a) Bullock CW, Jacob RB, McDougal OM, Hampikian G, Andersen T. BMC Res Notes. 2010;3:289–298. doi: 10.1186/1756-0500-3-289. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bullock C, Cornia N, Jacob R, Remm A, Peavey T, Weekes K, Mallory C, Oxford JT, McDougal OM, Andersen TL. J Chem Inf Model. 2013;53:2161–2170. doi: 10.1021/ci400047w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melo F, Sali A. Protein Sci. 2007;16:2412–2426. doi: 10.1110/ps.072895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.a) Laskowski RA. J Appl Cryst. 1993;26:283–291. [Google Scholar]; b) Bowie JU, Luthy R, Eisenberg D. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 39.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shon KJ, Koerber SC, Rivier JE, Olivera BM, McIntosh JM. Biochemistry. 1997;36:15693–15700. doi: 10.1021/bi971443r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.